

Abstract
Background
Cell therapy remains the most promising approach against ischemic heart injury. However, the poor survival of engrafted stem cells in the ischemic environment limits their therapeutic efficacy for cardiac repair post myocardial infarction (MI). C1q/tumor necrosis factor–related protein-9 (CTRP9) is a novel pro-survival cardiokine with significantly downregulated expression after MI. Here, we tested a hypothesis that CTRP9 might be a cardiokine required for a healthy microenvironment promoting implanted stem cell survival and cardioprotection.
Methods
Mice were subjected to MI and treated with adipose-derived mesenchymal stem cells (ADSCs, intramyocardial transplantation), CTRP9, or their combination. Survival, cardiac remodeling and function, cardiomyocytes apoptosis, and ADSCs engraftment were evaluated. Whether CTRP9 directly regulates ADSCs function was determined in vitro. Discovery-drive approaches followed by cause-effect analysis were employed to uncover the molecular mechanisms of CTRP9.
Results
Administration of ADSCs alone failed to exert significant cardioprotection. However, administration of ADSCs in addition to CTRP9 further enhanced the cardioprotective effect of CTRP9 (P<0.05 or P<0.01 vs. CTRP9 alone), suggesting a synergistic effect. Administration of CTRP9 at a dose recovering physiological CTRP9 levels significantly prolonged ADSCs retention/survival after implantation. Conversely, the number of engrafted ADSCs was significantly reduced in the CTRP9KO heart. In vitro study demonstrated that CTRP9 promoted ADSCs proliferation and migration, and protected ADSCs against hydrogen peroxide-induced cellular death. CTRP9 enhances ADSCs proliferation/migration by ERK1/2-MMP-9 signaling and promotes anti-apoptotic/cell survival via ERK-Nrf2/anti-oxidative protein expression. N-cadherin was identified as a novel CTRP9 receptor mediating ADSCs signaling. Blockade of either N-cadherin or ERK1/2 completely abolished the above noted CTRP9 effects. Although CTRP9 failed to promote ADSCs cardiogenic differentiation, CTRP9 promotes Sod-3 expression and secretion from ADSCs, protecting cardiomyocytes against oxidative stress-induced cell death.
Conclusion
We provide the first evidence that CTRP9 promotes ADSCs proliferation/survival, stimulates ADSCs migration, and attenuates cardiomyocyte cell death by previously unrecognized signaling mechanisms. These include binding with N-cadherin, activation of ERK/MMP-9 and ERK/Nrf2 signaling, and upregulation/secretion of anti-oxidative proteins. These results suggest that CTRP9 is a cardiokine critical in maintaining a healthy microenvironment facilitating stem cell engraftment in infarcted myocardial tissue, thereby enhancing stem cell therapeutic efficacy.
Keywords: Stem Cell, Ischemic Heart Injury, Cardiokines, N-cadherin
Introduction
Myocardial infarction results in compromised myocardial function and heart failure (HF), due to acute and chronic loss of cardiomyocytes1. Because adult mammalian hearts have very limited regenerative capacity, stem cell therapy is a potential strategy regenerating myocardium2. Multiple preclinical studies utilizing mesenchymal stem cells (MSCs) have been conducted, demonstrating favorable impact on left ventricle remodeling3–5. However, the favorable results observed in animal study have not been fully translated to patients with myocardial infarction (MI) and HF, due largely to the poor survival of engrafted MSCs in the ischemic environment6. Overcoming this limitation by either promoting MSCs survival or improving local environment may enhance the effectiveness of cell therapy for myocardial infarction7.
The cellular secretome influences the local microenvironment, controlling disease development. The secretomes produced by the heart, collectively known as ‘cardiokines’, are increasingly recognized as essential regulators of cardiac physiology and pathology. The C1q/TNF-related proteins (CTRPs) are a protein family consisting of fifteen (CTRP1-CTRP15) newly identified APN paralogs8. We have previously demonstrated that CTRP9 is not a typical adipokine, and actually functions as a cardiokine9. CTRP9 is highly expressed in the adult heart. The globular domain isoform of CTRP9 (gCTRP9) is generated via proteolytic cleavage of full length CTRP9 (fCTRP9) in cardiac tissue, and functions as the active cardioprotective isoform9, 10. A recent study confirmed our original finding and identified the heart as the organ expressing CTRP9 to the greatest degree, exceeding adipose tissue levels (where CTRP9 was originally discovered)11. Others and we have demonstrated that CTRP9 is a potent cardioprotective cardiokine; gCTRP9 supplementation protects the heart from ischemic injury, and attenuates adverse cardiac remodeling after ischemic myocardial infarction12–14. Moreover, several recent studies have demonstrated significantly reduced plasma CTRP9 levels, as well as myocardial CTRP9 (both mRNA and protein) expression levels, after MI12, 14. However, whether reduced cardiac CTRP9 expression may affect the microenvironment of the infarcted heart, adversely affecting the fate of engrafted stem cells, has never been studied.
Thus, the aims of this study were: 1) to determine whether cardiokine CTRP9 may regulate the microenvironment of infarcted heart, thus controlling the fate of engrafted adipose-derived mesenchymal stem cells (ADSCs) and their cardiac protective effect; and 2) if so, to determine the underlying mechanisms, including the specific receptor(s) and intracellular signaling pathways involved.
Methods
ADSCs isolation, identification, and in vitro studies
ADSCs were isolated from male EGFP TG mice (The Jackson Laboratory) or littermate C57BL/6J control mice as previously reported (Supplemental Figure 1A)15. Cells were cultured in DMEM-F12-10% FBS. Surface marker expression was evaluated by flow cytometry (BD LSRFortessa). For flow cytometry, 2×106 ADSCs were stained with the following fluorescent antibodies: CD105 (BD Biosciences), CD31 (BD Biosciences), CD90.2 (BD Biosciences), CD45 (BD Biosciences), and their PE isotype control. ADSCs multilineage potential was evaluated by adipogenic and osteogenic differentiation. ADSCs population viability was determined over time via Cell Counting Kit-8 (CCK-8) (Sigma). For migration assay, wound healing and transwell studies were performed. The conditioned medium (CM) of ADSCs was collected via a modified method16: Passage 2 ADSCs were grown to 90% confluence in 6-well dishes, and incubated for 24 hours with gCTRP9 (2.0μg/mL) or vehicle. The culture medium was washed and replaced by serum-free DMEM/F12 medium. After another 24 hours, CM was collected, and 1) used for cardiomyocyte treatment, or 2) stored at −80℃ for future use.
Animal study protocol
All experiments were performed in adherence to the National Institutes of Health Guidelines on the Use of Laboratory Animals and were approved by the Thomas Jefferson University Committee on Animal Care. Permanent myocardial infarction (MI) surgery was performed in adult male C57BL/6J mice and CTRP9 knock-out mice (provided by Dr. G. William Wong17) by ligating the left anterior descending coronary artery18. Immediately after MI, 1×105 EGFP-ADSCs suspended in 25 μL PBS (containing 0.2mM EDTA, pH=7.3) administered via intramyocardial injection to the infarct border zone at three different sites. 30 min after MI, globular CTRP9 (gCTRP9, 0.25 μg/g/d) or vehicle was administrated via peritoneal implant osmotic pumps for 2 weeks. The number of ADSCs engrafted to the heart 1, 3, 7, and 14 days after transplantation was determined via GFP immunostaining and quantitative PCR assessments of GFP DNA levels as previously reported6. Apoptosis was evaluated via terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) staining 3 days after MI. 4 weeks after MI, cardiac function was evaluated via echocardiography and LV catheterization. Fibrosis was evaluated by Masson’s trichrome staining.
Co-immunoprecipitation, Western blot analysis, immunohistochemistry, and quantitative PCR
For co-immunoprecipitation, ADSCs were treated with his-gCTRP9, his-fCTRP9, flag-fCTRP9, or vehicle. Cells were washed once with PBS, and lysed with cold 1×lysis buffer supplemented with a protease inhibitor cocktail. For in-tube assay, human recombinant CTRP9 (Novus) and human recombinant N-cadherin (LSBio) were mixed in cold 1×lysis buffer. The samples were immunoprecipitated with anti-CTRP9 antibody (provided by Dr. G. William Wong) or anti-flag M2 (Sigma) antibody.
For Western blot, total proteins were isolated from cells or heart tissues. Nuclear and cytoplasmic proteins were prepared by a Nuclear and Cytoplasmic Extraction Kit (Thermo Fisher Scientific). Protein samples (including samples immunoprecipitated with anti-CTRP9 antibody) were separated via gel electrophoresis, transferred to a poly-vinylidene fluoride membrane, and blocked with 5% milk for 1 hour. The membrane was incubated overnight with primary antibodies (4°C). The membranes were then incubated with secondary HRP-conjugated anti-mouse antibody or anti-rabbit antibody at room temperature for 2 hours, and exposed to enhanced chemiluminescent (ECL) substrate (Thermo Fisher Scientific). Western blot results were quantified by densitometry (Image Lab).
For immunohistochemistry, wax blocks were cut into 5 μm thick sections, and mounted on glass slides for staining. Slides were deparaffinized, and subjected to antigen retrieval in hot citric acid buffer. After cooling, slides were permeabilized with 0.2% Triton-100 for 15 minutes and were blocked with 1% BSA in PBS for 2 hours. Slides were incubated overnight with primary antibodies (4°C). Primary antibodies were visualized with secondary antibodies conjugated with Alexa Fluor 488 or Rhodamine (ThermoFisher scientific). Nuclei in both cells and embedded tissues were stained with 4′,6-diamidino-2-phenylindole (DAPI, Vector Laboratories, H-1200). Micrographs of all immunostains were acquired via Olympus BX51 Fluorescence Microscope (Japan) and Olympus DP72 camera (Japan). For qPCR, primers were purchased from Integrated DNA Technologies (listed in Supplemental Table 1) and qPCR was performed via 7900HT Fast Real-Time PCR System (Applied Biosystems).
Statistical Analysis
Data are reported as means±SEM. The Kaplan-Meier survival curves were analyzed by Gehan-Breslow-Wilcoxon test. For analysis of differences between two groups, unpaired student’s t test was performed. For multiple groups, one-way ANOVA was carried out followed by Bonferroni post-hoc test. For multiple groups over time, or for testing the interaction of CTRP9/ADSC therapy upon post-MI heart function, two-way ANOVA was performed followed by Bonferroni post-hoc test. P values <0.05 were considered significant. In this study, n is the number of animals or cell cultures tested.
Results
CTRP9 and ADSCs exerted synergistic protection against post-MI cardiac injury
The Passage 2 ADSCs were tested for surface marker expression and differentiation potential. They expressed mesenchymal stem cells markers, such as CD105 and CD90.2, but not hematopoietic lineage marker CD45 or endothelial cell marker CD31 (Supplemental Figures 1B and 1C). Their adipogenic and osteoblastic potential demonstrated their pluripotency (Supplemental Figure 1D). To determine whether MI-induced CTRP9 reduction may adversely impact the fate of ADSCs and their cardioprotective potential, we administered recombinant CTRP9 at a dose regiment fully restoring plasma and intracardiac CTRP9 to physiological levels (Supplemental Figure 2). 154 mice were subjected to 4 weeks of MI, and were randomized to one of the following groups: 1) MI+vehicle; 2) MI+gCTRP9 (0.25 μg/g/d via peritoneal implant osmotic pumps for 2 weeks); 3) MI+ADSC+vehicle (1×105 Passage 2 cells directly injected into the peri-infarct region immediately post-MI); or 4) MI+ADSC+gCTRP9. The degree of initial cardiac dysfunction after MI was similar in all groups 12 hours after MI (Supplemental Figures 3). An additional 12 mice were subjected to identical surgical procedures, except that no coronary artery ligature occurred (Sham MI group).
Of these 154 MI mice, 59 died at various time points during follow-up (Figure 1A). Administration of ADSC or gCTRP9 alone failed to significantly improve survival rate. However, combination of ADSC and gCTRP9 improved survival significantly greater than vehicle or ADSCs alone. Autopsy demonstrated that LV rupture was the main reason for post-MI death in this mouse MI model, consistent with our survival data. Combination gCTRP9/ADSC treatment resulted in a sharp reduction of LV rupture, although the difference was not statistically significant (Supplemental Figure 4A). No significant difference was observed in heart rate between vehicle-treated and the ADSC, gCTRP9, or combination ADSC/gCTRP9 treatment groups (Supplemental Figure 4B). However, gCTRP9/ADSC combination increased LVEF significantly greater than ADSCs or gCTRP9 treatment alone, and a positive interaction between gCTRP9 and ADSC was observed for this most important cardiac function measurement (p for interactions=0.0096) (Figures 1B and 1C). The gCTRP9/ADSC combination also increased ±dp/dt significantly greater than ADSCs or gCTRP9 treatment alone (Figures 1D–1F, Supplemental Figure 4C), although a statistically significant positive interaction was not reached (p for interactions>0.05). Taken together, these results provide clear evidence that ADSC/CTRP9 combination reduces rupture-related death, and significantly protects the ischemic heart against post-MI cardiac dysfunction in synergistic fashion.
Figure 1. CTRP9 and ADSCs synergistically improved cardiac functional recovery, and reduced cardiac fibrosis and cardiomyocyte loss after MI.
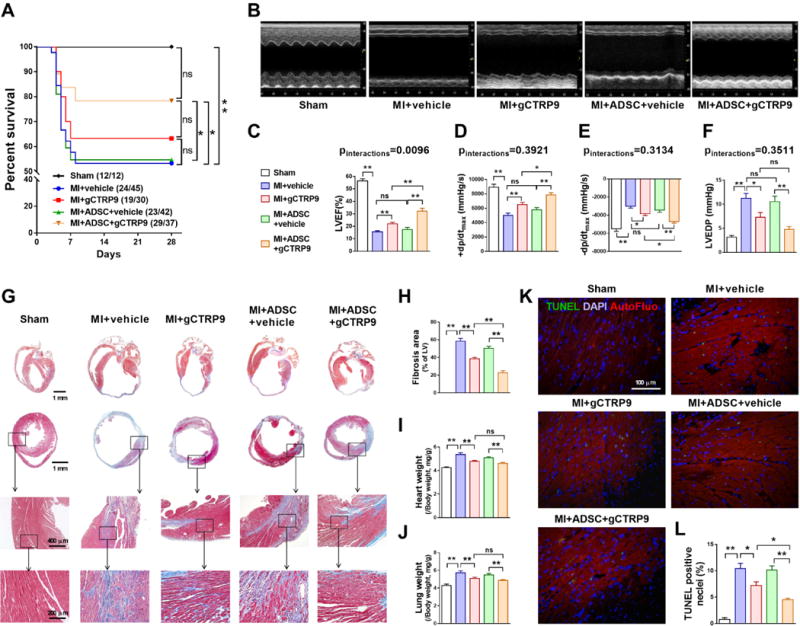
A. Survival curves. n=12–45. B. Representative echocardiographic images 4 weeks after MI. C. Left ventricular ejection fraction (LVEF). n=10–18. D–F. Hemodynamic analyses including +dp/dtmax, − dp/dtmax, and LV end diastolic pressure (LVEDP). n=10–19. G. Representative images of Masson’s trichrome staining of the transverse and coronal planes. H. Quantification of fibrotic area. n=6–9. I. Heart weight to body weight ratio (HW/BW). J. Lung weight to body weight ratio (LW/BW). n=10–18. K/L. Representative images and quantification of TUNEL positive cardiomyocytes in infarct border zone 3 days after MI. Cell nuclei were stained with 4′-6-diamidino-2-phenylindole (DAPI; blue). n=6. Data are mean ± SEM. *P<0.05, **P<0.01. NS, not significant. Abbreviation: AutoFluo, Autofluorescence. Survival curves were analyzed by Gehan-Breslow-Wilcoxon test. LVEF and ±dp/dt were analyzed with two-way ANOVA followed by Bonferroni post-hoc test. Other data were analyzed with one-way ANOVA followed by Bonferroni post-hoc test.
CTRP9 treatment atop ADSCs implantation significantly reduced fibrosis and cardiomyocyte apoptosis
Masson’s trichrome staining of the transverse and coronal plane revealed that the cardiac size and myocardial fibrotic area were slightly reduced by ADSCs transplantation, but were significantly reduced by gCTRP9 administration, compared to MI+vehicle (Figures 1G and 1H, Supplemental Figure 4D). Consistent with cardiac function data, gCTRP9/ADSCs combination reduced fibrotic area significantly greater than ADSCs or gCTRP9 treatment alone (p<0.01 vs. MI+ADSC+vehicle and MI+gCTRP9). Although a trend toward a better protection was observed, differences in heart weight (Figure 1I) and lung weight (Figure 1J) were not statistically significant between the gCTRP9 alone and gCTRP9+ADSC groups. TUNEL assay (infarct border zone 3 days after MI; Figures 1K and 1L) demonstrated that ADSCs did not significantly reduce cardiomyocyte apoptosis (p>0.05 vs. MI+vehicle). However, gCTRP9/ADSCs combination reduced cardiomyocyte apoptosis to an extent exceeding gCTRP9 alone (p<0.05) and markedly surpassing ADSCs treatment alone (p<0.01). Together, these data suggest that gCTRP9 and ADSCs combination treatment significantly attenuated post-MI fibrosis and apoptosis (the two most important pathological alterations during post-MI remodeling), contributing to their synergistic cardioprotection against ischemic cardiac injury.
Myocardial CTRP9 protein level is a critical factor determining ADSCs engraftment rate
To investigate the effects of myocardial CTRP9 upon engrafted ADSCs survival, we employed both gain-of-function and loss-of-function approaches. ADSCs retention 1, 3, 7, and 14 days after MI in CTRP9 knock-out (KO) mice and WT littermates treated with CTRP9 or vehicle was determined. Transplanted cells were tracked in myocardial tissues via immunofluorescence staining for GFP expression. Engraftment rate was calculated via quantitative PCR measurements of GFP DNA. Clusters of ADSCs were observed in the border zone of ischemia 1 day after transplantation (Figures 2A and 2B). Compared to WT, markedly fewer GFP+ cells were present in CTRP9KO hearts 3 days after MI. PCR results demonstrated that the engraftment/survival rate of ADSCs in the CTRP9KO group was significantly decreased compared to WT mice (Figure 2C). More importantly, gCTRP9 treatment significantly prolonged cell retention/survival in the infarct border zone (Figures 2A–2C). These results suggest that CTRP9 promotes the survival of implanted ADSCs and protects the ischemic heart, and that decreased myocardial CTRP9 contributes to low ADSCs engraftment in the infarcted heart.
Figure 2. CTRP9 increased ADSCs survival in peri-infarct area 1 and 3 days after MI.
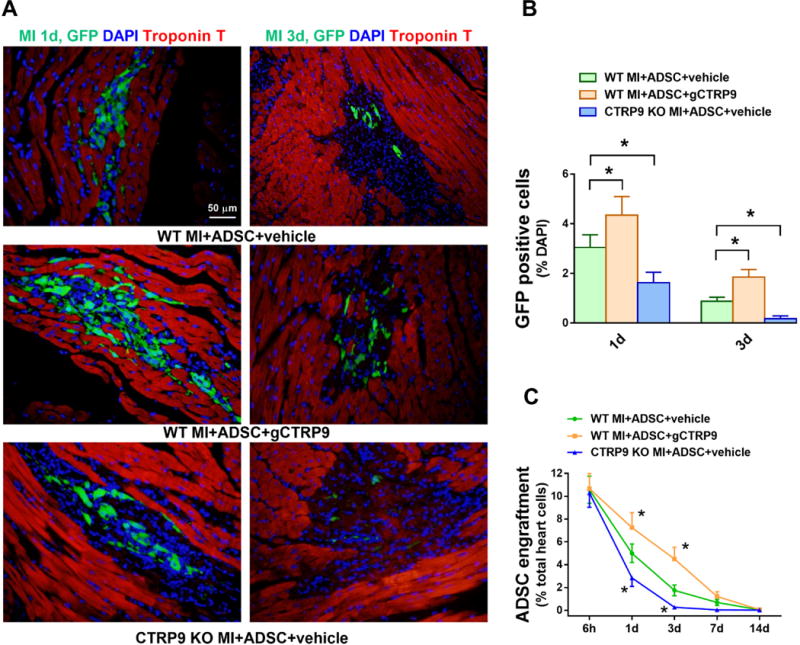
A. Representative images of EGFP-ADSCs in hearts 1 and 3 days after MI. Heart tissue was immunostained for GFP (green), DAPI (blue), and Troponin T (red). B. Quantification of EGFP-ADSCs in the peri-infarct area was determined by the number of GFP-positive cells per total nuclei. n=20 from 4 mice. C. ADSCs engraftment was quantified as the number of GFP-positive cells per 100 heart cells in apex region. n=4 mice. Data are mean ± SEM. *P<0.05 vs. WT MI+ADSC+vehicle. Statistical significance was determined with two-way ANOVA followed by Bonferroni post-hoc test.
CTRP9 promoted ADSCs proliferation, migration, and protected ADSCs from hydrogen peroxide-induced cell death
To determine whether CTRP9 directly or indirectly regulates ADSCs function, we examined the differentiation, proliferation, migration, and cell death of ADSCs treated with or without gCTRP9 in vitro. Real-time PCR demonstrated that CTRP9 failed to affect cardiogenic, vasculogenic, or fibrogenic differentiation of ADSCs after 7 days of co-culture with neonatal mouse ventricular cardiomyocytes (Supplemental Figure 5). However, the Cell Counting Kit-8 (CCK-8) assay demonstrated that, compared to vehicle, gCTRP9 increased ADSCs proliferation after 48 hours of treatment (Figure 3A). Wound healing and matrigel-coated-transwell assays demonstrated that ADSCs exhibited a markedly enhanced migratory capacity after 24 hours of gCTRP9 treatment (Figures 3B and 3C). Treatment of ADSCs with H2O2 increased LDH release, cleaved caspase-3 expression, cleaved caspase-3 activity, and TUNEL positive nuclei; gCTRP9 treatment significantly attenuated all these markers of apoptosis and necrosis (Figures 3D–3H). These in vitro results demonstrate that CTRP9 directly promotes ADSCs proliferation, migration, and survival.
Figure 3. CTRP9 promoted ADSCs proliferation, migration, and survival in vitro.
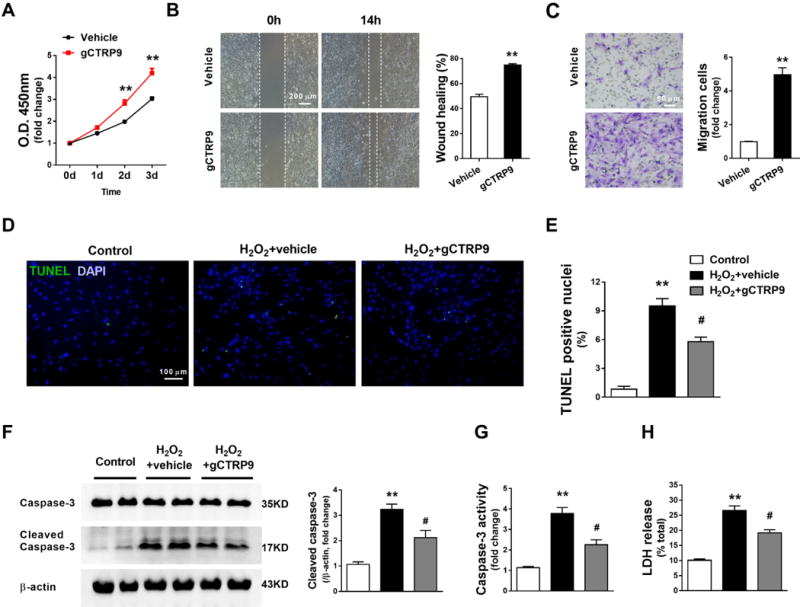
A. ADSCs proliferation curves. n=12. gCTRP9: 2μg/mL. **P<0.01 vs. Vehicle at indicated time. Statistical significance was determined with two-way ANOVA followed by Bonferroni post-hoc test. B/C. Wound healing assay and Matrigel pre-coated transwell assay were performed 24 hours after gCTRP9 (2μg/mL) or vehicle treatment. n=6. **P<0.01 vs. Vehicle. Statistical significance was determined with unpaired student’s t test. D–G. ADSCs apoptosis were determined by TUNEL staining, cleaved caspase-3 expression, and caspase-3 activity. n=6. H. ADSCs death was determined by LDH release. n=12. gCTRP9: 2μg/mL, 24 hours. H2O2: 200μM, 6 hours. Data are mean ± SEM. **P<0.01 vs. Control; #P<0.05 vs. H2O2+vehicle. Statistical significance was determined with one-way ANOVA followed by Bonferroni post-hoc test.
CTRP9 increased MMP-9 and anti-oxidant protein expression in ADSCs via ERK1/2 activation
To identify the molecular mechanisms underlying the effects of CTRP9 upon ADSCs proliferation, migration, and survival, we employed quantitative PCR to analyze mRNA levels in ADSCs with or without gCTRP9 treatment. 66 mRNAs involving proliferation, migration, death, paracrine function, angiogenesis, anti-oxidation, mitochondrial function, pluripotency, cardiogenic differentiation, and endothelial differentiation were tested. The gCTRP9-treated ADSCs profile exhibited significantly increased expression of genes controlling migration and anti-oxidation (Figure 4A). 7 genes were increased by gCTRP9 more than twofold (Figure 4B). Among them, Western blot analysis confirmed that gCTRP9 significantly increased protein expression of four anti-oxidants, including manganese superoxide dismutase 2 (MnSOD, Sod-2), extracellular superoxide dismutase 3 (EC-SOD, Sod-3), heme-oxygenase 1 (HO-1), and peroxiredoxin 1 (Prdx1) (Figures 4C and 4D). Moreover, both Western blot and In-gel zymography showed that CTRP9 significantly increased matrix metallopeptidase 9 (MMP-9) protein expression and activity in ADSCs (Figures 4C and 4D, Supplemental Figures 6A–6C).
Figure 4. CTRP9 increased MMP-9 and anti-oxidant protein expression in ADSCs via ERK1/2 activation.
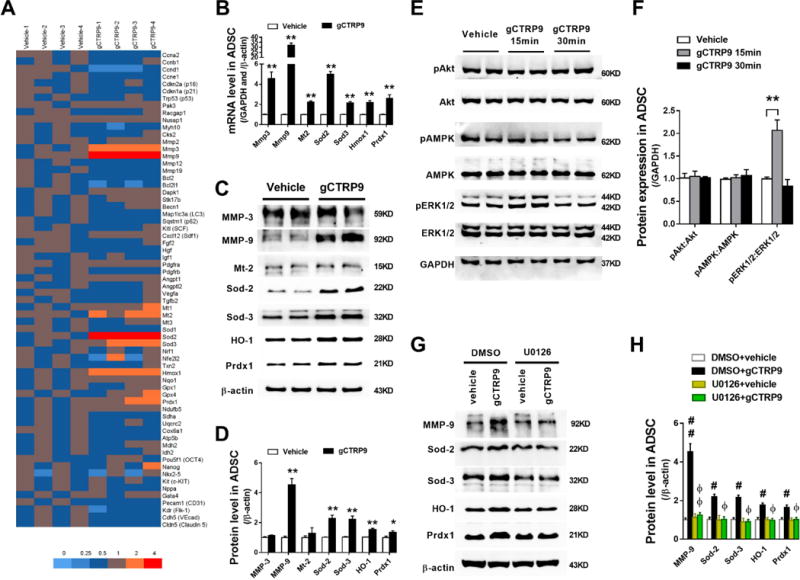
A. Gene expression profile of ADSCs treated with 2μg/mL gCTRP9 or vehicle for 24 hours, analyzed by qPCR. n=4. B. Statistical significance of mRNA expression for genes of interest. n=4–8. C/D. Western blots and quantification protein expression for genes of interest in ADSCs cell lysis. ADSCs were treated with 2μg/mL gCTRP9 or vehicle for 48 hours. n=6. Data are mean ± SEM. *P<0.05, **P<0.01 vs. Vehicle. E/F. Western blots and quantification demonstrate that ERK1/2, but not Akt or AMPK, was activated by 2μg/mL gCTRP9 for 15 minutes. n=4. Data are mean ± SEM. **P<0.01. G/H. Western blots and quantification of MMP-9, Sod-2, Sod-3, HO-1, and Prdx1 in ADSCs cell lysis demonstrate that U0126 (an ERK1/2 activation inhibitor) blocked gCTRP9-induced increase of MMP-9/antioxidant protein expression. U0126: 10μM, 2h before gCTRP9 treatment. n=4–5. Data are mean ± SEM. #P<0.05; ##P<0.01 vs. DMSO+vehicle; ΦP<0.05; ΦΦP<0.01 vs. DMSO+gCTRP9. Statistical significance of B and D was determined with unpaired student’s t test, other data were analyzed with one-way ANOVA followed by Bonferroni post-hoc test.
To identify the upstream signaling molecules leading to MMP-9 and anti-oxidant protein expression, the effect of CTRP9 upon several kinases known to regulate cellular proliferation and survival was determined. gCTRP9 treatment (15 minutes) significantly increased ERK1/2 phosphorylation (Figures 4E and 4F). However, signaling pathways (such as AMPK and Akt) activated by CTRP9 in other cell types were not activated in ADSCs (Figures 4E and 4F). To identify a cause-effect relationship between ERK1/2 activation and MMP-9/anti-oxidant protein expression after CTRP9 treatment, we administered U0126 (an ERK1/2 activation inhibitor, 10μM) 2 hours before gCTRP9 treatment. U0126 blocked gCTRP9-induced upregulation of phosphorylated ERK1/2 (Supplemental Figure 7). More importantly, gCTRP9-induced upregulation of MMP-9 and antioxidant proteins were completely blocked by U0126 (Figures 4G and 4H).
ERK1/2-MMP-9 and ERK1/2-Nrf2 signaling pathways are respectively responsible for CTRP9-induced ADSCs proliferation/migration and antioxidant/anti-apoptotic effects
MMP-9 is the most significantly upregulated protein among all proteins regulated by CTRP9 (>30 fold mRNA level, and >4 fold protein level). To determine whether the proliferation/migration and antioxidant/anti-apoptotic effects of CTRP9 are regulated by ERK1/2-MMP-9 signaling, ADSCs were treated with ERK1/2 activation inhibitor or MMP-9 blocking antibody before CTRP9 administration. Wound healing and matrigel-coated-transwell assays demonstrated both U0126 and the MMP-9 blocking antibody attenuated gCTRP9-enhanced migratory capacity (Figures 5A and 5B). The CCK-8 assay revealed both U0126 and MMP-9 blocking antibody inhibited gCTRP9-induced ADSCs proliferation (Figure 5C). Interestingly, the anti-apoptotic effect of CTRP9 was abolished by ERK1/2 activation inhibitor, but not by MMP-9 blocking antibody (Figures 5D and 5E).
Figure 5. ERK1/2-MMP-9 signaling is responsible for CTRP9-induced ADSCs proliferation and migration.
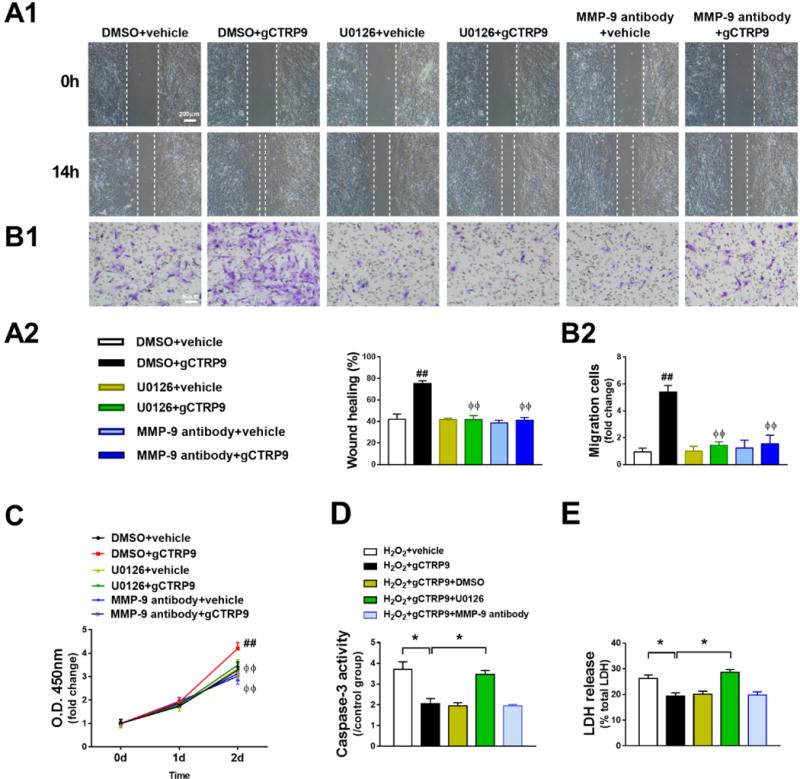
A–C. gCTRP9 promoted ADSCs migration (A1, A2, B1, and B2, n=4) and proliferation (C, n=12) were significantly inhibited by pretreatment with U0126 (10μM) or MMP-9 blocking antibody (5μg/mL, 2 hours before gCTRP9 treatment). Data are mean ± SEM. ##P<0.01 vs. DMSO+vehicle; ΦΦP<0.01 vs. DMSO+gCTRP9. D/E. ADSCs apoptosis and death were determined by caspase-3 activity (D, n=4) and LDH release (E, n=12). Data are mean ± SEM. *P<0.05. Statistical significance of C was determined with two-way ANOVA followed by Bonferroni post-hoc test, other data were analyzed with one-way ANOVA followed by Bonferroni post-hoc test.
Nrf2 (nuclear factor erythroid-derived 2-like 2) importantly defends against oxidative stress at the cellular level19. Expression of Sod-2, Sod-3, HO-1, and Prdx1 are regulated by Nrf220, 21. We therefore assessed nuclear Nrf2 levels to determine if the increase of these proteins resulted from stabilization of Nrf2 protein. As determined by immunostaining and protein expression in nuclear extracts, CTRP9 (2μg/ml for 24 hours) increased the nuclear Nrf2 level in ADSCs (Figures 6A and 6B). Nrf2 activation by CTRP9 was also completely abolished by ERK1/2 activation inhibitor (Figure 6C), suggesting Nrf2 is downstream of ERK1/2 in mediating the actions of CTRP9.
Figure 6. ERK1/2-Nrf2 signaling is responsible for CTRP9-induced antioxidant/anti-apoptotic effects.
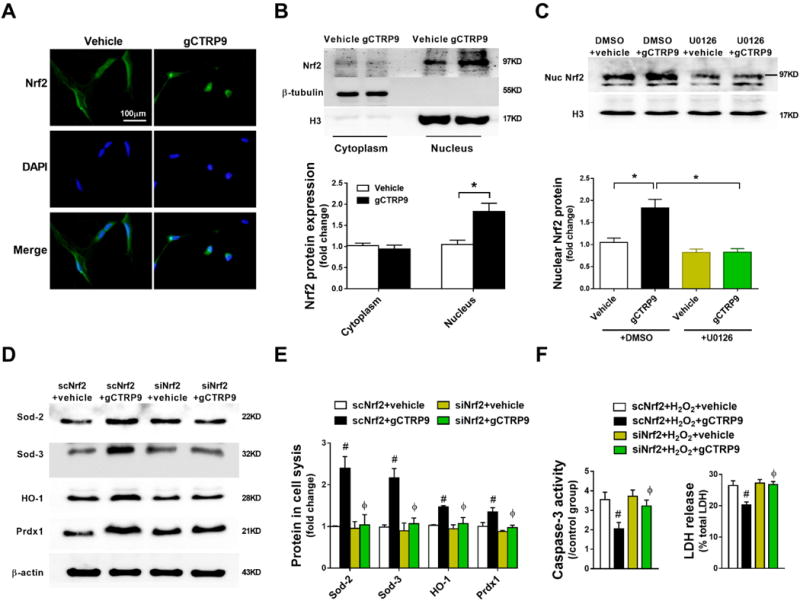
A. Representative images of Nrf2 immunostaining in ADSCs treated with vehicle or gCTRP9 (2μg/mL) for 24 hours. n=3. B. Western blots and quantification of cytoplasmic and nuclear Nrf2 protein in ADSCs demonstrate gCTRP9 (2μg/mL, 24 hours) significantly increased Nrf2 nuclear translocation in ADSCs. n=3. C. Western blots and quantification of nuclear Nrf2 protein in ADSCs. gCTRP9: 2μg/mL, 24 hours. U0126: 10μM, 2 hours before gCTRP9 treatment. n=4. Data are mean ± SEM.*P<0.05. D/E. Western blots and quantificationof Sod-2, Sod-3, HO-1, and Prdx1 in ADSCs cell lysis. n=4. Data are mean ± SEM. #P<0.05 vs. scNrf2+vehicle; ΦP<0.05 vs. scNrf2+gCTRP9. F. ADSCs apoptosis and death were determined by caspase-3 activity (left, n=4) and LDH release (right, n=12). Data are mean ± SEM. #P<0.05 vs. scNrf2+H2O2+vehicle; ΦP<0.05 vs. scNrf2+H2O2+gCTRP9. Abbreviations: scNrf2, Nrf2 scramble RNA; siNrf2, Nrf2 siRNA. Statistical significance of B was determined with unpaired student’s t test, other data were analyzed with one-way ANOVA followed by Bonferroni post-hoc test.
To establish a causative relationship between Nrf2 activation and anti-oxidant protein expression, Nrf2 expression was genetically inhibited by siRNA (Supplemental Figure 8). Nrf2 inhibition blocked CTRP9-induced anti-oxidant protein expression (Figures 6D and 6E). Moreover, Nrf2 knockdown virtually abolished the protective effects of CTRP9 against H2O2-induced ADSCs apoptosis and necrosis (Figure 6F). Collectively, these data demonstrate that the proliferation/migration effect of CTRP9 is mediated by ERK1/2-MMP-9 signaling, whereas the anti-apoptotic and cell survival effects of CTRP9 are mediated by ERK1/2-Nrf2 pathway activation.
N-cadherin is a novel CTRP9 receptor mediating CTRP9 effect in ADSCs
Several studies reported that adiponectin receptor 1 (AdipoR1) contributes to the cardioprotective and vasculoprotective actions of CTRP912, 14, 22. To analyze the involvement of AdipoR1 in the actions of CTRP9 upon ADSCs, we isolated ADSCs from the AdipoR1 knock-out (KO) mice. To our surprise, the effect of CTRP9 upon ADSCs proliferation, migration, cell survival, and antioxidant effect was fully intact in AdipoR1KO ADSCs (Supplemental Figure 9). Moreover, CTRP9 had no effect upon AMPK phosphorylation in ADSCs (Figures 4E and 4F), whereas the same CTRP9 concentration activates AMPK in an AdipoR1-dependent fashion in cardiomyocytes and endothelial cells12, 22. These results suggest a novel, unidentified receptor(s) exists in ADSCs mediating the effects of CTRP9.
In order to identify the CTRP9 receptor(s) of ADSCs, several discovery-driven approaches were undertaken. As the heart expresses the greatest levels of CTRP9, CTRP9-binding partner proteins in cardiac tissue samples were immunoprecipitated by anti-CTRP9 antibody. Irrelevant IgG immunoprecipitation served as negative control. After electrophoresis, silver staining was performed. Differential spots between irrelevant IgG and anti-CTRP9 antibody were analyzed by mass spectrometry (MS) (Supplemental Figure 10). MS analysis identified 8 membrane proteins co-immunoprecipitating with CTRP9 (Supplemental Table 2). As T-cadherin is a binding protein for adiponectin23, 24 and CTRP9 is a highly conserved adiponectin paralog, we thus reasoned that N-cadherin might be a potential CTRP9 receptor of ADSCs.
To obtain direct evidence supporting this notion, we performed several additional experiments. First, immunocytochemistry demonstrated co-localization of CTRP9 and N-cadherin in the cell membrane of CTRP9-treated ADSCs (Figure 7A). Second, ADSCs were treated with 5 μg/ml fCTRP9 or 2 μg/mL gCTRP9 for 30 minutes. Cells were harvested, homogenized, and samples were immunoprecipitated with antibody against CTRP9, followed by immunoblot with antibody against N-cadherin. A band representing N-cadherin is clearly visible in samples immunoprecipitated with anti-CTRP9 antibody (Figure 7B). Third, to eliminate the influence of endogenous CTRP9, ADSCs were treated with flag-fCTRP9, and samples were immunoprecipitated with anti-flag antibody. N-cadherin is co-immunoprecipitated with anti-flag antibody (Figure 7C). Fourth, to determine whether CTRP9 directly or indirectly interacts with N-cadherin, human recombinant CTRP9 (hrCTRP9) and human recombinant N-cadherin (hrN-cadherin) were incubated in a cell-free system. Samples were immunoprecipitated with antibody against CTRP9, and immunoblotted with antibody against N-cadherin. The band representing N-cadherin was clearly detected in CTRP9+N-cadherin sample (right lane), but not lysis buffer (left lane), immunoprecipitated with antibody against CTRP9 (Figure 7D). Finally, to clarify whether the CTRP9/N-cadherin interaction mediates CTRP9 biologic function in ADSCs, N-cadherin blocking peptide (Supplemental Figures 11A–11B) or N-cadherin siRNA (Supplemental Figure 11C) were utilized. In the presence of N-cadherin blocking peptide or N-cadherin siRNA, CTRP9 largely lost its ability to promote ADSCs proliferation/migration or inhibit H2O2-induced injury (Figures 7E–7G, Supplemental Figures 11D–11E). Similarly, the effects of CTRP9 upon ERK1/2 phosphorylation and its downstream molecule expression were abrogated by N-cadherin blocking peptide or N-cadherin siRNA (Supplemental Figure 12). More importantly, in the presence of gCTRP9 treatment, markedly less EGFP-ADSCs with N-cadherin knockdown were present 1 and 3 days after MI compared to EGFP-ADSCs with normal N-cadherin expression (Supplemental Figure 13).
Figure 7. N-cadherin is a novel CTRP9 receptor mediating CTRP9 effect in ADSCs.
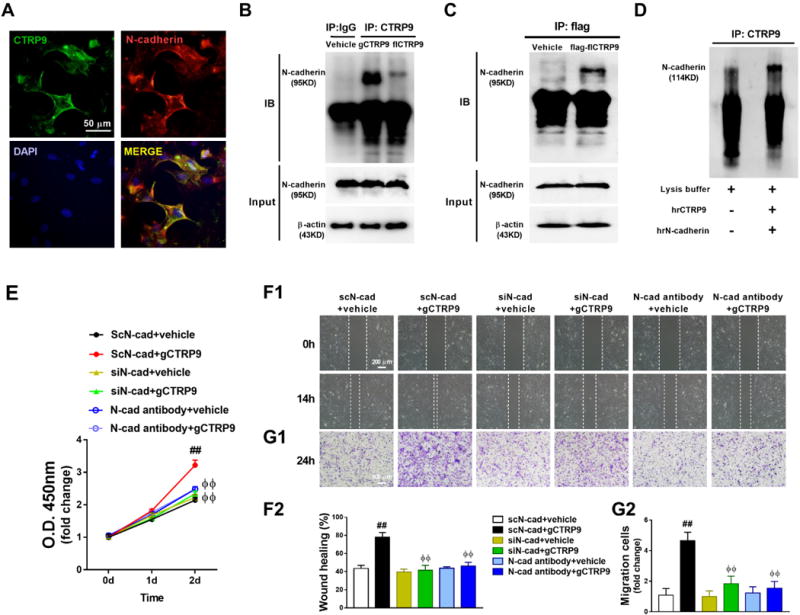
A. Co-immunofluorescent staining of CTRP9 (green) and N-cadherin (red) in ADSCs. ADSCs were treated with 2μg/mL gCTRP9 for 30 minutes before fixation. B. Co-immunoprecipitation (Co-IP) and immunoblot (IB) analysis of N-cadherin with his-gCTRP9 and his-fCTRP9 in ADSCs. ADSCs were treated with vehicle, his-gCTRP9 (2μg/mL), or his-fCTRP9 (5μg/mL) for 30 minutes before homogenization. C. Co-IP and IB analysis of N-cadherin with flag-fCTRP9 in ADSCs. ADSCs were treated with vehicle or flag-fCTRP9 (5μg/mL) for 30 minutes before homogenization. D. Co-IP and IB analysis of N-cadherin with CTRP9 in cell-free tubes. E–G. gCTRP9 promoted ADSCs proliferation (E, n=8) and migration (F1, F2, G1, and G2, n=4) were significantly inhibited by N-cadherin siRNA and N-cadherin blocking antibody. N-cadherin blocking antibody (5μg/mL) administered 2 hours before gCTRP9 treatment. Data are mean ± SEM. ##P<0.01 vs. scN-cad+vehicle; ΦΦP<0.01 vs. scN-cad+gCTRP9. Abbreviations: hrCTRP9, human recombinant CTRP9 (2μg); hrN-cadherin, human recombinant N-cadherin (2μg); scN-cad, N-cadherin scramble RNA; siN-cad, N-cadherin siRNA. Statistical significance of E was determined with two-way ANOVA followed by Bonferroni post-hoc test, other data were analyzed with one-way ANOVA followed by Bonferroni post-hoc test.
CTRP9/N-cadherin/ERK/Nrf2-dependent Sod-3 secretion is responsible for CTRP9-enhanced ADSCs-mediated cardiomyocyte protection
The above results demonstrate that CTRP9 promotes ADSCs survival via CTRP9/N-cadherin/ERK/Nrf2 signaling. However, CTRP9 fails to promote cardiogenic differentiation of ADSCs, and therefore cannot be responsible for the CTRP9/ADSCs synergistic cardioprotective effect. We performed in vitro experiments to determine whether gCTRP9 directly promotes ADSCs’ protective effects upon cardiomyocytes. Neonatal mouse ventricular cardiomyocytes (NMVCs) were treated with fresh medium (FM), conditioned medium of ADSCs pretreated with vehicle (veh-CM), or conditioned medium of ADSCs pretreated with gCTRP9 (gC-CM, gCTRP9-free, see Methods section for details). The effect of these different mediums upon 3-morpholinosydnonimine (SIN-1)-induced cardiomyocyte apoptosis was determined25. Treatment of NMVCs with 1 mM SIN-1 for 6 hours caused significant apoptosis, evidenced by increased TUNEL positive nuclei and cleaved caspase-3 levels (Figures 8A–8C). Compared to fresh medium, veh-CM moderately decreased TUNEL positive nuclei and cleaved caspase-3 expression in cardiomyocytes (p<0.05 vs. SIN-1+FM; Figures 8A–8C). Cardiomyocyte apoptosis was further decreased by gC-CM (p<0.05 or p<0.01 vs. SIN-1+veh-CM). These results suggest that gCTRP9 augments secretion of anti-apoptotic ADSCs paracrine factors protective of cardiomyocytes.
Figure 8. CTRP9/N-cadherin/ERK/Nrf2-dependent Sod-3 secretion is responsible for ADSCs-mediated cardiomyocyte protection.
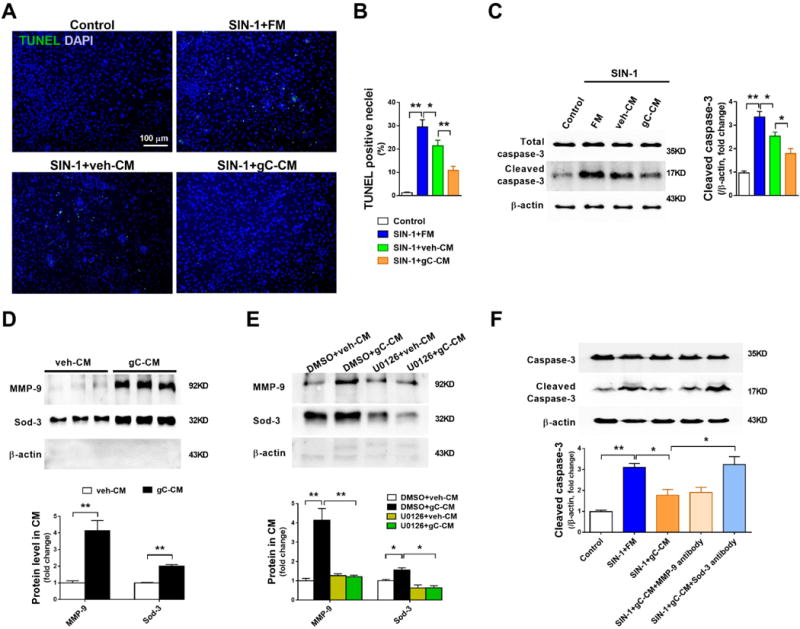
A/B. Representative images and quantification of TUNEL positive neonatal mouse ventricular cardiomyocytes (NMVCs) subjected to 1mM SIN-1 for 6 hours. n=6. C. Western blot analysis for cleaved caspase-3 in NMVCs. FM, veh-CM, or gC-CM was treated 15min before SIN-1 administration. D. Western blots and quantification of MMP-9 and Sod-3 protein expression in ADSCs conditioned medium (CM). n=4–5. E. Western blots and quantification of MMP-9 and Sod-3 in CM demonstrated that U0126, an ERK1/2 activation inhibitor, blocked gCTRP9-induced increased MMP-9 and Sod-3 secretion. U0126 (10μM) administered 2 hours before gCTRP9 treatment. n=4-5. F. NMVCs apoptosis was determined by cleaved caspase-3 expression. gC-CM was mixed with MMP-9 antibody (5μg/mL) or Sod-3 antibody (5μg/mL) 60min before gC-CM treatment. Data are mean ± SEM. *P<0.05; **P<0.01. Abbreviations: SIN-1: 3-morpholinosydnonimine, 1mM, 6h. FM: fresh medium. gC-CM: conditioned medium from gCTRP9 (2.0μg/ml) pre-treated ADSCs. MMP-9 antibody: MMP-9 blocking antibody. Sod-3 antibody: Sod-3 blocking antibody. n=4. Statistical significance of D was determined with unpaired student’s t test, other data were analyzed with one-way ANOVA followed by Bonferroni post-hoc test.
To determine the molecules present in conditioned medium from CTRP9-pretreated ADSCs responsible for cardiomyocyte protection, the levels of 5 molecules upregulated by CTRP9 were determined in conditioned medium. No significant difference in total protein concentration between veh-CM and gC-CM was observed (Supplemental Figure 14). Sod-2, HO-1, and Prdx1 were undetectable in either veh-CM or gC-CM. However, compared to veh-CM, MMP-9, and Sod-3 were significantly increased >4-fold and >2-fold in gC-CM (Figure 8D, Supplemental Figures 6D–6F). Furthermore, ERK1/2 inhibition with U0126, as well as N-cadherin knockdown or N-cadherin blocking peptide, completely blocked gCTRP9-induced extracellular secretion of MMP-9 and Sod-3 (Figure 8E, Supplemental Figure 15).
To determine the involvement of MMP-9, Sod-3, or both in ADSC-mediated cardiomyocyte protection, the effect of MMP-9 blocking antibody or Sod-3 blocking antibody pre-treatment upon gC-CM-mediated cardiomyocyte protection was determined. Pre-treatment with Sod-3 blocking antibody, but not MMP-9 blocking antibody, abolished the protective effect of gC-CM against SIN-1-induced cardiomyocyte apoptosis (Figure 8F). We also compared Sod-3 level in peri-infarcted heart tissue 1 day after MI (a time point where significant amount of injected ADSCs remain visible). There were no differences among sham, MI+vehicle, or MI+gCTRP9 groups. Compared to MI+vehicle, Sod-3 was slightly increased by ADSC (p>0.05). Importantly, ADSC+gCTRP9 further increased Sod-3 protein expression compared to ADSC alone (p<0.05, Supplemental Figure 4E). Collectively, these data suggest that CTRP9 stimulates Sod-3 secretion by ADSCs via N-cadherin/ERK/Nrf2 signaling, protecting cardiomyocytes from oxidative stress-induced cell death.
CTRP9 differentially regulates MMP-9 expression in ADSCs and cardiomyocytes, and inhibits leukocyte accumulation in peri-infarct region
MMP-9 overexpression in the post-MI heart promotes cardiac rupture, and is thereby deleterious26. However, our in vitro experiment demonstrated that gCTRP9 significantly increases MMP-9 expression/activation in ADSCs, but reduced cardiac rupture and protected the heart when administered in vivo. To clarify the underlying mechanism(s) for these seemingly contradictory results, three additional experiments were performed. First, the effect of in vivo administration of gCTRP9, ADSCs, and their combination upon MMP-9 expression in the peri-infarct area was determined. Consistent with previous reported results, MMP-9 was significantly increased (>10 fold) 1 day after MI. ADSC treatment did not significantly alter MMP-9 levels. However, treatment with either gCTRP9 alone or its combination with ADSCs significantly decreased MMP-9 levels in the peri-infarct area (Supplemental Figure 4E). Second, the effect of CTRP9 upon MMP-9 expression levels in cultured neonatal mouse ventricular cardiomyocytes was determined. Both gCTRP9 and fCTRP9 significantly decreased MMP-9 levels in cardiomyocytes (Supplemental Figure 4F), an effect opposite that observed in ADSCs. As cardiomyocytes are the predominant cell type in the heart, the inhibitory effect of gCTRP9 upon cardiomyocyte MMP-9 expression masked the stimulatory effect of ADSCs upon MMP-9 expression. A net inhibitory effect was observed. Third, the effect of gCTRP9, ADSCs, and their combination upon leukocyte accumulation in the peri-infarct region was determined. As illustrated in Supplemental Figure 16 (n=5 each group), treatment with ADSCs alone slightly reduced CD45+ positive leukocyte accumulation. Administration of gCTRP9 alone significantly reduced, and gCTRP9/ADSC combination further decreased CD45+ leukocyte infiltration in the post-MI heart. As inflammatory cells are the main/major source of MMPs27, reduction of leukocyte accumulation by gCTRP9 and gCTRP9/ADSC will reduce MMPs activity in the peri-infarct region. Taken together, these results suggest that the MMP-9 stimulatory effect of CTRP9 upon ADSCs may promote ADSC migration and proliferation. However, CTRP9’s inhibitory effect upon cardiomyocyte MMP-9 expression, in combination with its strong anti-inflammatory actions, results in an overall MMPs level reduction in the peri-infarct region, reducing cardiac rupture and protecting the heart.
Discussion
Stem cell therapy for the repair of damaged myocardium has evolved into a promising treatment for ischemic heart disease. MSC-based therapy, originating from BM-, adipose tissue-, or umbilical cord-cells, continues to gain consent and appeal, because of the large body of preclinical evidence supporting higher paracrine cardioreparative potential4. However, poor survival of donated stem cells and failure of stem cell engraftment in the damaged organ occurs within the first days after delivery, posing a significant challenge in the field1. A complete understanding of the mechanisms enhancing MSC migration and survival in the microenvironment of the injured myocardium is imperative, to improve both MSC repair capacity and therapeutic application.
In the present study, we have made several novel observations. First, we demonstrate that CTRP9 is a critical molecule determining the fate of implanted ADSCs. Specifically, we demonstrated that local CTRP9 levels are markedly reduced post-MI. CTRP9 supplementation at a dose recovering local CTRP9 level markedly improves ADSC engraftment, promotes their migration in the ischemic border zone, and facilitates ADSC-mediated cardiac repair after MI. In contrast, CTRP9 knock-out restricted migration of implanted ADSCs, and further accelerated their death. These results indicate that CTRP9 is an important molecule creating a healthy environment for ADSC survival and enhanced ADSC cardioreparative potential.
Second, we have uncovered new cellular and molecular mechanisms directly responsible for CTRP9 effect upon ADSCs. We identified that proliferation/pro-migration effects of gCTRP9 upon ADSCs were mediated by CTRP9-ERK1/2-MMP-9 signaling, as these effects were abolished when ERK1/2 or MMP-9 were inhibited genetically or pharmacologically. MMP-9 has relation to stem cell and cancer cell proliferation and migration28, 29. Promoting MMP-9 expression by gCTRP9 in ADSCs may thus facilitate their migration when implanted in infarcted heart tissue. Moreover, we provided direct evidence that the anti-apoptotic/cell survival effect of gCTRP9 upon ADSCs were mediated by the CTRP9-ERK1/2-Nrf2-anti-oxidative protein expression pathway. Four antioxidants downstream of the redox-sensitive transcription factor Nrf220, 21, 30 (including Sod-2, Sod-3, HO-1, and Prdx1) were significantly increased by gCTRP9 in ADSCs. ERK1/2 or Nrf2 knockdown blocked gCTRP9-induced anti-oxidant protein expression and abolished its anti-apoptotic/cell survival effect. These results further support our hypothesis that CTRP9 creates a microenvironment suitable for ADSC survival and migration.
Third, we have identified a novel receptor mediating the actions of CTRP9 upon ADSCs. Previous results demonstrated that CTRP9 exerts cardioprotective and vasculoprotective effects at least partially via AdipoR112–14, 22. However, we demonstrated that CTRP9 activates ERK1/2 and its downstream signaling in ADSCs in an AdipoR1-independent fashion. Employing several techniques, we successfully identified N-cadherin (also known as Cadherin-2, neural cadherin, or CD325) as the specific receptor for CTRP9 acting upon ADSCs. N-cadherin is a member of the cell surface glycoproteins mediating calcium-dependent adhesion31. Beyond its function in cell-cell adhesion, N-cadherin plays roles in differentiation, migration, invasion, and signal transduction32, 33. A cell surface marker of mesenchymal stem cells (MSCs), N-cadherin’s downstream signaling in MSCs is largely unknown. In the present study, we revealed CTRP9 co-localizes and directly interacts with N-cadherin in ADSCs. In agreement with previous reports that ERK1/2 activation is increased when N-cadherin is overexpressed in human umbilical cord blood-derived MSCs and mouse tumor cells33, 34, our results demonstrate that CTRP9 activation of ERK1/2-MMP-9 and ERK1/2-Nrf2-antioxidant signaling were abolished by N-cadherin siRNA and blocking antibody.
Finally, we have identified the molecule secreted by CTRP9-stimulated ADSCs that promotes cardiomyocyte survival. Stem cell transplantation for the treatment of cardiac disease is predicated on the hypothesis such cells engraft, differentiate, and replace damaged cardiac tissue, or secrete factors reducing tissue injury and/or enhancing tissue repair35. Among many characteristics, direct trans-differentiation and paracrine effector release are the two most important mechanisms by which implanted stem cells protect heart36. We provide no evidence in the present study that CTRP9 manipulates cardiogenic, vasculogenic, or fibrogenic differentiation of ADSCs (Supplemental Figure 5). This suggests that CTRP9 does not protect via direct trans-differentiation. However, the conditioned medium from CTRP9-pretreated ADSCs exerted potent protection against SIN-1-induced cardiomyocytes apoptosis, suggesting CTRP9 induced release of anti-apoptotic factors from ADSCs. Analysis of MMP-9 and the 4 antioxidants mentioned above (Sod-2, Sod-3, HO-1, and Prdx1) revealed CTRP9 significantly increased MMP-9 and Sod-3 in the conditioned medium of ADSCs, which was abolished by ERK1/2 activation inhibitor and N-cadherin knockdown/blocking antibody. However, the protective properties of conditioned medium from CTRP9-pretreated ADSCs upon cardiomyocytes were abolished by Sod-3 neutralizing antibody, but not by MMP-9 neutralizing antibody. These results demonstrate that CTRP9/N-cadherin/ERK/Nrf2-dependent Sod-3 secretion is responsible for CTRP9-enhanced ADSCs-mediated cardiomyocyte protection. It is worth noting that although ADSCs may not be the best cell type conducive for cardiac regeneration, ADSCs carry prominent advantage as a rich cellular source with prominent clinical applications.
In summary, we identify CTRP9 as a novel cardiokine regulating MSC engraftment in the infarcted heart via three major mechanisms. First, CTRP9 promotes ADSC survival by N-cadherin/ERK/Nrf2 signaling and subsequent upregulation of multiple anti-oxidative proteins. Second, CTRP9 promotes ADSC proliferation/migration via N-cadherin/ERK signaling and subsequent MMP-9 upregulation. Finally, CTRP9 stimulates ADSC Sod-3 production and secretion, protecting cardiomyocytes against oxidative stress-induced cell death. These results suggest that molecular interventions preserving CTRP9 production or exogenous CTRP9 supplementation may create a healthy microenvironment promoting stem cell survival and optimizing their cardioprotective effect against ischemic cardiac injury.
Limitations
Three limitations exist in the current study. First, cardiac dysfunction was determined by echocardiography at the end of observation period upon surviving animals. Serial echocardiography may have provided more information regarding CTRP9-mediated cardioprotection during the post-MI remodeling period. Second, we utilized short-axis M-mode echocardiography to analyze post-MI cardiac function, instead of longitudinal echocardiographic analysis, which is preferred over M-mode for MI models. Third, although we observed that gCTRP9+ADSC treatment caused a sharp reduction of LV rupture, the translational value of anti-rupture effect of gCTRP9+ADSC is somewhat limited, as LV rupture is not a frequent complication of MI in modern cardiology. However, we provided clear data demonstrating that gCTRP9+ADSC exerted synergistic protection against post-MI cardiac dysfunction, an effect that is of great clinical significant.
Supplementary Material
Clinical Perspective.
What is new?
This study demonstrates for the first time that CTRP9 is an important cardiokine governing the local environment in infarcted heart that determines the fate of implanted cells.
This study provides the first evidence that N-cadherin is the receptor mediating CTRP9-initiated signaling in ADSCs, filling a significant gap in the field concerning this important cardiokine.
ERK1/2-MMP9 and ERK1/2-Nrf2 signaling are respectively responsible for CTRP9-induced ADSC proliferation/migration and antioxidant/anti-apoptotic effects.
What are the clinical implications?
Strategies improving the local microenvironment in the ischemic heart may have great potential to enhance cell therapy efficacy against ischemic heart injury.
The combination therapy of CTRP9 with MSCs may be a novel strategy protecting against post-MI cardiac injury.
Targeting CTRP9/N-cadherin signaling in MSCs may improve the efficacy of cell therapy.
Acknowledgments
The authors thank Mr. Nadan Wang in the Center for Translational Medicine, Thomas Jefferson University, for his expertise in evaluating cardiac function by echocardiography. The authors thank Dr. Hsin-Yao Tang at the Wistar Institute Proteomics and Metabolomics Facility for his assistance in analyzing proteomics data.
Sources of Funding
This research was supported by National Institutes of Health grants HL-096686 and HL-123404, American Diabetes Association grant 1-15-BS-122 (XLM), and American Diabetes Association grant 1-14-BS-228 (YW).
Footnotes
Disclosures
None.
References
- 1.Mohsin S, Troupes CD, Starosta T, Sharp TE, Agra EJ, Smith S, Duran JM, Zalavadia N, Zhou Y, Kubo H, Berretta RM, Houser SR. Unique Features of Cortical Bone Stem Cells Associated With Repair of the Injured Heart. Circ Res. 2015;117:1024–1033. doi: 10.1161/CIRCRESAHA.115.307362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Golpanian S, Wolf A, Hatzistergos KE, Hare JM. Rebuilding the Damaged Heart: Mesenchymal Stem Cells, Cell-Based Therapy, and Engineered Heart Tissue. Physiol Rev. 2016;96:1127–1168. doi: 10.1152/physrev.00019.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hatzistergos KE, Saur D, Seidler B, Balkan W, Breton M, Valasaki K, Takeuchi LM, Landin AM, Khan A, Hare JM. Stimulatory Effects of Mesenchymal Stem Cells on cKit+ Cardiac Stem Cells Are Mediated by SDF1/CXCR4 and SCF/cKit Signaling Pathways. Circ Res. 2016;119:921–930. doi: 10.1161/CIRCRESAHA.116.309281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Broughton KM, Sussman MA. Empowering Adult Stem Cells for Myocardial Regeneration V2.0: Success in Small Steps. Circ Res. 2016;118:867–880. doi: 10.1161/CIRCRESAHA.115.305227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sanina C, Hare JM. Mesenchymal Stem Cells as a Biological Drug for Heart Disease: Where Are We With Cardiac Cell-Based Therapy? Circ Res. 2015;117:229–233. doi: 10.1161/CIRCRESAHA.117.306306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hu X, Xu Y, Zhong Z, Wu Y, Zhao J, Wang Y, Cheng H, Kong M, Zhang F, Chen Q, Sun J, Li Q, Jin J, Li Q, Chen L, Wang C, Zhan H, Fan Y, Yang Q, Yu L, Wu R, Liang J, Zhu J, Wang Y, Jin Y, Lin Y, Yang F, Jia L, Zhu W, Chen J, Yu H, Zhang J, Wang J. A Large-Scale Investigation of Hypoxia-Preconditioned Allogeneic Mesenchymal Stem Cells for Myocardial Repair in Nonhuman Primates: Paracrine Activity Without Remuscularization. Circ Res. 2016;118:970–983. doi: 10.1161/CIRCRESAHA.115.307516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Taylor DA, Chandler AM, Gobin AS, Sampaio LC. Maximizing Cardiac Repair: Should We Focus on the Cells or on the Matrix? Circ Res. 2017;120:30–32. doi: 10.1161/CIRCRESAHA.116.309959. [DOI] [PubMed] [Google Scholar]
- 8.Ouchi N, Walsh K. Cardiovascular and metabolic regulation by the adiponectin/C1q/tumor necrosis factor-related protein family of proteins. Circulation. 2012;125:3066–3068. doi: 10.1161/CIRCULATIONAHA.112.114181. [DOI] [PubMed] [Google Scholar]
- 9.Yuan Y, Lau WB, Su H, Sun Y, Yi W, Du Y, Christopher T, Lopez B, Wang Y, Ma XL. C1q-TNF-related protein-9, a novel cardioprotetcive cardiokine, requires proteolytic cleavage to generate a biologically active globular domain isoform. Am J Physiol Endocrinol Metab. 2015;308:E891–898. doi: 10.1152/ajpendo.00450.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Su H, Yuan Y, Wang XM, Lau WB, Wang Y, Wang X, Gao E, Koch WJ, Ma XL. Inhibition of CTRP9, a novel and cardiac-abundantly expressed cell survival molecule, by TNFalpha-initiated oxidative signaling contributes to exacerbated cardiac injury in diabetic mice. Basic Res Cardiol. 2013;108:315. doi: 10.1007/s00395-012-0315-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Appari M, Breitbart A, Brandes F, Szaroszyk M, Froese N, Korf-Klingebiel M, Mohammadi MM, Grund A, Scharf GM, Wang H, Zwadlo C, Fraccarollo D, Schrameck U, Nemer M, Wong GW, Katus HA, Wollert KC, Muller OJ, Bauersachs J, Heineke J. C1q-TNF-Related Protein-9 Promotes Cardiac Hypertrophy and Failure. Circ Res. 2017;120:66–77. doi: 10.1161/CIRCRESAHA.116.309398. [DOI] [PubMed] [Google Scholar]
- 12.Kambara T, Shibata R, Ohashi K, Matsuo K, Hiramatsu-Ito M, Enomoto T, Yuasa D, Ito M, Hayakawa S, Ogawa H, Aprahamian T, Walsh K, Murohara T, Ouchi N. C1q/Tumor Necrosis Factor-Related Protein 9 Protects against Acute Myocardial Injury through an Adiponectin Receptor I-AMPK-Dependent Mechanism. Mol Cell Biol. 2015;35:2173–2185. doi: 10.1128/MCB.01518-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sun Y, Yi W, Yuan Y, Lau WB, Yi D, Wang X, Wang Y, Su H, Wang X, Gao E, Koch WJ, Ma XL. C1q/tumor necrosis factor-related protein-9, a novel adipocyte-derived cytokine, attenuates adverse remodeling in the ischemic mouse heart via protein kinase A activation. Circulation. 2013;128:S113–120. doi: 10.1161/CIRCULATIONAHA.112.000010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kambara T, Ohashi K, Shibata R, Ogura Y, Maruyama S, Enomoto T, Uemura Y, Shimizu Y, Yuasa D, Matsuo K, Miyabe M, Kataoka Y, Murohara T, Ouchi N. CTRP9 protein protects against myocardial injury following ischemia-reperfusion through AMP-activated protein kinase (AMPK)-dependent mechanism. J Biol Chem. 2012;287:18965–18973. doi: 10.1074/jbc.M112.357939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Takahashi M, Suzuki E, Oba S, Nishimatsu H, Kimura K, Nagano T, Nagai R, Hirata Y. Adipose tissue-derived stem cells inhibit neointimal formation in a paracrine fashion in rat femoral artery. Am J Physiol Heart Circ Physiol. 2010;298:H415–423. doi: 10.1152/ajpheart.00391.2009. [DOI] [PubMed] [Google Scholar]
- 16.Wang WE, Yang D, Li L, Wang W, Peng Y, Chen C, Chen P, Xia X, Wang H, Jiang J, Liao Q, Li Y, Xie G, Huang H, Guo Y, Ye L, Duan DD, Chen X, Houser SR, Zeng C. Prolyl hydroxylase domain protein 2 silencing enhances the survival and paracrine function of transplanted adipose-derived stem cells in infarcted myocardium. Circ Res. 2013;113:288–300. doi: 10.1161/CIRCRESAHA.113.300929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wei Z, Lei X, Petersen PS, Aja S, Wong GW. Targeted deletion of C1q/TNF-related protein 9 increases food intake, decreases insulin sensitivity, and promotes hepatic steatosis in mice. Am J Physiol Endocrinol Metab. 2014;306:E779–790. doi: 10.1152/ajpendo.00593.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gao E, Lei YH, Shang X, Huang ZM, Zuo L, Boucher M, Fan Q, Chuprun JK, Ma XL, Koch WJ. A novel and efficient model of coronary artery ligation and myocardial infarction in the mouse. Circ Res. 2010;107:1445–1453. doi: 10.1161/CIRCRESAHA.110.223925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yoon DS, Choi Y, Lee JW. Cellular localization of NRF2 determines the self-renewal and osteogenic differentiation potential of human MSCs via the P53-SIRT1 axis. Cell Death Dis. 2016;7:e2093. doi: 10.1038/cddis.2016.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ashrafian H, Czibik G, Bellahcene M, Aksentijevic D, Smith AC, Mitchell SJ, Dodd MS, Kirwan J, Byrne JJ, Ludwig C, Isackson H, Yavari A, Stottrup NB, Contractor H, Cahill TJ, Sahgal N, Ball DR, Birkler RI, Hargreaves I, Tennant DA, Land J, Lygate CA, Johannsen M, Kharbanda RK, Neubauer S, Redwood C, de Cabo R, Ahmet I, Talan M, Gunther UL, Robinson AJ, Viant MR, Pollard PJ, Tyler DJ, Watkins H. Fumarate is cardioprotective via activation of the Nrf2 antioxidant pathway. Cell Metab. 2012;15:361–371. doi: 10.1016/j.cmet.2012.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dong J, Sulik KK, Chen SY. Nrf2-mediated transcriptional induction of antioxidant response in mouse embryos exposed to ethanol in vivo: implications for the prevention of fetal alcohol spectrum disorders. Antioxid Redox Signal. 2008;10:2023–2033. doi: 10.1089/ars.2007.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zheng Q, Yuan Y, Yi W, Lau WB, Wang Y, Wang X, Sun Y, Lopez BL, Christopher TA, Peterson JM, Wong GW, Yu S, Yi D, Ma XL. C1q/TNF-related proteins, a family of novel adipokines, induce vascular relaxation through the adiponectin receptor-1/AMPK/eNOS/nitric oxide signaling pathway. Arterioscler Thromb Vasc Biol. 2011;31:2616–2623. doi: 10.1161/ATVBAHA.111.231050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Denzel MS, Scimia MC, Zumstein PM, Walsh K, Ruiz-Lozano P, Ranscht B. T-cadherin is critical for adiponectin-mediated cardioprotection in mice. J Clin Invest. 2010;120:4342–4352. doi: 10.1172/JCI43464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hug C, Wang J, Ahmad NS, Bogan JS, Tsao TS, Lodish HF. T-cadherin is a receptor for hexameric and high-molecular-weight forms of Acrp30/adiponectin. Proc Natl Acad Sci U S A. 2004;101:10308–10313. doi: 10.1073/pnas.0403382101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tao L, Gao E, Hu A, Coletti C, Wang Y, Christopher TA, Lopez BL, Koch W, Ma XL. Thioredoxin reduces post-ischemic myocardial apoptosis by reducing oxidative/nitrative stress. Br J Pharmacol. 2006;149:311–318. doi: 10.1038/sj.bjp.0706853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Heymans S, Luttun A, Nuyens D, Theilmeier G, Creemers E, Moons L, Dyspersin GD, Cleutjens JP, Shipley M, Angellilo A, Levi M, Nube O, Baker A, Keshet E, Lupu F, Herbert JM, Smits JF, Shapiro SD, Baes M, Borgers M, Collen D, Daemen MJ, Carmeliet P. Inhibition of plasminogen activators or matrix metalloproteinases prevents cardiac rupture but impairs therapeutic angiogenesis and causes cardiac failure. Nat Med. 1999;5:1135–1142. doi: 10.1038/13459. [DOI] [PubMed] [Google Scholar]
- 27.Iyer RP, Jung M, Lindsey ML. MMP-9 signaling in the left ventricle following myocardial infarction. Am J Physiol Heart Circ Physiol. 2016;311:H190–198. doi: 10.1152/ajpheart.00243.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ingraham CA, Park GC, Makarenkova HP, Crossin KL. Matrix metalloproteinase (MMP)-9 induced by Wnt signaling increases the proliferation and migration of embryonic neural stem cells at low O2 levels. J Biol Chem. 2011;286:17649–17657. doi: 10.1074/jbc.M111.229427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lagares-Tena L, Garcia-Monclus S, Lopez-Alemany R, Almacellas-Rabaiget O, Huertas-Martinez J, Sainz-Jaspeado M, Mateo-Lozano S, Rodriguez-Galindo C, Rello-Varona S, Herrero-Martin D, Tirado OM. Caveolin-1 promotes Ewing sarcoma metastasis regulating MMP-9 expression through MAPK/ERK pathway. Oncotarget. 2016;7:56889–56903. doi: 10.18632/oncotarget.10872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gu J, Cheng Y, Wu H, Kong L, Wang S, Xu Z, Zhang Z, Tan Y, Keller BB, Zhou H, Wang Y, Xu Z, Cai L. Metallothionein Is Downstream of Nrf2 and Partially Mediates Sulforaphane Prevention of Diabetic Cardiomyopathy. Diabetes. 2017;66:529–542. doi: 10.2337/db15-1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li J, Patel VV, Kostetskii I, Xiong Y, Chu AF, Jacobson JT, Yu C, Morley GE, Molkentin JD, Radice GL. Cardiac-specific loss of N-cadherin leads to alteration in connexins with conduction slowing and arrhythmogenesis. Circ Res. 2005;97:474–481. doi: 10.1161/01.RES.0000181132.11393.18. [DOI] [PubMed] [Google Scholar]
- 32.Derycke LD, Bracke ME. N-cadherin in the spotlight of cell-cell adhesion, differentiation, embryogenesis, invasion and signalling. Int J Dev Biol. 2004;48:463–476. doi: 10.1387/ijdb.041793ld. [DOI] [PubMed] [Google Scholar]
- 33.Hulit J, Suyama K, Chung S, Keren R, Agiostratidou G, Shan W, Dong X, Williams TM, Lisanti MP, Knudsen K, Hazan RB. N-cadherin signaling potentiates mammary tumor metastasis via enhanced extracellular signal-regulated kinase activation. Cancer Res. 2007;67:3106–3116. doi: 10.1158/0008-5472.CAN-06-3401. [DOI] [PubMed] [Google Scholar]
- 34.Lee EJ, Choi EK, Kang SK, Kim GH, Park JY, Kang HJ, Lee SW, Kim KH, Kwon JS, Lee KH, Ahn Y, Lee HJ, Cho HJ, Choi SJ, Oh WI, Park YB, Kim HS. N-cadherin determines individual variations in the therapeutic efficacy of human umbilical cord blood-derived mesenchymal stem cells in a rat model of myocardial infarction. Mol Ther. 2012;20:155–167. doi: 10.1038/mt.2011.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lai RC, Chen TS, Lim SK. Mesenchymal stem cell exosome: a novel stem cell-based therapy for cardiovascular disease. Regen Med. 2011;6:481–492. doi: 10.2217/rme.11.35. [DOI] [PubMed] [Google Scholar]
- 36.Kishore R, Khan M. More Than Tiny Sacks: Stem Cell Exosomes as Cell-Free Modality for Cardiac Repair. Circ Res. 2016;118:330–343. doi: 10.1161/CIRCRESAHA.115.307654. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.