

Abstract
Agonism of the G protein-coupled Free-Fatty Acid receptor-4 (FFA4) has been shown to promote numerous anti-inflammatory effects in macrophages that arise due to interaction with β-arrestin partner proteins. Humans express functionally distinct short and long FFA4 splice variants, such that FFA4-S signals through Gαq/11 and β-arrestin, while FFA4-L is intrinsically biased solely towards β-arrestin signaling. Recently, we and others have shown that phosphorylation of the FFA4 C-terminal tail is responsible for β-arrestin interactability and signaling. Given the significance of β-arrestin in the anti-inflammatory function of FFA4, the goal of this study was to examine the role of the C-terminal β-arrestin phosphosensor in FFA4 signaling induced by PMA and LPS in murine Raw 264.7 macrophages. Our data reveal for the first time that both FFA4 isoforms modulate PMA-induced ROS generation, and that abolishment of the FFA4-S, but not FFA4-L C-terminal phosphosensor, is detrimental to this effect. Furthermore, we show that while both isoforms reduce PMA-induced expression of COX-2, removal of the FFA4-S phosphosensor significantly decreases this response, suggesting that these effects of FFA4-S are β-arrestin mediated. On the contrary, FFA4-S, as well as the truncated C-terminal congener lacking the β-arrestin phosphosensor were both able to reduce LPS-induced NF-κB activity and ERK1/2 phosphorylation. However, FFA4-L and its corresponding mutant were incapable of modulating either, suggesting that these responses are mediated by G protein coupling. Taken together, our data reveal important structure-function and signaling differences between the two FFA4 isoforms, and for the first time link FFA4 to modulation of ROS in macrophages.
Keywords: FFA4, GPR120, free-fatty acids, reactive oxygen species, COX-2
Graphical Abstract
1. INTRODUCTION
Macrophages are critical orchestrators of phagocytosis and innate immune function and play central roles in tissue repair via activation, maintenance, and resolution of inflammation within target tissues (1). This mixed-function modality of macrophages resides in their ability to produce cytokines, growth factors, reactive oxygen species (ROS), reactive nitrogen species (RNS), as well as eicosanoids, including prostaglandins (PG) (1). The rapid and transient generation of ROS in macrophages is dependent on NADPH oxidase (NOX) holoenzymes, which generate superoxide ions (2). ROS are intricately involved in regulation of macrophage biology, and along with RNS, facilitate macrophage-mediated antimicrobial responses. Importantly however, ROS also serve purposeful roles in signal transduction and are able to promote inflammatory responses via activation of NF-κB or Jak/STAT signaling in pro-inflammatory M1 polarized macrophages (2). In a variety of inflammomodulatory cell types, including macrophages, the consequences of this ROS signaling cascade includes upregulation of cyclooxygenase-2 (COX-2) and subsequent synthesis of PG, which in part, direct the inflammatory response (3–7). ROS signaling also promotes differentiation of M1 macrophages to the anti-inflammatory M2 phenotype, which is responsible for tissue healing and remodeling (8).
Long chained free-fatty acids (FFA), including the family of n-3 (i.e., omega-3) fatty acids, have long since been known to influence macrophage biology and inflammatory processes (reviewed in 9–11). Over the last decade, a subfamily of G protein-coupled receptors (GPCR) that is activated by endogenous and dietary FFA has been discovered and shown to mediate many of the well-described effects of FFA. These free-fatty acid receptors include FFA2 and FFA3, which are agonized by short-chained FFA, as well as FFA1 and FFA4, which are agonized by medium-to-long chained FFA. FFA4, previously termed GPR120, has recently drawn considerable attention due to its profound anti-inflammatory effects (reviewed in 12). Oh and colleagues recently demonstrated that FFA4 agonism can reverse LPS-induced activation of NF-κB and inhibit release of proinflammatory IL-6 and TNF–α from macrophages, in a manner dependent on interaction with β-arrestin-2 partner proteins (13). In addition, NLRP3 inflammasome assembly/activity, NF-κB translocation, and IL-1β secretion was inhibited via agonism of FFA4 with the n-3 fatty acid docosahexaenoic acid (DHA), in a manner dependent on both Gαq/11 and β-arrestin-2 (14). Importantly, FFA4 agonism has also been shown to reduce COX-2, but not COX-1, in a β-arrestin-dependent manner (15), and together, these results suggest a primary role for the FFA4/β-arrestin axis in modulating the anti-inflammatory effects of FFA in these cells.
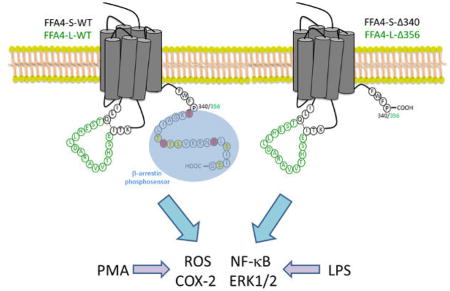
In humans, the FFA4 gene yields two protein isoforms, FFA4-L (long; 377 amino acids) and FFA4-S (short; 361 amino acids) (Fig 1), which are identical with the exception of the 16 additional amino acid sequence within the 3rd intracellular loop (16), a GPCR domain that typically contributes to partner-protein interactions, downstream signaling, and desensitization. Interestingly, in transfected HEK-293 cells, FFA4-S signals by way of both Gαq/11 and β-arrestin-2 cascades, whereas FFA4-L fails to couple to Gαq/11 and is intrinsically biased towards only β-arrestin-2 signaling (17). Since the GPCR-β-arrestin signaling axis is dependent on receptor phosphorylation by G protein-coupled receptor kinases (GRKs), we have previously studied and reported that upon agonism with DHA, FFA4-S is phosphorylated by GRK6 at Thr347, Ser350, Ser357 located within the C-terminal tail (18). In an elegant series of proteomic studies using the synthetic FFA4 agonist TUG-891, Butcher and colleagues confirmed phosphorylation of these sites, and also revealed agonist-mediated phosphorylation of nearby Thr349 and Ser360(19). Importantly, this work also demonstrated that three nearby acidic residues, Glu341, Asp348, and Asp355, work in concert with the 5 phosphorylation sites to comprise the negatively charged FFA4 phosphosensor that interacts with β-arrestin (19). Consistent with this observation, truncation of this C-terminal tail phosphosensor after residue 340 abolished β-arrestin interactability and signaling (19), demonstrating the importance of the FFA4 C-terminal tail towards its function.
Figure 1. Schematic representation of wild-type and C-terminal truncate mutant FFA4 receptors.

A, Wild-type FFA4-S and FFA4-L (green insert), depicting the differences between the isoforms at the 3rd ICL. The β-arrestin-phosphosensor (blue) is comprised of acidic residues Glu341, Asp348, Asp355 (red) and the phosphorylation sites Thr347, Thr349, Ser350, Ser357, and Ser360 (yellow). B, FFA4 C-terminal mutants used in this study were truncated after Pro340 (corresponding to 356 in FFA4-L), thereby eliminating the C-terminal β-arrestin phosphosensor.
Given the significance of β-arrestin signaling in FFA4 function and the seemingly intrinsic-bias of FFA4-L towards β-arrestin signaling, we wished to examine the role of the C-terminus of both FFA4 isoforms in macrophages. In the present study, we use Raw 264.7 mouse macrophages to show for the first time that FFA4 agonism significantly abrogates NOX-mediated ROS generation. We also assess the ability of FFA4 isoforms and their respective phosphosensor-null C-terminal mutants to modulate ROS generation, COX-2 expression, as well as LPS-mediated NF-κB activity and activation of MAPK. Our results reveal important structure-function differences between the two FFA4 isoforms, and also show that that the C-terminal tail of FFA4-S is critical to the activity of this isoform.
2. MATERIALS AND METHODS
2.1 Reagents and Chemicals
Wild-type (WT) flag-tagged FFA4-S and FFA4-L constructs were generated as previously described (16). Mutant receptors lacking the β-arrestin phosphosensor (19) were truncated after amino acid residue 340 in FFA4-S (FFA4-S-Δ340) and after residue 356 in FFA4-L (FFA4-L-Δ356) and created via overlapping PCR and cloned into pcDNA3.1, as we described previously (16). Phorbol 12-myristate 13-acetate (PMA), and bisindolylmaleimide II (BIMII) were obtained from Sigma-Aldrich (St. Louis, MO). PMA was used at a concentration of 1 μM throughout the study. 8-Amino-5-chloro-2,3-dihydro-7-phenyl-pyrido[3,4-d]pyridazine sodium salt (L-012) was obtained from Wako Chemicals USA (Richmond, VA). Initially, TUG-891 and AH7614 were purchased from Tocris (Minneapolis, MN) and Sigma-Aldrich, respectively, and were later synthesized as described in 2.2. All other chemicals used were obtained at the highest available purity from Thermo Fisher Scientific or Sigma-Aldrich.
2.2 Synthesis of TUG-891 and AH7614
The synthesis of TUG-891 or 3-(4-((4-fluoro-4′-methyl-[1,1′-biphenyl]-2-yl)methoxy)phenyl)propanoic acid was achieved via a five-step synthetic scheme, as previously described (20). Briefly, 5-fluoro-2-iodobenzoic acid was reduced to the primary alcohol derivative using lithium aluminum hydride in ether, followed by conversion to the acid chloride 2-(chloromethyl)-4-fluoro-1-iodobenzene using thionyl chloride in dichloromethane. The acid chloride was then coupled with methyl 3-(4-hydroxyphenyl)propanoate using potassium carbonate in acetonitrile. Suzuki cross-coupling was then performed on the resulted methyl 3-(4-((5-fluoro-2-iodobenzyl)oxy)phenyl)propanoate with p-tolylboronic acid in the presence of palladium acetate and triphenyl phosphine in anhydrous dimethylformamide to yield methyl 3-(4-((4-fluoro-4′-methyl-[1,1′-biphenyl]-2-yl)methoxy)phenyl) propanoate. The resulted methyl ester was hydrolyzed using lithium hydroxide in tetrahydrofuran to achieve the final compound 3-(4-((4-fluoro-4′-methyl-[1,1′-biphenyl]-2-yl)methoxy)phenyl)propanoic acid with an overall yield of 43%. High-resolution mass spectrometry [calculated for C23H21FO3Na (M+ Na+) 386.39 versus experimental 386.80] and 1H-NMR spectra were consistent with the published data (20). The synthesis of AH7614 or 4-methyl-N-(9H-xanthen-9-yl)benzenesulfonamide was obtained via a three-step scheme starting with the reductive amination of oanisidine and salicaldehyde in the presence of isopropyl alcohol at refluxing temperature to achieve 2-(((2-methoxyphenyl)imino)methyl)phenol, which was followed by its reaction with p-tolualdehyde in the presence of glacial acetic acid and acetic anhydride. The resulted product N-(2-hydroxybenzylidene)-4-methylbenzenesulfonamide was reacted with 2-(trimethylsilyl)phenyl trifluoromethanesulfonate in the presence of potassium fluoride, 18-crown ether and tetrahydrofuran to afford the final product in an overall yield of 58%. The 1H-NMR spectrum was consistent with the published data (21–22).
2.3 Cell culture, transfections, and selection
Raw 264.7 macrophages were obtained from ATCC (Manassas, VA) and cultured in 100 mm plates containing Dulbecco’s modified Eagle’s medium (DMEM), supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin (Life Technologies, Grand Island, NY). Transfections were performed using LipoD293 reagent (Signagen Laboratories, Gaithersburg, MD) and 8 μg of the respective plasmid DNA, according to the manufacturer’s directions. Forty-eight hours after transfection, stable cell selection was initiated and maintained with 1mg/ml G418-containing selection medium.
2.4 RNA extraction, PCR and quantitative real-time PCR
Total cellular RNA was extracted from cells using the RNeasy Mini Kit (Qiagen, Gaithersburg, MD), according to the manufacturer’s directions. Extracted RNA was reverse-transcribed to cDNA using high capacity cDNA Reverse Transcription (Applied Biosystems, Foster City, CA) and random primers. cDNA templates were amplified for 30 cycles by polymerase chain reaction (PCR) with primers corresponding to FFA4-S-WT, FFA4-L-WT and their corresponding mutants. Quantitative real-time PCR was performed using EvaGreen PCR master mix (Biotium, Fremont, CA). Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as a control housekeeping gene with the below primers: FFA4 (N-terminal region): 5′-ATGTCCCCTGAATGCGCGCGG-3′ (FWD) and 5′-ATGAGCACCAGCACGGTTGTCTCC-3′ (REV); Catalase: 5′-GCAGATACCTGTGAACTGTC-3′ (FWD) and 5′-GTAGAATGTCCGCACCTGAG-3′ (REV). GAPDH: 5′-ACCACAGTCCATGCCATCAC-3′ (FWD) and 5′-TCCACCACCCTGTTGCTGTA-3′ (REV). For data analysis, the ΔΔCt method was used.
2.5 ROS generation
The kinetic generation of ROS was induced by stimulation of cells with the superoxide and hydrogen peroxide generator Phorbol 12-myristate 13-acetate (PMA) and monitored by the ROS-sensitive luminol-enhanced chemiluminescent probe L-012 (23–25). Briefly, cells were treated with vehicle, TUG-891 (3μM), or AH7614 (1μM) for 1 h, washed with PBS to remove drugs, and detached with a cell scraper. Cells were suspended in HBSS containing 100 μM of L-012 and 100μl of suspended cells were transferred in triplicate into white 96-well plates, and basal chemiluminescence was recorded immediately in a Tecan Infinite M200. After 5 min, PMA (1μM) or vehicle (water) was injected to induce ROS generation and chemiluminescence was recorded for an additional 90 min.
2.6 Immunoblotting
Immunoblotting was performed as described previously (16,18). Briefly, cells were lysed in RIPA buffer (50 mM Tris-HCl, 150 mM NaCl, 5 mM EDTA, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS, 10 mM NaF, 10 mM Na2HPO4, pH 7.4) plus protease inhibitor cocktail for 20 min and cleared for insoluble debris by centrifugation at 15,000 × g for 15 minutes at 4°C. Protein concentrations were standardized using DC Protein Assay (Bio-Rad, Hercules, CA) and an aliquot of the lysate was denatured in 2X SDS-sample buffer and boiled for 5 minutes. Equivalent concentrations of lysates were resolved by SDS–PAGE, followed by transfer to PVDF membranes, and immunoblotted with the appropriate antibody. Blots were visualized with HRP-conjugated secondary antibody followed by ECL. Blots were stripped by incubation in glycine IgG stripping buffer for 30–45 minutes with agitation (25 mM Glycine, 1% SDS, 50°C, pH 2.0) for re-probing. Polyclonal anti-COX-2 antibody (160126) was purchased from Cayman Chemical (Ann Arbor, MI), rabbit anti-FFA4 antibody was purchased from Novus Biologicals (Littleton, CO), rabbit anti-phosphoThr202/Tyr204-p44/p42 MAPK (ERK1/2) and p44/42 MAPK (ERK1/2) antibodies were purchased from Cell Signaling Technologies (Beverly, MA), and anti-actin-HRP antibody was purchased from Santa Cruz Biotechnology (Santa Cruz, CA).
2.7 Cell viability
Cell viability and inhibition of cell proliferation was measured by reduction of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) to formazan (Thermo Fisher, Carlsbad, CA). Briefly, cells were seeded in 96-well plates and after overnight incubation, treated with PMA (1 μM) or vehicle (water) for 1 h after which, MTT reagent was added to each well and incubated for 4 h at 37°C. Reduced formazan crystals were dissolved in DMSO and absorbance was measured at 540 nm on a microliter plate reader. For assays that induced cytotoxicity via H2O2, 1 mM H2O2 was added to cells for 1 h, then fresh media was added for an additional 16 h prior to addition of MTT reagent and absorbance reading.
2.8 NF–κB reporter assay
NF–κB activity was assessed using the pNFκB-MetLuc2-Reporter assay (Clonetech Laboratories, Mountain View, CA). Briefly, cells were seeded in 100 mm plates and transiently transfected with 5 μg pNFκB-MetLuc2-Reporter or 1 μg pMetLuc2-Control (for normalization of transfection efficiency) vectors, according to manufacturer’s protocol. After 24 h, cells were plated in 24 well-plates and treated with TUG-891 (3 μM), 1h prior to treatment with LPS (100 ng/ml). The cell culture medium was collected after 16 h incubation and analyzed utilizing Ready-To-Glow Secreted Luciferase according to the manufacturer’s instructions. NF-κB metridia luciferase activity was normalized to control renilla luciferase values to account for transfection efficiency.
2.9 PGE2 production
PGE2 metabolites were assessed by commercially available ELISA (Cayman, Chemical, Mountain View, CA). Briefly, cells were plated in six-well plates and incubated with PMA for 4 h. Fifty microliters of supernatant of culture medium was collected for the determination of PGE2 metabolite concentration according to the manufacturer’s directions. Due to differences in absolute concentrations of PGE2 metabolite between experiments, results are expressed as the concentration of PGE2 metabolites normalized to the respective experiments untransfected control condition.
2.10 Data analysis
Autoradiographic data were quantified by densitometric analysis within the linear range using NIH Image J (Bethesda, MD) and graphed using Graphpad Prism 3.0 (San Diego, CA). Data are expressed as mean ± S.E.M for representative experiments repeated at least three independent times. Where not visible, error bars fall within the symbol size. Statistical analysis was performed in Graphpad Instat using one-way analysis of variance and post-hoc Tukey-Kramer analysis or Student’s t-test, as described in the figure legends. Statistical significance is represented as a single symbol for p < 0.05, a double symbol for p < 0.01, and a triple symbol for p < 0.001, as noted in the figure legends.
3. RESULTS
3.1 FFA4 agonism decreases PMA-induced ROS generation in Raw 264.7 macrophages
Since ROS serve primary functions in macrophage biology, we assessed the role of FFA4 in modulating ROS generation in murine Raw 264.7 macrophages. Phorbol-12-myristate 13-acetate (PMA) is known to facilitate the immediate and robust activation of the membrane-bound NADPH oxidase (NOX) machinery to rapidly generate endogenous ROS in superoxide-producing cells, including Raw 264.7 macrophages (26–28). True to this nature, PMA (1 μM) stimulated a rapid (within 1 min) and robust increase in ROS generation, which was transiently sustained over a timeframe of 90 min (Fig 2A). The effects of PMA on NOX and subsequent ROS generation are known to be mediated by protein kinase-C (PKC) (7,26–27), and our results confirm that PMA-induced ROS generation was abolished by pretreatment of cells with the PKC inhibitor bisindolylmaleimide II (BIMII) (10 μM, 20 min prior to PMA) (Fig 2B). Since Raw 264.7 macrophages have been shown to endogenously express low levels of FFA4 that are agonist-responsive (13–15), we next assessed PMA-induced ROS generation in cells treated with synthetic FFA4 modulators. Preincubation of cells for 1 h with the selective FFA4 agonist TUG-891 (3 μM) prior to PMA stimulation resulted in significant abrogation of ROS generatability (Fig 2C,F), but had no effect in the absence of PMA, demonstrating that agonism of FFA4 can negatively-modulate ROS generation. On the contrary, pretreatment of cells for 1 h with the selective FFA4 antagonist AH7614 (1 μM) (29) prior to PMA treatment facilitated a statistically significant increase in ROS generation compared to PMA alone (Fig 2D,F). Importantly, pretreatment of cells with TUG-891 (3 μM) in combination with AH7614 (1 μM) prior to PMA treatment lead to complete loss of the agonist’s effect, demonstrating that FFA4 is responsible for reversal of PMA-induced ROS generation (Fig 2E,F). In order to quantify the entire transient nature of the ROS cycle induced by PMA, and which is modulated by FFA4, we pooled all data and expressed them as the entirety of the area under the curve (AUC) ± SEM of each ROS tracing (Fig 2F). To ensure that the effects on ROS seen with TUG-891 were not simply due to the compounds ability to quench or scavenge ROS, we performed cell-free assays with TUG-891, AH7614, or both agents in the presence of exogenous ROS in the form of 100 μM H2O2. These results show that neither TUG-891 nor AH7614, alone or in combination, affect ROS detection or ROS levels in cell-free systems (Fig 2G). Finally, to ensure that the ROS generation seen here was not purely a property of TUG-891, we utilized the endogenous FFA4 agonist DHA as well as the FFA1/FFA4 dual agonist GW9508, which has been used as a selective FFA4 agonist in cells that lack FFA1, as Raw 264.7 macrophages do (13). DHA (100 μM) decreased PMA-induced ROS generation to a level similar to that seen with TUG-891 (Fig 2H), and this effect was again reversed to a level greater than PMA alone upon pretreatment with AH7614 (Fig 2H). Interestingly, others have previously shown that GW9508 facilitates an increase in ROS generation from mitochondrial sources and in a manner independent of FFA1/FFA4 (30). Our data support this observation as GW9508 (1 μM) significantly increased PMAinduced ROS generation (Fig 2H) and this effect was dose-dependent (data not shown). This result suggests that GW9508 would not be an acceptable agent for subsequent studies on FFA4-mediated ROS generation.
Figure 2. Effects of FFA4 agonism and antagonism on PMA-induced ROS generation in Raw 264.7 macrophages.
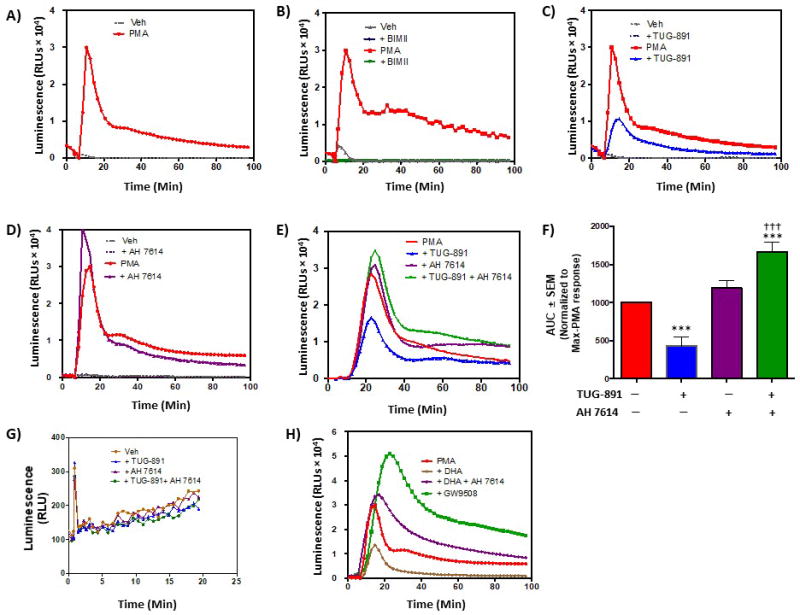
As described in Experimental Procedures, ROS generation was detected by suspending cells in HBSS containing the ROS-sensitive luminescent probe L-012 (100 μM) in the presence of the noted FFA4 modulators or their vehicles (veh). Basal luminescence was read for 5 min prior to addition of PMA (1 μM) or vehicle (H2O), after which, luminescence was recorded for an additional 90 min. In A-E, representative traces are shown for experiments repeated independently (n)-times. A, PMA induces rapid and robust ROS generation in Raw 264.7 cells. A representative trace is shown for this condition, which was included in all subsequent experiments (n = 24). B, The PKC inhibitor BIMII (10 μM, 20 min prior to PMA treatment) abolishes PMA-induced ROS generation (n = 3), demonstrating the PKC-dependency of NADPH-oxidase generation of ROS. C, The FFA4 agonist TUG-891 (3 μM, 1 h prior to PMA treatment) decreases PMA-induced ROS generation (n = 8). D, The FFA4 antagonist AH7614 (1 μM, 1 h prior to PMA treatment) increases PMA-induced ROS generation (n = 6). E, AH7614 blocks the ability of TUG-891 to reduce PMA-induced ROS generation, demonstrating that this effect is modulated directly by FFA4 receptor agonism (n > 5). F, Area under the curves (AUC) for all conditions induced by PMA were quantified (mean ± SEM) using Prism 3.0 and normalized to the maximal response elicited by PMA. G, Cell-free effects of TUG-891 and AH7614 on ROS shows no effects of either agent on ROS detection. Statistical analysis was performed by ANOVA and *** denotes p < 0.001 versus the maximal PMA-response, ††† denotes p < 0.001 versus the TUG-891-treated condition. H, The FFA4 agonist DHA (100 μM, 1 h prior to PMA treatment) decreases PMA-induced ROS generation (n = 3) and this effect was reversed by AH7614 (1 μM). The FFA4/FFA1 agonist GW9508, which has been shown to increase ROS generation via receptor-independent mechanisms robustly increases PMA-induced ROS generation.
3.2 The C-terminal tail of FFA4-S but not FFA4-L is critical in decreasing PMA-induced ROS generation
Next, we sought to better understand the role of FFA4 isoforms, as well as the importance of FFA4 phosphorylation/β-arrestin interactions in the modulation of ROS in macrophages. Since Raw 264.7 macrophages are difficult to transfect transiently and are known to have poor transfection efficiency, we created Raw 264.7 cell lines that stably expressed wild-type (WT) FFA4-S or FFA4-L, or the C-terminal truncated mutants (Fig 1), which lack the β-arrestin phosphosensor and are hence incapable of being phosphorylated or recruiting β-arrestin (18–19). Using primers specific to the C-terminal-FLAG epitope used in our constructs (16), Figure 3A demonstrates stable expression of FLAG-tagged receptor transcripts in FFA4-S-WT and FFA4-L-WT cells, as well as cells stably expressing the respective C-terminal mutants lacking the β-arrestin phosphosensor, FFA4-S–Δ340 and FFA4-L–Δ356. Using real-time PCR to detect an N-terminal region of FFA4 that is homologous to endogenous FFA4 as well as FFA4-S-WT, FFA4-L-WT, FFA4-S–Δ340 and FFA4-L–Δ356, we show that the stable cell lines expressed approximately 4–5-fold of the respective transcripts relative to endogenous FFA4 mRNA within untransfected Raw 264.7 cells (Fig 3B). Immunoblotting of whole cell lysates with an anti-FFA4 antibody revealed specific detection of an immunoreactive band at the expected size of 45 kDa in untransfected Raw 264.7 cells (UNTR) that was not present in HEK-293 cells [(−) CON)], which we and others have shown to lack endogenous FFA4 (16–17) (Fig 3C). Importantly, the FFA4 band was increased in the stable transfectants compared to untransfected cells (Fig 3C), and was not significantly different amongst transfected cells, confirming similar protein expression of FFA4 constructs in stably transfected cells. Next, we assessed ROS generation in each stable cell line and these results demonstrate that overexpression of FFA4-S-WT nearly abolishes the PMA-induced ROS generation seen in untransfected Raw 264.7 cells (Fig 3D,F–G), while the FFA4-S–Δ340 mutant that lacks amino acids 341–361 comprising the β-arrestin phosphosensor, only slightly albeit significantly, reduces the PMA effect (Fig 3D,F). Similarly, PMA-induced ROS generation was nearly abolished in FFA4-L-WT overexpressing cells, but not to the degree seen with FFA4-S-WT (Fig 3E–G). Interestingly, unlike that seen with FFA4-S–Δ340, overexpression of FFA4-L–Δ356 was able to decrease PMA-induced ROS generation to the same degree as FFA4-L-WT (Fig 3E–G), suggesting that the presence of the additional 16 residues in the longer isoform are able to compensate for the loss of the C-terminal tail. These results confirm that FFA4 can modulate NOX-induced ROS generation and importantly, the C-terminal tail of FFA4-S, but not FFA4-L, is to a large extent required for this effect.
Figure 3. Effects of FFA4 isoforms and β-arrestin phosphosensor-mutants on PMA-induced ROS generation in stably expressing Raw 264.7 macrophages.
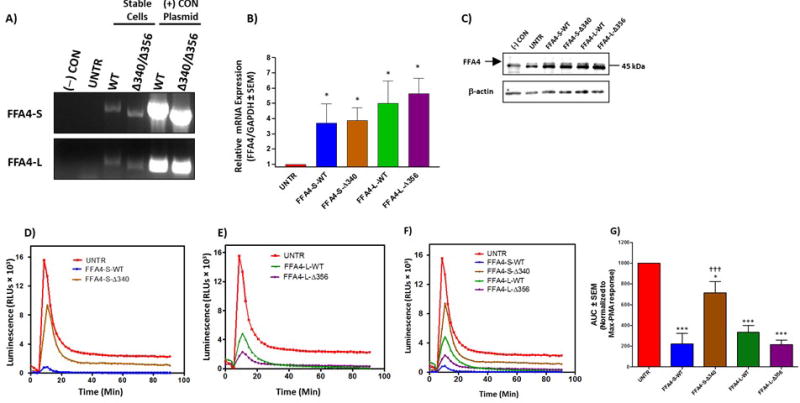
A, Raw 264.7 macrophages were stably transfected with FLAG-tagged FFA4-S, FFA4-L, or their respective β-arrestin phosphosensor truncated mutants, FFA4-S–Δ340, or FFA4-L–Δ356. RT-PCR was used to detect the FLAG-epitope after stable selection in G418, as described in the Experimental Procedures. (−) CON represents PCR condition lacking DNA template, UNTR represents PCR with template derived from untransfected Raw 264.7 cells. PCR amplicons represent full-length FFA4 sequence beginning with the initiating codon and terminating with the C-terminal FLAG epitope, and plasmids encoding for each condition were used as positive controls. B, real-time quantitative PCR to detect the level of endogenous FFA4 mRNA expression compared to stable cell lines shows an approximately 4–5-fold increase in FFA4 transcript compared to endogenous mRNA in untransfected cells. Statistical significance was determined by Student’s t-test, and all stable cell lines were significantly different (p < 0.05) compared to untransfected control, but not compared to each other. C, Immunoblotting of whole cell lysates with an anti-FFA4 antibody reveals the presence of two bands near the expected size of 45 kDa. The lower band is a non-specific immunoreactive band as it is also present in lysates of HEK-293 cells [(−) CON], which we and others have shown to lack specific FFA4 expression (16–17). The upper band, corresponding to FFA4, is as expected, detected in untransfected Raw 264.7 cells (UNTR) and is lacking in the (−) CON lane. FFA4-immunoreactivity is increased in stable cell lines compared to that seen in UNTR cells, and is of similar intensity in the transfectants. D, ROS generation induced by PMA in untransfected Raw 264.7 cells (UNTR), compared with cells stably expressing FFA4-S-WT, or FFA4-S–Δ340 reveals that PMA-induced ROS generation is almost completely abolished in cells expressing FFA4-S-WT and only slightly decreased in cells expressing FFA4-S–Δ340 (n ≥ 5). E, ROS generation induced by PMA in untransfected Raw 264.7 cells (UNTR), compared with cells stably expressing FFA4-L-WT, or FFA4-L–Δ356 reveals that PMA-induced ROS generation is similarly decreased in cells expressing both FFA4-L-WT and FFA4-L–Δ356 (n ≥ 5). F, Overview of the effects of FFA4-isoforms and mutants on PMA-induced ROS generation (n ≥ 5). G, Area under the curves (AUC) for all conditions induced by PMA were quantified (mean ± SEM) using Prism 3.0 and normalized to the maximal response elicited by PMA. Statistical analysis was performed by ANOVA and * denotes p < 0.05 versus the maximal PMA-response, *** denotes p < 0.001 versus the maximal PMA-response, ††† denotes p < 0.001 versus the FFA4-S-WT condition.
3.3 The C-terminal tail of FFA4-S, but not FFA4-L is required for antagonism of PMA-induced ROS generation
Next, we wished to assess the effects of TUG-891 and AH7614 on PMA-induced ROS generation in macrophages stably expressing the given FFA4 constructs. While as seen in Fig 3, FFA4-S-WT and FFA4-L-WT expressing cells demonstrated significantly lowered levels of PMA-induced ROS generation compared to untransfected Raw 264.7 cells, to our surprise, agonism with TUG-891 (3 μM, 1 h prior to PMA addition) in these stably overexpressing cells yielded no further reduction of the PMA-induced effect (Fig 4A–B), suggesting that receptor overexpression supplies the maximal activity, which is not sensitive to further agonism. On the contrary, receptor blockade by AH7614 (1 μM, 1 h prior to PMA addition) decreased the effect of both FFA4-S-WT and FFA4-L-WT, thereby inhibiting the receptors ability to reduce PMA-induced ROS generation (Fig 4A–B). Interestingly, this effect was completely lost in cells overexpressing the FFA4-S–Δ340 mutant (Fig 4C), but not in cells overexpressing FFA4-L–Δ356 (Fig 4D). These results confirm that the C-terminal tail of the shorter isoform is required for the ROS-reducing effect, and that the longer isoform overcomes the loss of the C-terminus due to its additional 16 residue segment. To insure that effects on diminished ROS generation in these studies were not simply due to cell toxicity, we assessed cell viability of all stable cell lines. Cell viability did not differ from untransfected Raw 264.7 cells for any of the stable expressants (Fig 5A), nor did stable expression of FFA4-S, -L, or mutants inhibit or promote the cytotoxicity induced by exogenously administered ROS in the form of H2O2 (1mM, 1 h) (Fig 5A). PMA has mitogenic properties that could promote cell proliferation or could induce toxicity via generation of ROS. To ensure that PMA treatment in ROS-generation assays did not induce cytotoxicity, or induce proliferation differently in our cell lines, we also performed cell viability assays in control and stable cells exposed to PMA (1 μM) for 4 h. As expected, these data reveal increased cell numbers in PMA treated cells that were not altered by FFA4 expression, and also show no adverse effect on cell viability in any of the stable expressants (Fig 5B).
Figure 4. Effects of TUG-891 and AH7614 on PMA-induced ROS generation in Raw 264.7 macrophages stably expressing FFA4 isoforms or β-arrestin phosphosensor-mutants.
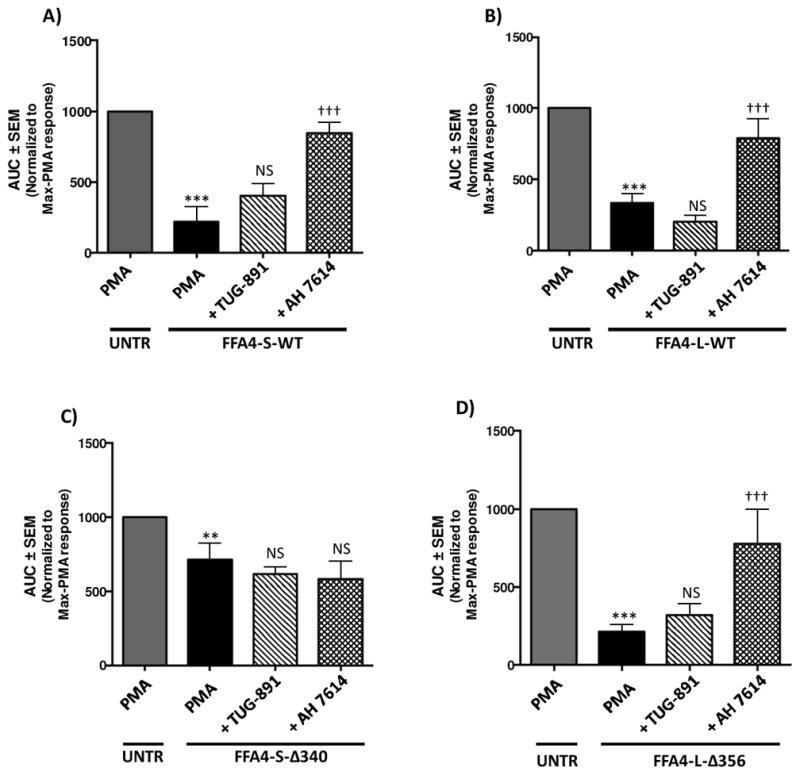
For all experiments, full traces were performed as described in the Experimental Procedures and as shown in Figures 2–3 and results are expressed as mean ± SEM of the area under the curves (AUC) for all conditions induced by PMA and are normalized to the maximal response elicited by PMA in untransfected cells (UNTR). Statistical analysis was performed by ANOVA. A, PMA-induced ROS generation was significantly reduced in cells expressing FFA4-S-WT compared to untransfected control Raw 264.7 cells (UNTR). Treatment of cells with TUG-891 (3 μM, 1 h prior to PMA addition) did not further alter PMA-induced ROS-generation, while treatment of cells with AH7614 (1 μM, 1 h prior to PMA addition) lead to increases in PMA-induced ROS generation compared to cells treated with PMA alone. *** denotes p < 0.001 versus the maximal PMA-response in untransfected cells, ††† denotes p < 0.001 versus the PMA-treated condition in FFA4-S-WT cells, NS denotes not statistically significant compared to PMA condition in FFA4-S-WT cells (n≥ 5). B, PMA-induced ROS generation was reduced in cells expressing FFA4-L-WT compared to untransfected cells. Treatment of cells with TUG-891 (3 μM, 1 h prior to PMA addition) did not further alter PMA-induced ROS-generation, while treatment of cells with AH7614 (1 μM, 1 h prior to PMA addition) lead to increases in PMA-induced ROS generation compared to cells treated with PMA alone. *** denotes p < 0.001 versus the maximal PMA-response in untransfected cells, ††† denotes p < 0.001 versus the PMA-treated condition in FFA4-L-WT cells, NS denotes not statistically significant compared to PMA condition in FFA4-L-WT cells (n ≥ 5). C, PMA-induced ROS generation was reduced in cells expressing FFA4-S–Δ340 compared to untransfected Raw 264.7 cells (UNTR). Treatment of cells with TUG-891 (3 μM, 1 h prior to PMA addition) or AH7614 (1 μM, 1 h prior to PMA addition) did not further alter PMA-induced ROS-generation compared to cells treated with PMA alone. ** denotes p < 0.01 versus the maximal PMA-response in untransfected cells, NS denotes not statistically significant compared to PMA condition in FFA4-S–Δ340 cells (n ≥ 5). D, PMA-induced ROS generation was reduced in cells expressing FFA4-L–Δ356 compared to untransfected Raw 264.7 cells (UNTR). Treatment of cells with TUG-891 (3 μM, 1 h prior to PMA addition) did not further alter PMA-induced ROS-generation, while treatment of cells with AH7614 (1 μM, 1 h prior to PMA addition) lead to increases in PMA-induced ROS generation compared to cells treated with PMA alone. *** denotes p < 0.001 versus the maximal PMA-response in untransfected cells, ††† denotes p < 0.001 versus the PMA-treated condition in FFA4-L–Δ356 cells, NS denotes not statistically significant compared to PMA condition in FFA4-L–Δ356 cells (n ≥ 5).
Figure 5. Viability of cells stably expressing FFA4 isoforms or β-arrestin phosphosensor-mutants.

Cell viability was assessed by MTT assay as described in Experimental Procedures. A, Cells stably expressing FFA4-S-WT, FFA4-L-WT, FFA4-S–Δ340, or FFA4-L–Δ356 display similar viability as untransfected Raw 264.7 cells (UNTR) (gray). All stable cells also display similar levels of cytotoxicity to UNTR when treated with exogenous H2O2 (1 mM for 1 h, removed, and assayed 16 h later). B, To ensure that PMA treatment does not induce cytotoxicity, experiments were repeated within our PMA-treatment paradigm and no significant differences in cytotoxicity was apparent in any of the stable cells compared to untransfected cells (UNTR).
3.4 The C-terminal tail of FFA4-S, but not FFA4-L modulates the receptors ability to reduce COX-2 expression
Since ROS are known to play a part in increasing expression of pro-inflammatory COX-2 in macrophages and other cell types (5–8), we next assessed the role of FFA4 in modulating COX-2 expression. In untransfected Raw 264.7 macrophages, PMA treatment (1 μM, 4 h) induced COX-2 expression via PKC, as BIMII (10 μM 1 h prior to PMA) completely abolished the effect (Fig 6A). Similarly, treatment of cells with TUG-891 (3 μM, 1 h prior to PMA) in untransfected macrophages significantly reduced PMA-induced COX-2 expression, an effect that was furthered by BIMII (10 μM, 1 h prior to PMA with TUG-891) (Fig 6A). Bacterial LPS treatment is also known to induce COX-2 expression in macrophages, an effect that is more robust, though not as rapid as that of PMA. Treatment of cells with LPS (100 ng/ml, 6 h) induced a potent upregulation of COX-2 expression, an effect that was not significantly altered by TUG-891, but was nearly fully blocked by BIMII (Fig 6A; same control as PMA blot). Based on these results, we then assessed PMA-induced COX-2 expression in untransfected Raw 264.7 macrophages and those stably expressing FFA4-S, FFA4-L, or their respective C-terminal truncates. The PMA-induced COX-2 upregulation seen in untransfected cells was significantly decreased in cells expressing FFA4-S-WT and to a lesser degree FFA4-L-WT, but was not significantly decreased in cells expressing either of the respective C-terminal mutants (Fig 6B). Here, the effect of FFA4-S–Δ340 was significantly less that that seen with FFA4-S-WT (Fig 6B), but the effect of FFA4-L-WT was only slightly not significantly different than that of FFA4-L–Δ356. These results also coincided with a significant decrease in PGE2 only in cells expressing FFA4-S-WT (Fig 6C), and taken together, demonstrate that FFA4-WT-S has a profound influence on PMA-induced COX-2 expression and subsequent PG synthesis, and the C-terminal phosphosensor plays a major role in this activity. Given the known linkage between ROS generation and COX-2 expression in macrophages, we gauged the role of ROS in the FFA4-mediated effect in the absence or presence of the ROS scavenger N-acetyl-L-cysteine (NAC). In untransfected Raw 264.7 cells, NAC treatment significantly reduced PMA-induced COX-2 expression, confirming the partial-ROS dependence of this effect (Fig 6D–F). While cells that stably expressed FFA4-S-WT or FFA4-L-WT had reductions in PMA-induced COX-2 expression (as in Fig 6B), there was no additive effect of NAC in these cells (Fig 6F), suggesting that the partial ROS-dependency of this effect was already maximally inhibited by receptor expression. Our data reveal a role for FFA4 in reduction of both ROS generation and COX-2 expression, yet, NAC had no additional effect in lowering COX-2 expression in FFA4-WT expressing cells. Hence, we hypothesized that FFA4 expression could modulate cellular adaptations that could explain the effects seen on ROS generation and COX-2 expression. As such, we examined expression of selected oxidant-relevant genes including superoxide dismutase, glutathione reductase, arginase, and catalase to assess potential differences in their expression that may have been induced by overexpression of FFA4 receptors. Using real-time PCR, our results show that Raw 264.7 macrophages which stably overexpressed FFA4-S-WT displayed nearly two-fold higher levels of catalase, the enzyme responsible for reduction of H2O2, compared to untransfected cells or those expressing FFA4-L-WT or either of the C-terminal truncates (Fig 6G). We did not note significant effects on any of the other transcripts within our panel. These results could explain the lack of ability of NAC to further reduce PMA-induced COX-2 expression in FFA4-S-WT expressing cells and may in part, mechanistically explain the role of FFA4-S-WT in reducing COX-2 expression.
Figure 6. Effects of FFA4 isoforms or β-arrestin phosphosensor-mutants on COX-2 expression in stably expressing Raw 264.7 macrophages.

A, In untransfected Raw 264.7 macrophages (UNTR), PMA treatment (1μM for 4 h) induces COX-2 expression, and this effect is decreased by TUG-891 (3 μM, 1 h prior to PMA treatment) and is fully blocked by the PKC inhibitor BIMII (10 μM, 1 h prior to PMA treatment). The effects of TUG-891 (3 μM, 1 h prior to LPS treatment) on LPS (100 ng/ml, 6 h) induced COX-2 expression are noticeably absent, despite partial dependence on PKC for the LPS-effect. The PMA blot (left) and LPS blot (right) in this figure were run on one blot, and thus share the same control (lane 1) and are separated for naming clarity. B, PMA (1μM for 4 h) induced COX-2 expression was substantially decreased in cells expressing FFA4-S-WT and to a lesser, albeit significant degree in cells expressing FFA4-L-WT, but not in cells expressing the respective β-arrestin phosphosensor-lacking mutants, FFA4-S–Δ340 and FFA4-L–Δ356. A representative immunoblot is shown, and data from at least three independent experiments was quantified as described in the Experimental Procedures and expressed as COX-2 over β-actin, which was used as a loading control in each. Statistical analysis was performed by ANOVA and * and *** denote p < 0.05 and p < 0.001, respectively, versus the PMA-response in untransfected control cells (UNTR), † denotes p < 0.05 versus the PMA-treated condition in FFA4-S-WT cells. C, PGE2 metabolites were assessed using ELISA as described in the Experimental Procedures and reveal that PMA (1μM for 4 h)-induced PGE2 biosynthesis was significantly decreased only in cells that expressed FFA4-S-WT. * denotes p < 0.05 versus the PMA-response in untransfected control cells (UNTR) (n ≥ 3). D–F, In untransfected Raw 264.7 cells (UNTR), PMA-induced COX-2 expression was significantly (ca. 50%), but not fully blocked by 16 h treatment of cells with the ROS scavenger NAC (1 mM) prior to PMA treatment (1μM for 4 h). This reduction by NAC was not seen in stable transfectants. A representative immunoblot is shown, and the experiments were performed independently three times and quantified (F). *, and ** denote p < 0.05 and p < 0.01, respectively, versus the untransfected condition stimulated with PMA. † denotes p < 0.05 versus the PMA-treated condition in FFA4-S-WT cells. ‡ denotes p < 0.05 versus the PMA and NAC-treated condition in FFA4-S-WT cells. (G) Real-time PCR was utilized to detect the expression of catalase in untransfected Raw 264.7 cells and in cells stably expressing FFA4 isoforms or mutants. Catalase mRNA expression was normalized to the housekeeping gene GAPDH, and is expressed as the mean ± SEM of three independent experiments. Statistical analysis was performed by Student’s t-test and * denotes p < 0.05 versus untransfected control cells.
3.5 FFA4-S but not FFA4-L reduces LPS-mediated NF–κB activity and LPS-mediated ERK1/2 phosphorylation
In Raw 264.7 macrophages, bacterial lipopolysaccharide is known to stimulate NF–κB promotor activity and transcription of inflammatory genes, and this effect is in part associated with mitogen-activated protein kinases, including the extracellular-signal-regulated kinases 1/2 (ERK1/2), phosphorylation and activation of which, plays integral roles in release of proinflammatory cytokines (31–33). Hence, we also wished to assess the role of the FFA4-S and FFA4-L β-arrestin phosphosensor on LPS-mediated effects to determine if there were similarities to that seen with PMA stimulation. To do so, we assessed NF-κB reporter activity and phosphorylation of ERK1/2. Agonism of endogenous FFA4-S on untransfected Raw 264.7 cells with TUG-891 facilitated reduction in NF-κB promoter activity as assessed by a luciferase-promotor assay (Fig 7A), consistent with previous reports (13). On the contrary to the observed PMA-mediated effects, overexpression of both FFA4-S-WT and FFA4-S–Δ340, but not FFA4-L-WT or FFA4-L–Δ356, significantly reduced LPS-mediated NF-κB activity (Fig 7B). Notably, while expression of FFA4-S–Δ340 reduced NF-κB activity, it did so to a significant degree less than that seen in cells expressing FFA4-S-WT (Fig 7B), suggesting that this component of receptor activity may be both β-arrestin and Gαq/11-dependent. Similar to results seen in ROS-generation assays, the addition of TUG-891 to cells overexpressing these constructs did not further the effect (data not shown), suggesting that heightened receptor numbers yield maximal activity that is not sensitive to further agonism. In Raw 264.7 macrophages, LPS-induced phosphorylation of ERK1/2, can be activated in part by Ca2+-dependent signals through PI3-Kinase, phospholipase-C, or PKC activity (33–35), as well as via TAK1 scaffolds that are upheld by β-arrestin-2 (13,36–37). Hence, we next examined the effects of FFA4 isoform and C-terminal mutant expression on LPS-mediated ERK1/2 phosphorylation. Similar to that seen with NF-κB activity, our results show that FFA4-S-WT and FFA4-S–Δ340, but not FFA4-L-WT or FFA4-L–Δ356, decrease LPS-mediated ERK1/2 phosphorylation, compared to untransfected Raw 264.7 cells (Fig 7C–F). Taken together, these data suggest that there are differences in PMA vs. LPS-mediated pathways targeted by FFA4 isoforms, and that while the C-terminal tail of FFA4-S is an important component of its ability to reverse PMA-mediated ROS-generation and COX-2 upregulation, it is not required for LPS-driven NF-κB activity or ERK1/2 phosphorylation. Meanwhile, though FFA4-L-WT decreased PMA-induced ROS generation and COX-2 expression, it had no effect in decreasing LPS-stimulated NF-κB activity or phosphorylation of ERK1/2, suggesting that the isoforms have distinct abilities to modulate differential signal transduction cascades.
Figure 7. Effects of FFA4 isoforms or β-arrestin phosphosensor-mutants on LPS-induced NF-κB activity and ERK1/2 phosphorylation.

A, LPS-stimulated NF-κB activity was assessed in untransfected Raw 264.7 cells stably expressing the pNF-κB –MetLuc2-reporter (UNTR) or the corresponding constitutively active positive control, as described in the Experimental Procedures. Cells expressing the reporter were stimulated with vehicle or TUG-891 (3 μM) for 1 h prior to overnight treatment with 100 ng/ml LPS. NF-κB metridia luciferase activity was normalized to control renilla luciferase values to account for transfection efficiency and results are expressed as mean of the maximal LPS-response ± SEM for three independent experiments. Statistical analysis was performed by ANOVA and * denotes p < 0.05 versus the LPS-response. B, Untransfected Raw 264.7 cells (UNTR) or those stably expressing FFA4 isoforms or mutants and transiently expressing the pNF-κB –MetLuc2-reporter or the corresponding constitutively active positive control, were stimulated with LPS (100 ng/ml) for 16 h as described in A. NF-κB metridia luciferase activity was normalized to control renilla luciferase values to account for transfection efficiency and results are expressed as mean of the maximal LPS-response ± SEM for three independent experiments. Results show that FFA4-S-WT and FFA4-S–Δ340, but not FFA4-L-WT and FFA4-L–Δ356, decrease LPS-induced NF-κB activity. Statistical analysis was performed by ANOVA and *** denotes p < 0.001 versus the LPS-response in untransfected Raw 264.7 cells while †† denotes p < 0.01 versus the LPS-response in cells expressing FFA4-S-WT. C–F, Phosphorylation of ERK1/2 was assessed by immunoblotting of phospho-ERK1/2, and ERK1/2 followed by quantification of the phospho-ERK1/2 over ERK1/2, as described in the Experimental Procedures. Untransfected control cells (UNTR), or cells expressing FFA4 isoforms or mutants were stimulated with vehicle or LPS (100 ng/ml) for 30 min. Results are expressed as a percentage of the maximal LPS-induced response. Results show that FFA4-S-WT (C) and FFA4-S–Δ340 (D), but not FFA4-L-WT (E) or FFA4-L–Δ356 (F) decrease LPS-induced ERK1/2 phosphorylation. Statistical analysis was performed by ANOVA and ** denotes p < 0.01 versus the LPS-response in untransfected Raw 264.7 cells.
4. DISCUSSION
In a previous study, we have shown that agonism of FFA4 with DHA facilitates its rapid phosphorylation at Thr347, Ser350, Ser357 located within the C-terminal tail (18). Butcher and colleagues subsequent work showed that TUG-891 induces phosphorylation at these sites, and also at nearby Thr349 and Ser360 (19), which together with neighboring acidic residues Glu341, Asp348, and Asp355 comprise an area of negative charge that defines the β-arrestin phosphosensor (19). Indeed, truncation of the C-terminal tail following Pro340 formed a mutant FFA4 receptor that failed to interact with, or signal through, β-arrestin-2 but was otherwise signal-capable (19). In this study, we examined the role that the β-arrestin phosphosensor plays in modulating activities of both FFA4 isoforms in Raw 264.7 macrophages. Our results show that agonism of endogenous FFA4 receptors on the cell-surface of native Raw 264.7 macrophages with TUG-891 significantly reduces PMA-induced ROS generation, an effect that is blocked by the FFA4 antagonist AH7614. Utilizing stable cell lines that express either FFA4 isoform or their respective phosphosensor-null mutant, our results demonstrate that both FFA4-S and FFA4-L can inhibit PMA-induced ROS generation, yet, only removal of the FFA4-S, but not the FFA4-L phosphosensor significantly altered this effect. This result is intriguing and suggests that the presence of the additional 16 amino acid sequence in the 3rd ICL of FFA4-L offsets the loss of its C-terminal tail in regards to signals that facilitate the blockade of NOX-generated ROS.
ROS have been shown to contribute in part to upregulation of COX-2 in a variety of cell types, including macrophages (3–7). Meanwhile, agonism of FFA4 with DHA has been shown to decrease COX-2 expression in Raw 264.7 cells via β-arrestin (15), prompting us to examine the role of loss of the β-arrestin phosphosensor towards this effect. Here, we show that PMA-induces upregulation of COX-2 and that expression of FFA4-S significantly abrogates this effect, while expression of FFA4-L was able to do so to a lesser, albeit still significant degree. The decrease in COX-2 expression translated to a significant reduction of PGE2 and its metabolites only in cells expressing FFA4-S, questioning the physiological significance of the FFA4-L induced reduction of COX-2. Notably, our results point to a definitive role for the FFA4-S phosphosensor towards decreasing COX-2 expression, and this result is in line with previous data showing that knockdown of β-arrestin-2 in Raw 264.7 cells abolishes the ability of DHA to reduce COX-2 via FFA4 endogenously expressed on these cells (15). Our results here also confirm the partial ROS dependence of COX-2 expression, as NAC decreased the PMA-induced effect in untransfected cells, yet this effect was lacking in cells expressing wild-type FFA4 isoforms, which inherently already reduce ROS levels. In these cells, PMA-induced COX-2 expression was not further reduced by NAC treatment, suggesting that the ROS-contribution to this axis was already maximally inhibited upon wild-type FFA4 overexpression. Our results showing upregulation of catalase in only cells which overexpress FFA-S-WT, but not other stable transfectants, confirms this hypothesis and could also mechanistically explain the near full abolishment of PMA-induced ROS generation by the shorter isoform. Expression of FFA4-L-WT also significantly reduced PMA-induced ROS generation, but yet, no significant upregulation in catalase was detected in these cells. Hence, even though the effect of FFA4-S on PMA-induced ROS generation was significantly more profound than that of FFA4-L, catalase upregulation likely only plays a partial role in absolving acute ROS generation. While canonical signaling of GPCRs has been shown to modulate COX-2 expression patterns (38–43), a recent study found that the C-terminal tail of the angiotensin-II type 1 GPCR regulated COX-2 expression in a manner independent of canonical signals, including phosphorylation of the C-terminal tail and β-arrestin recruitment (44). Rather, ATII-1R decreases COX-2 expression via direct enhancement of COX-2 ubiquitination and subsequent proteosomal degradation, an effect that was facilitated by the C-terminal tail in absence of the rest of the GPCR (44). In this regard, our results did not reveal any significant alterations to COX-2 ubiquitination induced by FFA4 isoforms or C-terminal truncates (data not shown), suggesting that the effects of FFA4 on COX-2 expression are not modulated by simple enhancement of degradation, distinct from that seen with ATII-1R.
FFA4-S agonism and overexpression has a clear role in decreasing ROS generation and COX-2 expression induced by PMA, yet in our studies, agonism of stably expressing cells with TUG-891 did not facilitate a further decrease in either of these effects. On the contrary, antagonism of FFA4 in stably expressing cells served to significantly inhibit the observed decreases in ROS generation. These results were surprising, but suggest that receptor overexpression in our stable cell lines likely encompasses heightened constitutive activity that has reached maximal efficacy and which is not sensitive to further activity upon receptor agonism. Despite a 4–5-fold increase in transcript expression in our stable cell lines, a level that should not signify artefactually massive overexpression, these observations were contradictory to those in untransfected Raw 264.7 cells, where agonism of the low levels of endogenous FFA4 (13) with TUG-891 did elicit both ROS and COX-2 responses. Interestingly, when we examined the effects of TUG-891 on PMA-induced ROS generation in primary intraperitoneal macrophages, which express FFA4 (13), we failed to detect a robust PMA-induced ROS generation (data not shown), making it difficult to interpret if FFA4 modulates this cascade in these cells. Nonetheless, in our stable Raw 264.7 cell lines, antagonism of stably expressed FFA4 isoforms with AH7614 was able to significantly reduce receptor constitutive activity, confirming the FFA4-mediated mechanism, and corroborating our hypothesis on the lack of further effect upon agonism.
Due to its more ubiquitous expression, much more work has been published on the shorter FFA4 isoform (12), while very little has been described regarding the function of FFA4-L. A study published in 2012 showed that agonism of FFA4-S transiently expressed in HEK-293 cells could signal via both Gαq/11 and β-arrestin-2 cascades, while importantly, agonism of FFA-L was incapable of promoting Ca2+-dependent signals via Gαq/11, yet could signal fully via β-arrestin-2 (17). Our results shed further light on differences in signal capabilities of the two FFA4 isoforms. While both isoforms were capable of significantly reducing PMA-induced ROS generation and COX-2 expression, only FFA4-S was able to significantly reduce LPS-stimulated NF-κB and ERK1/2 activity. This raises important questions regarding the signaling mechanisms leading to blockade of PKC-dependent functions such as PMA-induced activation of NOX and COX-2 expression, versus LPS/TLR4-dependent cascades. Furthermore, while expression of both FFA4-S-WT and FFA4-S–Δ340 was able to reduce LPS-stimulated NF-κB activity, the C-terminal truncate was significantly less robust at doing so, confirming previous results that showed a β-arrestin component to FFA4 modulation of NF-κB in Raw 264.7 cells (13). In that study, the FFA4-β-arrestin-2 axis was responsible for sequestration of TAB1 by β-arrestin-2, blocking the TAB1/TAK1 interaction that facilitates NF-κB activity (13). The ability of FFA4-β-arrestin-2 signaling to inhibit TAK1 effects are also of note given the importance of TAK1 in ROS-dependent upregulation of COX-2 (5), signifying that the noted effects of FFA4 on ROS generation may also be mediated, at least in part, by β-arrestin-2, a hypothesis that is confirmed by reduced ability of FFA4-S–Δ340 to decrease ROS generation. However, a curiosity of these data is the lack of effect of FFA4-L-WT, the purported β-arrestin-biased isoform, on inhibition of NF-κB activity, since neither FFA4-L-WT or FFA4-L–Δ356 were able to reduce LPS-mediated NF-κB or ERK1/2 activities, contrary to their ability to reduce PMA-mediated ROS generation.
In summary, we have found that agonism of FFA4 facilitates decreases in ROS generation and COX-2 expression, and that the C-terminal β-arrestin-phosphosensor of FFA4-S, but not FFA4-L, is in part required for this effect. Meanwhile, our results point to a partial contribution of the C-terminal phosphosensor of FFA4-S in modulating LPS-induced effects, which were completely lacking in the longer isoform.
Acknowledgments
This work was supported by NIH grant DK098730 to N.H.M and in part by two Diabetes Action Research and Educational Foundation grants to N.H.M.
The abbreviations used are
- FFA4
Free-fatty acid receptor-4
- ROS
reactive oxygen species
- COX-2
cyclooxygenase-2
- PMA
phorbol-12-myristate 13-acetate
Footnotes
Chemical compounds studied in this article: TUG-891 (Pubchem CID 57522038); AH7614 (PubChem CID 233085).
CONFLICT OF INTEREST
The authors declare that they have no conflicts of interest with the contents of this article.
AUTHOR CONTRIBUTIONS
AC designed the studies, performed all experiments, analyzed the results, and wrote the manuscript. ISS constructed expression vectors for truncated FFA4 receptors. JD and OG synthesized TUG-891 and AH7614. NHM conceived and oversaw the studies, analyzed the results, and wrote and edited the manuscript. All authors approved the final version of the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Chawla A, Nguygen KD, Goh YPS. Macrophage-mediated inflammation in metabolic disease. Nat Rev Immun. 2011;11:738–749. doi: 10.1038/nri3071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gloire E, Legrand-Poels S, Piette J. NF-KappaB activation by reactive oxygen species: fifteen years later. Biochem Pharmacol. 2006;72(11):1493–11505. doi: 10.1016/j.bcp.2006.04.011. [DOI] [PubMed] [Google Scholar]
- 3.Lee CW, Lin ZC, Hu SC, Chiang YC, Hsu LF, Lin YC, Lee IT, Tsai MH, Fang JY. Urban particulate matter down-regulates filaggrin via COX2 expression/PGE2 production leading to skin barrier dysfunction. Sci Rep. 2016;6:27995. doi: 10.1038/srep27995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Callaway DA, Jiang JX. Reactive oxygen species and oxidative stress in osteoclastogenesis, skeletal aging and bone diseases. J Bone Miner Metab. 2015;33(4):359–370. doi: 10.1007/s00774-015-0656-4. [DOI] [PubMed] [Google Scholar]
- 5.Onodera Y, Teramura T, Takehara T, Shigi K, Fukuda K. Reactive oxygen species induce Cox-2 expression via TAK1 activation in synovial fibroblast cells. FEBS Open Bio. 2015;5:492–501. doi: 10.1016/j.fob.2015.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Martínez-Revelles S, Avendaño MS, García-Redondo AB, Alvarez Y, Aguado A, Pérez-Girón JV, García-Redondo L, Esteban V, Redondo JM, Alonso MJ, Briones AM, Salaices M. Reciprocal relationship between reactive oxygen species and cyclooxygenase-2 and vascular dysfunction in hypertension. Antioxid Redox Signal. 2013;18(1):51–65. doi: 10.1089/ars.2011.4335. [DOI] [PubMed] [Google Scholar]
- 7.Cheng SE, Lee IT, Lin CC, Wu WL, Hsiao LD, Yang CM. ATP mediates NADPH oxidase/ROS generation and COX-2/PGE2 expression in A549 cells: role of P2 receptor-dependent STAT3 activation. PLoS One. 2013;8:e54125. doi: 10.1371/journal.pone.0054125. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 8.Zhang Y, Choksi S, Chen K, Pobezinskaya Y, Linnoila I, Liu ZG. ROS play a critical role in the differentiation of alternatively activated macrophages and the occurrence of tumor-associated macrophages. Cell Res. 2013;23(7):898–914. doi: 10.1038/cr.2013.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Calder PC. Omega-3 fatty acids and inflammatory processes. Nutrients. 2010;2(3):355–374. doi: 10.3390/nu2030355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Oliver E, McGillicuddy F, Phillips C, Toomey S, Roche HM. The role of inflammation and macrophage accumulation in the development of obesity-induced type 2 diabetes mellitus and the possible therapeutic effects of long-chain n-3 PUFA. Proc Nutr Soc. 2010;69(2):232–43. doi: 10.1017/S0029665110000042. [DOI] [PubMed] [Google Scholar]
- 11.Yacoubian S, Serhan CN. New endogenous anti-inflammatory and proresolving lipid mediators: implications for rheumatic diseases. Nat Clin Pract Rhematol. 2007;3(10):570–579. doi: 10.1038/ncprheum0616. [DOI] [PubMed] [Google Scholar]
- 12.Moniri NH. Free-fatty acid receptor-4 (GPR120): Cellular and molecular function and its role in metabolic disorders. Biochem Pharmacol. 2016;110–111:1–15. doi: 10.1016/j.bcp.2016.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Oh DY, Talukdar S, Bae EJ, Imamura T, Morinaga H, Fan W, Li P, Lu WJ, Watkins SM, Olefsky JM. GPR120 is an omega-3 fatty acid receptor mediating potent anti-inflammatory and insulin-sensitizing effects. Cell. 2010;142(5):687–698. doi: 10.1016/j.cell.2010.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Williams-Bey Y, Boularan C, Vural A, Huang NN, Hwang IY, Shan-Shi C, Kehrl JH. Omega-3 free fatty acids suppress macrophage inflammasome activation by inhibiting NF-κB activation and enhancing autophagy. PLoS One. 2014;9(6):e97957. doi: 10.1371/journal.pone.0097957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li X, Yu Y, Funk CD. Cyclooxygenase-2 induction in macrophages is modulated by docosahexaenoic acid via interactions with free fatty acid receptor 4 (FFA4) FASEB J. 2013;27(12):4987–4997. doi: 10.1096/fj.13-235333. [DOI] [PubMed] [Google Scholar]
- 16.Burns RN, Moniri NH. Agonism with the omega-3 fatty acids alpha-linolenic acid and docosahexaenoic acid mediates phosphorylation of both the short and long isoforms of the human GPR120 receptor. Biochem Biophys Res Commun. 2010;396(4):1030–1035. doi: 10.1016/j.bbrc.2010.05.057. [DOI] [PubMed] [Google Scholar]
- 17.Watson SJ, Brown AJ, Holliday ND. Differential signaling by splice variants of the human free fatty acid receptor GPR120. Mol Pharmacol. 2012;81(5):631–642. doi: 10.1124/mol.111.077388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Burns RN, Singh M, Senatorov IS, Moniri NH. Mechanisms of homologous and heterologous phosphorylation of FFA receptor 4 (GPR120): GRK6 and PKC mediate phosphorylation of Thr347, Ser350, and Ser357 in the C-terminal tail. Biochem Pharmacol. 2014;87(4):650–659. doi: 10.1016/j.bcp.2013.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Butcher AJ, Hudson BD, Shimpukade B, Alvarez-Curto E, Prihandoko R, Ulven T, Milligan G, Tobin AB. Concomitant action of structural elements and receptor phosphorylation determines arrestin-3 interaction with the free fatty acid receptor FFA4. J Biol Chem. 2014;289(26):18451–18465. doi: 10.1074/jbc.M114.568816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shimpukade B1, Hudson BD, Hovgaard CK, Milligan G, Ulven T. Discovery of a potent and selective GPR120 agonist. J Med Chem. 2012;55(9):4511–5. doi: 10.1021/jm300215x. [DOI] [PubMed] [Google Scholar]
- 21.Lu S, Lu C. Efficient Synthesis of N-(9-xanthyl)-p-toluenesulfonamides enabled by an addition-cyclization cascade of arynes. Synlett. 2013;24(5):640–644. [Google Scholar]
- 22.Sparks SM, Chen G, Collins JL, Danger D, Dock ST, Jayawickreme C, Jenkinson S, Laudeman C, et al. Identification of diarylsulfonamides as agonists of the free fatty acid receptor 4 (FFA4/GPR120) Bioorg Med Chem Lett. 2014;24(14):3100–3103. doi: 10.1016/j.bmcl.2014.05.012. [DOI] [PubMed] [Google Scholar]
- 23.Sohn HY, Gloe T, Keller M, Schoenafinger K, Pohl U. Sensitive superoxide detection in vascular cells by the new chemiluminescence dye L-012. J Vasc Res. 1999;36(6):456–464. doi: 10.1159/000025688. [DOI] [PubMed] [Google Scholar]
- 24.Martel-Gallegos G, Casas-Pruneda G, Ortega-Ortega F, Sánchez-Armass S, Olivares-Reyes JA, Diebold B, Pérez-Cornejo P, Arreola J. Oxidative stress induced by P2X7 receptor stimulation in murine macrophages is mediated by c-Src/Pyk2 and ERK1/2. Biochim Biophys Acta. 2013;1830(10):4650–9. doi: 10.1016/j.bbagen.2013.05.023. [DOI] [PubMed] [Google Scholar]
- 25.Zhao K, Huang Z, Lu H, Zhou J, Wei T. Induction of inducible nitric oxide synthase increases the production of reactive oxygen species in RAW264.7 macrophages. Biosci Rep. 2010;30:233–241. doi: 10.1042/BSR20090048. [DOI] [PubMed] [Google Scholar]
- 26.Gieche J, Mehlhase J, Licht A, Zacke T, Sitte N, Grune T. Protein oxidation and proteolysis in RAW264.7 macrophages: effects of PMA activation. Biochim Biophys Acta. 2001;1538(2–3):321–328. doi: 10.1016/s0167-4889(01)00083-0. [DOI] [PubMed] [Google Scholar]
- 27.Palmer CD, Rahmanm FZ, Sewell GW, Ahmed A, Ashcroft M, Bloom SL, Segal AW, Smith AM. Diminished macrophage apoptosis and reactive oxygen species generation after phorbol ester stimulation in Crohn’s disease. PLoS One. 2009;4(11):e7787. doi: 10.1371/journal.pone.0007787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Qiao S, Li W, Tsubouchi R, Haneda M, Murakami K, Takeuchi F, Nisimoto Y, Yoshino M. Rosmarinic acid inhibits the formation of reactive oxygen and nitrogen species in RAW264.7 macrophages. Free Radic Res. 2005;39(9):995–1003. doi: 10.1080/10715760500231836. [DOI] [PubMed] [Google Scholar]
- 29.Aga M, Watters JJ, Pfeiffer ZA, Wiepz GJ, Sommer JA, Bertics PJ. Evidence for nucleotide receptor modulation of cross talk between MAP kinase and NF-κB signaling pathways in murine RAW 264.7 macrophages. 2004;286(4):923–930. doi: 10.1152/ajpcell.00417.2003. [DOI] [PubMed] [Google Scholar]
- 30.Philippe C, Wauquier F, Léotoing L, Coxam V, Wittrant Y. GW9508, a free fatty acid receptor agonist, specifically induces cell death in bone resorbing precursor cells through increased oxidative stress from mitochondrial origin. Exp Cell Res. 2013;319(19):3035–41. doi: 10.1016/j.yexcr.2013.08.013. [DOI] [PubMed] [Google Scholar]
- 31.Zong Y, Sun L, Liu B, Deng YS, Zhan D, Chen YL, He Y, Liu J, Zhang ZJ, Sun J, Lu D. Resveratrol inhibits LPS-induced MAPKs activation via activation of the phosphatidylinositol 3-kinase pathway in murine RAW 264.7 macrophage cells. PLoS One. 2012;7(8):e44107. doi: 10.1371/journal.pone.0044107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schröfelbauer B, Raffetseder J, Hauner M, Wolkerstorfer A, Ernst W, Szolar OH. Glycyrrhizin, the main active compound in liquorice, attenuates pro-inflammatory responses by interfering with membrane-dependent receptor signalling. Biochem J. 2009;421(3):473–82. doi: 10.1042/BJ20082416. [DOI] [PubMed] [Google Scholar]
- 33.Meja KK, Seldon PM, Nasuhara Y, Ito K, Barnes PJ, Lindsay MA, Giembycz MA. p38 MAP kinase and MKK-1 co-operate in the generation of GM-CSF from LPS-stimulated human monocytes by an NF-kappa B-independent mechanism. Br J Pharmacol. 2000;131(6):1143–53. doi: 10.1038/sj.bjp.0703684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fenton MJ, Golenbock DT. LPS-binding proteins and receptors. J Leukoc Biol. 1998;64(1):25–32. doi: 10.1002/jlb.64.1.25. [DOI] [PubMed] [Google Scholar]
- 35.Lee SH, Kwak CH, Lee SK, Ha SH, Park J, Chung TW, et al. Anti-Inflammatory Effect of Ascochlorin in LPS-Stimulated RAW 264.7 Macrophage Cells Is Accompanied With the Down-Regulation of iNOS, COX-2 and Proinflammatory Cytokines Through NF-κB, ERK1/2, and p38 Signaling Pathway. J Cell Biochem. 2016;117(4):978–87. doi: 10.1002/jcb.25383. [DOI] [PubMed] [Google Scholar]
- 36.Endale M, Kim TH, Kwak Y, Kim NM, Kim SH, Cho JY, Yun BS, Rhee MH. Torilin Inhibits Inflammation by Limiting TAK1-Mediated MAP Kinase and NF-κB Activation. Mediators Inflamm. 2017 doi: 10.1155/2017/7250968. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang Y, Tu Q, Yan W, Xiao D, Zeng Z, Ouyang Y, Huang L, Cai J, Zeng X, Chen Y, Liu A. CXC195 suppresses proliferation and inflammatory response in LPS-induced human hepatocellular carcinoma cells via regulating TLR4-MyD88-TAK1-mediated NF-κB and MAPK pathway. Biochem Biophys Res Commun. 2015;456(1):373–379. doi: 10.1016/j.bbrc.2014.11.090. [DOI] [PubMed] [Google Scholar]
- 38.Oyesanya RA, Lee ZP, Wu J, Chen J, Song Y, Mukherjee A, Dent P, Kordula T, Zhou H, Fang X. Transcriptional and post-transcriptional mechanisms for lysophosphatidic acid-induced cyclooxygenase-2 expression in ovarian cancer cells. FASEB J. 2008;22(8):2639–2651. doi: 10.1096/fj.07-101428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Paruchuri S, Jiang Y, Feng C, Francis SA, Plutzky J, Boyce JA. Leukotriene E4 activates peroxisome proliferator-activated receptor gamma and induces prostaglandin D2 generation by human mast cells. J Biol Chem. 2008;283(24):16477–16487. doi: 10.1074/jbc.M705822200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Deacon K, Knox AJ. Endothelin-1 (ET-1) increases the expression of remodeling genes in vascular smooth muscle through linked calcium and cAMP pathways: role of a phospholipase A(2)(cPLA(2))/cyclooxygenase-2 (COX-2)/prostacyclin receptor-dependent autocrine loop. J Biol Chem. 2010;285(34):25913–25927. doi: 10.1074/jbc.M110.139485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dusaban S, Purcell NH, Rockenstein E, Masliah E, Cho MK, Smrcka AV, Brown JH. Phospholipase C epsilon links G protein-coupled receptor activation to inflammatory astrocytic responses. Proc Natl Acad Sci U S A. 2013;110(9):3609–3614. doi: 10.1073/pnas.1217355110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yoo J, Rodriguez Perez CE, Nie W, Sinnett-Smith J, Rozengurt E. BMC Gastroenterol. 2013;13:90. doi: 10.1186/1471-230X-13-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xiong SL, Liu X, Yi GH. High-density lipoprotein induces cyclooxygenase-2 expression and prostaglandin I-2 release in endothelial cells through sphingosine kinase-2. Mol Cell Biochem. 2014;389(1–2):197–207. doi: 10.1007/s11010-013-1941-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sood R, Minzel W, Rimon G, Tal S, Barki-Harrington L. Down-regulation of cyclooxygenase-2 by the carboxyl tail of the angiotensin II type 1 receptor. J Biol Chem. 2014;289(45):31473–31479. doi: 10.1074/jbc.M114.587576. [DOI] [PMC free article] [PubMed] [Google Scholar]