

Abstract
Inflammation is associated with a variety of diseases. The hallmark of inflammation is leukocyte infiltration at disease sites in response to pathogen- or damage-associated chemotactic molecular patterns (PAMPs and MAMPs), which are recognized by a superfamily of seven transmembrane, Gi-protein-coupled receptors (GPCRs) on cell surface. Chemotactic GPCRs are composed of two major subfamilies: the classical GPCRs and chemokine GPCRs. Formyl-peptide receptors (FPRs) belong to the classical chemotactic GPCR subfamily with unique properties that are increasingly appreciated for their expression on diverse host cell types and the capacity to interact with a plethora of chemotactic PAMPs and MAMPs. Three FPRs have been identified in human: FPR1–FPR3, with putative corresponding mouse counterparts. FPR expression was initially described in myeloid cells but subsequently in many non-hematopoietic cells including cancer cells. Accumulating evidence demonstrates that FPRs possess multiple functions in addition to controlling inflammation, and participate in the processes of many pathophysiologic conditions. They are not only critical mediators of myeloid cell trafficking, but are also implicated in tissue repair, angiogenesis and protection against inflammation-associated tumorigenesis. A series recent discoveries have greatly expanded the scope of FPRs in host defense which uncovered the essential participation of FPRs in step-wise trafficking of myeloid cells including neutrophils and dendritic cells (DCs) in host responses to bacterial infection, tissue injury and wound healing. Also of great interest is the FPRs are exploited by malignant cancer cells for their growth, invasion and metastasis. In this article, we review the current understanding of FPRs concerning their expression in a vast array of cell types, their involvement in guiding leukocyte trafficking in pathophysiological conditions, and their capacity to promote the differentiation of immune cells, their participation in tumor-associated inflammation and cancer progression. The close association of FPRs with human diseases and cancer indicates their potential as targets for the development of therapeutics.
Introduction
Formyl-peptide receptors (FPRs) are a family of seven transmembrane domains, Gi-protein-coupled receptors (GPCRs). In human, there are 3 FPRs, FPR1, FPR2 and FPR3. FPR1 and FPR2 were originally identified based on their capacity to recognize N-formyl peptides produced in nature by degradation of either bacterial (1–4) or host cell mitochondrial proteins, which represent major proinflammatory products (5,6). Activation of FPR1 and FPR2 by chemotactic agonists elicits a cascade of signaling events leading to myeloid cell migration, mediator release, increased phagocytosis and new gene transcription (7). But for FPR3, although it is expressed in monocytes and dendritic cells (DCs), the overall function remains unclear. Compared to FPR1 and FPR2, FPR3 is highly phosphorylated (a signal for receptor inactivation and internalization) and more localized to small intracellular vesicles (8). This suggests that FPR3 rapidly internalizes after binding its ligands and thereby may serve as a “decoy” receptor to reduce the binding of its ligands to other receptors (8,9). Interestingly, FPR3 does not interact with formylated chemoattract peptides, nor shares ligands with FPR1 or FPR2. Therefore, FPR3 may have its own unique functional significance. The mouse FPR (mFPR or Fpr) gene family consists of at least 8 members including Fpr1, Fpr2, Fpr-rs1, Fpr-rs3, Fpr-rs4, Fpr-rs5, Fpr-rs6, and Fpr-rs7 (4). Fpr1 is considered as an orthologue of human FPR1, whereas Fpr2 is structurally and functionally like human FPR2 (10). The mouse counterpart of human FPR3 is not well defined. Since Fpr2 shares a human FPR3 ligand (11,12), Fpr2 was suggested to act as a counterpart of both FPR2 and FPR3. The other 6 murine Fpr genes are expressed in leukocytes, but the identity of their encoded receptors remain unknown (3).
FPRs are mainly expressed in leukocytes (Table 1) including neutrophils (13), monocytes/macrophages (4,14), natural killer (NK) cells (15,16), and DCs (17,18). Recently, FPR2 was detected in naive CD4+ T cells (CD3+CD4+CD45RA+CD45RO−CCR7+), human tonsillar follicular helper T cells, Th1 cells, Th2 cells, and Th17 cells (16,19). FPR2 is also expressed in follicular DCs and B cells. In B cells located in the germinal center (GC) of Peyer’s patches, FPR2 is activated by an endogenous agonist LL-37 (20). Importantly, FPRs are also expressed in a variety of non-immune cells (Table 2) including endothelial cells, endothelial progenitor cells (21,22), synovial fibroblasts (23,24), keratinocytes (25), intestinal epithelial cells (26), bone marrow-derived mesenchymal stem cells (MSCs) (27,28), and hepatocytes (29), suggesting a broader spectrum of biological function of these receptors.
Table 1.
The expression of FPRs in immune cells
| Cells | FPR members | Functions | Refs |
|---|---|---|---|
| Neutrophils | FPR1, FPR2 | Chemotaxis | 3,13 |
| Monocytes/Macrophages | FPR1, FPR2, FPR3 | Chemotaxis | 3, 13, 17 |
| Natural killer (NK) cells | FPR1, FPR2 | Chemotaxis, production of IFN-γ | 15 |
| Dendritic cells | FPR1, FPR2, FPR3 | Chemotaxis | 17 |
| Naive CD4 T-cells (CD3+CD4+CD4SRA+CD4SRO−CCR7+) | FPR2 | Increase in IFN-γ production | 16 |
| Th 1 cells | FPR2 | N/A | 16, 19 |
| Th 2 cells | FPR2 | N/A | 16, 19 |
| Th 17 cells | FPR2 | N/A | 16, 19 |
| Follicular DCs | FPR2 | 8 cell activation in the germinal center of Peyer’s patch | 20 |
| Tonsillar follicular helper T cells | FPR2 | N/A | 16, 19 |
N/A: not clear
Table 2.
Expression of FPRs in non-immune cells
| Cells | Expression of FPRs | Functions | Refs |
|---|---|---|---|
| Endothelial cells | FPR2 | N/A | 21, 22 |
| Endothelial progenitor cells | FPR2 | N/A | 21, 22 |
| Synovial Fibroblasts | FPB2 | N/A | 23, 24 |
| Keratinocytes | FPR2 | Cells proliferation and proinflammatory response | 25 |
| Intestinal epithelial cells | FPR1, FPR2 | Cell proliferation migration | 26 |
| Mesenchymal stem cells (MSCs) | FPR2 | Chemotaxis | 27, 28 |
| Hepatocytes | FPR | Chemotaxis angiogenesis factor production | 29 |
N/A: not clear
Some malignant human tumors cells also express FPRs and respond to bacterial or endogenous agonists by increased motility and growth. For instance, FPRs expressed by human gastric cancer cells, mediate epithelial-mesenchymal transition, cell proliferation, migration, and resistance to apoptosis (30). The prototype FPR, FPR1, selectively expressed by highly malignant glioblastoma multiforme (GBM) cells, responds to an endogenous chemotactic ligand anexin 1 (ANXA1) released by necrotic GBM cells (31). Activated FPR1 cooperates with the epidermal growth factor receptor (EGFR) to enhance the survival, invasiveness, and production of antigenic factors by GBM cells (2,32–35). Human breast cancer cells also express FPR1 and FPR2, which interact with a shared ligand ANXA1 to enhance tumor cell proliferation (36). In human liver cancer cells, FPR1 was implicated in promoting cell invasion, proliferation, and production of angiogenic factors (2). A recent study shows that FPR2 is utilized by human colon cancer cells for their growth advantage (37). Thus, FPRs are suggested to be hijacked by tumor cells for their benefits.
In addition to their expression in diverse cell types, FPRs are notorious for ligand promiscuity, by interacting with both pathogen-associated chemotactic molecular patterns (PAMPs) and damage-associated chemotactic molecular patterns (DAMPs). During the past few years, with the availability of genetically engineered mouse strains deficient in one or more Fprs, the critical roles of FPRs (Fprs) in disease progression are increasingly recognized (38). A lot of studies revealed that FPRs not only mediate leukocyte trafficking but also promote myeloid cell differentiation, colon epithelial homeostasis, and cancer progression. Therefore, a better understanding of the biologic significance of FPRs (Fprs) should have important clinical relevance. This review will focus on the role of FPRs in the regulation of inflammatory responses as a complement to other excellent reviews of more facets of FPRs (2,3,19,39,40).
1. FPRs guide leukocyte trafficking in pathophysiological conditions
Leukocyte infiltration is a hallmark in inflammation, immune responses and cancer progression. Both physiological and pathological trafficking of leukocytes in vivo is mediated by multiple chemoattractant GPCRs on the cell surface sequentially regulated by differentiation or maturation signals in the microenvironment. FPRs (Fprs) actively participate in the process of leukocyte sensing of chemotactic cues established by chemotactic PAMPs or DAMPs. Using of genetically engineered mice or inhibitors of selected chemoattractant GPCRs ligands enable establishment of several models of leukocyte trafficking in bacterial infection, immune responses and wound healing.
1.1. Fpr1/Fpr2-CXCR2-mediated neutrophil recruitment in bacterial infection
Listeria monocytogenes is an opportunistic pathogen that causes severe infection in immunocompromised individuals (41). The lethality rate is as high as 30% in infected patients although the overall incidence of listeriosis in human is low (2). Listeria infect and replicate inside host cells thus escape immune surveillance (42,43). Rapid neutrophil accumulation at the site of infection is a critical step in host resistance to infection by Listeria. Fprs on neutrophil antecede chemokine GPCRs in directly recognizing bacteria-derived chemotactic PAMPs to initiate neutrophil accumulation (44).
In a mouse i.v. Listeria infection model, a large number of neutrophils appear in the liver of wild type (WT) mice as early as less than 30 min after bacteria administration, with the peak at 4 h. Neutrophil-specific chemokines CXCL1/2, ligands interacting with the GPCR CXCR2 on neutrophils, to mediate cell migration (45–51), are barely detectable in the liver of mice 30 min after infection. The appearance of CXCL1/2 in the liver starts at 4 h after infection, a time point beyond the maximal neutrophil infiltration. Interestingly, in either Fpr1- or Frp2-deficient mice, although the production of CXCL1/2 in Listeria infected mouse liver showed kinetics and magnitude similar to that in WT mice, the early phase neutrophil recruitment into the liver is markedly reduced. There is an almost complete absence of early phase neutrophils in the liver of infected mice deficient in both Fpr1 and Fpr2 (44). Further studies reveal that Listeria produces chemotactic agonists for both Fpr1 and Fpr2, consistent with findings that synthetic peptides based on the putative Listeria product sequences are potent neutrophil chemoattractants by interacting with both human and mouse FPRs (52). Consequently, Fpr-deficient mice bear increased bacterial load in the liver with markedly reduced production of H2O2 by neutrophils in response to bacterial challenge, in association with compromised pathogen killing and greatly increased mortality. Those findings challenge previously existing paradigm of host defense against Listeria in which the pattern recognition receptor TLR2 on host cells is activated by bacterial lipoprotein followed by production of CXCR2 ligands to initiate neutrophil accumulation. In fact, Fprs antecede CXCR2 to rapidly recruit the first wave of neutrophils into the infected liver followed by a CXCR2-mediated second wave of neutrophil accumulation (Fig. 1).
Figure 1. FPRs control the first wave neutrophil infiltration in Listeria infection.
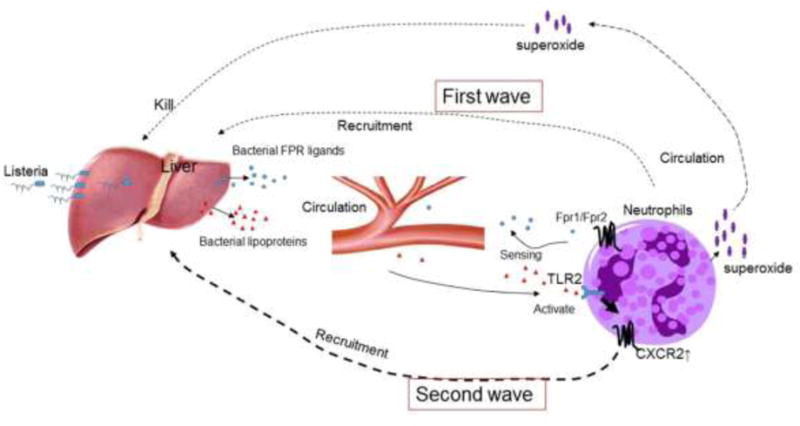
Both Fpr1 and Fpr2 expressed by mouse neutrophils sense bacteria-derived chemotactic PAMPs to mediate a rapid neutrophil influx into the liver of listeria-infected mice. Listeria lipoproteins stimulate TLR2 on hepatocytes and leukocytes to trigger the production of CXCR2-specific chemokines to mobilize a late wave neutrophil recruitment in the liver. Neutrophils activated by bacterial FPR (Fpr) ligands produce superoxide which is critical for pathogen elimination.
1.2. Fpr2 mediates neutrophil recruitment in other bacterial infection models
FPRs also mediate neutrophil recruitment in other infection models presumably by recognizing a broad range of chemotactic PAMPs (2). For example, FPRs (Fprs) have been implicated in the onset of pneumococcal meningitis (53) by eliciting early neutrophil infiltration in response to bacteria-derived chemoattractants. In DSS-induced mouse colitis, the number of neutrophils in the gut lesion areas was significantly reduced in Toll-interacting-protein (Tollip) deficient mice, in which reduced neutrophil infiltration in intestinal lesions in the presence of gut microflora is correlated with reduced expression of Fpr2 by neutrophil (54). In acute sepsis, neutrophils in both human and mouse show skewed expression of a chemokine GPCR CCR5 with reduced FPR2 (55,56). As a consequence, septic neutrophils are directed toward sterile tissue sites by CCR5, but fail to move to bacterial infection sites due to reduced FPR2. Such “inflamed” yet “incompetent” neutrophils may place the host to higher morbidity and mortality associated with septic insult (54,57), emphasizing the importance of FPRs in the frontier of anti-microbial host defense.
1.3. Cooperation of Fpr1/Fpr2 with other GPCRs in wound healing
Tissue injury is associated with rapid accumulation of neutrophils which constitute nearly 50% of all cells at the wound site (58). The accumulation of neutrophils is controlled by multiple chemoattractants (58,59), such as IL-8 (CXCL8), CXCL7 (60) and CXCL1 (61,62), which use CXCR2 expressed by neutrophils. However, neutrophils also express FPR1 (Fpr1) and FPR2 (Fpr2) in human and mice. In a mouse skin-wound healing model, Fpr1/Fpr2 act as the first player in sensing chemotaxis signals, resulting in rapid neutrophil infiltration (63). Activation of FPR1/FPR2 (Fpr1/Fpr2) by agonists produced at the site of injury elicits a signaling cascade that culminates in neutrophil migration, increased phagocytosis and release of superoxide. In Fpr1/2−/− mice, healing of the skin wound was significantly delayed with markedly reduced neutrophil infiltration in the dermis before the production of neutrophil-specific CXC chemokines by injured tissue as compared with WT mice, indicating that Fpr1/Fpr2 are critical for normal healing process of the sterile skin wound (63).
1.4. Fpr2 in stepwise trafficking of DCs in allergic airway inflammation
Asthma is a chronic disorder characterized by airway inflammation and hyperresponsiveness. The disease is mediated by increased levels of T-helper 2 (Th2) cytokines, IL-4, IL-5, and IL-13 and elevated serum IgE (64). The expression of a Fpr2 agonist, the anti-microbial peptide CRAMP, is also up-regulated in the lung during asthmatic syndrome (65,66). Currently glucocorticoids still are regarded as the most effective treatment for asthma and is a recommended first line medicine (65). However, glucocorticoids, such as budesonide suppresses pulmonary antibacterial host defense in asthmatic mouse model and inhibits the function of lung epithelial cells because of the down-regulation of CRAMP (65). Therefore, CRAMP produced by airway epithelial cells and inflammatory cells protects airway immune responses. In addition to the antimicrobial properties of CRAMP and its human ortholog, LL-37, these two endogenous peptides induce the migration of neutrophils, monocytes/macrophages, eosinophils, and mast cells (67) by activating Fpr2 (FPR2) (68,69). Injection of CRAMP into air pouches of mouse skin induces accumulation of neutrophils and monocytes, confirming the chemoattractant nature of CRAMP (70).
DCs are critical in airway immune responses (71,72). Upon viral infection, allergen challenge, or endotoxin inhalation, CD11b+ monocyte-derived DC precursors are rapidly recruited from the circulation into the airway to initiate and eventually determine the severity of allergic airway responses (72,73). In mice, Ly6Chigh conventional monocytes are CX3CR1low, CCR2+, CD62L+, and CCR5−. Under inflammatory conditions, these cells differentiate into inflammatory DCs and acquire the capacity to prime T cell-mediated immune responses in draining lymph nodes (74). In the lung, exposure to inflammatory stimuli, such as TLR agonist PAMPs or environmental pollutants, triggers the production of chemokines that were implicated in the recruitment of inflammatory DCs in a CCR2-dependent manner (72,73,75,76). It is therefore not surprising that mice deficient in CCR2 (CCR2−/−) showed defective trafficking of antigen-loaded lung DCs, resulting in reduced Th2 responses stimulated by OVA (77–80). However, recently, Fpr2 has been found also to be involved in DC trafficking in mouse model of asthma as Fpr2−/− mice show reduced severity in OVA-induced allergic airway inflammation, in association with diminished recruitment of CD11c+ DCs into the peribronchiolar regions of the inflamed lung (81). Mechanistically, the endogenous Fpr2 ligand CRAMP was found to control DC trafficking from the perivascular regions to the area surrounding bronchioles in the inflamed lungs (82). These observations establish a paradigm of DC trafficking sequentially mediated by GPCRs initiated by CCR2 followed by Fpr2 and finally CCR7 to complete the journey of DCs to draining LN for Th2 priming. The tightly orchestrated DC trafficking model in allergic airway inflammation illustrates the necessity for CCR2 to mobilize Ly6Chigh monocytic DC precursors from bone marrow (BM) into the circulation (80,83), where the cells extravasate into the perivascular regions of the inflamed lung and become immature (i) DCs upon exposure to DAMPs present in the airway (84). The iDCs then lose the functional CCR2, but gain high-level expression of Fpr2, which guides the cells into the peribronchiolar regions in response to a host-derived chemotactic DAMP—CRAMP (84). CRAMP not only forms a chemotactic gradient cue for DC trafficking in the inflamed lung, but also is capable of promoting DC maturation stimulated by TLR agonistic PAMPs (18). A shift of the chemoattractant GPCR expression occurs again as DCs mature when the Fpr2 is downregulated with highly elevated expression of the DC homing chemokine GPCR, CCR7, enabling matured DCs to be directed into lymphatic organs. Thus, DC trafficking in the inflammatory airway consists of a fine-tuned cooperation of chemoattractant GPCRs on the cell surface, initiating from CCR2, with Fpr2 as an intermediate, and CCR7 as the final player to accomplish the last segment of homing (Fig. 2).
Figure 2. Sequential CCR2-Fpr2-CCR7 signals mediate DC trafficking in allergic airway inflammation.
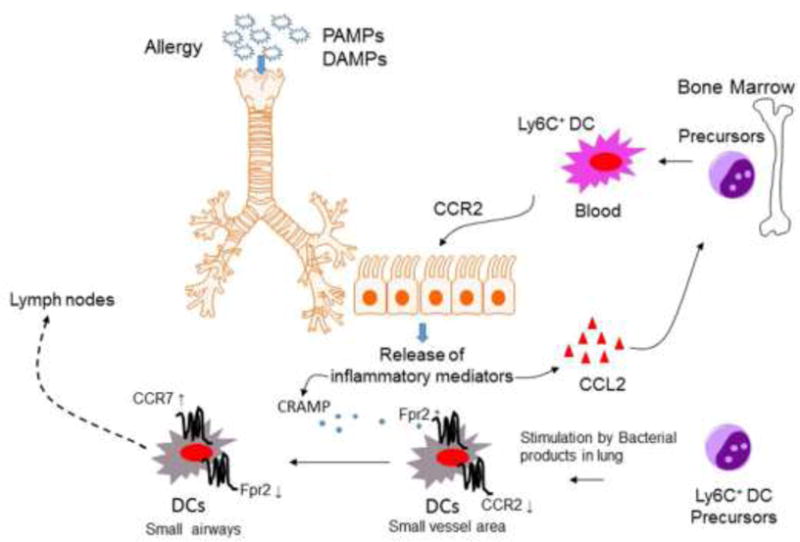
The chemokine receptor CCR2 mobilizes inflammatory DC precursors from the circulation to the perivascular regions of the inflamed lung in response to the cognate ligand CCL2. Once in the perivascular regions, immature DCs (iDCs) express high levels of Fpr2 with down-regulation of CCR2 after exposure to bacterial PAMPs or environmental stimuli. Fpr2 then mediates DC trafficking to the peribronchiolar regions in response to the endogenous chemotactic ligand CRAMP, which is increased in the inflamed lungs. In the peribronchiolar areas, inflammatory iDCs undergo maturation in association with the expression of high levels of the chemokine GPCR CCR7 for further homing into draining lymph nodes to initiate adaptive immune responses.
2. FPRs promote the differentiation and proliferation of immune cells
2.1. Fpr2 promotes DC maturation
Inflammatory DCs are BM–derived, specialized antigen-presenting cells (APCs) and consist of heterogeneous populations. DCs are abundantly distributed in body surface tissues such as the skin, naso-pharynx, upper esophagus, vagina, ectocervix and anus as well as so called internal or mucosal surfaces including respiratory and gastrointestinal systems. Such broad distribution increases the opportunity for DCs to capture antigens from the environment even when there is no overt infection or inflammation, which may allow for silencing of the immune system in the presence of harmless environmental antigens. In response to danger signals such as bacterial and viral infection, DCs undergo a complex process of maturation, a ‘metamorphosis’ from antigen-capturing cells into APCs. This process includes changes in phenotype such as up-regulation of costimulatory molecules such as CD40, CD80, and CD86 on the cell surface, translocation of MHC class II and production of cytokines that differentiate and polarize effector cells (85,86). Maturation of DCs is initiated primarily by signals transduced via pattern recognition receptors (PRRs) expressed by iDCs, which respond to PAMPs of microbes, parasites, and viruses (85,87), recognized by PRR family members: Toll-like receptors (TLRs), cell surface C-type lectin receptors (CLRs), intracytoplasmic nucleotide oligomerization domain (NOD)-like receptors (NLRs) and retinoic acid inducible gene I (RIG-I)-like receptors (RLRs). PRRs also interact with host-derived DAMPs. The signals transduced by PRRs convert iDCs into mature (m)DCs capable of promoting the expansion and differentiation of pathogen-specific effector T cells (88–90). PAMPs and DAMPs also activate PRRs expressed on a variety of cells, such as epithelial cells, fibroblasts and endothelial cell to release cytokines assisting the differentiation and activation of DCs, (91,92).
CRAMP as a chemoattractant and anti-microbial peptide does not directly promote the differentiation of iDCs into mDCs. But rather, it increases the response of immature DCs to differentiation and maturation signals. BM-derived iDCs from wild type (WT) mice produce CRAMP and express its receptor Fpr2 (18). The interaction of Fpr2 with CRAMP via an autocrine and or paracrine loop primes iDCs to exhibit enhanced sensitivity to maturation stimulators such as LPS. This was demonstrated by the fact that in DC culture, neutralization of either Fpr2 or CRAMP decreased DC maturation in response to LPS. Also, LPS-treated DCs from Fpr2−/− mice did not express normal levels of maturation markers with reduced production of IL-12 and diminished chemotaxis in response to the DC homing chemokine CCL21 mediated by CCR7. Further, DCs from Fpr2−/− mice failed to induce a robust allogeneic T-cell proliferation in vitro and DC recruitment into the T-cell zones of the spleen in Fpr2−/− mice was reduced after antigen immunization. The involvement of CRAMP via Fpr2 in DC maturation was confirmed by the observation that addition of exogenous CRAMP to immature Fpr2−/− DC culture failed to increase cell sensitivity to LPS stimulation. After treatment with LPS, mDCs differentiated from CRAMP−/− mice showed a considerably lower level expression of the mDC markers CD86, CD80, and MHC II as compared with the cells from WT mice. The addition of exogenous CRAMP restored the responses of iDCs from CRAMP−/− mice to LPS. Mechanistically, there was a rapid phosphorylation of p38 MAPK in iDCs from WT mice stimulated by a low level of LPS. In contrast, much higher levels of LPS were required to stimulate a delayed phosphorylation of p38 in iDCs from Fpr2−/− mice, albeit the cells from both mice showed comparable levels of ERK1/2MAPK phosphorylation. Therefore, both CRAMP and Fpr2 are important partners in promoting DC differentiation and maturation (Fig. 3).
Figure 3. Fpr2/CRAMP interaction increases the sensitivity of immature DCs to maturation signals.
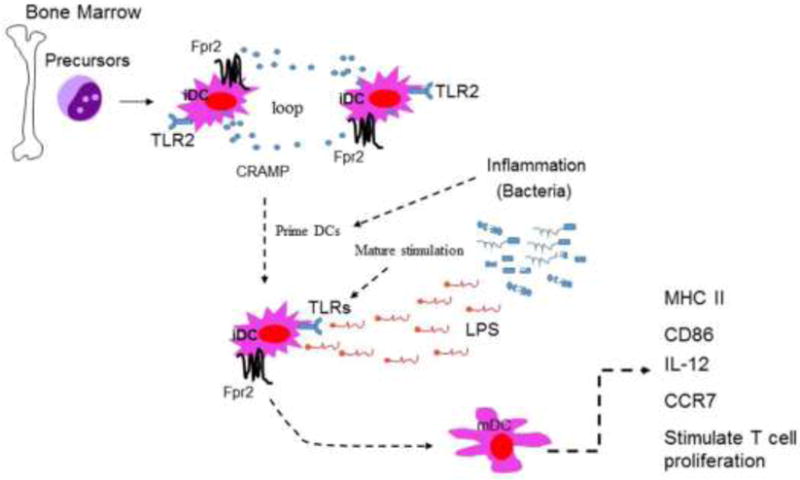
DC precursors from mouse bone marrow (BM) express both CRAMP and its receptor Fpr2. The interaction of Fpr2 with CRAMP via an autocrine or paracrine loop primes iDCs to enhance the sensitivity to maturation stimulants such as LPS.
2.2. Fpr2 contributes to follicular DC-triggered B cell activation in the germinal centers of Peyer’s patch (PP)
Peyer’s patches (PPs) are major mucosal immune inductive sites, and germinal centers (GCs) in PPs determine the quality of the antibodies produced. PP GCs are continuously induced by gut microbiota and their maintenance is required for the induction of potent IgA responses to Ags. Follicular DCs (FDCs) as key organizers of B cell follicles and GCs in the mucosal immunity express Fpr2, which promotes the expression of Cxcl13 and B cell activating factors to stimulate B cell proliferation and activation. Therefore, Fpr2 signaling in FDCs maintains GCs in PPs and is critical in promoting Ag-specific IgA responses in the gut mucosal compartment (20) (Fig. 4).
Figure 4. Fpr2 signaling in follicular DCs activates B cells in Peyer’s patch (PP) germinal centers.
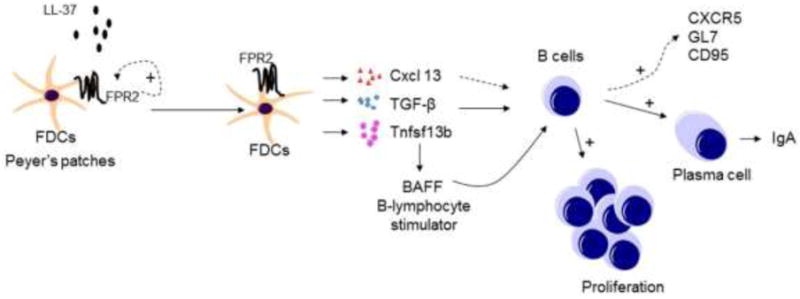
Follicular DCs (FDCs) express FPR2, which increase by ~60-fold in response to LL-37. LL-37/FPR2 signaling stimulates FDCs to produce Cxcl 13, TGF-β1, and Tnfsfl 3b to activate B cells in germinal centers (GCs) for proliferation, production of IgA, and up-regulation of CXCR5, CD95 and GL7.
3. FPRs participate in the control tumor-associated inflammation and immune responses
Chronic inflammation is an aberrantly prolonged form of a protective host response to the loss of tissue homeostasis (93), which has been recognized also a causative factor of many cancer. In fact, inflammation and neoplasia co-develop into “wounds that do not heal” (93). Leukocytes, such as neutrophils, monocytes, macrophages, and eosinophils, provide soluble factors including metabolites of arachidonic acid, cytokines, chemokines, and free radicals, that may benefit the development of inflammation-associated cancer (94). A wide array of chronic inflammatory conditions predispose susceptible cells to neoplastic transformation, Most of which are of epithelial cell origin (carcinomas) such as colon cancer linked to inflammatory bowel disease (IBD, chronic ulcerative colitis and Crohn’s disease), esophageal adenocarcinoma associated with reflux esophagitis (Barrett’s esophagus), hepatitis predisposing to cirrhosis and subsequent liver cancer, schistosomiasis associated with bladder and colon cancer, and Helicobacter infection leading to cancer of the stomach (93,94). The contribution of FPRs to inflammation-associated cancer has been increasingly recognized, which is illustrated in following chapters.
3.1. Fpr2 protects mice from chronic inflammation-associated tumors in the colon
Clinically, patients with chronic ulcerative colitis or Crohn’s disease have a five-to seven-fold increased risk of developing colorectal carcinoma, as a result of persistence of colon inflammation (94). Colon carcinogenesis begins with focal proliferation of dysplastic cells, the formation of benign adenomatous polyps, and progression to malignant adenocarcinomas. In a mouse model of chronic colitis, Fpr2 is found to play an important role in promoting mucosal restitution as demonstrated by the fact that Fpr2 deficiency resulted in a prolonged form of host response to a lost tissue homeostasis including greatly shortened colon, increased foci of ulcers in the lost mucosa and exacerbated inflammatory cell infiltration in ulcerative lesions (26). This is associated with markedly increased number of adenomas (26). Thus, Fpr2 is essential for limit the susceptibility of mouse colon to chronic inflammation-induced tumors.
3.2. LL-37, a FPR2 ligand, suppresses the progression of colon cancer
LL-37 is also known as hCAP-18, FALL-39 or CAMP—human cationic antimicrobial peptide (95,96). The mouse homologue of LL37 is termed cathelin-related antimicrobial peptide (CRAMP). LL-37 is expressed by various cells and tissues such as bone marrow (BM) myeloid cells, neutrophils, macrophages and epithelial cells and can be detected in the skin, gastrointestinal tract, urinary tract and the lung (97–111). CRAMP is expressed abundantly by mouse granulocytes and BM cells of the myeloid lineage (95,112–114). In adult mice, CRAMP is detected in testis, spleen, stomach, lung and intestine but not in the brain, liver, heart, or skeletal muscle (112). LL-37/CRAMP uses FPR2 (Fpr2) as a receptor to mediate leukocyte chemotaxis and pro-angiogenic activities (69,70,115).
It is interesting to note that unlike normal human colon epithelial cells, most human colon cancer cells did not express LL-37 (116). Therefore, the expression of low levels of LL-37 is used as a biomarker of colon cancer (117). However, FK-16, a fragment of LL-37 corresponding to the residues 17 to 32, induces the death of colon cancer cell by activating caspase-independent apoptosis and autophagy (118), suggesting an anti-colon cancer activity of LL-37. The colon carcinoma cell line HCT116 treated in vitro with FF/CAP18, a 27-residue analogue of LL-37, exhibits a marked loss in major metabolic profiles beneficial for the proliferation of cancer cells (119). In addition, LL-37 inhibits epithelial-mesenchymal transition (EMT) of colon cells, and fibroblast-supported colon cancer cell proliferation (120), mouse CRAMP reduces AOM-DSS-induced colon cancer by inhibiting vimentin positive fibroblasts to express collagen and disrupting tubulin distribution in fibroblasts (120). These studies indicate that LL-37/CRAMP directly inhibits tumorigenesis in colon. However, it is unclear whether the receptor FPR2/Fpr2 is utilized to trigger pro-apoptotic cascade in tumor cells and fibroblasts.
3.3. The FPR2 ligand, serum amyloid A (SAA), promotes lung carcinogenesis associated with chronic obstructive pulmonary disease (COPD)
Cigarette smoking is the predominant risk factor for chronic obstructive pulmonary disease (COPD) characterized by progressive airflow obstruction and association with abnormal and chronic inflammatory responses of the lung to noxious particles and gases. The pathology of COPD is heterogeneous including chronic obstructive bronchiolitis, fibrotic small airways, mucus gland hypertrophy, emphysema, and destruction of lung parenchyma. An important pathogenic process of COPD is excessive and uncompensated oxidative stress may initiate lung tumourigenesis (121). Oxidative stress directly attenuate the activity of the tumor suppressor gene, PTEN (122), and oxidative DNA damage often occurs in COPD lungs (123).
COPD is associated with persistent recruitment and activation of innate immune cells including neutrophils and macrophages. Uncontrolled release of free radicals and proteases neutrophils not only causes DNA damage but also has tumor promoting effect. Airway macrophages in COPD polarized to an M2 phenotype also release factors in support of tumor growth. In COPD, T-cell anergy may also lead to tumor cell evasion of immune surveillance. In COPD, non-conventional T-cells are an important source of IL-17A which is increasingly recognized as a mediator of tumor growth.
Serum myeloid A (SAA) is increased in the circulation of COPD patients. Increased level of SAA in the lungs with COPD associated lung cancer contributes to tumor growth by stimulating M2-like alternative macrophages (121). There is evidence for production of SAA within lung cancer microenvironment (124) by tumor-associated macrophages (TAM) and there was a positive correlation between the number of neutrophils and the content of SAA in resections of lung cancer with COPD (125). As an endogenous FPR2 agonist, SAA promotes immune evasion of cancer cells by enhancing the suppressive capacity of myeloid-derived suppressor cells (MDSCs) (126), stimulating lung metastasis of primary tumor cells under inflammatory condition (127). Although activation of FPR2 in macrophages has been reported to polarize the cells into a M1 phenotype, careful consideration must be given in dissections of ligand/FPR2 interaction under different pathophysiological conditions, due to a multitude of agonists for the receptor, that may interact with diverse domains in the receptor, hence distinct signaling cascade (128).
3.4. SAA/Fpr2 signaling promotes differentiation of IL-10-secreting neutrophils in melanoma
Some myeloid cell populations such as MDSCs have been found to exacerbate tumor progression either directly by inhibiting tumor-specific immune responses or indirectly by promoting angiogenesis, tumor cell proliferation and tissue remodeling (129). TAMs (130,131) and MDSCs are among such myeloid cells (132). Surprisingly, tumor cells are capable of impairing specific host immune responses by harnessing FPR2/SAA signaling pathway to promote the proliferation of immunosuppressive IL-10-secreting neutrophils (133). There are large numbers of IL-10-secreting neutrophils in the blood of patients with melanoma, which may account for the suppression of proliferation of melan-A-specific CD8+ T cells (26–35). SAA, as an acute-phase reactant secreted during responses to infection and injury, was present in the plasma and primary tumors of patients with melanoma (134). SAA induces the differentiation of IL-10-secreting neutrophils via FPR2 to suppress antigen-specific T cell responses. SAA also promotes the interaction of neutrophils with invariant natural killer T cells (iNKT cells) that restores T cell proliferation by abolishing IL-10 secretion. The end results are dependent on an equilibrium of the antigen-presenting molecule CD1d and costimulatory molecule CD40 that reduces the production of IL-10 but enhances IL-12 secreting, thus limiting the suppressive activity of neutrophils (133,135) (Fig. 5). These findings emphasize the plasticity of neutrophils with both pro- and anti-inflammatory properties and highlight the role of iNKT cells as important regulators of inflammatory and anti-tumor responses. Such a complex and fine-tuned mechanism designed to control excessive inflammation is exploited by tumors to topple anti-tumor host immunity via an FPR2/SAA axis. Thus, harnessing iNKT cells and interfering with FPR2/SSA pathway may diminish the frequency of immunosuppressive neutrophils that may have important implications for vaccination strategies.
Figure 5. Fpr2 promotes the differentiation of IL-10-secreting neutrophils.
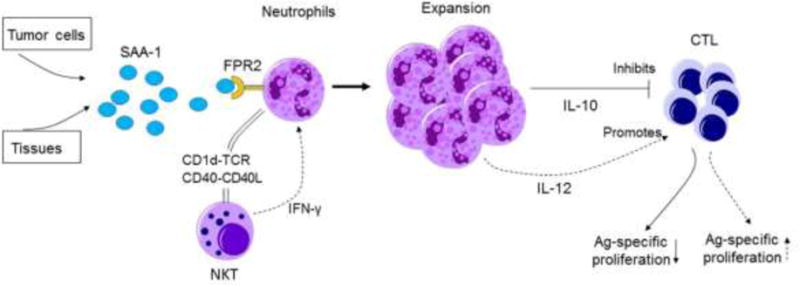
Serum amyloid A 1 (SAA-1) produced by tumor and stromal cells interacts with FPR2 on neutrophils to induce differentiation and expansion of an IL-10-producing population. CD1d- and CD40-dependent crosstalk between neutrophils and NKT cells results in the inhibition of IL-10, but promotion of IL-12, production by neutrophils. The process was enhanced by interferon-γ (IFN-γ) produced by NKT cells after CD1d ligation. The immunosuppressive activity of neutrophils is then reversed and the proliferation of antigen-specific cytotoxic T lymphocytes restored.
3.5. Fpr2 promotes antitumor host defense by supporting M1 macrophage polarization
TAMs constitute a major stromal component within the tumor and in general exhibit functions resembling M2 macrophages of the M2d subtype (136,137) (Fig. 6). During tumor development, infiltrating Ml macrophages, characterized by an IL-12high IL-10low phenotype, promote immune responses against tumor cells. During tumor progression, TAMs frequently switch to an IL-12low IL-10high M2-like phenotype with low tumoricidal activity (136). Such TAMs have been shown to provide a favorable microenvironment for tumor cell survival, angiogenesis, progression and metastasis (138,139).
Figure 6. Fpr2 in macrophage polarization.
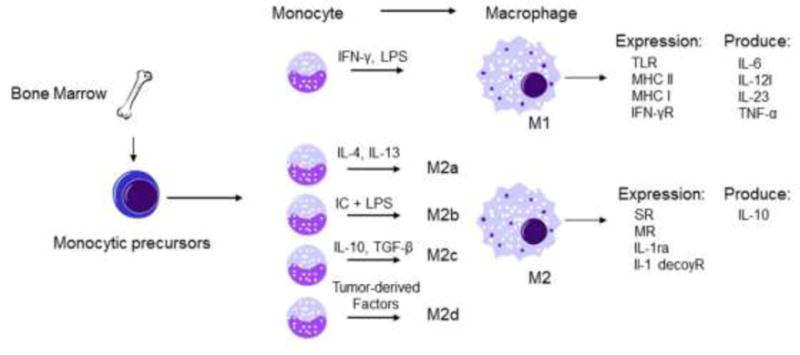
Tissue macrophages derived from circulating monocytes acquire either a Ml or alternative M2 phenotype after encounter microenvironmental stimuli. Ml macrophages are polarized by IFN-γ and LPS, and produce high levels of the pro-inflammatory cytokines such as IL-6, IL-12, IL-23, and TNF-α. M2 macrophages are subdivided into M2a, M2b, M2c, and M2d, and produce high levels of IL-10 with expression of scavenger receptors, mannose receptors, IL-1 receptor antagonist, and IL-1Rα decoy receptor. Ml macrophages mediate pro-inflammatory, cytotoxic and antitumor responses, while M2 macrophages promotes angiogenesis, immunosuppression, and tumor progression. LPS: lipopolysaccharide; IC, immune complex; GC, glucocorticoid; SR, scavenger receptor; MR, mannose receptor; IL-lra, IL-1 receptor antagonist; TLR, Toll-like receptor; MHC, major histocompatibility complex.
In a mouse Lewis lung cancer (LLC) implantation model, Fpr2−/− mice bearing subcutaneously implanted LLC tumors exhibited significantly shortened survival than did WT mice because of the more rapidly growing tumors. In contrast, in transgenic mice overexpressing Fpr2, subcutaneously implanted LLC tumors grew more slowly than those in WT littermates. Investigation of tumor tissues revealed more TAMs in tumors grown in Fpr2−/− mice and the macrophages isolated from Fpr2−/− mice showed a more-potent chemotactic response to LLC-derived supernatant, which could be neutralized by an antibody against the chemokine CCL2, suggesting the notion that CCL2 is the major chemoattractant for TAM infiltration into the microenvironment of LLC tumor. The increased chemotaxis of Fpr2−/− mouse macrophages in response to LLC supernatant was due to their higher level expression of CCR4, a chemokine GPCR that recognizes the ligand CCL2 (140,141). Interestingly, treatment with Fpr2 antagonists increased the chemotactic response of WT mouse macrophages to CCL2, in association with increased expression of CCR4. LLC tumor cell supernatant and Fpr2 agonists were capable of polarizing WT macrophages toward an Ml phenotype (142). Therefore, Fpr2 appears to increase host defense against implanted LLC by favoring polarization of macrophages toward Ml phenotype with more-potent antitumor activities (Fig. 7). However, a caveat may exist in generalizing this LLC model because not all tumors produce copious levels of CCL2 and the nature of LLC-derived Fpr2 agonists is unknown. Also, certain FPR2 agonist such as SAA has been shown to directly stimulate tumor cell growth as discussed early. Possible interpretation includes the diverse nature of FPR2 agonist to trigger biased intracellular signaling or FPR2 expressed by tumor cells versus myeloid cells may transduce opposing signals for tumor growth as exemplified by the action of SAA on neutrophils. More studies are required to elucidate the precise mechanisms by which Fpr2 promotes macrophage polarization into an M1 subtype to limit tumor growth and whether this property of Fpr2 could be exploited to develop antitumor therapies.
Figure 7. Fpr2 promotes antitumor host defense.
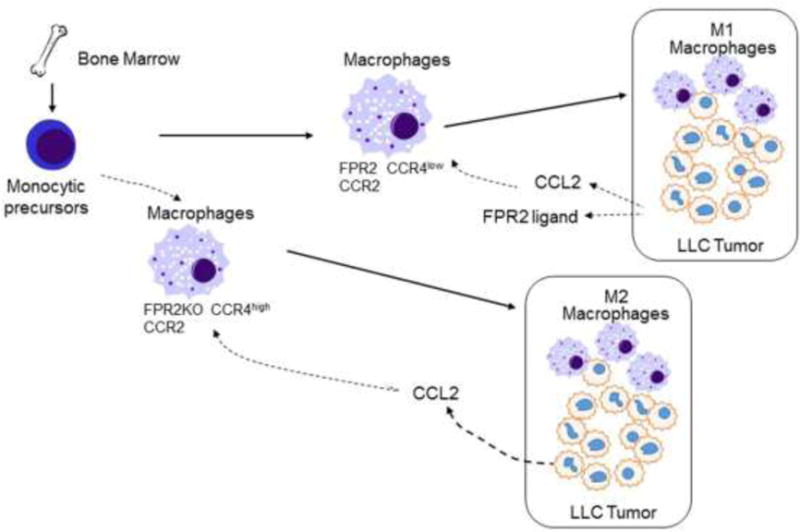
Mouse macrophages expressing both CCR2 and Fpr2 infiltrate implanted Lewis lung cancer (LLC) where macrophages undergo M1 polarization in response to tumor-derived Fpr2 ligands. Macrophages deficient in Fpr2 express high levels of CCR4, which synergizes with CCR2 to recruit TAM in response to tumor-derived CCL2, followed by polarization into M2 cells in response to as-yet-undefined tumor microenvironment factors.
4. Detrimental effects of FPRs hijacked by tumor cells
In addition to controlling leukocyte trafficking and activation, FPRs expressed by tumor cells very often are utilized for their growth advantage.
4.1. FPR2 in human colon cancer cells
High levels of FPR2 mRNA and protein were expressed by a considerable proportion of human colon cancer cell lines. FPR2 ligands induce colon cancer cell migration and proliferation. Colon cancer cells expressing high levels of FPR2 also formed more rapidly growing tumors in immunocompromised mice as compared with cell lines expressing lower levels of FPR2. Knocking down of FPR2 in colon cancer cells aberrantly expressing FPR2 reduced their tumorigenicity. Clinically, FPR2 is more highly expressed in progressive primary human colorectal cancer, associated with poorer patient prognosis (37). These data suggest that FPR2 can be utilized by colon cancer cells to accelerate their growth and invasion. It is interesting to note that some human colon cancer cells also either express FPR1 or both FPRs. However, the role of FPR1 in human colon cancer remains to be determined.
4.2. FPR2 promotes invasion and metastasis of gastric cancer cells
Gastric cancer (GC) is the fifth most common malignancy, and the third leading cause of cancer-related deaths worldwide (143). Although remarkable progress has been achieved in surgical and other therapeutic options, the overall 5-year survival rate of GC patients remains low (144). The main obstacle is the difficulties in early diagnosis of the disease. Recently, the levels of FPR2 expression in GC were reported to be correlated with their invasion depth, lymph node metastasis, overall survival of patients. FPR2 expression was an independent prognostic marker for GC patients. FPR2-knockdown significantly reduced the tumorigenic and metastatic capabilities of GC cells in immunocompromised mice. Mechanistically, stimulation with FPR2 ligands resulted in down-regulation of E-cadherin and up-regulation of vimentin in GC cells, implying the potential for FPR2 to mediate epithelial-mesenchymal transition (EMT) of GC cells. Therefore, FPR2 is functionally involved in the invasion and metastasis of human GC (145). However, it is unclear whether FPR2 on GC cells senses any endogenously produced agonists to trigger cellular functions similar to motile myeloid cells.
4.3. FPR1 in human gastric cancer
In addition to FPR2, FPR1 was detected in human GC specimens and its levels are correlated with more aggressive submucosal and serosal invasion accompanied by poorer outcome of patients (146). However, it is interesting that FPR1 silencing in GC cells significantly enhanced their tumorigenicity in mice because of augmented vessel density and tumor cell proliferation with high levels of HIF-1α and VEGF mRNA in xenografts tumor. Thus, FPR1 functions as GC suppressor by inhibiting tumor-induced angiogenesis (30). These observations, in contrast to those with FPR2 in GC, call for more careful evaluation of individual receptors in tumor growth, particularly in the context of microenvironment whether complex composition of tumor and stroma may determine the outcome of FPR signaling.
4.4. Annexin 1, in tumor cell growth
Annexin 1 (ANXA1), a 37-kDa member of the annexin superfamily, is a steroid-regulated protein that is implicated in mediating beneficial effects of glucocorticoids (147), including the regulation of cell proliferation (148), differentiation (149) and metastasis (150) by FPR1 (151). Dysregulation of ANXA1 has been reported in multiple neoplasms, suggesting an important position of ANXA1 in tumor development and progression (152). ANXA1 is overexpressed in cancers of the breast (153), liver (154), and pancreases (155), but is markedly low in cancers of esophagus (156,157) prostate (158) and stomach (159). ANXA1 activates FPR1 to promote the growth and invasion of tumor cells. For example, removal of ANXA1 released by necrotic human glioblastoma cells markedly reduced the chemotactic activity of tumor cell supernatant for live tumor cells, with diminished capacity to induce tumor cell growth, invasion, and colony formation. ANXA1 knockdown significantly reduced the tumorigenicity of glioblastoma cells in nude mice and deletion of both FPR1 and ANXA1 almost completely abolished the capacity of glioblastoma cells to form xenografts tumors (31). Therefore, ANXA1 and FPR1 form an auto- and paracrine loop in glioblastoma to promote their malignancy.
Perspectives
FPRs, as a subfamily of classic chemoattractant GPCRs, have one of the most diverse collections of ligands and they are expressed by a great variety of cell types, including cancer cells. Recent studies have shown essential roles of FPRs not only in leukocyte trafficking but also in colon mucosal homeostasis (26) and tumorigenesis. One of the major advances in understanding the pathophysiological functions of FPRs (Fprs) is their important positions in controlling leukocyte trafficking and immune responses. This is confirmed by studies of a variety of animal models and human diseases in which FPR1 was shown to be crucial for host responses to mitochondrial elements in sepsis (9) in mediating final leg of neutrophil accumulation in the center of necrotic liver lesions (160) and a partnership of FPR2, other chemoattractant GPCRs mediating neutrophil swarm in skin wounds and in bacterial infection (161).
FPRs also play an important role in inflammation associated tumorigenesis. Fpr2 deficiency impairs restoration of the colonic epithelial layer after injury, forming aberrant “wounds that do not heal” status, thereby increasing the susceptibility to chronic inflammation-associated tumors. Also, many tumor cells express FPRs to stimulate tumor cell proliferation, metastasis and angiogenesis, presumably by interact with endogenous ligands released into the microenvironments such as ANXA1.
These novel findings greatly expanded the scope of FPR biology whose pathophysiologic relevance had remained obscure since their discovery many years ago. However, much more needs to be learned with rigorous exploration of the participation of FPRs in diseases, which, fortunately, has become increasingly feasible with engineered mice with altered expression of FPR (Fpr) and/or ligand genes. It is thus plausible that future studies focusing on the regulation, signal transduction, structural basis for ligand recognition, and more importantly, on participation in pathophysiologic processes of FPRs in human, should yield novel insight into their potential as therapeutic targets for diseases.
Table 3.
Expression of FPRs in tumor cells
| Tumor cells | Expression of FPRs | Functions | Refs |
|---|---|---|---|
| Gastric cancer cells | FPRs | Epithelial–mesenchymal transition proliferation, migration, and cell resistance to apoptosis | 30 |
| Glioblastoma multiforme(GBM) | FPR1, FPR2 | Responding to an endogenous chemotactic ligand anexin 1 (Anx A1) released by necrotic GBM cells; Cooperating with EGFR to enhance the survival invasiveness and production of angiogenic factors by GBM cells | 2, 31–35 |
| Breast cancer cells | FPR1, FPR2 | Enhancing tumor cell proliferation | 36 |
| Liver cancer cells | FPR1 | Enhances cell chemotaxis invasion, proliferation and production of angiogenic factors | 2 |
| Colon cancer cells | FPR2 | Cell proliferation chemotaxis | 37 |
Highlight.
FPRs, as a subfamily of classic chemoattractant GPCRs, have one of the most diverse collections of ligands and they are expressed by a great variety of cell types, including cancer cells.
One of the major advances in understanding the pathophysiological functions of FPRs (Fprs) is their important positions in controlling leukocyte trafficking and immune responses.
FPRs also play an important role in inflammation associated tumorigenesis. Fpr2 deficiency impairs restoration of the colonic epithelial layer after injury, forming aberrant “wounds that do not heal” status, thereby increasing the susceptibility to chronic inflammation-associated tumor
Many tumor cells express FPRs to stimulate tumor cell proliferation, metastasis and angiogenesis, presumably by interact with endogenous ligands released into the microenvironments such as ANXA1.
Acknowledgments
1. We thank Ms. S. Livingstone and Ms. C. Lamb for secretarial assistance.
2. This project was funded in part by federal funds from the National Cancer Institute, National Institutes of Health, under Contract No. HHSN261200800001E and was supported in part by the Intramural Research Program of the NCI, NIH.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
CONFLICT OF INTEREST
The authors declare that they have no conflicts of interest with the contents of this article.
It is our great honor to dedicate this article to Dr. Alberto Mantovani who has made outstanding contributions to the advancement of the understanding of the mechanistic basis for inflammation, immune responses and cancer progression. We are grateful to Dr. Alberto Mantovani for his decades of care, training and support, without which, our success in scientific career is unconceivable.
References
- 1.Huang J, Chen K, Gong W, Dunlop NM, Wang JM. G-protein coupled chemoattractant receptors and cancer. Front Biosci. 2008;13:3352–3363. doi: 10.2741/2930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Li L, Chen K, Xiang Y, Yoshimura T, Su S, Zhu J, Bian XW, Wang JM. New development in studies of formyl-peptide receptors: critical roles in host defense. J Leukoc Biol. 2016;99:425–435. doi: 10.1189/jlb.2RI0815-354RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Le Y, Murphy PM, Wang JM. Formyl-peptide receptors revisited. Trends Immunol. 2002;23:541–548. doi: 10.1016/s1471-4906(02)02316-5. [DOI] [PubMed] [Google Scholar]
- 4.Ye RD, Boulay F, Wang JM, Dahlgren C, Gerard C, Parmentier M, Serhan CN, Murphy PM. International Union of Basic and Clinical Pharmacology. LXXIII. Nomenclature for the formyl peptide receptor (FPR) family. Pharmacological reviews. 2009;61:119–161. doi: 10.1124/pr.109.001578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chadwick VS, Mellor DM, Myers DB, Selden AC, Keshavarzian A, Broom MF, Hobson CH. Production of peptides inducing chemotaxis and lysosomal enzyme release in human neutrophils by intestinal bacteria in vitro and in vivo. Scandinavian journal of gastroenterology. 1988;23:121–128. doi: 10.3109/00365528809093861. [DOI] [PubMed] [Google Scholar]
- 6.Schiffmann E, Corcoran BA, Wahl SM. N-formylmethionyl peptides as chemoattractants for leucocytes. Proc Natl Acad Sci U S A. 1975;72:1059–1062. doi: 10.1073/pnas.72.3.1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cattaneo F, Parisi M, Ammendola R. Distinct signaling cascades elicited by different formyl peptide receptor 2 (FPR2) agonists. Int J Mol Sci. 2013;14:7193–7230. doi: 10.3390/ijms14047193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rabiet MJ, Macari L, Dahlgren C, Boulay F. N-formyl peptide receptor 3 (FPR3) departs from the homologous FPR2/ALX receptor with regard to the major processes governing chemoattractant receptor regulation, expression at the cell surface, and phosphorylation. J Biol Chem. 2011;286:26718–26731. doi: 10.1074/jbc.M111.244590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dorward DA, Lucas CD, Chapman GB, Haslett C, Dhaliwal K, Rossi AG. The role of formylated peptides and formyl peptide receptor 1 in governing neutrophil function during acute inflammation. Am J Pathol. 2015;185:1172–1184. doi: 10.1016/j.ajpath.2015.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hartt JK, Barish G, Murphy PM, Gao JL. N-formylpeptides induce two distinct concentration optima for mouse neutrophil chemotaxis by differential interaction with two N-formylpeptide receptor (FPR) subtypes. Molecular characterization of FPR2, a second mouse neutrophil FPR. J Exp Med. 1999;190:741–747. doi: 10.1084/jem.190.5.741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gao JL, Lee EJ, Murphy PM. Impaired antibacterial host defense in mice lacking the N-formylpeptide receptor. J Exp Med. 1999;189:657–662. doi: 10.1084/jem.189.4.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Devosse T, Guillabert A, D’Haene N, Berton A, De Nadai P, Noel S, Brait M, Franssen JD, Sozzani S, Salmon I, Parmentier M. Formyl peptide receptor-like 2 is expressed and functional in plasmacytoid dendritic cells, tissue-specific macrophage subpopulations, and eosinophils. J Immunol. 2009;182:4974–4984. doi: 10.4049/jimmunol.0803128. [DOI] [PubMed] [Google Scholar]
- 13.Le Y, Oppenheim JJ, Wang JM. Pleiotropic roles of formyl peptide receptors. Cytokine & growth factor reviews. 2001;12:91–105. doi: 10.1016/s1359-6101(01)00003-x. [DOI] [PubMed] [Google Scholar]
- 14.Boulay F, Tardif M, Brouchon L, Vignais P. Synthesis and use of a novel N-formyl peptide derivative to isolate a human N-formyl peptide receptor cDNA. Biochemical and biophysical research communications. 1990;168:1103–1109. doi: 10.1016/0006-291x(90)91143-g. [DOI] [PubMed] [Google Scholar]
- 15.Kim SD, Kim JM, Jo SH, Lee HY, Lee SY, Shim JW, Seo SK, Yun J, Bae YS. Functional expression of formyl peptide receptor family in human NK cells. Journal of immunology. 2009;183:5511–5517. doi: 10.4049/jimmunol.0802986. [DOI] [PubMed] [Google Scholar]
- 16.Nagaya T, Kawata K, Kamekura R, Jitsukawa S, Kubo T, Kamei M, Ogasawara N, Takano KI, Himi T, Ichimiya S. Lipid mediators foster the differentiation of T follicular helper cells. Immunol Lett. 2017;181:51–57. doi: 10.1016/j.imlet.2016.11.006. [DOI] [PubMed] [Google Scholar]
- 17.Yang D, Chen Q, Gertz B, He R, Phulsuksombati M, Ye RD, Oppenheim JJ. Human dendritic cells express functional formyl peptide receptor-like-2 (FPRL2) throughout maturation. J Leukoc Biol. 2002;72:598–607. [PubMed] [Google Scholar]
- 18.Chen K, Xiang Y, Huang J, Gong W, Yoshimura T, Jiang Q, Tessarollo L, Le Y, Wang JM. The formylpeptide receptor 2 (Fpr2) and its endogenous ligand cathelin-related antimicrobial peptide (CRAMP) promote dendritic cell maturation. J Biol Chem. 2014;289:17553–17563. doi: 10.1074/jbc.M113.535674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee HY, Lee M, Bae YS. Formyl Peptide Receptors in Cellular Differentiation and Inflammatory Diseases. Journal of cellular biochemistry. 2017 doi: 10.1002/jcb.25877. [DOI] [PubMed] [Google Scholar]
- 20.Kim SH, Kim YN, Jang YS. Cutting Edge: LL-37-Mediated Formyl Peptide Receptor-2 Signaling in Follicular Dendritic Cells Contributes to B Cell Activation in Peyer’s Patch Germinal Centers. J Immunol. 2017;198:629–633. doi: 10.4049/jimmunol.1600886. [DOI] [PubMed] [Google Scholar]
- 21.Lee SY, Lee MS, Lee HY, Kim SD, Shim JW, Jo SH, Lee JW, Kim JY, Choi YW, Baek SH, Ryu SH, Bae YS. F2L, a peptide derived from heme-binding protein, inhibits LL-37-induced cell proliferation and tube formation in human umbilical vein endothelial cells. FEBS letters. 2008;582:273–278. doi: 10.1016/j.febslet.2007.12.015. [DOI] [PubMed] [Google Scholar]
- 22.Heo SC, Kwon YW, Jang IH, Jeong GO, Yoon JW, Kim CD, Kwon SM, Bae YS, Kim JH. WKYMVm-induced activation of formyl peptide receptor 2 stimulates ischemic neovasculogenesis by promoting homing of endothelial colony-forming cells. Stem cells. 2014;32:779–790. doi: 10.1002/stem.1578. [DOI] [PubMed] [Google Scholar]
- 23.Lee MS, Yoo SA, Cho CS, Suh PG, Kim WU, Ryu SH. Serum amyloid A binding to formyl peptide receptor-like 1 induces synovial hyperplasia and angiogenesis. J Immunol. 2006;177:5585–5594. doi: 10.4049/jimmunol.177.8.5585. [DOI] [PubMed] [Google Scholar]
- 24.Tagoe CE, Marjanovic N, Park JY, Chan ES, Abeles AM, Attur M, Abramson SB, Pillinger MH. Annexin-1 mediates TNF-alpha-stimulated matrix metalloproteinase secretion from rheumatoid arthritis synovial fibroblasts. J Immunol. 2008;181:2813–2820. doi: 10.4049/jimmunol.181.4.2813. [DOI] [PubMed] [Google Scholar]
- 25.Yu N, Zhang S, Lu J, Li Y, Yi X, Tang L, Su L, Ding Y. Serum amyloid A, an acute phase protein, stimulates proliferative and proinflammatory responses of keratinocytes. Cell proliferation. 2016 doi: 10.1111/cpr.12320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen K, Liu M, Liu Y, Yoshimura T, Shen W, Le Y, Durum S, Gong W, Wang C, Gao JL, Murphy PM, Wang JM. Formylpeptide receptor-2 contributes to colonic epithelial homeostasis, inflammation, and tumorigenesis. J Clin Invest. 2013;123:1694–1704. doi: 10.1172/JCI65569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim MK, Min do S, Park YJ, Kim JH, Ryu SH, Bae YS. Expression and functional role of formyl peptide receptor in human bone marrow-derived mesenchymal stem cells. FEBS letters. 2007;581:1917–1922. doi: 10.1016/j.febslet.2007.03.078. [DOI] [PubMed] [Google Scholar]
- 28.Shin MK, Jang YH, Yoo HJ, Kang DW, Park MH, Kim MK, Song JH, Kim SD, Min G, You HK, Choi KY, Bae YS, Min do S. N-formyl-methionyl-leucyl-phenylalanine (fMLP) promotes osteoblast differentiation via the N-formyl peptide receptor 1-mediated signaling pathway in human mesenchymal stem cells from bone marrow. J Biol Chem. 2011;286:17133–17143. doi: 10.1074/jbc.M110.197772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McCoy R, Haviland DL, Molmenti EP, Ziambaras T, Wetsel RA, Perlmutter DH. N-formylpeptide and complement C5a receptors are expressed in liver cells and mediate hepatic acute phase gene regulation. J Exp Med. 1995;182:207–217. doi: 10.1084/jem.182.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Prevete N, Liotti F, Visciano C, Marone G, Melillo RM, de Paulis A. The formyl peptide receptor 1 exerts a tumor suppressor function in human gastric cancer by inhibiting angiogenesis. Oncogene. 2015;34:3826–3838. doi: 10.1038/onc.2014.309. [DOI] [PubMed] [Google Scholar]
- 31.Yang Y, Liu Y, Yao X, Ping Y, Jiang T, Liu Q, Xu S, Huang J, Mou H, Gong W, Chen K, Bian X, Wang JM. Annexin 1 released by necrotic human glioblastoma cells stimulates tumor cell growth through the formyl peptide receptor 1. Am J Pathol. 2011;179:1504–1512. doi: 10.1016/j.ajpath.2011.05.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yao XH, Ping YF, Chen JH, Chen DL, Xu CP, Zheng J, Wang JM, Bian XW. Production of angiogenic factors by human glioblastoma cells following activation of the G-protein coupled formylpeptide receptor FPR. Journal of neuro-oncology. 2008;86:47–53. doi: 10.1007/s11060-007-9443-y. [DOI] [PubMed] [Google Scholar]
- 33.Huang J, Hu J, Bian X, Chen K, Gong W, Dunlop NM, Howard OM, Wang JM. Transactivation of the epidermal growth factor receptor by formylpeptide receptor exacerbates the malignant behavior of human glioblastoma cells. Cancer Res. 2007;67:5906–5913. doi: 10.1158/0008-5472.CAN-07-0691. [DOI] [PubMed] [Google Scholar]
- 34.Huang J, Chen K, Chen J, Gong W, Dunlop NM, Howard OM, Gao Y, Bian XW, Wang JM. The G-protein-coupled formylpeptide receptor FPR confers a more invasive phenotype on human glioblastoma cells. British journal of cancer. 2010;102:1052–1060. doi: 10.1038/sj.bjc.6605591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huang J, Chen K, Huang J, Gong W, Dunlop NM, Howard OM, Bian X, Gao Y, Wang JM. Regulation of the leucocyte chemoattractant receptor FPR in glioblastoma cells by cell differentiation. Carcinogenesis. 2009;30:348–355. doi: 10.1093/carcin/bgn266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Khau T, Langenbach SY, Schuliga M, Harris T, Johnstone CN, Anderson RL, Stewart AG. Annexin-1 signals mitogen-stimulated breast tumor cell proliferation by activation of the formyl peptide receptors (FPRs) 1 and 2. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2011;25:483–496. doi: 10.1096/fj.09-154096. [DOI] [PubMed] [Google Scholar]
- 37.Xiang Y, Yao X, Chen K, Wang X, Zhou J, Gong W, Yoshimura T, Huang J, Wang R, Wu Y, Shi G, Bian X, Wang J. The G-protein coupled chemoattractant receptor FPR2 promotes malignant phenotype of human colon cancer cells. Am J Cancer Res. 2016;6:2599–2610. [PMC free article] [PubMed] [Google Scholar]
- 38.Sun T, Rojas OL, Li C, Philpott DJ, Gommerman JL. Hematopoietic LTbetaR deficiency results in skewed T cell cytokine profiles during a mucosal viral infection. J Leukoc Biol. 2016;100:103–110. doi: 10.1189/jlb.4MAB0715-294R. [DOI] [PubMed] [Google Scholar]
- 39.Prevete N, Liotti F, Marone G, Melillo RM, de Paulis A. Formyl peptide receptors at the interface of inflammation, angiogenesis and tumor growth. Pharmacol Res. 2015;102:184–191. doi: 10.1016/j.phrs.2015.09.017. [DOI] [PubMed] [Google Scholar]
- 40.Murphy PM. The molecular biology of leukocyte chemoattractant receptors. Annu Rev Immunol. 1994;12:593–633. doi: 10.1146/annurev.iy.12.040194.003113. [DOI] [PubMed] [Google Scholar]
- 41.Stavru F, Archambaud C, Cossart P. Cell biology and immunology of Listeria monocytogenes infections: novel insights. Immunol Rev. 2011;240:160–184. doi: 10.1111/j.1600-065X.2010.00993.x. [DOI] [PubMed] [Google Scholar]
- 42.Cossart P, Toledo-Arana A. Listeria monocytogenes, a unique model in infection biology: an overview. Microbes Infect. 2008;10:1041–1050. doi: 10.1016/j.micinf.2008.07.043. [DOI] [PubMed] [Google Scholar]
- 43.Hamon M, Bierne H, Cossart P. Listeria monocytogenes: a multifaceted model. Nat Rev Microbiol. 2006;4:423–434. doi: 10.1038/nrmicro1413. [DOI] [PubMed] [Google Scholar]
- 44.Liu M, Chen K, Yoshimura T, Liu Y, Gong W, Wang A, Gao JL, Murphy PM, Wang JM. Formylpeptide receptors are critical for rapid neutrophil mobilization in host defense against Listeria monocytogenes. Sci Rep. 2012;2:786. doi: 10.1038/srep00786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chapman RW, Minnicozzi M, Celly CS, Phillips JE, Kung TT, Hipkin RW, Fan X, Rindgen D, Deno G, Bond R, Gonsiorek W, Billah MM, Fine JS, Hey JA. A novel, orally active CXCR1/2 receptor antagonist, Sch527123, inhibits neutrophil recruitment, mucus production, and goblet cell hyperplasia in animal models of pulmonary inflammation. J Pharmacol Exp Ther. 2007;322:486–493. doi: 10.1124/jpet.106.119040. [DOI] [PubMed] [Google Scholar]
- 46.Rose JJ, Foley JF, Murphy PM, Venkatesan S. On the mechanism and significance of ligand-induced internalization of human neutrophil chemokine receptors CXCR1 and CXCR2. J Biol Chem. 2004;279:24372–24386. doi: 10.1074/jbc.M401364200. [DOI] [PubMed] [Google Scholar]
- 47.Cunha TM, Barsante MM, Guerrero AT, Verri WA, Jr, Ferreira SH, Coelho FM, Bertini R, Di Giacinto C, Allegretti M, Cunha FQ, Teixeira MM. Treatment with DF 2162, a non-competitive allosteric inhibitor of CXCR1/2, diminishes neutrophil influx and inflammatory hypernociception in mice. Br J Pharmacol. 2008;154:460–470. doi: 10.1038/bjp.2008.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Reichel CA, Khandoga A, Anders HJ, Schlondorff D, Luckow B, Krombach F. Chemokine receptors Ccr1, Ccr2, and Ccr5 mediate neutrophil migration to postischemic tissue. J Leukoc Biol. 2006;79:114–122. doi: 10.1189/jlb.0605337. [DOI] [PubMed] [Google Scholar]
- 49.Chintakuntlawar AV, Chodosh J. Chemokine CXCL1/KC and its receptor CXCR2 are responsible for neutrophil chemotaxis in adenoviral keratitis. J Interferon Cytokine Res. 2009;29:657–666. doi: 10.1089/jir.2009.0006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang L, Ran L, Garcia GE, Wang XH, Han S, Du J, Mitch WE. Chemokine CXCL16 regulates neutrophil and macrophage infiltration into injured muscle, promoting muscle regeneration. Am J Pathol. 2009;175:2518–2527. doi: 10.2353/ajpath.2009.090275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Burdon PC, Martin C, Rankin SM. The CXC chemokine MIP-2 stimulates neutrophil mobilization from the rat bone marrow in a CD49d-dependent manner. Blood. 2005;105:2543–2548. doi: 10.1182/blood-2004-08-3193. [DOI] [PubMed] [Google Scholar]
- 52.Southgate EL, He RL, Gao JL, Murphy PM, Nanamori M, Ye RD. Identification of formyl peptides from Listeria monocytogenes and Staphylococcus aureus as potent chemoattractants for mouse neutrophils. J Immunol. 2008;181:1429–1437. doi: 10.4049/jimmunol.181.2.1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Oldekamp S, Pscheidl S, Kress E, Soehnlein O, Jansen S, Pufe T, Wang JM, Tauber SC, Brandenburg LO. Lack of formyl peptide receptor 1 and 2 leads to more severe inflammation and higher mortality in mice with of pneumococcal meningitis. Immunology. 2014;143:447–461. doi: 10.1111/imm.12324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.diaoDiao N, Zhang Y, Chen K, Yuan R, Lee C, Geng S, Kowalski E, Guo W, Xiong H, Li M, Li L. Deficiency in Toll-interacting protein (Tollip) skews inflamed yet incompetent innate leukocytes in vivo during DSS-induced septic colitis. Sci Rep. 2016;6:34672. doi: 10.1038/srep34672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gobbetti T, Coldewey SM, Chen J, McArthur S, le Faouder P, Cenac N, Flower RJ, Thiemermann C, Perretti M. Nonredundant protective properties of FPR2/ALX in polymicrobial murine sepsis. Proc Natl Acad Sci U S A. 2014;111:18685–18690. doi: 10.1073/pnas.1410938111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Speyer CL, Gao H, Rancilio NJ, Neff TA, Huffnagle GB, Sarma JV, Ward PA. Novel chemokine responsiveness and mobilization of neutrophils during sepsis. Am J Pathol. 2004;165:2187–2196. doi: 10.1016/S0002-9440(10)63268-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Alves-Filho JC, Spiller F, Cunha FQ. Neutrophil paralysis in sepsis. Shock. 2010;34(Suppl 1):15–21. doi: 10.1097/SHK.0b013e3181e7e61b. [DOI] [PubMed] [Google Scholar]
- 58.Engelhardt E, Toksoy A, Goebeler M, Debus S, Brocker EB, Gillitzer R. Chemokines IL-8, GROalpha, MCP-1, IP-10, and Mig are sequentially and differentially expressed during phase-specific infiltration of leukocyte subsets in human wound healing. Am J Pathol. 1998;153:1849–1860. doi: 10.1016/s0002-9440(10)65699-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Foxman EF, Campbell JJ, Butcher EC. Multistep navigation and the combinatorial control of leukocyte chemotaxis. J Cell Biol. 1997;139:1349–1360. doi: 10.1083/jcb.139.5.1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Brandt E, Petersen F, Ludwig A, Ehlert JE, Bock L, Flad HD. The beta-thromboglobulins and platelet factor 4: blood platelet-derived CXC chemokines with divergent roles in early neutrophil regulation. J Leukoc Biol. 2000;67:471–478. doi: 10.1002/jlb.67.4.471. [DOI] [PubMed] [Google Scholar]
- 61.Goebeler M, Yoshimura T, Toksoy A, Ritter U, Brocker EB, Gillitzer R. The chemokine repertoire of human dermal microvascular endothelial cells and its regulation by inflammatory cytokines. J Invest Dermatol. 1997;108:445–451. doi: 10.1111/1523-1747.ep12289711. [DOI] [PubMed] [Google Scholar]
- 62.Gillitzer R, Ritter U, Spandau U, Goebeler M, Brocker EB. Differential expression of GRO-alpha and IL-8 mRNA in psoriasis: a model for neutrophil migration and accumulation in vivo. J Invest Dermatol. 1996;107:778–782. doi: 10.1111/1523-1747.ep12371803. [DOI] [PubMed] [Google Scholar]
- 63.Liu M, Chen K, Yoshimura T, Liu Y, Gong W, Le Y, Gao JL, Zhao J, Wang JM, Wang A. Formylpeptide receptors mediate rapid neutrophil mobilization to accelerate wound healing. PLoS One. 2014;9:e90613. doi: 10.1371/journal.pone.0090613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Finn PW, Bigby TD. Innate immunity and asthma. Proceedings of the American Thoracic Society. 2009;6:260–265. doi: 10.1513/pats.200807-064RM. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wang P, Wang X, Yang X, Liu Z, Wu M, Li G. Budesonide suppresses pulmonary antibacterial host defense by down-regulating cathelicidin-related antimicrobial peptide in allergic inflammation mice and in lung epithelial cells. BMC immunology. 2013;14:7. doi: 10.1186/1471-2172-14-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Liu W, Chen Y, Golan MA, Annunziata ML, Du J, Dougherty U, Kong J, Musch M, Huang Y, Pekow J, Zheng C, Bissonnette M, Hanauer SB, Li YC. Intestinal epithelial vitamin D receptor signaling inhibits experimental colitis. J Clin Invest. 2013;123:3983–3996. doi: 10.1172/JCI65842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Agier J, Efenberger M, Brzezinska-Blaszczyk E. Cathelicidin impact on inflammatory cells. Central-European journal of immunology/Polish Society for Immunology and eleven other Central-European immunological societies. 2015;40:225–235. doi: 10.5114/ceji.2015.51359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tjabringa GS, Ninaber DK, Drijfhout JW, Rabe KF, Hiemstra PS. Human cathelicidin LL-37 is a chemoattractant for eosinophils and neutrophils that acts via formyl-peptide receptors. International archives of allergy and immunology. 2006;140:103–112. doi: 10.1159/000092305. [DOI] [PubMed] [Google Scholar]
- 69.De Y, Chen Q, Schmidt AP, Anderson GM, Wang JM, Wooters J, Oppenheim JJ, Chertov O. LL-37, the neutrophil granule- and epithelial cell-derived cathelicidin, utilizes formyl peptide receptor-like 1 (FPRL1) as a receptor to chemoattract human peripheral blood neutrophils, monocytes, and T cells. J Exp Med. 2000;192:1069–1074. doi: 10.1084/jem.192.7.1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kurosaka K, Chen Q, Yarovinsky F, Oppenheim JJ, Yang D. Mouse cathelin-related antimicrobial peptide chemoattracts leukocytes using formyl peptide receptor-like 1/mouse formyl peptide receptor-like 2 as the receptor and acts as an immune adjuvant. J Immunol. 2005;174:6257–6265. doi: 10.4049/jimmunol.174.10.6257. [DOI] [PubMed] [Google Scholar]
- 71.Nathan C. Points of control in inflammation. Nature. 2002;420:846–852. doi: 10.1038/nature01320. [DOI] [PubMed] [Google Scholar]
- 72.Ley K, Laudanna C, Cybulsky MI, Nourshargh S. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat Rev Immunol. 2007;7:678–689. doi: 10.1038/nri2156. [DOI] [PubMed] [Google Scholar]
- 73.Chtanova T, Schaeffer M, Han SJ, van Dooren GG, Nollmann M, Herzmark P, Chan SW, Satija H, Camfield K, Aaron H, Striepen B, Robey EA. Dynamics of neutrophil migration in lymph nodes during infection. Immunity. 2008;29:487–496. doi: 10.1016/j.immuni.2008.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Peters NC, Egen JG, Secundino N, Debrabant A, Kimblin N, Kamhawi S, Lawyer P, Fay MP, Germain RN, Sacks D. In vivo imaging reveals an essential role for neutrophils in leishmaniasis transmitted by sand flies. Science. 2008;321:970–974. doi: 10.1126/science.1159194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bruns S, Kniemeyer O, Hasenberg M, Aimanianda V, Nietzsche S, Thywissen A, Jeron A, Latge JP, Brakhage AA, Gunzer M. Production of extracellular traps against Aspergillus fumigatus in vitro and in infected lung tissue is dependent on invading neutrophils and influenced by hydrophobin RodA. PLoS Pathog. 2010;6:e1000873. doi: 10.1371/journal.ppat.1000873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yipp BG, Petri B, Salina D, Jenne CN, Scott BN, Zbytnuik LD, Pittman K, Asaduzzaman M, Wu K, Meijndert HC, Malawista SE, de Boisfleury Chevance A, Zhang K, Conly J, Kubes P. Infection-induced NETosis is a dynamic process involving neutrophil multitasking in vivo. Nat Med. 2012;18:1386–1393. doi: 10.1038/nm.2847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lambrecht BN, Hammad H. Biology of lung dendritic cells at the origin of asthma. Immunity. 2009;31:412–424. doi: 10.1016/j.immuni.2009.08.008. [DOI] [PubMed] [Google Scholar]
- 78.GeurtsvanKessel CH, Lambrecht BN. Division of labor between dendritic cell subsets of the lung. Mucosal immunology. 2008;1:442–450. doi: 10.1038/mi.2008.39. [DOI] [PubMed] [Google Scholar]
- 79.Robays LJ, Maes T, Lebecque S, Lira SA, Kuziel WA, Brusselle GG, Joos GF, Vermaelen KV. Chemokine receptor CCR2 but not CCR5 or CCR6 mediates the increase in pulmonary dendritic cells during allergic airway inflammation. Journal of immunology. 2007;178:5305–5311. doi: 10.4049/jimmunol.178.8.5305. [DOI] [PubMed] [Google Scholar]
- 80.Provoost S, Maes T, Joos GF, Tournoy KG. Monocyte-derived dendritic cell recruitment and allergic T(H)2 responses after exposure to diesel particles are CCR2 dependent. J Allergy Clin Immunol. 2012;129:483–491. doi: 10.1016/j.jaci.2011.07.051. [DOI] [PubMed] [Google Scholar]
- 81.Toulabi L, Wu X, Cheng Y, Mao Y. Identification and structural characterization of a Legionella phosphoinositide phosphatase. The Journal of biological chemistry. 2013;288:24518–24527. doi: 10.1074/jbc.M113.474239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Chen D, Fang L, Li H, Tang MS, Jin C. Cigarette smoke component acrolein modulates chromatin assembly by inhibiting histone acetylation. The Journal of biological chemistry. 2013;288:21678–21687. doi: 10.1074/jbc.M113.476630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Serbina NV, Pamer EG. Monocyte emigration from bone marrow during bacterial infection requires signals mediated by chemokine receptor CCR2. Nat Immunol. 2006;7:311–317. doi: 10.1038/ni1309. [DOI] [PubMed] [Google Scholar]
- 84.Chen K, Liu M, Liu Y, Wang C, Yoshimura T, Gong W, Le Y, Tessarollo L, Wang JM. Signal relay by CC chemokine receptor 2 (CCR2) and formylpeptide receptor 2 (Fpr2) in the recruitment of monocyte-derived dendritic cells in allergic airway inflammation. J Biol Chem. 2013;288:16262–16273. doi: 10.1074/jbc.M113.450635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ueno H, Klechevsky E, Morita R, Aspord C, Cao T, Matsui T, Di Pucchio T, Connolly J, Fay JW, Pascual V, Palucka AK, Banchereau J. Dendritic cell subsets in health and disease. Immunological reviews. 2007;219:118–142. doi: 10.1111/j.1600-065X.2007.00551.x. [DOI] [PubMed] [Google Scholar]
- 86.Trombetta ES, Mellman I. Cell biology of antigen processing in vitro and in vivo. Annu Rev Immunol. 2005;23:975–1028. doi: 10.1146/annurev.immunol.22.012703.104538. [DOI] [PubMed] [Google Scholar]
- 87.Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 88.Schreibelt G, Tel J, Sliepen KH, Benitez-Ribas D, Figdor CG, Adema GJ, de Vries IJ. Toll-like receptor expression and function in human dendritic cell subsets: implications for dendritic cell-based anti-cancer immunotherapy. Cancer Immunol Immunother. 59:1573–1582. doi: 10.1007/s00262-010-0833-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Fritz JH, Girardin SE, Fitting C, Werts C, Mengin-Lecreulx D, Caroff M, Cavaillon JM, Philpott DJ, Adib-Conquy M. Synergistic stimulation of human monocytes and dendritic cells by Toll-like receptor 4 and NOD1- and NOD2-activating agonists. Eur J Immunol. 2005;35:2459–2470. doi: 10.1002/eji.200526286. [DOI] [PubMed] [Google Scholar]
- 90.van Vliet SJ, den Dunnen J, Gringhuis SI, Geijtenbeek TB, van Kooyk Y. Innate signaling and regulation of Dendritic cell immunity. Curr Opin Immunol. 2007;19:435–440. doi: 10.1016/j.coi.2007.05.006. [DOI] [PubMed] [Google Scholar]
- 91.Caux C, Dezutter-Dambuyant C, Schmitt D, Banchereau J. GM-CSF and TNF-alpha cooperate in the generation of dendritic Langerhans cells. Nature. 1992;360:258–261. doi: 10.1038/360258a0. [DOI] [PubMed] [Google Scholar]
- 92.Caux C, Favre C, Saeland S, Duvert V, Durand I, Mannoni P, Banchereau J. Potentiation of early hematopoiesis by tumor necrosis factor-alpha is followed by inhibition of granulopoietic differentiation and proliferation. Blood. 1991;78:635–644. [PubMed] [Google Scholar]
- 93.Elinav E, Nowarski R, Thaiss CA, Hu B, Jin C, Flavell RA. Inflammation-induced cancer: crosstalk between tumours, immune cells and microorganisms. Nat Rev Cancer. 2013;13:759–771. doi: 10.1038/nrc3611. [DOI] [PubMed] [Google Scholar]
- 94.Shacter E, Weitzman SA. Chronic inflammation and cancer. Oncology (Williston Park) 2002;16:217–226. 229. discussion 230-212. [PubMed] [Google Scholar]
- 95.Dorschner RA, Pestonjamasp VK, Tamakuwala S, Ohtake T, Rudisill J, Nizet V, Agerberth B, Gudmundsson GH, Gallo RL. Cutaneous injury induces the release of cathelicidin anti-microbial peptides active against group A Streptococcus. J Invest Dermatol. 2001;117:91–97. doi: 10.1046/j.1523-1747.2001.01340.x. [DOI] [PubMed] [Google Scholar]
- 96.Tomasinsig L, Zanetti M. The cathelicidins–structure, function and evolution. Curr Protein Pept Sci. 2005;6:23–34. doi: 10.2174/1389203053027520. [DOI] [PubMed] [Google Scholar]
- 97.Bals R, Wang X, Zasloff M, Wilson JM. The peptide antibiotic LL-37/hCAP-18 is expressed in epithelia of the human lung where it has broad antimicrobial activity at the airway surface. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:9541–9546. doi: 10.1073/pnas.95.16.9541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Frohm M, Agerberth B, Ahangari G, Stahle-Backdahl M, Liden S, Wigzell H, Gudmundsson GH. The expression of the gene coding for the antibacterial peptide LL-37 is induced in human keratinocytes during inflammatory disorders. The Journal of biological chemistry. 1997;272:15258–15263. doi: 10.1074/jbc.272.24.15258. [DOI] [PubMed] [Google Scholar]
- 99.Menard S, Forster V, Lotz M, Gutle D, Duerr CU, Gallo RL, Henriques-Normark B, Putsep K, Andersson M, Glocker EO, Hornef MW. Developmental switch of intestinal antimicrobial peptide expression. J Exp Med. 2008;205:183–193. doi: 10.1084/jem.20071022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Chromek M, Slamova Z, Bergman P, Kovacs L, Podracka L, Ehren I, Hokfelt T, Gudmundsson GH, Gallo RL, Agerberth B, Brauner A. The antimicrobial peptide cathelicidin protects the urinary tract against invasive bacterial infection. Nat Med. 2006;12:636–641. doi: 10.1681/01.asn.0000926856.92699.53. [DOI] [PubMed] [Google Scholar]
- 101.Tjabringa GS, Aarbiou J, Ninaber DK, Drijfhout JW, Sorensen OE, Borregaard N, Rabe KF, Hiemstra PS. The antimicrobial peptide LL-37 activates innate immunity at the airway epithelial surface by transactivation of the epidermal growth factor receptor. J Immunol. 2003;171:6690–6696. doi: 10.4049/jimmunol.171.12.6690. [DOI] [PubMed] [Google Scholar]
- 102.Tjabringa GS, Rabe KF, Hiemstra PS. The human cathelicidin LL-37: a multifunctional peptide involved in infection and inflammation in the lung. Pulmonary pharmacology & therapeutics. 2005;18:321–327. doi: 10.1016/j.pupt.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 103.Kovach MA, Ballinger MN, Newstead MW, Zeng X, Bhan U, Yu FS, Moore BB, Gallo RL, Standiford TJ. Cathelicidin-related antimicrobial peptide is required for effective lung mucosal immunity in Gram-negative bacterial pneumonia. J Immunol. 2012;189:304–311. doi: 10.4049/jimmunol.1103196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Hase K, Murakami M, Iimura M, Cole SP, Horibe Y, Ohtake T, Obonyo M, Gallo RL, Eckmann L, Kagnoff MF. Expression of LL-37 by human gastric epithelial cells as a potential host defense mechanism against Helicobacter pylori. Gastroenterology. 2003;125:1613–1625. doi: 10.1053/j.gastro.2003.08.028. [DOI] [PubMed] [Google Scholar]
- 105.Hase K, Eckmann L, Leopard JD, Varki N, Kagnoff MF. Cell differentiation is a key determinant of cathelicidin LL-37/human cationic antimicrobial protein 18 expression by human colon epithelium. Infect Immun. 2002;70:953–963. doi: 10.1128/iai.70.2.953-963.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Frew L, Makieva S, McKinlay AT, McHugh BJ, Doust A, Norman JE, Davidson DJ, Stock SJ. Human cathelicidin production by the cervix. PLoS One. 2014;9:e103434. doi: 10.1371/journal.pone.0103434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Reinholz M, Ruzicka T, Schauber J. Cathelicidin LL-37: an antimicrobial peptide with a role in inflammatory skin disease. Ann Dermatol. 2012;24:126–135. doi: 10.5021/ad.2012.24.2.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Agerberth B, Charo J, Werr J, Olsson B, Idali F, Lindbom L, Kiessling R, Jornvall H, Wigzell H, Gudmundsson GH. The human antimicrobial and chemotactic peptides LL-37 and alpha-defensins are expressed by specific lymphocyte and monocyte populations. Blood. 2000;96:3086–3093. [PubMed] [Google Scholar]
- 109.Malm J, Sorensen O, Persson T, Frohm-Nilsson M, Johansson B, Bjartell A, Lilja H, Stahle-Backdahl M, Borregaard N, Egesten A. The human cationic antimicrobial protein (hCAP-18) is expressed in the epithelium of human epididymis, is present in seminal plasma at high concentrations, and is attached to spermatozoa. Infect Immun. 2000;68:4297–4302. doi: 10.1128/iai.68.7.4297-4302.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Murakami M, Ohtake T, Dorschner RA, Gallo RL. Cathelicidin antimicrobial peptides are expressed in salivary glands and saliva. J Dent Res. 2002;81:845–850. doi: 10.1177/154405910208101210. [DOI] [PubMed] [Google Scholar]
- 111.Sorensen O, Cowland JB, Askaa J, Borregaard N. An ELISA for hCAP-18, the cathelicidin present in human neutrophils and plasma. J Immunol Methods. 1997;206:53–59. doi: 10.1016/s0022-1759(97)00084-7. [DOI] [PubMed] [Google Scholar]
- 112.Gallo RL, Kim KJ, Bernfield M, Kozak CA, Zanetti M, Merluzzi L, Gennaro R. Identification of CRAMP, a cathelin-related antimicrobial peptide expressed in the embryonic and adult mouse. J Biol Chem. 1997;272:13088–13093. doi: 10.1074/jbc.272.20.13088. [DOI] [PubMed] [Google Scholar]
- 113.Gallo RL, Ono M, Povsic T, Page C, Eriksson E, Klagsbrun M, Bernfield M. Syndecans, cell surface heparan sulfate proteoglycans, are induced by a proline-rich antimicrobial peptide from wounds. Proc Natl Acad Sci U S A. 1994;91:11035–11039. doi: 10.1073/pnas.91.23.11035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Shi J, Ross CR, Chengappa MM, Blecha F. Identification of a proline-arginine-rich antibacterial peptide from neutrophils that is analogous to PR-39, an antibacterial peptide from the small intestine. Journal of leukocyte biology. 1994;56:807–811. doi: 10.1002/jlb.56.6.807. [DOI] [PubMed] [Google Scholar]
- 115.Koczulla R, von Degenfeld G, Kupatt C, Krotz F, Zahler S, Gloe T, Issbrucker K, Unterberger P, Zaiou M, Lebherz C, Karl A, Raake P, Pfosser A, Boekstegers P, Welsch U, Hiemstra PS, Vogelmeier C, Gallo RL, Clauss M, Bals R. An angiogenic role for the human peptide antibiotic LL-37/hCAP-18. The Journal of clinical investigation. 2003;111:1665–1672. doi: 10.1172/JCI17545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ren SX, Cheng AS, To KF, Tong JH, Li MS, Shen J, Wong CC, Zhang L, Chan RL, Wang XJ, Ng SS, Chiu LC, Marquez VE, Gallo RL, Chan FK, Yu J, Sung JJ, Wu WK, Cho CH. Host immune defense peptide LL-37 activates caspase-independent apoptosis and suppresses colon cancer. Cancer Res. 2012;72:6512–6523. doi: 10.1158/0008-5472.CAN-12-2359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Lim R, Lappas M, Riley C, Borregaard N, Moller HJ, Ahmed N, Rice GE. Investigation of human cationic antimicrobial protein-18 (hCAP-18), lactoferrin and CD163 as potential biomarkers for ovarian cancer. J Ovarian Res. 2013;6:5. doi: 10.1186/1757-2215-6-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Ren SX, Shen J, Cheng AS, Lu L, Chan RL, Li ZJ, Wang XJ, Wong CC, Zhang L, Ng SS, Chan FL, Chan FK, Yu J, Sung JJ, Wu WK, Cho CH. FK-16 derived from the anticancer peptide LL-37 induces caspase-independent apoptosis and autophagic cell death in colon cancer cells. PLoS One. 2013;8:e63641. doi: 10.1371/journal.pone.0063641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Kuroda K, Fukuda T, Isogai H, Okumura K, Krstic-Demonacos M, Isogai E. Antimicrobial peptide FF/CAP18 induces apoptotic cell death in HCT116 colon cancer cells via changes in the metabolic profile. Int J Oncol. 2015;46:1516–1526. doi: 10.3892/ijo.2015.2887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Cheng M, Ho S, Yoo JH, Tran DH, Bakirtzi K, Su B, Tran DH, Kubota Y, Ichikawa R, Koon HW. Cathelicidin suppresses colon cancer development by inhibition of cancer associated fibroblasts. Clin Exp Gastroenterol. 2015;8:13–29. doi: 10.2147/CEG.S70906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Bozinovski S, Vlahos R, Anthony D, McQualter J, Anderson G, Irving L, Steinfort D. COPD and squamous cell lung cancer: aberrant inflammation and immunity is the common link. Br J Pharmacol. 2016;173:635–648. doi: 10.1111/bph.13198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Leslie NR, Bennett D, Lindsay YE, Stewart H, Gray A, Downes CP. Redox regulation of PI 3-kinase signalling via inactivation of PTEN. EMBO J. 2003;22:5501–5510. doi: 10.1093/emboj/cdg513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Pastukh VM, Zhang L, Ruchko MV, Gorodnya O, Bardwell GC, Tuder RM, Gillespie MN. Oxidative DNA damage in lung tissue from patients with COPD is clustered in functionally significant sequences. Int J Chron Obstruct Pulmon Dis. 2011;6:209–217. doi: 10.2147/COPD.S15922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Bozinovski S, Uddin M, Vlahos R, Thompson M, McQualter JL, Merritt AS, Wark PA, Hutchinson A, Irving LB, Levy BD, Anderson GP. Serum amyloid A opposes lipoxin A(4) to mediate glucocorticoid refractory lung inflammation in chronic obstructive pulmonary disease. Proc Natl Acad Sci U S A. 2012;109:935–940. doi: 10.1073/pnas.1109382109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Anthony D, Seow HJ, Uddin M, Thompson M, Dousha L, Vlahos R, Irving LB, Levy BD, Anderson GP, Bozinovski S. Serum amyloid A promotes lung neutrophilia by increasing IL-17A levels in the mucosa and gammadelta T cells. Am J Respir Crit Care Med. 2013;188:179–186. doi: 10.1164/rccm.201211-2139OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Lee JM, Kim EK, Seo H, Jeon I, Chae MJ, Park YJ, Song B, Kim YS, Kim YJ, Ko HJ, Kang CY. Serum amyloid A3 exacerbates cancer by enhancing the suppressive capacity of myeloid-derived suppressor cells via TLR2-dependent STAT3 activation. Eur J Immunol. 2014;44:1672–1684. doi: 10.1002/eji.201343867. [DOI] [PubMed] [Google Scholar]
- 127.Hiratsuka S, Watanabe A, Sakurai Y, Akashi-Takamura S, Ishibashi S, Miyake K, Shibuya M, Akira S, Aburatani H, Maru Y. The S100A8-serum amyloid A3-TLR4 paracrine cascade establishes a pre-metastatic phase. Nat Cell Biol. 2008;10:1349–1355. doi: 10.1038/ncb1794. [DOI] [PubMed] [Google Scholar]
- 128.Cooray SN, Gobbetti T, Montero-Melendez T, McArthur S, Thompson D, Clark AJ, Flower RJ, Perretti M. Ligand-specific conformational change of the G-protein-coupled receptor ALX/FPR2 determines proresolving functional responses. Proc Natl Acad Sci U S A. 2013;110:18232–18237. doi: 10.1073/pnas.1308253110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature. 2008;454:436–444. doi: 10.1038/nature07205. [DOI] [PubMed] [Google Scholar]
- 130.Martinez FO, Helming L, Gordon S. Alternative activation of macrophages: an immunologic functional perspective. Annu Rev Immunol. 2009;27:451–483. doi: 10.1146/annurev.immunol.021908.132532. [DOI] [PubMed] [Google Scholar]
- 131.Liu Y, Cao X. The origin and function of tumor-associated macrophages. Cell Mol Immunol. 2015;12:1–4. doi: 10.1038/cmi.2014.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Gabrilovich DI, Bronte V, Chen SH, Colombo MP, Ochoa A, Ostrand-Rosenberg S, Schreiber H. The terminology issue for myeloid-derived suppressor cells. Cancer Res. 2007;67:425. doi: 10.1158/0008-5472.CAN-06-3037. author reply 426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.De Santo C, Arscott R, Booth S, Karydis I, Jones M, Asher R, Salio M, Middleton M, Cerundolo V. Invariant NKT cells modulate the suppressive activity of IL-10-secreting neutrophils differentiated with serum amyloid A. Nat Immunol. 2010;11:1039–1046. doi: 10.1038/ni.1942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Su SB, Gong W, Gao JL, Shen W, Murphy PM, Oppenheim JJ, Wang JM. A seven-transmembrane, G protein-coupled receptor, FPRL1, mediates the chemotactic activity of serum amyloid A for human phagocytic cells. J Exp Med. 1999;189:395–402. doi: 10.1084/jem.189.2.395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Mattarollo SR, Smyth MJ. A novel axis of innate immunity in cancer. Nat Immunol. 2010;11:981–982. doi: 10.1038/ni1110-981. [DOI] [PubMed] [Google Scholar]
- 136.Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest. 2012;122:787–795. doi: 10.1172/JCI59643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Hao NB, Lu MH, Fan YH, Cao YL, Zhang ZR, Yang SM. Macrophages in tumor microenvironments and the progression of tumors. Clin Dev Immunol. 2012;2012:948098. doi: 10.1155/2012/948098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Pollard JW. Tumour-educated macrophages promote tumour progression and metastasis. Nat Rev Cancer. 2004;4:71–78. doi: 10.1038/nrc1256. [DOI] [PubMed] [Google Scholar]
- 139.Chanmee T, Ontong P, Konno K, Itano N. Tumor-associated macrophages as major players in the tumor microenvironment. Cancers (Basel) 2014;6:1670–1690. doi: 10.3390/cancers6031670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Yadav A, Saini V, Arora S. MCP-1: chemoattractant with a role beyond immunity: a review. Clin Chim Acta. 2010;411:1570–1579. doi: 10.1016/j.cca.2010.07.006. [DOI] [PubMed] [Google Scholar]
- 141.Frossard JL, Lenglet S, Montecucco F, Steffens S, Galan K, Pelli G, Spahr L, Mach F, Hadengue A. Role of CCL-2, CCR-2 and CCR-4 in cerulein-induced acute pancreatitis and pancreatitis-associated lung injury. J Clin Pathol. 2011;64:387–393. doi: 10.1136/jcp.2010.088500. [DOI] [PubMed] [Google Scholar]
- 142.Liu Y, Chen K, Wang C, Gong W, Yoshimura T, Liu M, Wang JM. Cell surface receptor FPR2 promotes antitumor host defense by limiting M2 polarization of macrophages. Cancer Res. 2013;73:550–560. doi: 10.1158/0008-5472.CAN-12-2290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D, Bray F. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136:E359–386. doi: 10.1002/ijc.29210. [DOI] [PubMed] [Google Scholar]
- 144.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66:7–30. doi: 10.3322/caac.21332. [DOI] [PubMed] [Google Scholar]
- 145.Hou XL, Ji CD, Tang J, Wang YX, Xiang DF, Li HQ, Liu WW, Wang JX, Yan HZ, Wang Y, Zhang P, Cui YH, Wang JM, Bian XW, Liu W. FPR2 promotes invasion and metastasis of gastric cancer cells and predicts the prognosis of patients. Sci Rep. 2017;7:3153. doi: 10.1038/s41598-017-03368-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Cheng TY, Wu MS, Lin JT, Lin MT, Shun CT, Hua KT, Kuo ML. Formyl Peptide receptor 1 expression is associated with tumor progression and survival in gastric cancer. Anticancer Res. 2014;34:2223–2229. [PubMed] [Google Scholar]
- 147.Sena A, Grishina I, Thai A, Goulart L, Macal M, Fenton A, Li J, Prindiville T, Oliani SM, Dandekar S, Goulart L, Sankaran-Walters S. Dysregulation of anti-inflammatory annexin A1 expression in progressive Crohns Disease. PLoS One. 2013;8:e76969. doi: 10.1371/journal.pone.0076969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Babbin BA, Lee WY, Parkos CA, Winfree LM, Akyildiz A, Perretti M, Nusrat A. Annexin I regulates SKCO-15 cell invasion by signaling through formyl peptide receptors. J Biol Chem. 2006;281:19588–19599. doi: 10.1074/jbc.M513025200. [DOI] [PubMed] [Google Scholar]
- 149.Flower RJ, Rothwell NJ. Lipocortin-1: cellular mechanisms and clinical relevance. Trends Pharmacol Sci. 1994;15:71–76. doi: 10.1016/0165-6147(94)90281-x. [DOI] [PubMed] [Google Scholar]
- 150.Wu W, Tang X, Hu W, Lotan R, Hong WK, Mao L. Identification and validation of metastasis-associated proteins in head and neck cancer cell lines by two-dimensional electrophoresis and mass spectrometry. Clin Exp Metastasis. 2002;19:319–326. doi: 10.1023/a:1015515119300. [DOI] [PubMed] [Google Scholar]
- 151.Saito N, Qiao H, Yanagi T, Shinkuma S, Nishimura K, Suto A, Fujita Y, Suzuki S, Nomura T, Nakamura H, Nagao K, Obuse C, Shimizu H, Abe R. An annexin A1-FPR1 interaction contributes to necroptosis of keratinocytes in severe cutaneous adverse drug reactions. Sci Transl Med. 2014;6:245ra295. doi: 10.1126/scitranslmed.3008227. [DOI] [PubMed] [Google Scholar]
- 152.Jorge YC, Mataruco MM, Araujo LP, Rossi AF, de Oliveira JG, Valsechi MC, Caetano A, Miyazaki K, Fazzio CS, Thome JA, Rahal P, Oliani SM, Silva AE. Expression of annexin-A1 and galectin-1 anti-inflammatory proteins and mRNA in chronic gastritis and gastric cancer. Mediators Inflamm. 2013;2013:152860. doi: 10.1155/2013/152860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Ahn SH, Sawada H, Ro JY, Nicolson GL. Differential expression of annexin I in human mammary ductal epithelial cells in normal and benign and malignant breast tissues. Clin Exp Metastasis. 1997;15:151–156. doi: 10.1023/a:1018452810915. [DOI] [PubMed] [Google Scholar]
- 154.Masaki T, Tokuda M, Ohnishi M, Watanabe S, Fujimura T, Miyamoto K, Itano T, Matsui H, Arima K, Shirai M, Maeba T, Sogawa K, Konishi R, Taniguchi K, Hatanaka Y, Hatase O, Nishioka M. Enhanced expression of the protein kinase substrate annexin in human hepatocellular carcinoma. Hepatology. 1996;24:72–81. doi: 10.1053/jhep.1996.v24.pm0008707286. [DOI] [PubMed] [Google Scholar]
- 155.Bai XF, Ni XG, Zhao P, Liu SM, Wang HX, Guo B, Zhou LP, Liu F, Zhang JS, Wang K, Xie YQ, Shao YF, Zhao XH. Overexpression of annexin 1 in pancreatic cancer and its clinical significance. World J Gastroenterol. 2004;10:1466–1470. doi: 10.3748/wjg.v10.i10.1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Paweletz CP, Ornstein DK, Roth MJ, Bichsel VE, Gillespie JW, Calvert VS, Vocke CD, Hewitt SM, Duray PH, Herring J, Wang QH, Hu N, Linehan WM, Taylor PR, Liotta LA, Emmert-Buck MR, Petricoin EF., 3rd Loss of annexin 1 correlates with early onset of tumorigenesis in esophageal and prostate carcinoma. Cancer Res. 2000;60:6293–6297. [PubMed] [Google Scholar]
- 157.Xia SH, Hu LP, Hu H, Ying WT, Xu X, Cai Y, Han YL, Chen BS, Wei F, Qian XH, Cai YY, Shen Y, Wu M, Wang MR. Three isoforms of annexin I are preferentially expressed in normal esophageal epithelia but down-regulated in esophageal squamous cell carcinomas. Oncogene. 2002;21:6641–6648. doi: 10.1038/sj.onc.1205818. [DOI] [PubMed] [Google Scholar]
- 158.Kang JS, Calvo BF, Maygarden SJ, Caskey LS, Mohler JL, Ornstein DK. Dysregulation of annexin I protein expression in high-grade prostatic intraepithelial neoplasia and prostate cancer. Clin Cancer Res. 2002;8:117–123. [PubMed] [Google Scholar]
- 159.Hippo Y, Yashiro M, Ishii M, Taniguchi H, Tsutsumi S, Hirakawa K, Kodama T, Aburatani H. Differential gene expression profiles of scirrhous gastric cancer cells with high metastatic potential to peritoneum or lymph nodes. Cancer Res. 2001;61:889–895. [PubMed] [Google Scholar]
- 160.McDonald B, Pittman K, Menezes GB, Hirota SA, Slaba I, Waterhouse CC, Beck PL, Muruve DA, Kubes P. Intravascular danger signals guide neutrophils to sites of sterile inflammation. Science. 2010;330:362–366. doi: 10.1126/science.1195491. [DOI] [PubMed] [Google Scholar]
- 161.de Oliveira S, Rosowski EE, Huttenlocher A. Neutrophil migration in infection and wound repair: going forward in reverse. Nat Rev Immunol. 2016;16:378–391. doi: 10.1038/nri.2016.49. [DOI] [PMC free article] [PubMed] [Google Scholar]