

Abstract
Mutant mice deficient in hyaluronan (HA) have an epileptic phenotype. HA is one of the major constituents of the brain extracellular matrix. HA has a remarkable hydration capacity, and lack of HA causes reduced extracellular space (ECS) volume in the brain. Reducing ECS volume can initiate or exacerbate epileptiform activity in many in vitro models of epilepsy. There is both in vitro and in vivo evidence of a positive feedback loop between reduced ECS volume and synchronous neuronal activity. Reduced ECS volume promotes epileptiform activity primarily via enhanced ephaptic interactions and increased extracellular potassium concentration; however, the epileptiform activity in many models, including the brain slices from hyaluronan synthase-3 knockout mice, may still require glutamate-mediated synaptic activity. In brain slice epilepsy models, hyperosmotic solution can effectively shrink cells and thus increase ECS volume and block epileptiform activity. However, in vivo, intravenous administration of hyperosmotic solution shrinks both brain cells and brain ECS volume. On the other hand, manipulations that increase the synthesis of high-molecular-weight HA or decrease its breakdown may be used in the future to increase brain ECS volume and prevent seizures in patients with epilepsy. Prevention of epileptogenesis is also a future target of HA manipulation. Head trauma, ischemic stroke, and other brain insults that initiate epileptogenesis are known to be associated with an early decrease in high-molecular-weight HA, and preventing that decrease in HA may prevent the epileptogenesis.
Keywords: anticonvulsant, ephaptic, epilepsy, extracellular matrix, hyaluronic acid, potassium
Introduction
The extracellular matrix (ECM) of the brain resides in the narrow spaces between the cells. The key feature of brain ECM is the abundance of hyaluronan (HA) and hyaluronan-binding proteoglycans, which are synthesized by both neurons and glia (Fig. 1). In the past few years, increasing attention has been focused on the role of matrix molecules in the regulation of physiological functions in the adult brain. In a recent paper (Arranz et al., 2014), we investigated the role of HA using mutant mice deficient in each of the three hyaluronan synthase genes (Has1, Has2, Has3). Interestingly, we found that all of these mutant mice develop spontaneous epileptic seizures, with Has3 knockout (KO) mice exhibiting the strongest phenotype (Fig. 2A). Our multidisciplinary analyses revealed that deficiency of HA results in a reduction in the volume of the extracellular space (ECS) in the hippocampal CA1 pyramidal cell body layer (Fig. 2B,C), and our experiments pointed to a causal relationship between this reduced ECS volume and epileptiform activity.
Figure 1.
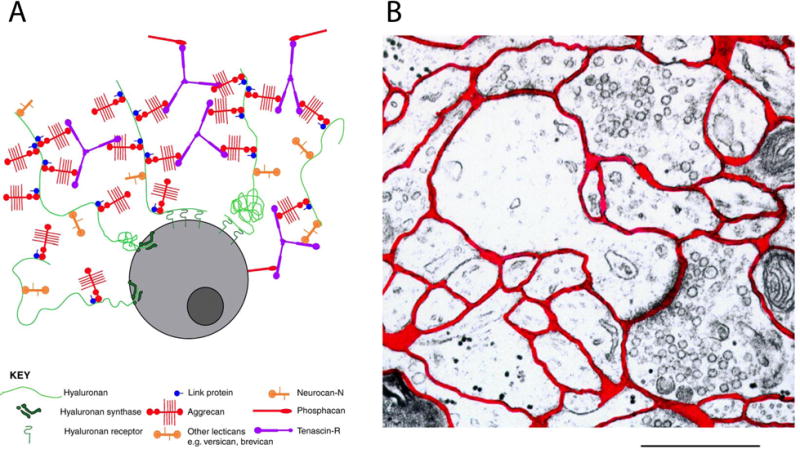
Extracellular matrix and extracellular space.
(A) Schematic of ECM around a brain cell. Hyaluronan (HA) provides a backbone to which some other ECM components, such as lecticans, are attached. HA is extruded into the ECS as it is synthesized, and can remain attached to the cell via the HA synthase or can bind to HA receptors on the cell surface, such as CD44. Some HA appears to be free floating in the ECS. (Reprinted from Galtrey and Fawcett, 2007, with permission). (B) Electron micrograph of rat neocortex. The ECS is labeled red. Scale bar is 1 μm. (Adapted from Nicholson and Sykova, 1998, with permission.)
Figure 2.
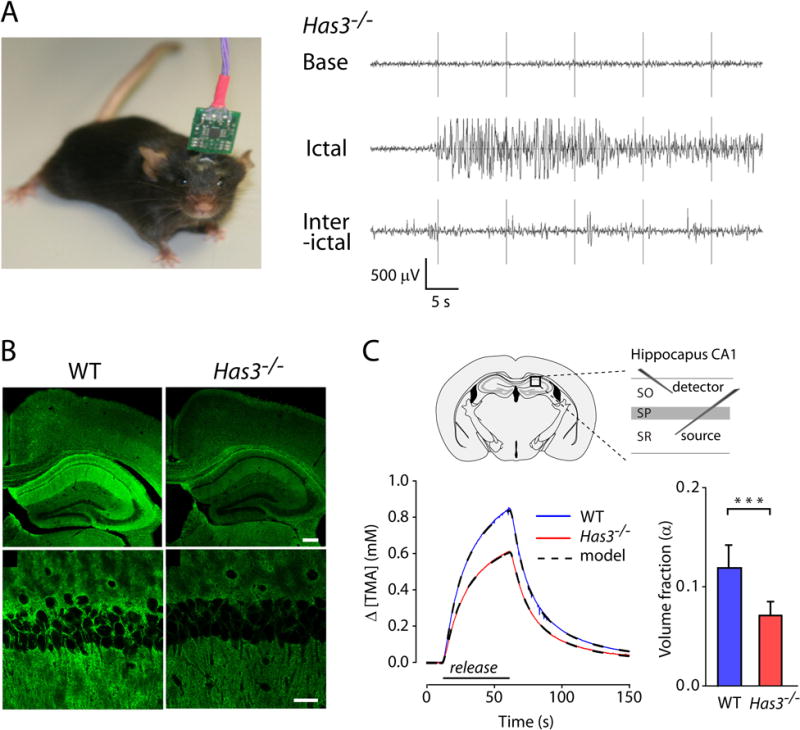
Spontaneous epileptic seizures and reduced extracellular space in the Has3−/− mice.
(A) Left, Photograph of a mouse with surgically implanted electrodes for 24 h EEG monitoring session. Right, Representative recordings of EEG activity in Has3−/− mice showing baseline activity (Base), electrographic seizure (Ictal) and interictal epileptic activity (Interictal). (B) Staining with hyaluronic acid binding protein shows HA reduction in the hippocampus of Has3−/− mice in comparison with wild type (WT) mice. Top row, Low-magnification images of hippocampus, neocortex and corpus callosum. Scale bar is 200 μm. Bottom row, High-magnification images of the CA1 hippocampal region. Scale bar is 30 μm. (C) Left, Representative RTI diffusion curves obtained in WT and Has3−/− mice are shown together with fitted multilayer model curves. Placements of iontophoretic microelectrode (source of TMA) and ion-selective microelectrode (detector of TMA) in the CA1 hippocampal region are indicated in the diagram. SO, stratum oriens, SP, stratum pyramidale, SR, stratum radiatum. Right, ECS volume fraction was reduced by 40% in the SP of the CA1 region in Has3−/− mice. Diffusion permeability was not changed (not shown). (Adapted from Figures 1, 2 and 7 in Arranz et al., 2014.)
In this review, we will discuss HA, brain ECS, and epilepsy, and focus on the causal relationship between reduced HA and reduced ECS volume and between reduced ECS volume and epilepsy. To wrap up the review, we will speculate about potential antiepileptic therapies targeted at increasing ECS volume, with a focus on increasing HA in the brain.
Hyaluronan
Structure
Hyaluronan (HA), which is also known as hyaluronic acid or hyaluronate, is a glycosaminoglycan found in nearly all extracellular matrices. HA is the largest polysaccharide found in vertebrates, and is composed of unbranched, repeating disaccharide units of N-acetylglucosamine (GlcNAc) and glucuronic acid (GlcUA) (Weissmann and Meyer, 1954; Weissmann et al., 1954). In normal physiological conditions, a single HA chain can be 2,000 to 25,000 disaccharides long, with a molecular mass of 106 to 107 Da, and an extended length of 2–25 μm (Toole, 2004). The carboxyl groups on GlcUA residues are negatively charged under physiological conditions, making HA polyanionic. In dilute solutions of HA, each HA molecule takes on a coil-like shape similar to a contracted 4-fold helix (Almond et al. 2006) and occupies a very large volume due in part to mutual repulsion between the carboxyl groups (Balazs, 1974; Toole, 2004). The hydroxyl, carboxyl, and amido groups on HA can form hydrogen bonds with groups on the adjacent saccharide in the chain or with water; a single water molecule or a dimer of water molecules can form a water bridge between adjacent saccharides in the chain (Fig. 3A; Almond, 2005; Almond et al., 2006; Prusova et al., 2010a). The intramolecular hydrogen bonds can dynamically interchange with hydrogen bonds to water, which lends some flexibility to the HA chain (Fig. 3B; Almond et al., 2006; Almond, 2007). In concentrated solutions of HA, the individual strands of HA intertwine and form a meshwork (Toole, 2004); furthermore, two different HA molecules can form hydrogen bonds with the same water molecule, lending additional structure to the HA-water solution (Prusova et al., 2010a).
Figure 3.
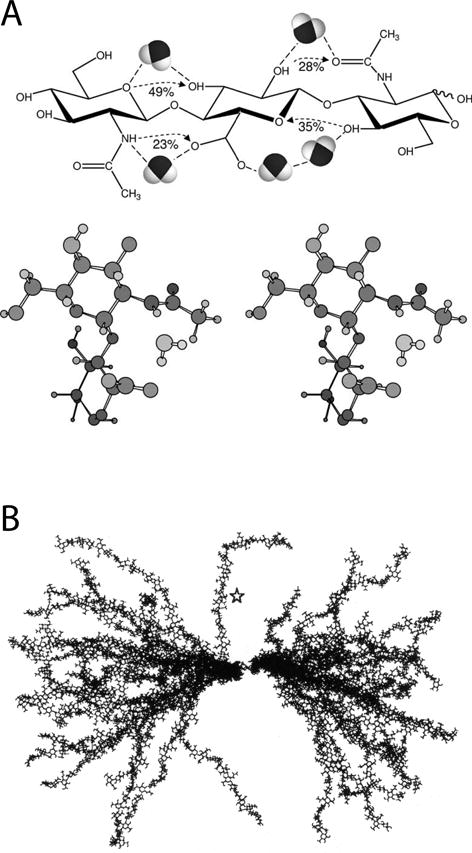
Structure of hyaluronan and its interaction with water.
(A) Top: Hyaluronan trisaccharide GlcNAc-GlcUA-GlcNAc shown with predicted intramolecular hydrogen bonds (with % occupancy) and water bridges. Bottom: Stereo image of a water molecule bridging the amide of GlcNAc and the carboxyl of GlcUA. (Reprinted from Almond, 2005, with permission.)
(B) Molecular dynamics simulations were used to construct 25 likely conformations in solution of an HA chain with 50 sugar residues. The molecules were overlaid on the central two sugar residues. The star indicates a relatively straight portion of a chain which exhibits a four-fold helical structure. (Reprinted from Almond et al., 2006, with permission.)
Distribution
HA is a major component of extracellular matrices, and it is ubiquitous in various organs and tissues. In mammals, the highest HA concentrations are found in connective tissues such as skin, synovial fluid, umbilical cord and the vitreous body. In addition, notable amounts are present in brain, lung, kidney and muscle (Fraser et al., 1997). In the adult brain, HA is widely distributed throughout the ECS (Fig. 1), but it is particularly found surrounding neuronal cell bodies, forming perineuronal nets (PNNs; Fig. 4), and localized around myelinated fibers (Sherman et al., 2002).
Figure 4.
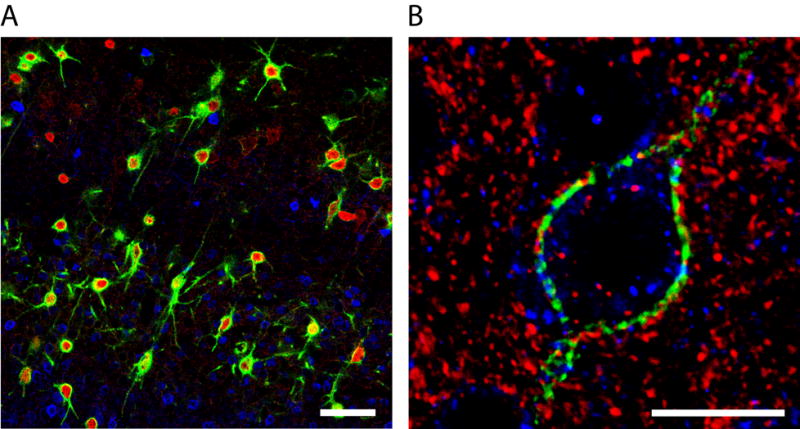
Perineuronal nets.
(A) Confocal image showing PNNs (Wisteria floribunda agglutinin staining, green) associated primarily with parvalbumin-positive inhibitory neurons (red) in the mouse neocortex while calbindin-positive neurons (blue) lack PNNs. Scale bar is 50 μm. (B) Confocal image showing the fine structure of a PNN (Wisteria floribunda agglutinin staining, green) surrounding a neuron in mouse neocortex. Anti-vesicular glutamate transporter-1 (red) and anti-vesicular inhibitory amino acid transporter (blue) antibodies are used to detect excitatory and inhibitory synapses, respectively. Scale bar is 25 μm. (Modified from Cover in Arranz et al., 2014.)
Hyaluronan synthases and biosynthesis of HA
HA belongs to the family of glycosaminoglycans that also includes chondroitin sulfate, heparan sulfate, and keratan sulfate. The synthesis of all glycosaminoglycans other than HA requires multiple different enzymes. That synthesis begins with the enzymatic attachment of the initiating saccharide to a serine residue on a core protein in the endoplasmic reticulum or Golgi (Uyama et al., 2007). The core protein then translocates through the Golgi, where the growing saccharide chain is selectively epimerized and sulfated (Uyama et al., 2007). The core protein with glycosaminoglycans attached is then secreted into the ECS. In contrast, HA synthesis requires only a single enzymatic step, mediated by hyaluronan synthase (HAS). HA is synthesized at the inner surface of the plasma membrane by HAS and directly extruded into the ECS as a chain of disaccharides, without epimerization or sulfation (Weigel, 2015). Vertebrate animals possess three hyaluronan synthases, namely HAS1, HAS2, and HAS3, which are encoded by independent genes. All HAS proteins are multipass transmembrane proteins which are believed to form a pore in the plasma membrane, through which nascent HA is extruded as it is polymerized. Each HAS possesses dual N-acetylglucosaminyltransferase and glucuronyltransferase activities, and the enzymatic active sites are located in a major intracellular loop (DeAngelis and Weigel, 1994; Weigel, 2015).
Creation of Has KO mice has made significant contributions to our understanding of the role of HA in development. Lack of the Has2 gene leads to embryonic lethality at E9.5–10 due to cardiac and vascular abnormalities (Camenisch et al., 2000). In contrast to Has2 KO mice, Has1 and Has3 mutants are viable and fertile. The expression patterns of the three HAS enzymes vary during development (Spicer and McDonald, 1998; Tien and Spicer, 2005). Northern blot analysis of whole mouse embryos (Spicer and McDonald, 1998) reveals that HAS1 and HAS2 transcripts are expressed at embryonic day E7.5, but then HAS1 mRNA decreases to marginally detectable levels; whereas HAS2 mRNA has increased by E11.5 and remains high thereafter. HAS3 expression, on the other hand, is undetectable in the mouse embryo before E10.5, detected in just a few locations at E10.5 and E12.5, and detected more strongly and widely from E15.5 onward (Spicer and McDonald 1998; Tien and Spicer 2005). In adult mouse brain tissue, HAS1 is weakly expressed, whereas HAS2 and HAS3 are expressed at higher levels (Spicer and McDonald, 1998; Tien and Spicer, 2005).
Hyaluronidases and degradation of HA
HA is degraded by a set of catabolic enzymes known as hyaluronidases. In humans there are six hyaluronidase genes in the HYAL family, including one pseudogene, arranged in two sets of three contiguous genes located on two chromosomes (Csoka et al., 2001). The cluster on chromosome 3p21.3 is composed of HYAL1, HYAL2 and HYAL3, and the cluster on chromosome 7q31.3 consists of HYALP1, HYAL4 and SPAM1. These genes encode hyaluronidases with different properties and cellular locations. Other than the HYAL family of molecules, two putative hyaluronidases, which are thought to act in the extracellular space, have been identified. The first is the cell migration-inducing hyaluronan-binding protein (CEMIP), also known as KIAA1199, which is a secretory protein that has HA-binding and -degrading activities (Yoshida et al., 2013). More recently, transmembrane protein 2 (TMEM2) has been identified as a novel cell surface hyaluronidase (Yamamoto et al., 2017). TMEM2 is a type II transmembrane protein and its hyaluronidase activity resides in its extracellular domain. With regards to hyaluronidases in the brain, mRNA for HYAL3 was detected in brain tissue via Northern blot analysis (Csoka et al., 1999), and HYAL2 expression was recently revealed in endothelial cells of blood vessels in the brain and in ciliated epithelial cells of the choroid plexus using immunohistochemistry (Chowdhury et al., 2016). In addition, transcripts of both TMEM2 and CEMIP are present in adult mouse brain (Yamamoto et al., 2017).
HA degradation occurs both locally and after transport by lymph to lymph nodes and by blood to the liver and spleen (Fraser et al., 1997; Jadin et al., 2012). It has been estimated that 20–30% of HA degradation is local in tissues such as skin and joints (Fraser et al., 1997). Whether locally or after transport of the HA, hyaluronidases at the cell surface start degrading the HA chains. Then, cell-surface receptors, most importantly the hyaluronan receptor for endocytosis (HARE; Zhou et al., 2002), interact with HA fragments in the ECS and internalize them. After endocytosis, HA fragments are transported to lysosomes that contain hyaluronidases for complete degradation to monosaccharides.
HA turnover and formation of HA-rich pericellular coats
There is a dynamic balance between HA synthesis and degradation in the body. The half-life of HA in blood is brief (2–5 min), and the half-life of HA in tissues in the mature animal ranges from several hours (skin, synovial fluid; Brown et al., 1991; Laurent and Fraser, 1991) to 2 or 3 days according to Fraser et al. (1997). As far as we know, HA turnover rate per se in normal brain tissue has not been reported (but see the information on perineuronal net turnover below). HA levels are locally regulated during development and regeneration via regulation of HAS and hyaluronidases (e.g., Brecht et al., 1986; Contreras et al., 2009; Toole and Trelstad, 1971). In addition, tissue injury can cause a fast increase in HA that is due to increased HAS activity; for example, an HA increase was measured within 15 min after tissue injury in a rat keratinocyte organotypic culture model (Monslow et al., 2009).
Surrounding many cells which are undergoing differentiation or division, there is a HA-rich hydrated zone, or coat, typically 5–10 μm thick, which is critical to the differentiation or division process (Toole, 2001). In cell culture this HA-rich pericellular coat can be visualized by the particle exclusion method (Underhill and Toole, 1982). The HA-rich coat can form quickly; for example, the coat forms within 14 min on vascular smooth muscle cells in culture immediately prior to mitotic cell rounding (Evanko et al., 1999). The HA-rich coat is apparently created because HA molecules extruded into the ECS can remain attached to the HAS which synthesized them, tethering them to the cell (Evanko et al., 1999; Heldin and Pertoft, 1993). Additionally, HA-rich coats can also be created by HA binding to the cell surface receptor CD44 (Siiskonen et al., 2014; Qu et al., 2014).
Function of HA in the central nervous system
Numerous functions have been associated with HA. Some can be attributed to its ability to generate cell-free spaces upon hydration (see later section), some to its signaling via cell surface receptors (see below in this section), and others to its ability to organize and modify the overall structure of the ECM surrounding cells.
In the central nervous system (CNS), HA is a major component of the ECM and perineuronal nets (PNNs; Figs. 1 and 4). PNNs are specialized, mesh-like, ECM structures that form around certain neuronal cell bodies and proximal dendrites, leaving holes in the mesh at the sites of synaptic boutons (Oohashi et al., 2015). The major matrix components of PNNs include HA and chondroitin sulfate proteoglycans, such as the lecticans (brevican, neurocan, versican, and aggrecan) and phosphacan, along with tenascin-R and various link proteins (Yamaguchi, 2000; Deepa et al., 2006). In the neocortex and hippocampus, PNNs are primarily associated with parvalbumin-positive GABAergic inhibitory interneurons (Fig. 4A; Celio, 1993; Celio and Chiquet-Ehrismann, 1993), but they are present around various types of excitatory and inhibitory neurons throughout the CNS (Frischknecht and Gundelfinger, 2012). PNNs play an important role in synaptic stabilization and plasticity. For example, disruption of PNNs restores ocular dominance plasticity in the adult visual cortex (Pizzorusso et al., 2002) and facilitates recovery from early monocular deprivation (Pizzorusso et al., 2006). In addition, creation of PNNs seems to be directly involved in closing the critical period in barrel cortex (McRae et al., 2007; Nowicka et al., 2009). Furthermore, damage to the peripheral vestibular labyrinth of adult rats causes PNN reduction in the lateral vestibular nuclei of the brainstem in conjunction with remodeling of afferents, followed by restoration of PNNs when vestibular compensation is complete (Faralli et al., 2016). In that study it was shown that PNNs break down over the first 3 to 6 days, and have recovered significantly by day 12 and completely by day 24 (Faralli et al., 2016).
Other potential roles of HA in brain are related to its ability to signal through specific cell-surface receptors, such as CD44 and RHAMM. There are multiple CD44 isoforms that arise by alternative splicing, but all forms contain the HA-binding domain within their primary sequence (Thorne et al., 2004). CD44 expression occurs in glia and some neurons, and HA-CD44 interactions are involved in glial cell differentiation and migration and in post-traumatic brain repair (Dzwonek and Wilczynski, 2015). RHAMM, the receptor for HA-mediated motility, is alternatively spliced as well, and the different forms of the resulting protein are widely found both intracellularly and on the surface of neurons and glia (Lynn et al., 2001a; Lynn et al., 2001b). HA-RHAMM interaction has been implicated in astrocyte and microglial motility and in neurite extension (Nagy et al., 1995; Turley et al., 1994).
HA and its associated molecules are involved in other important processes in the central nervous system as well. In many cases, the role of hyaluronan has been examined by using exogenous hyaluronidase to enzymatically digest the hyaluronan in the ECM. For example, Förster and colleagues (2001) showed that hyaluronidase treatment disrupts axonal fiber segregation in organotypic slice cultures of hippocampus. In addition, hyaluronidase treatment increases lateral diffusion of AMPA-type glutamate receptors in the cell membranes of cultured primary hippocampal neurons and reduces paired pulse depression (Frischknecht el al., 2009). Hyaluronidase application to hippocampal brain slices reduces activation of voltage-gated L-type calcium channels and reduces long-term potentiation (LTP; Kochlamazashvili et al., 2010). Furthermore, disruption of the ECM in the hippocampus or medial prefrontal cortex with both hyaluronidase and chondroitinase results in impaired fear conditioning (Hylin et al., 2013). Although it has several advantages, application of hyaluronidase has its limitations. Perhaps most importantly, commercially available hyaluronidases contain other contaminating enzymatic activities, such as chondroitinase and protease; for example, mammalian testicular hyaluronidase preparations invariably possess chondroitinase activity. In addition, exogenous hyaluronidase will create HA digestion products, which themselves can have biological activity (Sherman et al., 2015). Rather than using hyaluronidase, our group took a genetic approach to investigate the role of hyaluronan in the CNS (Arranz et al., 2014). We showed that genetic ablation of HA synthesis is epileptogenic: mutant mice deficient in Has genes, especially Has3 KO mice, exhibit epileptic seizures and altered neuronal activity in the hippocampus. Moreover, these Has3 KO mice showed a significant reduction of both HA and ECS volume in the CA1 stratum pyramidale (SP), suggesting this epileptic phenotype is likely caused by a reduction in brain ECS volume (Arranz et al., 2014).
Brain extracellular space
The ECS is a system of contiguous narrow spaces demarcated by cellular membranes, and it resembles by electron microscopy a syncytium of continuous channels separating the outer membranes of neurons, glia and vascular elements (Fig. 1B). Individual intercellular channels are only about 30–60 nm wide (Thorne and Nicholson, 2006; Xiao et al., 2008), and they are filled with ionic solution and macromolecules of the ECM, predominantly proteoglycans and glycosaminoglycans. The ECS facilitates transport of neurotransmitters, neuromodulators, nutrients, metabolites, and therapeutics. The ECS also provides a reservoir of ions that are essential for neuronal activity. Substances released into the ECS spread predominantly by diffusion because the ECS lacks any active transport mechanism.
Diffusion can be exploited experimentally to quantify macroscopic properties of the ECS in live brain tissue. Diffusion experiments employing small extracellular probe molecules quantify two parameters of the ECS structure: volume fraction and diffusion permeability. The volume fraction (α) represents the proportion of tissue volume occupied by the ECS. Diffusion permeability (θ) quantifies the hindrance imposed on the diffusion process by the tissue relative to an obstacle-free medium. Diffusion permeability is defined as the ratio of the effective diffusion coefficient in the brain tissue (D*, cm2/s) to the free diffusion coefficient in a media without obstructions (D, cm2/s) (Hrabe et al., 2004). Alternatively, diffusion hindrance can be expressed as tortuosity, which is defined as (D/D*)1/2 (Nicholson and Phillips, 1981; Nicholson, 2001). In isotropic healthy brain, α is about 0.2 (i.e., about 20% of brain tissue volume resides in the ECS) and θ is about 0.4 (i.e., diffusion of a small extracellular probe molecule in brain tissue is slowed down about 2.5 times) (Sykova and Nicholson, 2008). These two ECS parameters are typically obtained with the Real-Time Iontophoretic (RTI) method (Nicholson and Phillips, 1981), in which an extracellular probe molecule, such as the small cation tetramethylammonium (TMA; 74 MW), is iontophoretically released from a point source and detected with an ion-selective microelectrode positioned about 100 – 200 micrometers away. The RTI method has been used to quantify α and θ in brain tissue under physiological control conditions and in animal models of neurological disorders (for review see Sykova and Nicholson, 2008).
The structure of brain ECS dynamically changes during various physiological conditions such as neuronal activation, sleep, and osmotic challenge (Sykova and Nicholson, 2008; Xie et al., 2013). A recent study from Nedergaard’s group (Xie et al., 2013) reported that ECS constricts (α decreases) during an awake state and widens during the sleep state, and hypothesizes that the enlargement during sleep is necessary for removal of toxins and metabolites from brain tissue. The ECS structure also changes, often permanently, during pathological conditions, such as brain trauma and disease. For example, diffusion is significantly hindered and the volume of the ECS is reduced in many neuropathological states associated with cellular edema (Sykova and Nicholson, 2008).
A growing body of work indicates that the ECM plays a significant role in the control of ECS properties. For example, genetic ablation of the glycoprotein tenascin-R reduces α in the cortical and hippocampal regions (Sykova et al., 2005), while genetic ablation of the link protein Bral-1 decreases θ in the white matter (Bekku et al., 2010). Furthermore, our group (Arranz et al., 2014) showed that absence of HA reduces α in the SP of the CA1 region of the hippocampus in Has3 KO mice (see below).
Evidence for reduced extracellular space in Has3 KO mice
Histological analysis of the forebrain of Has3 KO mice using biotinylated hyaluronic acid binding protein (HABP) revealed a pronounced reduction in the level of HA in the hippocampus proper (Arranz et al., 2014; Fig. 2B). This was in contrast to wild type (WT) mice, in which the hippocampus proper showed the most intense expression of HA. We also noticed that cell bodies in the CA1 SP, but not CA3 SP, of Has3 KO mice appeared to be more tightly packed than in WT mice (Fig. 2B). Morphometric analysis in both fixed hippocampal sections (processed by an ECS preserving fixation method; Cragg, 1980) and live slices revealed that the thickness of the CA1 SP was decreased by nearly 15% in the Has3 KO mice compared with WT mice. Further inspection of the CA1 SP in fixed sections showed 15% higher cell density in Has3 KO mice compared with WT mice, but no difference in the number of pyramidal cells or their size. Taken together, our histological studies suggested that a pronounced HA-deficiency in Has3 KO mice causes a reduction in the size of the ECS in CA1 SP.
Even under normal conditions in rat brain, the CA1 SP has significantly lower ECS volume than the two neighboring layers, stratum oriens (SO) and stratum radiatum (SR; McBain et al., 1990; Saghyan et al., 2012). Saghyan and colleagues (2012) showed that the diffusion of substances across the SP is significantly hindered because of lower ECS volume and lower diffusion permeability in this layer in comparison with adjacent SR and SO. To determine the impact of HA deficiency in Has3 KO mice on ECS properties in the CA1 SP, we designed two experiments to study ECS properties in the CA1 region of brain slices taken from Has3 KO mice (Arranz et al., 2014). First, we imaged spread of fluorophore-labeled dextran (which is confined to the ECS) through CA1 SP after its release in the SR. Second, we employed the RTI method (Nicholson and Phillips, 1981) with multilayer analysis (Saghyan et al., 2012) to quantify ECS volume and diffusion permeability (Fig. 2C). The imaging experiment revealed a significant reduction in dextran spread across the CA1 SP in Has3 KO mice, and the RTI experiment found a 40% reduction of ECS volume fraction in the CA1 SP of Has3 KO mice in comparison with WT mice, without significant change in diffusion permeability. Taken together, our diffusion analyses confirmed that there was a significant reduction of ECS volume in CA1 SP in Has3 KO mice.
Why might absence of HA cause reduced extracellular space volume?
Our study in Has3 KO mice provided the first evidence for the physiological role of HA in the maintenance of brain ECS volume. However, HA has been known to play a role in maintaining cartilage volume for some time (Servaty et al., 2001). In addition, HA is widely used as a cosmetic for plumping skin and minimizing wrinkles (Kablik et al., 2009; Pavicic et al., 2011). Those attributes of HA are due to its remarkable hydration capacity. There are three shells of water extending from the surface of a molecule of HA (Prusova et al., 2010b). The first shell is composed of bound water, which is also called non-freezing water because it is so strongly bound to HA that it cannot crystallize when cooled down (Prusova et al., 2010a, 2010b). There are 8–17 bound (non-freezing) water molecules per HA disaccharide unit (Prusova et al., 2010b). The second shell is composed of partially bound water, which is also called freezing-bound water. This water freezes, but at a lower temperature than bulk water (Prusova et al., 2010b). Liu and Cowman (2000), using differential scanning calorimetry data, calculated that there is 73 times more freezing-bound water than non-freezing water associated with each HA disaccharide unit. The third shell is composed of free water. The mobility of the water in the third shell is hindered somewhat by its interaction with the water of the second shell (Prusova et al., 2010a), but it freezes at approximately 0°C (Prusova et al., 2010b).
HA is synthesized by the HAS that is situated in the cell membrane, and as it is being synthesized, it is exuded through a pore in the enzyme into the ECS (Weigel, 2015). As it enters the ECS, the HA molecule imbibes water and expands (Toole, 2001). Based on observations of proliferating and migrating cells and on the association between HA extrusion and cell proliferation/migration, it has been hypothesized that this expansion of the HA molecule in the ECS can actually push cells and fibrous barriers away from the cell which is extruding the HA, creating cell-free space (Toole, 2001; Toole, 2004). Based on the hydration capacity of HA and its ability to create cell-free space, it was not surprising that absence of HA in the Has3 KO mice resulted in reduced volume of the brain ECS and more tightly packed cell bodies in the CA1 region of the hippocampus (Arranz et al., 2014).
Epilepsy
Has3 KO mice have epilepsy (Fig. 2A). Epilepsy is a disorder in which the brain experiences recurrent, abnormal, synchronous, excitatory activity of a population of neurons that manifests as behavioral seizures. The location of this abnormal activity and its degree of spread determine the behavioral phenotype of each seizure. Epileptic activity recorded in temporal lobe epilepsy in vivo can be divided into ictal activity and interictal activity. Ictal activity is the abnormal synchronous activity that occurs during a behavioral seizure. There is also abnormal synchronous activity that occurs when an epileptic person or animal is not having a seizure; these events are too short to cause a behavioral seizure and are termed interictal, to indicate that they occur during the time period between seizures. When recording epileptiform activity in vitro, it is the duration, and sometimes also the pattern, of the epileptiform event which is used to classify it as “ictal-like” or “interictal-like” (e.g. Salah and Perkins, 2011). Epileptiform activity is generally considered to be dependent upon chemical synaptic transmission (but see sections below for exceptions). Temporal lobe epilepsy is a common type of epilepsy, and abnormal activity in the hippocampus is often involved. In the hippocampus, the population of synchronously active cells that generate an epileptiform event are typically glutamatergic principal neurons – pyramidal cells in the CA1 or CA3 regions, or granule cells in the dentate gyrus.
Extracellular matrix disruption and epilepsy
There have been mixed results in experiments testing the effect of ECM disruption on neuronal excitability and seizure susceptibility. For example, mice deficient in the ECM glycoprotein tenascin-R show increased neuronal excitability in cortex and hippocampus (Brenneke et al., 2004; Gurevicius et al., 2004) but not increased susceptibility to seizures (Brenneke et al., 2004); in fact, kindling progression is actually retarded in tenascin-R –deficient mice (Hoffmann et al., 2009). On the other hand, an early study involving injection of hyaluronidase into the right lateral ventricle of the brain of cats resulted in epileptiform activity on EEG (Young, 1963). In our study (Arranz et al., 2014), Has3 KO mice showed both ictal and interictal activity on EEG, and the ictal activity was coincident with behavioral seizures.
Atypical features of the epileptiform activity in brain slices from Has3 KO mice
Somewhat surprisingly, brain slices made from Has3 KO mice show epileptiform activity without manipulation. The most prevalent activity is rhythmic interictal-like activity, but the slices also show occasional ictal-like events that are several seconds long. This spontaneous activity in the Has3 KO slices is surprising because typically, when brain slices are made from brain tissue from patients with intractable epilepsy, or from rodents with spontaneous seizures, the brain slice does not have spontaneous epileptiform activity (e.g., Avoli et al., 1995), or else has spontaneous interictal-like activity but not spontaneous ictal-like activity (Huberfeld et al., 2007; Köhling et al., 1998; LeDuigou et al., 2008). Typically some manipulation is required in order to induce epileptiform activity in these brain slices (Avoli et al., 1987; Avoli and Olivier, 1989; Gabriel et al., 2004; LeDuigou et al., 2008).
The CA1 region of the hippocampus is the site of the epileptiform activity in brain slices from Has3 KO mice. In contrast, the CA3 region of the hippocampus is the driver of spontaneous epileptiform activity in many in vitro epilepsy models. Compared to the CA1 region, the CA3 region has far more recurrent excitatory connections among pyramidal cells, which have been shown to promote synchronization (Traub and Wong, 1982). In these models, cutting the Schaffer collateral pathway or excising the CA3 region typically blocks spontaneous, rhythmic epileptiform activity in the CA1 region (Schwartzkroin and Prince, 1978; Miles et al., 1984; Traynelis and Dingledine, 1988; Mody et al., 1987; Perreault and Avoli, 1992; Walther et al., 1986). In contrast to these models in which the CA3 region acts as the “pacemaker” for epileptiform activity, brain slices from Has3 KO mice show very little or no synchronous activity in the CA3 region and yet simultaneously exhibit robust, rhythmic, ionotropic glutamate receptor-dependent, interictal-like events and occasional ictal-like events in the CA1 region (Arranz et al., 2014). In addition, this epileptiform activity in the CA1 region is unaffected by removal of the CA3 region, which shows that intact excitatory connections from the CA3 region to the CA1 region are not necessary for the expression, or even the rhythmic triggering, of epileptiform activity in the CA1 region of the Has3 KO mice (Arranz et al., 2014).
Ictal-like activity in the CA1 region of hippocampal slices- Susceptibility caused by small ECS volume?
Even though it is true that the CA3 region tends to be the pacemaker for rhythmic epileptiform activity in the hippocampus, the CA1 region may be more likely to generate longer epileptiform events. Typically, the individual epileptiform events recorded in the CA3 region of hippocampal slices are less than 1 s long, which makes them more akin to interictal activity than to the longer ictal events which underlie behavioral seizures. Notably, many published recordings of ictal-like events in the CA regions of hippocampal slices from adult animals are from the CA1 region, not the CA3 region (Bikson et al., 2002; Jensen and Yaari, 1988; Jiruska et al., 2010; Karnup and Stelzer, 2001; Ziburkus et al., 2006). Even when CA3 is acting as the pacemaker, the epileptiform events may be more ictal-like in the CA1 region, as in the high K+ model (Traynelis and Dingledine, 1988). Why is the CA1 region prone to ictal-like activity? Could it be because CA1 has a smaller ECS volume than CA3, thus enhancing nonsynaptic mechanisms of synchronization? Indeed, McBain et al. (1990) used the RTI method (Nicholson and Phillips, 1981) to show that the CA1 SP has a lower extracellular volume fraction than the CA3 SP (0.12 vs. 0.18). Further supporting this hypothesis, nonsynaptic mechanisms of synchronization that are enhanced in conditions of reduced ECS volume have been identified, as detailed in the next section.
Nonsynaptic mechanisms of epilepsy that might be enhanced in conditions of reduced extracellular space volume
Epileptiform activity is generally considered to be dependent upon chemical synaptic transmission, but there is ample evidence that nonsynaptic mechanisms also play a role; there are even instances in which synchronous excitatory activity has been recorded in the absence of chemical synaptic transmission (see next section). Reduced ECS may amplify the contribution of nonsynaptic mechanisms. Nonsynaptic mechanisms that have been proposed to play a role in enhancing epileptiform activity are 1) build-up of extracellular K+, 2) ephaptic interactions/field effects and 3) gap junctions (electrical synapses).
Potassium
Seizure activity causes K+ to leave cells and build up in the ECS (Fertziger and Ranck, 1970), plateauing at a concentration of 8–11 mM in neocortex (Fisher et al., 1976; Machado et al., 2016; Moody et al., 1974; Lux, 1974; Sypert and Ward, 1974) and 5–10 mM in the CA1 pyramidal cell layer in hippocampus in vivo (Fisher et al., 1976). In their 1970 paper, Fertziger and Ranck proposed a regenerative model in which neuronal activity increases extracellular K+ concentration ([K+]o) and high [K+]o in turn increases neuronal activity, leading to a seizure. A reduced ECS volume would be expected to enhance the increase in [K+]o since the K+ that is exiting from active neurons would be accumulating in a smaller volume. Furthermore, the reduction in ECS volume that accompanies activity (see below) may further concentrate the potassium.
It has been demonstrated that a moderate increase in [K+]o can enhance existing epileptiform activity (Hablitz and Lundervold, 1981; Wang et al., 2016), induce seizures in vivo (Zuckermann and Glaser, 1968), and induce ictal-like activity in the CA1 region of hippocampus in vitro (Traynelis and Dingledine, 1988). Several mechanisms are involved in the increased excitability associated with increased [K+]o. First of all, a rise in [K+]o to 5–10 mM can make postsynaptic cells more excitable by depolarizing them so that they are closer to firing threshold (Balestrino et al., 1986; Hablitz and Lundervold, 1981; cf. Rausche et al., 1990). This build-up of extracellular K+ can also increase transmitter release in response to action potential invasion of the axon terminal (Hablitz and Lundervold, 1981). Elevated [K+]o (10 mM) also potentiates the persistent Na+ current in CA1 pyramidal cells, providing a sustained depolarization that would be expected to enhance ictal-like activity (Somjen and Muller, 2000). An additional contributing mechanism may be a reduction in the efficacy of GABA-mediated inhibition caused by a depolarizing shift in the reversal potential of GABA-A-mediated IPSPs caused by a high [K+]o-induced suppression or reversal of the K+/Cl− cotransporter KCC2 (Korn et al., 1987; Thompson and Gahwiler, 1989; DeFazio et al., 2000; Bihi et al., 2005). In summary, lower ECS volume will have a concentrating effect on K+ released from firing cells and thus may be expected to promote epileptiform activity.
Ephaptic interactions/field effects
Ephaptic interactions/field effects are also expected to be enhanced in circumstances of reduced ECS volume (reviewed by Weiss and Faber, 2010). In these conditions, cells are closer to one another, which increases the likelihood that a depolarization in one cell will affect a neighbor. In addition, reducing ECS volume increases extracellular resistance. After current leaves a depolarized cell, it will preferentially take the paths of least resistance, and thus will typically primarily travel in the ECS (Fig. 5A, top panel). If extracellular resistance is higher than usual, a greater proportion of the current will instead travel across the membrane of neighboring cells and cause a depolarizing voltage deflection in those neighboring cells, which may cause them to fire (Fig. 5A, middle panel). The shape and layout of neurons in the CA1 hippocampus, in which pyramidal cells are densely packed and their long apical dendrites are oriented in parallel, makes this region particularly susceptible to field effects. Indeed, applied electrical fields were able to entrain the population activity in the CA1 region at an order of magnitude weaker field intensity than that required to do the same in the CA3 region (Francis et al., 2003). Durand and colleagues demonstrated via computer simulation and hippocampal slice experiments that epileptiform events not only can propagate across the slice using field effects, but also propagate faster when cell membranes of neighboring cells are closer together (Qiu et al., 2015; Zhang et al., 2014).
Figure 5.
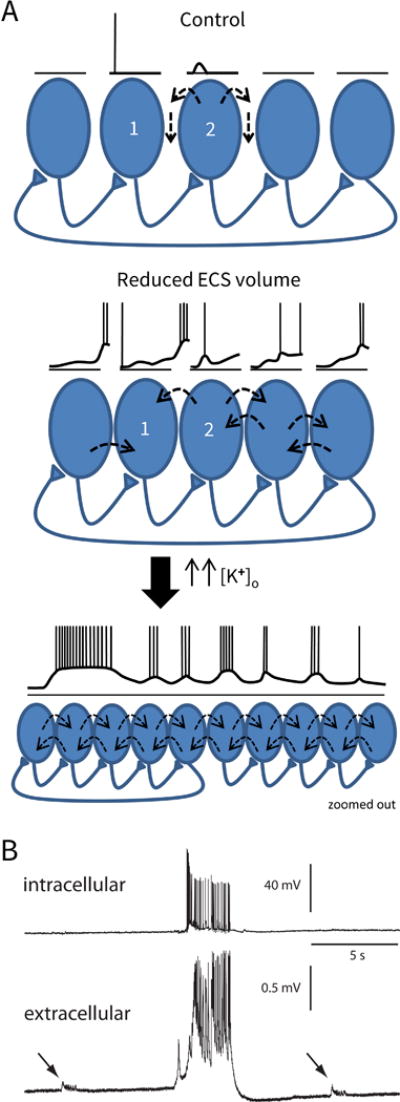
Generation of epileptic activity in the condition of reduced ECS volume.
(A) Model. Top panel is the control situation. CA1 pyramidal cell 1 fires an action potential which causes an epsp in the postsynaptic cell (2), but cell 2 does not fire. Current leaving cell 2 takes the paths of least resistance, and thus mostly travels in the ECS, and not across the cell membranes of the neighboring cells. (Dashed arrows depict current flow.) In the middle panel, the ECS volume is reduced (as in Has3−/− mice). There is a small increase in [K+]o because the K+ which leaves firing cells is diluted less in the smaller ECS volume. This small increase in [K+]o causes a small baseline depolarization of the cells, and now when cell 1 fires, the epsp in cell 2 triggers an action potential. Because the cells are closer together and extracellular resistance is larger, some of the current leaving cell 2 travels across the membranes of neighboring cells, causing them to depolarize (ephaptic interactions). Epsps in cells further down the chain overlap with depolarizations caused by ephaptic interactions, producing larger responses. (The longest voltage sequence is shown for cell 1.) Interictal activity will begin to occur as the depolarizations synchronize among the cells, which will cause a further rise in [K+]o. In the lower panel, a larger population of cells has been recruited. Both the rise in [K+]o and a fall in [Ca+]o will increase cell excitability and enhance the persistent Na+ current, which, in this condition of enhanced ephaptic interactions, will enable the recruitment of a larger population and allow ictal-like events to occur. (The view is zoomed out. The drawn voltage trace applies for all of the cells.) (B) Actual voltage traces recorded in the CA1 region of a Has3−/− brain slice. Top trace is an intracellular recording from a single pyramidal cell. Bottom trace is a simultaneous recording of the population field potential. The tiny interictal-like events (arrows) are occurring in a population of cells that does not include the neuron from which the intracellular recording is taken. However, the large, ictal-like event recruits a larger population of neurons, including the one from which the intracellular recording is taken. (B is modified from Fig. 4C in Arranz et al., 2014).
Gap junctions
Gap junctions between neurons allow synchronization of activity in a population of cells (Bennett and Zukin, 2004). The best characterized neuronal gap junctions are among GABAergic interneurons (Fukuda, 2007), but there is some evidence of gap junctions between glutamatergic cells in the brain (Hamzei-Sichani et al., 2007). As far as ECS volume is concerned, it is not outrageous to suggest that bringing cell membranes of adjacent neurons close together in a condition of reduced ECS might increase the formation of gap junctions between them. In fact, new gap junctions do seem to form between astrocytes in a condition of reduced ECS volume (hypo-osmolar solution, Scemes and Spray, 1998); however, there is no evidence that we are aware of that a similar phenomenon occurs in neurons.
Role of extracellular space volume and nonsynaptic mechanisms in epilepsy
Synchronous excitatory activity in the CA1 region in the low Ca2+ model
The foremost model in which epileptiform activity can be recorded in the absence of synaptic transmission is the low extracellular Ca2+ concentration ([Ca2+]o) model. This model is relevant because 1) hypocalcemia can cause seizures in humans (Kinoshita et al., 2010; Milman and Epstein, 2011; Singh et al., 2016; Tsai et al., 2009), 2) [Ca2+]o is known to fall during an ictal event (Heinemann et al., 1977), and 3) examining the model can shed light on nonsynaptic mechanisms involved in epilepsy, and in particular on the contribution of reduced ECS volume. In this model, which has several variations, spontaneous synchronous activity can occur among CA1 pyramidal cells of the hippocampus during complete block of synaptic transmission (Dudek et al., 1990; Jefferys and Haas, 1982; Jiruska et al., 2010; Yaari et al., 1983; Taylor and Dudek, 1982). Simulation experiments show that if cells display sufficiently high excitability, then that feature, in the presence of a sufficiently high extracellular resistance, can enable the synchronization of a population of neighboring cells via ephaptic interactions (Traub et al., 1985).
The CA1 pyramidal cells in these low [Ca2+]o experiments exhibit enhanced excitability in response to membrane depolarization, and spontaneous firing in these neurons tends to be as multiple rather than single action potentials (Taylor and Dudek, 1982; Su et al., 2001). Low [Ca2+]o is thought to increase cellular excitability primarily by reducing the charge screening at the outer surface of the cell membrane (Hille, 2001); however, the presence of extracellular Mn2+ in the experiments of Taylor and Dudek (1982) points to an alternate or additional mechanism of enhanced excitability, probably explained by the increase in persistent Na+ current that occurs in low Ca2+ solutions, even when the Ca2+ is replaced with Mn2+ (Su et al., 2001; Shuai et al., 2003; Golomb et al., 2006). Reduction of [Ca2+]o also causes a modest tonic rise in [K+]o, which will depolarize cells and thus may also contribute to the enhanced excitability in low [Ca2+]o; phasic rises in [K+]o caused by burst-firing of cells likely also contribute a positive feedback effect (Yaari et al., 1986).
Investigations using this low [Ca2+]o model showed that low osmolar solution increased the epileptiform activity even in the presence of antagonists of glutamate-mediated synaptic transmission, and high osmolar solution blocked the epileptiform activity (Dudek et al., 1990; Roper et al., 1992). The low osmolar solution is thought to have its effect by causing cells to swell, which reduces the volume of the ECS, bringing cell membranes of neighboring cells in closer proximity and increasing the resistivity of the ECS, which will both enhance ephaptic interactions. The high osmolar solution is thought to have its suppressive effect by causing cells to shrink, which increases the volume of the ECS. Interestingly, in the low [Ca2+]o model, epileptiform activity is far more likely to occur in the CA1 region, which has a lower ECS volume fraction, than in either the CA3 region or the dentate gyrus (Jefferys and Haas, 1982; Snow and Dudek, 1984; Roper et al., 1992). Hypo-osmolar solution can actually convert the absent or weak epileptiform activity in the dentate gyrus into robust events that are very similar to those seen in the CA1 region (Roper et al., 1992).
Role of extracellular space volume in other epilepsy models
In addition to investigations in the low calcium model that were discussed above, changing the osmolarity of the extracellular solution has been used to investigate the effect of changes in ECS volume in multiple different in vitro epilepsy models that do require chemical synaptic transmission: the high K+ (8.5 mM) model (Traynelis and Dingledine, 1989a), the low Mg2+ model (Andrew et al., 1989; Shahar et al., 2009), the 4-aminopyridine (4-AP) model (Zhang et al., 2014; Qiu et al., 2015), and the low Mg2+ + 4-AP model (Kilb et al., 2006). Hypo-osmolar solution, which causes cells to swell and thus reduces ECS volume, enhances ongoing epileptiform activity in these models as evidenced by an increase in the magnitude of the epileptiform field potentials, the duration of ictal-like events, and/or the propagation speed of ictal-like events. Hyperosmolar solution, on the other hand, suppresses epileptiform events or even blocks them altogether. Although osmolarity changes can also affect synaptic transmission (see Somjen, 2004, Ch. 4), their effect on epileptiform activity is thought to be primarily mediated via a change in ECS volume, not an effect on synaptic transmission; this conclusion was reached largely for three reasons: 1) osmolarity changes had the same pro-convulsant/anticonvulsant effects in the low Ca2+ model, (see above), in which epileptiform activity occurs independent of excitatory chemical synaptic transmission, 2) hypo-osmolarity boosts inhibitory as well as excitatory synaptic transmission (Huang and Somjen, 1997), and 3) the increase in spontaneous excitatory synaptic transmission occurs on a slower time scale than the increase in field potential magnitude (Saly and Andrew, 1993).
Dependence of ictal events on nonsynaptic mechanisms
Somjen (2004) has proposed that while ictal-length events may be initially triggered by chemical synaptic transmission, they depend on nonsynaptic mechanisms to reach ictal length; that is, they require a sustained active depolarization of the cell body along with ephaptic interactions or electrical synapses to provide synchronization. Neuronal activity coupled with reduced ECS volume can provide the conditions needed. Neuronal activity causes a rise in [K+]o, which will be more pronounced in the condition of reduced ECS volume. Neuronal activity also causes a drop in [Ca2+]o. Both the rise in [K+]o and the drop in [Ca2+]o can enhance the persistent sodium current and thus promote the sustained depolarization of the pyramidal cell. High [K+]o and low [Ca2+]o also both enhance cellular excitability; high [K+]o depolarizes the neurons via a Nernst effect and low [Ca2+]o reduces the charge screening at the membrane and causes cells to fire in multiple rather than single spikes. The closeness of neighboring cells and the increase in extracellular resistivity associated with reduced ECS volume will enhance ephaptic interactions among neurons, which can synchronize the activity of a population of neurons during an ictal-like event, even when synaptic activity is suppressed by falling [Ca2+]o. An in vivo version of these ictal epileptic events has been recorded in the neocortex of baboons with photosensitive reflex epilepsy. As the epileptic event progresses in these animals, the [Ca2+]o falls to such a low level that synaptic transmission must have ceased or nearly ceased, yet the events continue robustly, sustained by what must be nonsynaptic mechanisms (Pumain et al., 1985).
Positive Feedback loop between reduced extracellular space and epileptic activity
Traynelis and Dingledine (1988, 1989a) and Dudek et al. (1990) have proposed a positive feedback loop between synchronous neuronal activity and reduced ECS volume: synchronous neuronal activity causes a reduction in ECS volume (see below), and reduced ECS volume enhances ephaptic interactions and the build-up of extracellular K+ caused by neuronal activity, both of which are proconvulsant. ECS volume can be measured by measuring the concentration of an impermeant ion, or a decrease in ECS volume can be inferred from an increase in extracellular resistivity. Using the impermeant ion tetra-methyl-ammonium (TMA), Dietzel and colleagues (1980) measured a reduction in ECS volume in cat sensorimotor cortex in vivo in response to neuronal activity triggered by an electrical stimulus. Traynelis and Dingledine (1989a) recorded an increase in extracellular resistivity associated with epileptiform activity in vitro using the high K+ model. Fox et al. (2004) measured increases in extracellular resistivity during epileptiform bursts using the low Ca2+ model. By measuring the concentration of the impermeant ion tetra-ethyl-ammonium (TEA), Buchheim et al. (1999) recorded a decrease in ECS volume coincident with epileptiform activity in three different models in hippocampal-entorhinal cortex slices – low Mg2+, 4-AP, and low Ca2+.
Neuronal activity causes a reduction in ECS volume presumably due to cell swelling
The reduction in ECS volume that is associated with neuronal activity is presumably due to cell swelling. There are several different proposed mechanisms underlying cell swelling caused by neuronal activity. In nearly all cases, the cell swelling is presumably due to water entering cells from the ECS via osmosis (but see below for a case of secondary active transport). Even at rest, neurons are at steady state, not at equilibrium. The steady state is maintained by the activity of the Na+/K+ ATPase, which must pump out 3 Na+ ions for every 2 K+ ions pumped in. Without the pump activity, neurons would presumably swell, as other cells do, due to net entry of cations followed by water entry (Hoffmann and Simonsen, 1989). During periods of heavy activity, an increased number of Na+ ions enter the cell and an increased number of K+ ions leave the cell. This increased movement of ions across the cell membrane necessitates increased activity of the Na+/K+ ATPase to keep the cell at steady state, and thus increased demand for energy. If heavy activity depletes the ATP reserve so that the ATP-dependent Na+/K+ pump cannot keep the cell at steady state, the cell will swell (see Somjen, 2004, Ch. 19). An additional mechanism of cell swelling may result from this increased demand for energy: the increased demand for energy results in increased intracellular metabolic breakdown of substances into a greater number of osmotically active particles (Dietzel et al., 1982); for example, ATP is broken down into ADP + inorganic phosphate, and then phosphocreatine supplies its phosphate to re-make the ATP, resulting in a net increase in unbound inorganic phosphate; water would then enter the cells via osmosis.
K+ may play a key role in the cell swelling associated with neuronal activity. Dietzel et al. (1980) found a correlation between the rise in [K+]o and the magnitude of the shrinkage of the ECS in cat sensorimotor cortex in vivo. McBain et al. (1990) found that exposure of hippocampal slices to high [K+]o caused a reduction in the extracellular volume fraction in the pyramidal cell layer in CA1 even when epileptiform activity in CA1 was blocked by cutting the Schaffer collateral pathway. Glial cell swelling may account for some of this K+o-induced reduction in the ECS volume (Dietzel et al., 1989; Østby et al., 2009). However, compared to neurons, glial cell bodies and processes make up only a minor volume of the pyramidal cell layer in hippocampus (Ogata and Kosaka, 2002); therefore it is controversial whether swelling of glial cells could account for the reduction in ECS volume in the pyramidal cell layer (Haj-Yasein et al., 2012).
Glial cell swelling is associated with periods of high neuronal activity most likely because the glial cells take up K+ released from the neurons, and water follows the K+ via osmosis (Dietzel et al., 1980; Dietzel et al., 1989; Walz, 2000). Water may cross glial cell membranes via aquaporin 4 (AQ4), which forms water channels in the cell membrane. AQ4 is expressed in astrocyte cell membranes in the brain, particularly the astrocytes that abut capillaries (Jung et al., 1994; Nielsen et al., 1997), but also including the astrocytes of the CA1 region of the hippocampus (Hsu et al., 2011). Proposed mechanisms for glial cell swelling during neuronal activity include 1) K+ entry through K+ channels, particularly Kir4.1, followed by water entry through AQ4 (Nagelhus et al., 2004), 2) high [K+]o -induced depolarization activating transmembrane ion transport via the Na+/HCO3− (NBC) cotransporter, followed by water entry through AQ4 (Nagelhus et al., 2004), and 3) ion entry via the Na+, K+, Cl− cotransporter NKCC1 (Su et al., 2002; MacVicar et al., 2002) accompanied by water entry through the same transporter (MacAulay and Zeuthen, 2012; Zeuthen and MacAulay, 2012). The K+ channel/AQ4 hypothesis has become less appealing after two recent papers which showed that knockout of Kir4.1 (Haj-Yasein et al., 2011) or AQ4 (Haj-Yasein et al., 2012) had no effect on the reduction in ECS volume associated with neuronal activity. The proposed mechanisms of activity-induced cell swelling were also tested in a recent paper by Larsen et al. (2014), who eloquently demonstrated cell swelling in cultured astrocytes in response to an isosmotic increase in [K+]o which was almost completely blocked by the NKCC antagonist bumetanide. On the other hand, the same group showed that bumetanide had no effect on the shrinkage of the ECS volume in SR of CA1 hippocampus caused by stimulus-evoked neuronal activity, nor did barium, which blocks the Kir4.1 potassium channel (Larsen et al., 2014), arguing against both the NKCC1 mechanism and the Kir4.1/AQ4 mechanism. Detailed modelling work done by Østby et al (2009) suggests that increased activity of both the glial NKCC1 and NBC cotransporters accounts for the levels of ECS shrinkage observed in experiments. In that paper the authors suggested that increased activity of one of those two cotransporters may be able to compensate for a lack of the other.
As noted above, the pyramidal cell layer ECS is known to shrink in response to neuronal activity or high [K+]o and yet glia occupy only a tiny fraction of the volume of that layer. If the reduction in the volume of the ECS in that layer is not due to glial swelling, it is presumably due to neuronal swelling, which would presumably be caused by water entry across the cell membrane. The mechanism by which water might cross neuronal cell membranes is unclear. Indeed, some data suggest that in situ, an osmotic pressure difference across the cell membrane may cause water to enter glia but not neurons (Andrew et al., 2007, Risher et al., 2009). Thus far, aquaporins have not been found in the cell membranes of brain neurons, with the exception of the aquaglyceroporin AQ9, which has been found in catecholaminergic neurons and a few other neurons (Papadopoulos and Verkman, 2013; Badaut et al., 2004; Mylonakou et al., 2009; Arciénega et al., 2010). It is possible that certain cotransporters and uniporters in the neuronal cell membrane provide a pathway for water entry. There are three types of water transport through cotransporters (e.g., NKCC1, KCC4) or uniporters (e.g., GLUT1) that have been identified in non-neuronal cells: 1) passive water transport independent of transport of the primary substrate, 2) passive water transport dependent on ongoing transport of substrate, 3) and secondary active water transport (MacAulay and Zeuthen, 2010). Combining data from several papers, it may be true that most brain neurons in situ swell in response to a rise in [K+]o, but not in response to a moderate change in osmolarity (Haj-Yasein et al., 2012; Andrew et al., 2007; McBain et al., 1990; Andrew and MacVicar, 1994; Andrew et al., 1997). Furthering that hypothesis, it is intriguing to consider the possibility that neuronal water entry occurs via a cotransporter protein and is dependent upon ongoing ion transport via that same cotransporter.
Mechanism of generation of epileptic events in the Has3 KO mouse
As noted above, the Has3 KO mice have reduced ECS volume in the CA1 pyramidal cell layer in the hippocampus. In addition, in slices of hippocampus from these mice, epileptiform activity is blocked by increasing ECS volume via exposure to hypertonic saline (Arranz et al., 2014). On the other hand, the epileptiform events can also be blocked by the AMPA/kainite receptor antagonist NBQX, indicating a requirement for glutamate-mediated synaptic transmission (Arranz et al., 2014). However, as noted above, cutting off the CA3 region has no effect on the frequency of the epileptiform events recorded in the CA1 region of brain slices from Has 3 KO mice (Arranz et al., 2014), indicating that these epileptiform events are not triggered by CA3-driven glutamate release from Schaffer collaterals (cf. high K+ model, Traynelis and Dingledine, 1988, 1989a). Interestingly, in slices of hippocampus from WT mice, epileptiform activity can be induced by reducing ECS volume fraction via exposure to hypotonic saline, and the epileptiform activity in these slices is also independent from CA3 yet dependent upon intact glutamate-mediated synaptic transmission (Arranz et al., 2014).
This dependence upon glutamate-mediated synaptic transmission but independence from the CA3 region presumably means that recurrent excitatory connections among CA1 pyramidal cells are required for some aspect of the generation of the epileptiform activity. Unlike the CA3 region, which shows many recurrent excitatory connections among pyramidal cells (Christian and Dudek, 1988a; Miles and Wong, 1986; Miles and Wong, 1987) there are only sparse recurrent excitatory connections among CA1 pyramidal cells (Christian and Dudek, 1988b; Deuchars and Thomson, 1996). The fact that the CA1 region in WT slices also generates spontaneous epileptiform activity when exposed to hypotonic saline suggests that Has3−/− brain slices do not have increased recurrent connections in CA1 that account for the epileptiform activity. Could it be that the reduced ECS itself is enhancing synaptic transmission? Reduced ECS would decrease the dilution of neurotransmitter in the synaptic cleft, which could be expected to enhance excitatory transmission. This mechanism is unlikely in the Has3 KO mice, however, because the location of axon terminals of recurrent excitatory connections in CA1 is in the basal dendrites in SO (Deuchars and Thomson, 1996), not in the pyramidal cell layer where the reduced ECS volume fraction is localized (Arranz et al., 2014). Since the requirement is for ionotropic and not metabotropic glutamate receptors, presumably the glutamatergic transmission is needed for the trigger and/or synchronization mechanisms rather than to provide a sustained depolarization of the cell (see discussion in Salah and Perkins, 2008).
Figure 5A illustrates a possible mechanism for the generation of epileptic activity in the CA1 region of Has3 KO mice. We propose that sparse recurrent synaptic connections among nearby CA1 pyramidal cells, coupled with enhanced ephaptic interactions, would allow an action potential initiated in one or a few cells to trigger localized synchronous excitatory activity in a small core of cells. First of all, there is increased neuronal excitability in the condition of reduced ECS volume; that is, the reduced ECS volume has a concentrating effect on the K+ released from active neurons, making it more likely for an excitatory postsynaptic potential to cause an action potential or burst of action potentials in the postsynaptic cell (see above). Secondly, the significantly reduced ECS volume in the pyramidal cell layer of CA1— by making cells closer together and by increasing extracellular resistivity— will clearly encourage current leaving one cell to pass across the cell membrane of neighboring cells to cause a depolarization, allowing the synchronized depolarization of the small core of cells and enabling the spread to a larger population, creating interictal epileptic activity. This epileptic activity will further increase [K+]o and also decrease [Ca2+]o, both of which will enhance excitability and further promote epileptic activity in a positive feedback loop. Furthermore, the high [K+]o and low [Ca2+]o will both enhance the persistent Na+ current, providing a sustained active depolarization of the neurons, which together with the enhanced ephaptic interactions, will promote the generation of ictal activity (Fig. 5A, lower panel). Figure 5B illustrates the recruitment of a formerly inactive pyramidal cell into an ictal-like event in a brain slice from a Has3 KO mouse.
We would be remiss if we did not acknowledge that reduced HA will have effects other than just a reduction in ECS volume. Many of these effects were discussed already in the Function of HA in the central nervous system section of this paper, and some may alter the tendency of the brain to produce epileptiform activity. For example, increased migration of AMPA receptors in the cell membrane was associated with reduced paired pulse depression (Frischknecht et al., 2009). Vedunova et al. (2013), using cultured neurons, saw increased Ca2+ currents and increased network burst-firing after hyaluronidase treatment; however, as already discussed, hyaluronidase has been reported to reduce activation of voltage-gated L-type calcium channels and reduce LTP in brain slices (Kochlamazashvili et al., 2010). A third proposed mechanism by which reduced HA may promote epileptiform activity is via a disruption of PNNs. Theoretically, these PNNs, which form around interneurons in neocortex and hippocampus (Celio, 1993; Celio and Chiquet-Ehrismann, 1993), could be needed for proper function of the inhibitory interneuron network, and their disruption could lead to epilepsy (McRae and Porter, 2012). Our work in the Has3 KO mouse model, however, indicated no disruption of the PNNs in that model despite the epileptic phenotype. While it is true that reduced HA might affect synaptic efficacy, the primary mechanism by which reduced HA causes epilepsy seems to be via a reduction in ECS volume.
Could treatment of epilepsy be as simple as just manipulating osmolarity?
Given the importance of reduced ECS volume in promoting seizures, could we stop a seizure by delivering hyperosmotic solution to the seizing patient to pull water from brain cells and increase the ECS volume fraction in the brain? Unlike the experimental situation of the brain slice, wherein application of hyperosmolar solution shrinks cells and increases ECS volume, we have to consider the blood-brain barrier (BBB) when we are talking about patients. The BBB consists of a tight capillary endothelium surrounded by astrocyte endfeet (Nagelhus and Ottersen, 2013). In many ways, the BBB has permeability characteristics more like that of a cell membrane than like that of a typical systemic capillary. Salts do not normally cross the BBB on an acute time scale. On the other hand, the BBB is permeable to water (Jung et al., 1994; Nielsen et al., 1997). Raising plasma osmolarity by intravenous administration of hypertonic saline or mannitol pulls water out of the brain across the blood-brain barrier down its osmotic gradient within a matter of minutes (Shawkat et al., 2012), which causes the brain to shrink. However, unlike the case for the brain slice, when the brain shrinks following intravenous administration of hyperosmolar solution, we expect water to be lost both from cells and from the ECS. If the permeability of neuronal and glial cell membranes to water were high, then on an acute time scale, water would be lost from the brain in such a way as to maintain an equal osmolarity inside and outside cells with no change in ECS volume fraction. One may predict, however, that the drop in cellular volume may lag behind the drop in ECS volume due to low permeability of neuronal cell membranes to water, which would mean that ECS volume fraction would actually first decrease upon intravenous administration of hyperosmolar solution, which is opposite of the desired effect.
Despite the arguments presented above, mannitol has shown efficacy as an anticonvulsant in vivo (Baran et al., 1987; Haglund and Hochman, 2005), but in these cases, mannitol’s action was not via an increase in ECS volume fraction. In the first case (Baran et al., 1987), mannitol likely worked because it shrank a swollen brain that was pressing against the skull, and in the second case (Haglund and Hochman, 2005), resistance measurements indicated no correlation between brain ECS resistance and suppression of epileptic activity following intravenous mannitol.
Instead of initiating an acute change in body fluid osmolarity in an attempt to stop a seizure, what about maintaining a chronic condition of increased body fluid osmolarity as a preventive measure? Unfortunately for the epileptic patient, chronic hyperosmolarity would not cause shrinkage of brain cells with an accompanying increase in ECS volume. Instead, brain tissue actually accumulates electrolytes and organic solutes over time so that the brain and the brain cells maintain their original volume despite the chronic hyperosmolarity (Strange, 1992; Verbalis, 2010). On the other hand, if we could manipulate HA levels in vivo, we might be able to increase the brain ECS volume fraction.
HA as a possible therapy in epilepsy prevention or suppression
In discussing possible HA-related therapy, there are three issues to consider: 1) congenital deficiency in brain HA, 2) the suppression of epileptic activity via an increase in HA, 3) manipulation of HA levels in order to prevent epileptogenesis following an insult to the brain.
HA and congenital epilepsy
The fraction of human epilepsy cases that are inherited is thought to be “tiny” (McNamara et al., 2006). The known inherited mutations in humans with epilepsy involve voltage-gated or ligand-gated ion channels (McNamara et al., 2006; Villa and Combi, 2016), but it is possible that some small number of inherited cases could be HA-related. The majority of epilepsy cases in humans are classified as idiopathic (Herman, 2002). It is possible that some portion of these cases might be due to a sporadic mutation in one of the HAS or hyaluronidases, in one of the enzymes that provide the building blocks for HA, or in one of the other proteins involved in the regulation of HA synthesis or degradation, causing a deficiency of HA in the brain. In the future, targeted replacement of HA might be beneficial in epilepsy patients with congenital HA deficiency. Interestingly, application of exogenous HA has been shown to reverse the effects caused by hyaluronidase in an in vitro preparation (Kochlamazashvili et al., 2010) and to rescue cardiac morphogenesis in explants from Has2−/− mouse embryos (Camenisch et al., 2001).
Manipulation of HA levels as a potential anticonvulsant therapy
There are many patients with temporal lobe epilepsy who continue to have frequent seizures despite having tried currently available anticonvulsants (Schmidt and Löscher, 2005). The fact that increasing ECS volume reduces epileptiform activity in many different in vitro models of epilepsy (see above), not just in Has3 KO mice, suggests that an increase in brain HA might be beneficial in epilepsy patients even if the initiating cause of the epilepsy was not a deficiency of HA. There are reasons to believe that increasing the amount of brain HA would increase brain ECS volume. First of all, the knockout of Has3 decreased brain HA and decreased brain ECS volume (Arranz et al., 2014). Secondly, HA has a remarkable hydration capacity, a feature that is, for example, being exploited in dermatologic applications (Pavicic et al., 2011). While direct application of HA to the brains of epilepsy patients may not be feasible, we could potentially manipulate the balance between endogenous production and degradation of HA. The two obvious ways to regulate the amount of hyaluronic acid are via regulation of HA synthesis (Tammi et al., 2011; Vigetti et al., 2014) or via regulation of hyaluronidase activity (Sherman et al., 2015).
The synthesis of HA can be enhanced by increasing the supply of the building blocks of HA, the disaccharides UDP-GlcUA and UDP-GlcNAc (Vigetti et al., 2006; Vigetti et al., 2012). One way to increase the availability of these disaccharides is by increasing the supply of glucose and glucosamine to the cell cytosol, particularly if their levels are low (Jokela et al., 2008; Vigetti et al., 2012). Another way, which has been demonstrated in Xenopus, is via overexpression of UDP-glucose dehydrogenase, which increases the amount of UDP-GlcUA and in turn the amount of HA produced (Vigetti et al., 2006). Not only does increased UDP-GlcNAc increase HA levels by functioning as a building block, it can also increase HA levels by increasing the half-life of HAS: increased levels of UDP-GlcNAc will cause O-GlcNAcylation of serine 221 in the cytoplasmic loop of the HAS, which stabilizes the enzyme in the membrane (Vigetti et al., 2012). In the case of HAS2 in smooth muscle, which normally has a very short half-life, the half-life is increased from about 17 minutes to 5 hours, which results in increased levels of the synthase and increased HA synthesis (Vigetti et al., 2012).
Regulatory mechanisms increasing HA synthesis, other than those mediated by UDP-GlcUA and UDP-GlcNAc, have also been reported. For example, the rate of transcription of HAS can be affected by alterations in binding of various transcription factors to the promoter region of the HAS DNA (Tammi et al., 2011). In addition, post-translational modifications may also affect activity of HAS (Tammi et al., 2011). For example, alkaline phosphatase has been shown to increase HAS activity (Vigetti et al., 2009).
The activity of hyaluronidase could also potentially be altered. Inhibition of hyaluronidase would be expected to increase the level of high-molecular-weight HA. In vitro studies have successfully used the hyaluronidase inhibitor ascorbate 6-hexadecanoate, or 6-O-palmitoyl-L-ascorbic acid, to reduce the digestion products of hyaluronidase and promote remyelination in a model of a demyelinating lesion (Sloane et al., 2010; Preston et al., 2013). Poly styrene-4-sulfonate, which inhibits hyaluronidase via both competitive and noncompetitive mechanisms (Isoyama et al., 2006), has been used to suppress astrocyte proliferation and inhibit glioma growth in vitro (Daginakatte and Gutmann, 2007). Sulfated HA, which also acts via both competitive and noncompetitive mechanisms (Isoyama et al., 2006), has been successfully used as a hyaluronidase inhibitor both in vitro and in vivo (Benitez et al., 2011). For example, intraperitoneal injection of sulfated HA was used to successfully inhibit tumor growth in mice (Benitez et al., 2011).
HA and Epileptogenesis
Dityatev (2010) has proposed to use inhibitors of HAS to prevent epileptogenesis, with the idea being that HA encourages sprouting of axons and the creation of abnormal connections among neurons (Bausch, 2006). Based on our investigations of Has3 KO mice (Arranz et al., 2014), we propose the opposite, that is, to increase the synthesis of HA or to inhibit its degradation in order to inhibit epileptogenesis.
Epileptogenesis is the process by which normal brain tissue is changed to become epileptic, that is, to spontaneously produce epileptic activity. Brain insults such as stroke, head trauma, and CNS infection can be the trigger for epileptogenesis (Herman, 2002). In humans, there is a latent period of weeks to months or even several years between the initial insult and the onset of spontaneous recurrent seizures (Herman, 2002). There is hope that a treatment will be developed which can be administered during the latent period which would stop the process of epileptogenesis and prevent the onset of spontaneous seizures. In analyzing results that suggest that HA may be epileptogenic or antiepileptogenic, it is worth pointing out that the scientific community is lately becoming increasingly aware that both high- and low-molecular-weight HA have biological effects, that those effects are often not the same, and that some studies measuring changes in HA content have not differentiated between HA of different sizes (Cowman et al., 2015; Sherman et al., 2015).
Our finding in the Has3 KO mouse suggests that lack of HA leads to epileptogenesis. Might a reduction in the production of HA and/or an increase in the breakdown of HA be epileptogenic in the mature brain? We know that an initiating insult such as an episode of status epilepticus, ischemia, or mechanical trauma can set off the process of epileptogenesis. In many cases of brain insult, it seems that there is first a reduction and then an increase in high-molecular-weight HA (Sherman et al., 2015). Data indicate that the initial decrease in high-molecular-weight HA may be due either to an increase in HA breakdown or a decrease in HA synthesis, or both. For example, it has been shown that status epilepticus in rats causes a decrease in high-molecular-weight HA and a decrease in HAS3 (McRae et al., 2012). Ischemic stroke in humans or middle cerebral artery occlusion in rats is associated with elevated hyaluronidase activity and increases in HA breakdown products (Al’Qteishat et al., 2006a, 2006b). It has also been shown that an acute CNS injury leads to the generation of reactive oxygen species (Gilgun-Sherki et al., 2002), which cause HA degradation (Bates et al., 1984). Furthermore, it has been shown that low energy levels in the cell, which might occur due to status epilepticus or ischemic stroke, can lead to activation of AMP-activated protein kinase (AMPK), which is activated by an increase in the AMP:ATP ratio (Towler and Hardie, 2007). Activated AMPK phosphorylates threonine 110 on the cytoplasmic loop of HAS, causing a dramatic inhibition of HAS activity (Vigetti et al., 2014). Sherman et al. (2015) suggests that the initial decrease in high-molecular-weight HA that occurs following various CNS injuries may be the key step that should be targeted to promote a better outcome. Following that reasoning and considering the data presented above, we suggest that inhibitors of hyaluronidases, perhaps combined with stimulators of HAS, might prevent epileptogenesis if administered during the latent period.
Conclusions
HA has a remarkable hydration capacity and thus maintains ECS volume. Mice deficient in HA in the hippocampus have reduced ECS volume and an epileptic phenotype. Causing an increase in ECS volume is anticonvulsant in many different in vitro models of epilepsy, including brain slices from Has3 KO mice. An increase in ECS volume is antiepileptic because it reduces ephaptic interactions and activity-related increases in [K+]o. Promoting an increase in high-molecular-weight HA in the brain offers hope as a future anticonvulsant therapy, and prevention of the initial drop in high molecular weight HA that occurs after brain insults may inhibit epileptogenesis.
Acknowledgments
This work was supported by National Institutes of Health, National Institute of Neurological Disorders and Stroke grant R01 NS047557.
References
- Almond A. Towards understanding the interaction between oligosaccharides and water molecules. Carbohydr Res. 2005;340:907–920. doi: 10.1016/j.carres.2005.01.014. [DOI] [PubMed] [Google Scholar]
- Almond A. Hyaluronan. Cell Mol Life Sci. 2007;64:1591–1596. doi: 10.1007/s00018-007-7032-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almond A, DeAngelis PL, Blundell CD. Hyaluronan: the local solution conformation determined by NMR and computer modeling is close to a contracted left-handed 4-fold helix. J Mol Biol. 2006;358:1256–1269. doi: 10.1016/j.jmb.2006.02.077. [DOI] [PubMed] [Google Scholar]
- Al’Qteishat A, Gaffney J, Krupinski J, Rubio F, West D, Kumar S, Kumar P, Mitsios N, Slevin M. Changes in hyaluronan production and metabolism following ischaemic stroke in man. Brain. 2006a;129:2158–2176. doi: 10.1093/brain/awl139. [DOI] [PubMed] [Google Scholar]
- Al’Qteishat A, Gaffney JJ, Krupinski J, Slevin M. Hyaluronan expression following middle cerebral artery occlusion in the rat. Neuroreport. 2006b;17:1111–1114. doi: 10.1097/01.wnr.0000227986.69680.20. [DOI] [PubMed] [Google Scholar]
- Andrew RD, Fagan M, Ballyk BA, Rosen AS. Seizure susceptibility and the osmotic state. Brain Res. 1989;498:175–180. doi: 10.1016/0006-8993(89)90417-4. [DOI] [PubMed] [Google Scholar]
- Andrew RD, Labron MW, Boehnke SE, Carnduff L, Kirov SA. Physiological evidence that pyramidal neurons lack functional water channels. Cereb Cortex. 2007;17:787–802. doi: 10.1093/cercor/bhk032. [DOI] [PubMed] [Google Scholar]
- Andrew RD, Lobinowich ME, Osehobo EP. Evidence against volume regulation by cortical brain cells during acute osmotic stress. Exp Neurol. 1997;143:300–312. doi: 10.1006/exnr.1996.6375. [DOI] [PubMed] [Google Scholar]
- Andrew RD, MacVicar BA. Imaging cell volume changes and neuronal excitation in the hippocampal slice. Neuroscience. 1994;62:371–383. doi: 10.1016/0306-4522(94)90372-7. [DOI] [PubMed] [Google Scholar]
- Arciénega II, Brunet JF, Bloch J, Badaut J. Cell locations for AQP1, AQP4 and 9 in the non-human primate brain. Neuroscience. 2010;167:1103–1114. doi: 10.1016/j.neuroscience.2010.02.059. [DOI] [PubMed] [Google Scholar]
- Arranz AM, Perkins KL, Irie F, Lewis DP, Hrabe J, Xiao F, Itano N, Kimata K, Hrabetova S, Yamaguchi Y. Hyaluronan deficiency due to Has3 knock-out causes altered neuronal activity and seizures via reduction in brain extracellular space. J Neurosci. 2014;34:6164–6176. doi: 10.1523/JNEUROSCI.3458-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avoli M, Louvel J, Drapeau C, Pumain R, Kurcewicz I. GABAA-mediated inhibition and in vitro epileptogenesis in the human neocortex. J Neurophysiol. 1995;73:468–484. doi: 10.1152/jn.1995.73.2.468. [DOI] [PubMed] [Google Scholar]
- Avoli M, Louvel J, Pumain R, Olivier A. Seizure-like discharges induced by lowering [Mg2+]o in the human epileptogenic neocortex maintained in vitro. Brain Res. 1987;417:199–203. doi: 10.1016/0006-8993(87)90201-0. [DOI] [PubMed] [Google Scholar]
- Avoli M, Olivier A. Electrophysiological properties and synaptic responses in the deep layers of the human epileptogenic neocortex in vitro. J Neurophysiol. 1989;61:589–606. doi: 10.1152/jn.1989.61.3.589. [DOI] [PubMed] [Google Scholar]
- Badaut J, Petit JM, Brunet JF, Magistretti PJ, Charriaut-Marlangue C, Regli L. Distribution of Aquaporin 9 in the adult rat brain: preferential expression in catecholaminergic neurons and in glial cells. Neuroscience. 2004;128:27–38. doi: 10.1016/j.neuroscience.2004.05.042. [DOI] [PubMed] [Google Scholar]
- Balazs EA. The physical properties of synovial fluid and the special role of hyaluronic acid. In: Helfet A, editor. Disorders of the Knee. Philadelphia: T.B. Lippincott Co.; 1974. pp. 63–75. [Google Scholar]
- Balestrino M, Aitken PG, Somjen GG. The effects of moderate changes of extracellular K+ and Ca2+ on synaptic and neural function in the CA1 region of the hippocampal slice. Brain Res. 1986;377:229–239. doi: 10.1016/0006-8993(86)90863-2. [DOI] [PubMed] [Google Scholar]
- Baran H, Lassmann H, Sperk G, Seitelberger F, Hornykiewicz O. Effect of mannitol treatment on brain neurotransmitter markers in kainic acid-induced epilepsy. Neuroscience. 1987;21:679–684. doi: 10.1016/0306-4522(87)90029-7. [DOI] [PubMed] [Google Scholar]
- Bates EJ, Harper GS, Lowther DA, Preston BN. Effect of oxygen-derived reactive species on cartilage proteoglycan-hyaluronate aggregates. Biochem Int. 1984;8:629–637. [PubMed] [Google Scholar]
- Bausch SB. Potential roles for hyaluronan and CD44 in kainic acid-induced mossy fiber sprouting in organotypic hippocampal slice cultures. Neuroscience. 2006;143:339–350. doi: 10.1016/j.neuroscience.2006.07.037. [DOI] [PubMed] [Google Scholar]
- Bekku Y, Vargova L, Goto Y, Vorisek I, Dmytrenko L, Narasaki M, Ohtsuka A, Fassler R, Ninomiya Y, Sykova E, Oohashi T. Bral1: its role in diffusion barrier formation and conduction velocity in the CNS. J Neurosci. 2010;30:3113–3123. doi: 10.1523/JNEUROSCI.5598-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benitez A, Yates TJ, Lopez LE, Cerwinka WH, Bakkar A, Lokeshwar VB. Targeting hyaluronidase for cancer therapy: antitumor activity of sulfated hyaluronic acid in prostate cancer cells. Cancer Res. 2011;71:4085–4095. doi: 10.1158/0008-5472.CAN-10-4610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett MV, Zukin RS. Electrical coupling and neuronal synchronization in the mammalian brain. Neuron. 2004;41:495–511. doi: 10.1016/s0896-6273(04)00043-1. [DOI] [PubMed] [Google Scholar]
- Bihi RI, Jefferys JG, Vreugdenhil M. The role of extracellular potassium in the epileptogenic transformation of recurrent GABAergic inhibition. Epilepsia. 2005;46(s 5):64–71. doi: 10.1111/j.1528-1167.2005.01011.x. [DOI] [PubMed] [Google Scholar]
- Bikson M, Id Bihi R, Vreugdenhil M, Köhling R, Fox JE, Jefferys JG. Quinine suppresses extracellular potassium transients and ictal epileptiform activity without decreasing neuronal excitability in vitro. Neuroscience. 2002;115:251–261. doi: 10.1016/s0306-4522(02)00320-2. [DOI] [PubMed] [Google Scholar]
- Brecht M, Mayer U, Schlosser E, Prehm P. Increased hyaluronate synthesis is required for fibroblast detachment and mitosis. Biochem J. 1986;239:445–450. doi: 10.1042/bj2390445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenneke F, Bukalo O, Dityatev A, Lie AA. Mice deficient for the extracellular matrix glycoprotein tenascin-r show physiological and structural hallmarks of increased hippocampal excitability, but no increased susceptibility to seizures in the pilocarpine model of epilepsy. Neuroscience. 2004;124:841–855. doi: 10.1016/j.neuroscience.2003.11.037. [DOI] [PubMed] [Google Scholar]
- Brown TJ, Laurent UB, Fraser JR. Turnover of hyaluronan in synovial joints: elimination of labelled hyaluronan from the knee joint of the rabbit. Exp Physiol. 1991;76:125–134. doi: 10.1113/expphysiol.1991.sp003474. [DOI] [PubMed] [Google Scholar]
- Buchheim K, Schuchmann S, Siegmund H, Gabriel HJ, Heinemann U, Meierkord H. Intrinsic optical signal measurements reveal characteristic features during different forms of spontaneous neuronal hyperactivity associated with ECS shrinkage in vitro. Eur J Neurosci. 1999;11:1877–1882. doi: 10.1046/j.1460-9568.1999.00606.x. [DOI] [PubMed] [Google Scholar]
- Camenisch TD, Spicer AP, Brehm-Gibson T, Biesterfeldt J, Augustine ML, Calabro A, Jr, Kubalak S, Klewer SE, McDonald JA. Disruption of hyaluronan synthase-2 abrogates normal cardiac morphogenesis and hyaluronan-mediated transformation of epithelium to mesenchyme. J Clin Invest. 2000;106:349–360. doi: 10.1172/JCI10272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camenisch TD, Biesterfeldt J, Brehm-Gibson T, Bradley J, McDonald JA. Regulation of cardiac cushion development by hyaluronan. Exp Clin Cardiol. 2001;6:4–10. [PMC free article] [PubMed] [Google Scholar]
- Celio MR. Perineuronal nets of extracellular matrix around parvalbumin-containing neurons of the hippocampus. Hippocampus. 1993;3(S1):55–60. [PubMed] [Google Scholar]
- Celio MR, Chiquet-Ehrismann R. ‘Perineuronal nets’ around cortical interneurons expressing parvalbumin are rich in tenascin. Neurosci Lett. 1993;162:137–140. doi: 10.1016/0304-3940(93)90579-a. [DOI] [PubMed] [Google Scholar]
- Chowdhury B, Hemming R, Faiyaz S, Triggs-Raine B. Hyaluronidase 2 (HYAL2) is expressed in endothelial cells, as well as some specialized epithelial cells, and is required for normal hyaluronan catabolism. Histochem Cell Biol. 2016;145:53–66. doi: 10.1007/s00418-015-1373-8. [DOI] [PubMed] [Google Scholar]
- Christian EP, Dudek FE. Characteristics of local excitatory circuits studied with glutamate microapplication in the CA3 area of rat hippocampal slices. J Neurophysiol. 1988a;59:90–109. doi: 10.1152/jn.1988.59.1.90. [DOI] [PubMed] [Google Scholar]
- Christian EP, Dudek FE. Electrophysiological evidence from glutamate microapplications for local excitatory circuits in the CA1 area of rat hippocampal slices. J Neurophysiol. 1988b;59:110–123. doi: 10.1152/jn.1988.59.1.110. [DOI] [PubMed] [Google Scholar]
- Contreras EG, Gaete M, Sánchez N, Carrasco H, Larraín J. Early requirement of Hyaluronan for tail regeneration in Xenopus tadpoles. Development. 2009;136:2987–2996. doi: 10.1242/dev.035501. [DOI] [PubMed] [Google Scholar]
- Cowman MK, Lee HG, Schwertfeger KL, McCarthy JB, Turley EA. The content and size of hyaluronan in biological fluids and tissues. Front Immunol. 2015;6:261. doi: 10.3389/fimmu.2015.00261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cragg B. Preservation of extracellular space during fixation of the brain for electron microscopy. Tissue Cell. 1980;12:63–72. doi: 10.1016/0040-8166(80)90052-x. [DOI] [PubMed] [Google Scholar]
- Csoka AB, Scherer SW, Stern R. Expression analysis of six paralogous human hyaluronidase genes clustered on chromosomes 3p21 and 7q31. Genomics. 1999;60:356–361. doi: 10.1006/geno.1999.5876. [DOI] [PubMed] [Google Scholar]
- Csoka AB, Frost GI, Stern R. The six hyaluronidase-like genes in the human and mouse genomes. Matrix Biol. 2001;20:499–508. doi: 10.1016/s0945-053x(01)00172-x. [DOI] [PubMed] [Google Scholar]
- Daginakatte GC, Gutmann DH. Neurofibromatosis-1 (Nf1) heterozygous brain microglia elaborate paracrine factors that promote Nf1-deficient astrocyte and glioma growth. Hum Mol Gen. 2007;16:1098–1112. doi: 10.1093/hmg/ddm059. [DOI] [PubMed] [Google Scholar]
- DeAngelis PL, Weigel PH. Immunochemical confirmation of the primary structure of streptococcal hyaluronan synthase and synthesis of high molecular weight product by the recombinant enzyme. Biochemistry. 1994;33:9033–9039. doi: 10.1021/bi00197a001. [DOI] [PubMed] [Google Scholar]
- Deepa SS, Carulli D, Galtrey C, Rhodes K, Fukuda J, Mikami T, Sugahara K, Fawcett JW. Composition of Perineuronal Net Extracellular Matrix in Rat Brain. J Biol Chem. 2006;281:17789–17800. doi: 10.1074/jbc.M600544200. [DOI] [PubMed] [Google Scholar]
- DeFazio RA, Keros S, Quick MW, Hablitz JJ. Potassium-coupled chloride cotransport controls intracellular chloride in rat neocortical pyramidal neurons. J Neurosci. 2000;20:8069–8076. doi: 10.1523/JNEUROSCI.20-21-08069.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deuchars J, Thomson AM. CA1 pyramid-pyramid connections in rat hippocampus in vitro: dual intracellular recordings with biocytin filling. Neuroscience. 1996;74:1009–1018. doi: 10.1016/0306-4522(96)00251-5. [DOI] [PubMed] [Google Scholar]
- Dietzel I, Heinemann U, Hofmeier G, Lux HD. Transient changes in the size of the extracellular space in the sensorimotor cortex of cats in relation to stimulus-induced changes in potassium concentration. Exp Brain Res. 1980;40:432–439. doi: 10.1007/BF00236151. [DOI] [PubMed] [Google Scholar]
- Dietzel I, Heinemann U, Hofmeier G, Lux HD. Stimulus-induced changes in extracellular Na+ and Cl− concentration in relation to changes in the size of the extracellular space. Exp Brain Res. 1982;46:73–84. doi: 10.1007/BF00238100. [DOI] [PubMed] [Google Scholar]
- Dietzel I, Heinemann U, Lux HD. Relations between slow extracellular potential changes, glial potassium buffering, and electrolyte and cellular volume changes during neuronal hyperactivity in cat brain. Glia. 1989;2:25–44. doi: 10.1002/glia.440020104. [DOI] [PubMed] [Google Scholar]
- Dityatev A. Remodeling of extracellular matrix and epileptogenesis. Epilepsia. 2010;51(s3):61–65. doi: 10.1111/j.1528-1167.2010.02612.x. [DOI] [PubMed] [Google Scholar]
- Dzwonek J, Wilczynski GM. CD44: molecular interactions, signaling and functions in the nervous system. Front Cell Neurosci. 2015;9:175. doi: 10.3389/fncel.2015.00175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudek FE, Obenaus A, Tasker JG. Osmolality-induced changes in extracellular volume alter epileptiform bursts independent of chemical synapses in the rat: importance of non-synaptic mechanisms in hippocampal epileptogenesis. Neurosci Lett. 1990;120:267–270. doi: 10.1016/0304-3940(90)90056-f. [DOI] [PubMed] [Google Scholar]
- Evanko SP, Angello JC, Wight TN. Formation of hyaluronan-and versican-rich pericellular matrix is required for proliferation and migration of vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 1999;19:1004–1013. doi: 10.1161/01.atv.19.4.1004. [DOI] [PubMed] [Google Scholar]
- Faralli A, Dagna F, Albera A, Bekku Y, Oohashi T, Albera R, Rossi F, Carulli D. Modifications of perineuronal nets and remodelling of excitatory and inhibitory afferents during vestibular compensation in the adult mouse. Brain Struct Funct. 2016;221:3193–3209. doi: 10.1007/s00429-015-1095-7. [DOI] [PubMed] [Google Scholar]
- Fertziger AP, Ranck JB., Jr Potassium accumulation in interstitial space during epileptiform seizures. Exp Neurol. 1970;26:571–585. doi: 10.1016/0014-4886(70)90150-0. [DOI] [PubMed] [Google Scholar]
- Fisher RS, Pedley TA, Moody WJ, Jr, Prince DA. The role of extracellular potassium in hippocampal epilepsy. Arch Neurol. 1976;33:76–83. doi: 10.1001/archneur.1976.00500020004002. [DOI] [PubMed] [Google Scholar]
- Förster E, Zhao S, Frotscher M. Hyaluronan-associated adhesive cues control fiber segregation in the hippocampus. Development. 2001;128:3029–3039. doi: 10.1242/dev.128.15.3029. [DOI] [PubMed] [Google Scholar]
- Fox JE, Bikson M, Jefferys JG. Tissue resistance changes and the profile of synchronized neuronal activity during ictal events in the low-calcium model of epilepsy. J Neurophysiol. 2004;92:181–188. doi: 10.1152/jn.00123.2004. [DOI] [PubMed] [Google Scholar]
- Francis JT, Gluckman BJ, Schiff SJ. Sensitivity of neurons to weak electric fields. J Neurosci. 2003;23:7255–7261. doi: 10.1523/JNEUROSCI.23-19-07255.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser JR, Laurent TC, Laurent UB. Hyaluronan: its nature, distribution, functions and turnover. J Intern Med. 1997;242:27–33. doi: 10.1046/j.1365-2796.1997.00170.x. [DOI] [PubMed] [Google Scholar]
- Frischknecht R, Heine M, Perrais D, Seidenbecher CI, Choquet D, Gundelfinger ED. Brain extracellular matrix affects AMPA receptor lateral mobility and short-term synaptic plasticity. Nat Neurosci. 2009;12:897–904. doi: 10.1038/nn.2338. [DOI] [PubMed] [Google Scholar]
- Frischknecht R, Gundelfinger ED. The brain’s extracellular matrix and its role in synaptic plasticity. Adv Exp Med Biol. 2012;970:153–171. doi: 10.1007/978-3-7091-0932-8_7. [DOI] [PubMed] [Google Scholar]
- Fukuda T. Structural organization of the gap junction network in the cerebral cortex. Neuroscientist. 2007;13:199–207. doi: 10.1177/1073858406296760. [DOI] [PubMed] [Google Scholar]
- Gabriel S, Njunting M, Pomper JK, Merschhemke M, Sanabria ER, Eilers A, Kivi A, Zeller M, Meencke HJ, Cavalheiro EA, Heinemann U, Lehmann TN. Stimulus and potassium-induced epileptiform activity in the human dentate gyrus from patients with and without hippocampal sclerosis. J Neurosci. 2004;24:10416–10430. doi: 10.1523/JNEUROSCI.2074-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galtrey CM, Fawcett JW. The role of chondroitin sulfate proteoglycans in regeneration and plasticity in the central nervous system. Brain Res Rev. 2007;54:1–18. doi: 10.1016/j.brainresrev.2006.09.006. [DOI] [PubMed] [Google Scholar]
- Gilgun-Sherki Y, Rosenbaum Z, Melamed E, Offen D. Antioxidant therapy in acute central nervous system injury: current state. Pharmacol Rev. 2002;54:271–284. doi: 10.1124/pr.54.2.271. [DOI] [PubMed] [Google Scholar]
- Golomb D, Yue C, Yaari Y. Contribution of persistent Na+ current and M-type K+ current to somatic bursting in CA1 pyramidal cells: combined experimental and modeling study. J Neurophysiol. 2006;96:1912–1926. doi: 10.1152/jn.00205.2006. [DOI] [PubMed] [Google Scholar]
- Gurevicius K, Gureviciene I, Valjakka A, Schachner M, Tanila H. Enhanced cortical and hippocampal neuronal excitability in mice deficient in the extracellular matrix glycoprotein tenascin-R. Mol Cell Neurosci. 2004;25:515–523. doi: 10.1016/j.mcn.2003.12.001. [DOI] [PubMed] [Google Scholar]
- Hablitz JJ, Lundervold A. Hippocampal excitability and changes in extracellular potassium. Exp Neurol. 1981;71:410–420. doi: 10.1016/0014-4886(81)90099-6. [DOI] [PubMed] [Google Scholar]
- Haglund MM, Hochman DW. Furosemide and mannitol suppression of epileptic activity in the human brain. J Neurophysiol. 2005;94:907–918. doi: 10.1152/jn.00944.2004. [DOI] [PubMed] [Google Scholar]
- Haj-Yasein NN, Jensen V, Østby I, Omholt SW, Voipio J, Kaila K, Ottersen OP, Hvalby Ø, Nagelhus EA. Aquaporin-4 regulates extracellular space volume dynamics during high-frequency synaptic stimulation: a gene deletion study in mouse hippocampus. Glia. 2012;60:867–874. doi: 10.1002/glia.22319. [DOI] [PubMed] [Google Scholar]
- Haj-Yasein NN, Jensen V, Vindedal GF, Gundersen GA, Klungland A, Ottersen OP, Hvalby Ø, Nagelhus EA. Evidence that compromised K+ spatial buffering contributes to the epileptogenic effect of mutations in the human Kir4 1 gene (KCNJ10) Glia. 2011;59:1635–1642. doi: 10.1002/glia.21205. [DOI] [PubMed] [Google Scholar]
- Hamzei-Sichani F, Kamasawa N, Janssen WG, Yasumura T, Davidson KG, Hof PR, Wearne SL, Stewart MG, Young SR, Whittington MA, Rash JE. Gap junctions on hippocampal mossy fiber axons demonstrated by thin-section electron microscopy and freeze–fracture replica immunogold labeling. Proc Natl Acad Sci USA. 2007;104:12548–12553. doi: 10.1073/pnas.0705281104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinemann U, Lux HD, Gutnick MJ. Extracellular free calcium and potassium during paroxysmal activity in the cerebral cortex of the cat. Exp Brain Res. 1977;27:237–243. doi: 10.1007/BF00235500. [DOI] [PubMed] [Google Scholar]
- Heldin P, Pertoft H. Synthesis and assembly of the hyaluronan-containing coats around normal human mesothelial cells. Exp Cell Res. 1993;208:422–429. doi: 10.1006/excr.1993.1264. [DOI] [PubMed] [Google Scholar]
- Herman ST. Epilepsy after brain insult: targeting epileptogenesis. Neurology. 2002;59(9 S5):S21–S26. doi: 10.1212/wnl.59.9_suppl_5.s21. [DOI] [PubMed] [Google Scholar]
- Hille B. Ion Channels of Excitable Membranes. Sunderland, MA: Sinauer Associates, Inc.; 2001. chapter 20. [Google Scholar]
- Hoffmann K, Sivukhina E, Potschka H, Schachner M, Löscher W, Dityatev A. Retarded kindling progression in mice deficient in the extracellular matrix glycoprotein tenascin-R. Epilepsia. 2009;50:859–869. doi: 10.1111/j.1528-1167.2008.01774.x. [DOI] [PubMed] [Google Scholar]
- Hoffmann EK, Simonsen LO. Membrane mechanisms in volume and pH regulation in vertebrate cells. Physiol Rev. 1989;69:315–382. doi: 10.1152/physrev.1989.69.2.315. [DOI] [PubMed] [Google Scholar]
- Hrabe J, Hrabetova S, Segeth K. A model of effective diffusion and tortuosity in the extracellular space of the brain. Biophys J. 2004;87:1606–1617. doi: 10.1529/biophysj.103.039495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu MS, Seldin M, Lee DJ, Seifert G, Steinhäuser C, Binder DK. Laminar-specific and developmental expression of aquaporin-4 in the mouse hippocampus. Neuroscience. 2011;178:21–32. doi: 10.1016/j.neuroscience.2011.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang R, Somjen GG. Effects of hypertonia on voltage-gated ion currents in freshly isolated hippocampal neurons, and on synaptic currents in neurons in hippocampal slices. Brain Res. 1997;748:157–167. doi: 10.1016/s0006-8993(96)01294-2. [DOI] [PubMed] [Google Scholar]
- Huberfeld G, Wittner L, Clemenceau S, Baulac M, Kaila K, Miles R, Rivera C. Perturbed chloride homeostasis and GABAergic signaling in human temporal lobe epilepsy. J Neurosci. 2007;27:9866–9873. doi: 10.1523/JNEUROSCI.2761-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hylin MJ, Orsi SA, Moore AN, Dash PK. Disruption of the perineuronal net in the hippocampus or medial prefrontal cortex impairs fear conditioning. Learn Mem. 2013;20:267–273. doi: 10.1101/lm.030197.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isoyama T, Thwaites D, Selzer MG, Carey RI, Barbucci R, Lokeshwar VB. Differential selectivity of hyaluronidase inhibitors toward acidic and basic hyaluronidases. Glycobiology. 2006;16:11–21. doi: 10.1093/glycob/cwj036. [DOI] [PubMed] [Google Scholar]
- Jadin L, Bookbinder LH, Frost GI. A comprehensive model of hyaluronan turnover in the mouse. Matrix Biol. 2012;31:81–89. doi: 10.1016/j.matbio.2011.11.002. [DOI] [PubMed] [Google Scholar]
- Jefferys JGR, Haas HL. Synchronized bursting of CA1 hippocampal pyramidal cells in the absence of synaptic transmission. Nature. 1982;300:448–450. doi: 10.1038/300448a0. [DOI] [PubMed] [Google Scholar]
- Jensen MS, Yaari Y. The relationship between interictal and ictal paroxysms in an in vitro model of focal hippocampal epilepsy. Ann Neurol. 1988;24:591–598. doi: 10.1002/ana.410240502. [DOI] [PubMed] [Google Scholar]
- Jiruska P, Csicsvari J, Powell AD, Fox JE, Chang WC, Vreugdenhil M, Li X, Palus M, Bujan AF, Dearden RW, Jefferys JG. High-frequency network activity, global increase in neuronal activity, and synchrony expansion precede epileptic seizures in vitro. J Neurosci. 2010;30:5690–5701. doi: 10.1523/JNEUROSCI.0535-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jokela TA, Jauhiainen M, Auriola S, Kauhanen M, Tiihonen R, Tammi MI, Tammi RH. Mannose inhibits hyaluronan synthesis by down-regulation of the cellular pool of UDP-N-acetylhexosamines. J Biol Chem. 2008;283:7666–7673. doi: 10.1074/jbc.M706001200. [DOI] [PubMed] [Google Scholar]
- Jung JS, Bhat RV, Preston GM, Guggino WB, Baraban JM, Agre P. Molecular characterization of an aquaporin cDNA from brain: candidate osmoreceptor and regulator of water balance. Proc Natl Acad Sci USA. 1994;91:13052–13056. doi: 10.1073/pnas.91.26.13052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kablik J, Monheit GD, Yu L, Chang G, Gershkovich J. Comparative physical properties of hyaluronic acid dermal fillers. Dermatol Surg. 2009;35(s1):302–312. doi: 10.1111/j.1524-4725.2008.01046.x. [DOI] [PubMed] [Google Scholar]
- Karnup S, Stelzer A. Seizure-like activity in the disinhibited CA1 minislice of adult guinea-pigs. J Physiol. 2001;532:713–730. doi: 10.1111/j.1469-7793.2001.0713e.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilb W, Dierkes PW, Syková E, Vargová L, Luhmann HJ. Hypoosmolar conditions reduce extracellular volume fraction and enhance epileptiform activity in the CA3 region of the immature rat hippocampus. J Neurosci Res. 2006;84:119–129. doi: 10.1002/jnr.20871. [DOI] [PubMed] [Google Scholar]
- Kinoshita H, Kokudo T, Ide T, Kondo Y, Mori T, Homma Y, Yakushiji F. A patient with DiGeorge syndrome with spina bifida and sacral myelomeningocele, who developed both hypocalcemia-induced seizure and epilepsy. Seizure. 2010;19:303–305. doi: 10.1016/j.seizure.2010.04.005. [DOI] [PubMed] [Google Scholar]
- Kochlamazashvili G, Henneberger C, Bukalo O, Dvoretskova E, Senkov O, Lievens PM, Westenbroek R, Engel AK, Catterall WA, Rusakov DA, Schachner M, Dityatev A. The extracellular matrix molecule hyaluronic acid regulates hippocampal synaptic plasticity by modulating postsynaptic L-type Ca2+ channels. Neuron. 2010;67:116–128. doi: 10.1016/j.neuron.2010.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köhling R, Lücke A, Straub H, Speckmann EJ, Tuxhorn I, Wolf P, Pannek H, Oppel F. Spontaneous sharp waves in human neocortical slices excised from epileptic patients. Brain. 1998;121:1073–1087. doi: 10.1093/brain/121.6.1073. [DOI] [PubMed] [Google Scholar]
- Korn SJ, Giacchino JL, Chamberlin NL, Dingledine R. Epileptiform burst activity induced by potassium in the hippocampus and its regulation by GABA-mediated inhibition. J Neurophysiol. 1987;57:325–340. doi: 10.1152/jn.1987.57.1.325. [DOI] [PubMed] [Google Scholar]
- Larsen BR, Assentoft M, Cotrina ML, Hua SZ, Nedergaard M, Kaila K, Voipio J, MacAulay N. Contributions of the Na⁺/K⁺-ATPase, NKCC1, and Kir4.1 to hippocampal K⁺ clearance and volume responses. Glia. 2014;62:608–622. doi: 10.1002/glia.22629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurent TC, Fraser JRE. Catabolism of hyaluronan. In: Henriksen JH, editor. Degradation of bioactive substances: physiology and pathophysiology. Boca Raton, FL: CRC Press; 1991. pp. 249–265. [Google Scholar]
- Le Duigou C, Bouilleret V, Miles R. Epileptiform activities in slices of hippocampus from mice after intra-hippocampal injection of kainic acid. J Physiol. 2008;586:4891–4904. doi: 10.1113/jphysiol.2008.156281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Cowman MK. Thermal analysis of semi-dilute hyaluronan solutions. J Therm Anal Calorim. 2000;59:547–557. [Google Scholar]
- Lux HD. The kinetics of extracellular potassium: relation to epileptogenesis. Epilepsia. 1974;15:375–393. doi: 10.1111/j.1528-1157.1974.tb04015.x. [DOI] [PubMed] [Google Scholar]
- Lynn BD, Li X, Cattini PA, Turley EA, Nagy JI. Identification of sequence, protein isoforms, and distribution of the hyaluronan-binding protein RHAMM in adult and developing rat brain. J Comp Neurol. 2001a;439:315–330. doi: 10.1002/cne.1353. [DOI] [PubMed] [Google Scholar]
- Lynn BD, Turley EA, Nagy JI. Subcellular distribution, calmodulin interaction, and mitochondrial association of the hyaluronan-binding protein RHAMM in rat brain. J Neurosci Res. 2001b;65:6–16. doi: 10.1002/jnr.1122. [DOI] [PubMed] [Google Scholar]
- MacAulay N, Zeuthen T. Water transport between CNS compartments: contributions of aquaporins and cotransporters. Neuroscience. 2010;168:941–956. doi: 10.1016/j.neuroscience.2009.09.016. [DOI] [PubMed] [Google Scholar]
- MacAulay N, Zeuthen T. Glial K+ clearance and cell swelling: key roles for cotransporters and pumps. Neurochem Res. 2012;37:2299–2309. doi: 10.1007/s11064-012-0731-3. [DOI] [PubMed] [Google Scholar]
- Machado R, Soltani N, Dufour S, Salam MT, Carlen PL, Genov R, Thompson M. Biofouling-Resistant Impedimetric Sensor for Array High-Resolution Extracellular Potassium Monitoring in the Brain. Biosensors (Basel) 2016;6:53. doi: 10.3390/bios6040053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacVicar BA, Feighan D, Brown A, Ransom B. Intrinsic optical signals in the rat optic nerve: role for K+ uptake via NKCC1 and swelling of astrocytes. Glia. 2002;37:114–123. doi: 10.1002/glia.10023. [DOI] [PubMed] [Google Scholar]
- McBain CJ, Traynelis SF, Dingledine R. Regional variation of extracellular space in the hippocampus. Science. 1990;249:674–677. doi: 10.1126/science.2382142. [DOI] [PubMed] [Google Scholar]
- McNamara JO, Huang YZ, Leonard AS. Molecular signaling mechanisms underlying epileptogenesis. Sci STKE. 2006;356:re12. doi: 10.1126/stke.3562006re12. [DOI] [PubMed] [Google Scholar]
- McRae PA, Rocco MM, Kelly G, Brumberg JC, Matthews RT. Sensory deprivation alters aggrecan and perineuronal net expression in the mouse barrel cortex. J Neurosci. 2007;27:5405–5413. doi: 10.1523/JNEUROSCI.5425-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McRae PA, Baranov E, Rogers SL, Porter BE. Persistent decrease in multiple components of the perineuronal net following status epilepticus. Eur J Neurosci. 2012;36:3471–3482. doi: 10.1111/j.1460-9568.2012.08268.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McRae PA, Porter BE. The perineuronal net component of the extracellular matrix in plasticity and epilepsy. Neurochem Int. 2012;61:963–972. doi: 10.1016/j.neuint.2012.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miles R, Wong RKS. Excitatory synaptic interactions between CA3 neurones in the guinea-pig hippocampus. J Physiol. 1986;373:397–418. doi: 10.1113/jphysiol.1986.sp016055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miles R, Wong RKS. Inhibitory control of local excitatory circuits in the guinea-pig hippocampus. J Physiol. 1987;388:611–629. doi: 10.1113/jphysiol.1987.sp016634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miles R, Wong RKS, Traub RD. Synchronized afterdischarges in the hippocampus: contribution of local synaptic interactions. Neuroscience. 1984;12:1179–1189. doi: 10.1016/0306-4522(84)90012-5. [DOI] [PubMed] [Google Scholar]
- Milman S, Epstein EJ. Proton pump inhibitor-induced hypocalcemic seizure in a patient with hypoparathyroidism. Endocr Pract. 2011;17:104–107. doi: 10.4158/EP10241.CR. [DOI] [PubMed] [Google Scholar]
- Mody I, Lambert JD, Heinemann U. Low extracellular magnesium induces epileptiform activity and spreading depression in rat hippocampal slices. J Neurophysiol. 1987;57:869–888. doi: 10.1152/jn.1987.57.3.869. [DOI] [PubMed] [Google Scholar]
- Monslow J, Sato N, Mack JA, Maytin EV. Wounding-induced synthesis of hyaluronic acid in organotypic epidermal cultures requires the release of heparin-binding egf and activation of the EGFR. J Invest Dermatol. 2009;129:2046–2058. doi: 10.1038/jid.2009.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moody WJ, Futamachi KJ, Prince DA. Extracellular potassium activity during epileptogenesis. Exp Neurol. 1974;42:248–263. doi: 10.1016/0014-4886(74)90023-5. [DOI] [PubMed] [Google Scholar]
- Mylonakou MN, Petersen PH, Rinvik E, Rojek A, Valdimarsdottir E, Zelenin S, Zeuthen T, Nielsen S, Ottersen OP, Amiry-Moghaddam M. Analysis of mice with targeted deletion of AQP9 gene provides conclusive evidence for expression of AQP9 in neurons. J Neurosci Res. 2009;87:1310–1322. doi: 10.1002/jnr.21952. [DOI] [PubMed] [Google Scholar]
- Nagelhus EA, Mathiisen TM, Ottersen OP. Aquaporin-4 in the central nervous system: cellular and subcellular distribution and coexpression with Kir4.1. Neuroscience. 2004;129:905–913. doi: 10.1016/j.neuroscience.2004.08.053. [DOI] [PubMed] [Google Scholar]
- Nagelhus EA, Ottersen OP. Physiological roles of aquaporin-4 in brain. Physiol Rev. 2013;93:1543–1562. doi: 10.1152/physrev.00011.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy JI, Hacking J, Frankenstein UN, Turley EA. Requirement of the hyaluronan receptor RHAMM in neurite extension and motility as demonstrated in primary neurons and neuronal cell lines. J Neurosci. 1995;15:241–252. doi: 10.1523/JNEUROSCI.15-01-00241.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson C. Diffusion and related transport mechanisms in brain tissue. Rep Prog Phys. 2001;64:815–884. [Google Scholar]
- Nicholson C, Phillips JM. Ion diffusion modified by tortuosity and volume fraction in the extracellular microenvironment of the rat cerebellum. J Physiol. 1981;321:225–257. doi: 10.1113/jphysiol.1981.sp013981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson C, Syková E. Extracellular space structure revealed by diffusion analysis. Trends Neurosci. 1998;21:207–215. doi: 10.1016/s0166-2236(98)01261-2. [DOI] [PubMed] [Google Scholar]
- Nielsen S, Nagelhus EA, Amiry-Moghaddam M, Bourque C, Agre P, Ottersen OP. Specialized membrane domains for water transport in glial cells: high-resolution immunogold cytochemistry of aquaporin-4 in rat brain. J Neurosci. 1997;17:171–180. doi: 10.1523/JNEUROSCI.17-01-00171.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowicka D, Soulsby S, Skangiel-Kramska J, Glazewski S. Parvalbumin-containing neurons, perineuronal nets and experience-dependent plasticity in murine barrel cortex. Eur J Neurosci. 2009;30:2053–2063. doi: 10.1111/j.1460-9568.2009.06996.x. [DOI] [PubMed] [Google Scholar]
- Ogata K, Kosaka T. Structural and quantitative analysis of astrocytes in the mouse hippocampus. Neuroscience. 2002;113:221–233. doi: 10.1016/s0306-4522(02)00041-6. [DOI] [PubMed] [Google Scholar]
- Oohashi T, Edamatsu M, Bekku Y, Carulli D. The hyaluronan and proteoglycan link proteins: Organizers of the brain extracellular matrix and key molecules for neuronal function and plasticity. Exp Neurol. 2015;274:134–144. doi: 10.1016/j.expneurol.2015.09.010. [DOI] [PubMed] [Google Scholar]
- Østby I, Øyehaug L, Einevoll GT, Nagelhus EA, Plahte E, Zeuthen T, Lloyd CM, Ottersen OP, Omholt SW. Astrocytic mechanisms explaining neural-activity-induced shrinkage of extraneuronal space. PLOS Comput Biol. 2009;5:e1000272. doi: 10.1371/journal.pcbi.1000272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadopoulos MC, Verkman AS. Aquaporin water channels in the nervous system. Nat Rev Neurosci. 2013;14:265–277. doi: 10.1038/nrn3468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavicic T, Gauglitz GG, Lersch P, Schwach-Abdellaoui K, Malle B, Korting HC, Farwick M. Efficacy of cream-based novel formulations of hyaluronic acid of different molecular weights in anti-wrinkle treatment. J Drugs Dermatol. 2011;10:990–1000. [PubMed] [Google Scholar]
- Perreault P, Avoli M. 4-aminopyridine-induced epileptiform activity and a GABA-mediated long-lasting depolarization in the rat hippocampus. J Neurosci. 1992;12:104–115. doi: 10.1523/JNEUROSCI.12-01-00104.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pizzorusso T, Medini P, Berardi N, Chierzi S, Fawcett JW, Maffei L. Reactivation of ocular dominance plasticity in the adult visual cortex. Science. 2002;298:1248–1251. doi: 10.1126/science.1072699. [DOI] [PubMed] [Google Scholar]
- Pizzorusso T, Medini P, Landi S, Baldini S, Berardi N, Maffei L. Structural and functional recovery from early monocular deprivation in adult rats. Proc Natl Acad Sci USA. 2006;103:8517–8522. doi: 10.1073/pnas.0602657103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preston M, Gong X, Su W, Matsumoto SG, Banine F, Winkler C, Foster S, Xing R, Struve J, Dean J, Baggenstoss B. Digestion products of the PH20 hyaluronidase inhibit remyelination. Ann Neurol. 2013;73:266–280. doi: 10.1002/ana.23788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prusova A, Conte P, Kucerík J, Alonzo G. Dynamics of hyaluronan aqueous solutions as assessed by fast field cycling NMR relaxometry. Anal Bioanal Chem. 2010a;397:3023–3028. doi: 10.1007/s00216-010-3855-9. [DOI] [PubMed] [Google Scholar]
- Prusova A, Smejkalova D, Chytil M, Velebny V, Kucerik J. An alternative DSC approach to study hydration of hyaluronan. Carbohydr Polym. 2010b;82:498–503. doi: 10.1016/j.carbpol.2013.07.049. [DOI] [PubMed] [Google Scholar]
- Pumain R, Menini C, Heinemann U, Louvel J, Silva-Barrat C. Chemical synaptic transmission is not necessary for epileptic seizures to persist in the baboon. Papio papio Exp Neurol. 1985;89:250–258. doi: 10.1016/0014-4886(85)90280-8. [DOI] [PubMed] [Google Scholar]
- Qiu C, Shivacharan RS, Zhang M, Durand DM. Can Neural Activity Propagate by Endogenous Electrical Field? J Neurosci. 2015;35:15800–15811. doi: 10.1523/JNEUROSCI.1045-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu C, Rilla K, Tammi R, Tammi M, Kröger H, Lammi MJ. Extensive CD44-dependent hyaluronan coats on human bone marrow-derived mesenchymal stem cells produced by hyaluronan synthases HAS1, HAS2 and HAS3. Int J Biochem Cell Biol. 2014;48:45–54. doi: 10.1016/j.biocel.2013.12.016. [DOI] [PubMed] [Google Scholar]
- Rausche G, Igelmund P, Heinemann U. Effects of changes in extracellular potassium, magnesium and calcium concentration on synaptic transmission in area CA1 and the dentate gyrus of rat hippocampal slices. Pflugers Arch. 1990;415:588–593. doi: 10.1007/BF02583510. [DOI] [PubMed] [Google Scholar]
- Risher WC, Andrew RD, Kirov SA. Real-time passive volume responses of astrocytes to acute osmotic and ischemic stress in cortical slices and in vivo revealed by two-photon microscopy. Glia. 2009;57:207–221. doi: 10.1002/glia.20747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roper SN, Obenaus A, Dudek FE. Osmolality and nonsynaptic epileptiform bursts in rat CA1 and dentate gyrus. Ann Neurol. 1992;31:81–85. doi: 10.1002/ana.410310115. [DOI] [PubMed] [Google Scholar]
- Saghyan A, Lewis DP, Hrabe J, Hrabetova S. Extracellular diffusion in laminar brain structures exemplified by hippocampus. J Neurosci Methods. 2012;205:110–118. doi: 10.1016/j.jneumeth.2011.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salah A, Perkins KL. Effects of subtype-selective group I mGluR antagonists on synchronous activity induced by 4-aminopyridine/CGP 55845 in adult guinea pig hippocampal slices. Neuropharmacology. 2008;55:47–54. doi: 10.1016/j.neuropharm.2008.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salah A, Perkins KL. Persistent ictal-like activity in rat entorhinal/perirhinal cortex following washout of 4-aminopyridine. Epilepsy Res. 2011;94:163–176. doi: 10.1016/j.eplepsyres.2011.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saly V, Andrew RD. CA3 neuron excitation and epileptiform discharge are sensitive to osmolality. J Neurophysiol. 1993;69:2200–2208. doi: 10.1152/jn.1993.69.6.2200. [DOI] [PubMed] [Google Scholar]
- Scemes E, Spray DC. Increased intercellular communication in mouse astrocytes exposed to hyposmotic shocks. Glia. 1998;24:74–84. [PMC free article] [PubMed] [Google Scholar]
- Schmidt D, Loscher W. Drug resistance in Epilepsy: Putative Neurobiologic and Clinical Mechanisms. Epilepsia. 2005;46:858–877. doi: 10.1111/j.1528-1167.2005.54904.x. [DOI] [PubMed] [Google Scholar]
- Schwartzkroin PA, Prince DA. Cellular and field potential properties of epileptogenic hippocampal slices. Brain Res. 1978;147:117–130. doi: 10.1016/0006-8993(78)90776-x. [DOI] [PubMed] [Google Scholar]
- Servaty R, Schiller J, Binder H, Arnold K. Hydration of polymeric components of cartilage—an infrared spectroscopic study on hyaluronic acid and chondroitin sulfate. Int J Biol Macromol. 2001;28:121–127. doi: 10.1016/s0141-8130(00)00161-6. [DOI] [PubMed] [Google Scholar]
- Shahar E, Derchansky M, Carlen PL. The role of altered tissue osmolality on the characteristics and propagation of seizure activity in the intact isolated mouse hippocampus. Clin Neurophysiol. 2009;120:673–678. doi: 10.1016/j.clinph.2009.01.014. [DOI] [PubMed] [Google Scholar]
- Shawkat H, Westwood MM, Mortimer A. Mannitol: a review of its clinical uses. Continuing Education in Anaesthesia, Critical Care & Pain. 2012;12:82–85. [Google Scholar]
- Sherman LS, Matsumoto S, Su W, Srivastava T, Back SA. Hyaluronan synthesis, catabolism, and signaling in neurodegenerative diseases. Int J Cell Biol. 2015;2015:368584. doi: 10.1155/2015/368584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman LS, Struve JN, Rangwala R, Wallingford NM, Tuohy TM, Kuntz C., 4th Hyaluronate-based extracellular matrix: keeping glia in their place. Glia. 2002;38:93–102. doi: 10.1002/glia.10053. [DOI] [PubMed] [Google Scholar]
- Shuai J, Bikson M, Hahn PJ, Lian J, Durand DM. Ionic mechanisms underlying spontaneous CA1 neuronal firing in Ca2+-free solution. Biophys J. 2003;84:2099–2111. doi: 10.1016/S0006-3495(03)75017-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siiskonen H, Kärnä R, Hyttinen JM, Tammi RH, Tammi MI, Rilla K. Hyaluronan synthase 1 (HAS1) produces a cytokine-and glucose-inducible, CD44-dependent cell surface coat. Exp Cell Res. 2014;320:153–163. doi: 10.1016/j.yexcr.2013.09.021. [DOI] [PubMed] [Google Scholar]
- Singh TS, Mehta B, Trivedi A. Hypocalcemia presenting with multifocal seizure in a baby with osteopetrosis. J Paediatr Child Health. 2016;52:246–246. doi: 10.1111/jpc.12648. [DOI] [PubMed] [Google Scholar]
- Sloane JA, Batt C, Ma Y, Harris ZM, Trapp B, Vartanian T. Hyaluronan blocks oligodendrocyte progenitor maturation and remyelination through TLR2. Proc Natl Acad Sci USA. 2010;107:11555–11560. doi: 10.1073/pnas.1006496107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snow RW, Dudek FE. Synchronous epileptiform bursts without chemical transmission in CA2, CA3 and dentate areas of the hippocampus. Brain Res. 1984;298:382–385. doi: 10.1016/0006-8993(84)91443-4. [DOI] [PubMed] [Google Scholar]
- Somjen GG. Ions in the brain: normal function, seizures, and stroke. New York, NY: Oxford University Press Inc.; 2004. [Google Scholar]
- Somjen GG, Müller M. Potassium-induced enhancement of persistent inward current in hippocampal neurons in isolation and in tissue slices. Brain Res. 2000;885:102–110. doi: 10.1016/s0006-8993(00)02948-6. [DOI] [PubMed] [Google Scholar]
- Spicer AP, McDonald JA. Characterization and molecular evolution of a vertebrate hyaluronan synthase gene family. J Biol Chem. 1998;273:1923–1932. doi: 10.1074/jbc.273.4.1923. [DOI] [PubMed] [Google Scholar]
- Strange K. Regulation of solute and water balance and cell volume in the central nervous system. J Am Soc Nephrol. 1992;3:12–27. doi: 10.1681/ASN.V3112. [DOI] [PubMed] [Google Scholar]
- Su H, Alroy G, Kirson ED, Yaari Y. Extracellular calcium modulates persistent sodium current-dependent burst-firing in hippocampal pyramidal neurons. J Neurosci. 2001;21:4173–4182. doi: 10.1523/JNEUROSCI.21-12-04173.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su G, Kintner DB, Flagella M, Shull GE, Sun D. Astrocytes from Na+-K+-Cl− cotransporter-null mice exhibit absence of swelling and decrease in EAA release. Am J Physiol Cell Physiol. 2002;282:C1147–1160. doi: 10.1152/ajpcell.00538.2001. [DOI] [PubMed] [Google Scholar]
- Sykova E, Vorisek I, Mazel T, Antonova T, Schachner M. Reduced extracellular space in the brain of tenascin-R- and HNK-1-sulphotransferase deficient mice. Eur J Neurosci. 2005;22:1873–1880. doi: 10.1111/j.1460-9568.2005.04375.x. [DOI] [PubMed] [Google Scholar]
- Sykova E, Nicholson C. Diffusion in brain extracellular space. Physiol Rev. 2008;88:1277–1340. doi: 10.1152/physrev.00027.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sypert GW, Ward AA., Jr Changes in extracellular potassium activity during neocortical propagated seizures. Exp Neurol. 1974;45:19–41. doi: 10.1016/0014-4886(74)90097-1. [DOI] [PubMed] [Google Scholar]
- Tammi RH, Passi AG, Rilla K, Karousou E, Vigetti D, Makkonen K, Tammi MI. Transcriptional and post-translational regulation of hyaluronan synthesis. FEBS J. 2011;278:1419–1428. doi: 10.1111/j.1742-4658.2011.08070.x. [DOI] [PubMed] [Google Scholar]
- Taylor CP, Dudek FE. Synchronous neural afterdischarges in rat hippocampal slices without active chemical synapses. Science. 1982;218:810–812. doi: 10.1126/science.7134978. [DOI] [PubMed] [Google Scholar]
- Thompson SM, Gähwiler BH. Activity-dependent disinhibition. II. Effects of extracellular potassium, furosemide, and membrane potential on ECl- in hippocampal CA3 neurons. J Neurophysiol. 1989;61:512–523. doi: 10.1152/jn.1989.61.3.512. [DOI] [PubMed] [Google Scholar]
- Thorne RF, Legg JW, Isacke CM. The role of the CD44 transmembrane and cytoplasmic domains in co-ordinating adhesive and signalling events. J Cell Sci. 2004;117:373–380. doi: 10.1242/jcs.00954. [DOI] [PubMed] [Google Scholar]
- Thorne RG, Nicholson C. In vivo diffusion analysis with quantum dots and dextrans predicts the width of brain extracellular space. Proc Natl Acad Sci USA. 2006;103:5567–5572. doi: 10.1073/pnas.0509425103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tien JY, Spicer AP. Three vertebrate hyaluronan synthases are expressed during mouse development in distinct spatial and temporal patterns. Dev Dyn. 2005;233:130–141. doi: 10.1002/dvdy.20328. [DOI] [PubMed] [Google Scholar]
- Toole BP. Hyaluronan in morphogenesis. Semin Cell Dev Biol. 2001;12:79–87. doi: 10.1006/scdb.2000.0244. [DOI] [PubMed] [Google Scholar]
- Toole BP. Hyaluronan: from extracellular glue to pericellular cue. Nat Rev Cancer. 2004;4:528–539. doi: 10.1038/nrc1391. [DOI] [PubMed] [Google Scholar]
- Toole BP, Trelstad RL. Hyaluronate production and removal during corneal development in the chick. Dev Biol. 1971;26:28–35. doi: 10.1016/0012-1606(71)90104-7. [DOI] [PubMed] [Google Scholar]
- Towler MC, Hardie DG. AMP-activated protein kinase in metabolic control and insulin signaling. Circ Res. 2007;100:328–341. doi: 10.1161/01.RES.0000256090.42690.05. [DOI] [PubMed] [Google Scholar]
- Traub RD, Dudek FE, Taylor CP, Knowles WD. Simulation of hippocampal afterdischarges synchronized by electrical interactions. Neuroscience. 1985;14:1033–1038. doi: 10.1016/0306-4522(85)90274-x. [DOI] [PubMed] [Google Scholar]
- Traub RD, Wong RKS. Cellular mechanism of neuronal synchronization in epilepsy. Science. 1982;216:745–747. doi: 10.1126/science.7079735. [DOI] [PubMed] [Google Scholar]
- Traynelis SF, Dingledine R. Potassium-induced spontaneous electrographic seizures in the rat hippocampal slice. J Neurophysiol. 1988;59:259–276. doi: 10.1152/jn.1988.59.1.259. [DOI] [PubMed] [Google Scholar]
- Traynelis SF, Dingledine R. Role of extracellular space in hyperosmotic suppression of potassium-induced electrographic seizures. J Neurophysiol. 1989a;61:927–938. doi: 10.1152/jn.1989.61.5.927. [DOI] [PubMed] [Google Scholar]
- Traynelis SF, Dingledine R. Modification of potassium-induced interictal bursts and electrographic seizures by divalent cations. Neurosci Lett. 1989b;98:194–199. doi: 10.1016/0304-3940(89)90509-0. [DOI] [PubMed] [Google Scholar]
- Tsai PL, Lian LM, Chen WH. Hypocalcemic seizure mistaken for idiopathic epilepsy in two cases of DiGeorge syndrome (chromosome 22q11 deletion syndrome) Acta Neurol Taiwan. 2009;18:272–275. [PubMed] [Google Scholar]
- Turley EA, Hossain MZ, Sorokan T, Jordan LM, Nagy JI. Astrocyte and microglial motility in vitro is functionally dependent on the hyaluronan receptor RHAMM. Glia. 1994;12:68–80. doi: 10.1002/glia.440120109. [DOI] [PubMed] [Google Scholar]
- Underhill CB, Toole BP. Transformation-dependent loss of the hyaluronate-containing coats of cultured cells. J Cell Physiol. 1982;110:123–128. doi: 10.1002/jcp.1041100204. [DOI] [PubMed] [Google Scholar]
- Uyama T, Kitagawa H, Sugahara K. Biosynthesis of glycosaminoglycans and proteoglycans. In: Kamerling JP, editor. Comprehensive Glycoscience. Vol. 3. Elsevier; 2007. pp. 79–104. [Google Scholar]
- Vedunova M, Sakharnova T, Mitroshina E, Perminova M, Pimashkin A, Zakharov Y, Dityatev A, Mukhina I. Seizure-like activity in hyaluronidase-treated dissociated hippocampal cultures. Front Cell Neurosci. 2013;7:149. doi: 10.3389/fncel.2013.00149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verbalis JG. Brain volume regulation in response to changes in osmolality. Neuroscience. 2010;168:862–870. doi: 10.1016/j.neuroscience.2010.03.042. [DOI] [PubMed] [Google Scholar]
- Vigetti D, Deleonibus S, Moretto P, Karousou E, Viola M, Bartolini B, Hascall VC, Tammi M, De Luca G, Passi A. Role of UDP-N-acetylglucosamine (GlcNAc) and O-GlcNAcylation of hyaluronan synthase 2 in the control of chondroitin sulfate and hyaluronan synthesis. J Biol Chem. 2012;287:35544–35555. doi: 10.1074/jbc.M112.402347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigetti D, Genasetti A, Karousou E, Viola M, Clerici M, Bartolini B, Moretto P, De Luca G, Hascall VC, Passi A. Modulation of hyaluronan synthase activity in cellular membrane fractions. J Biol Chem. 2009;284:30684–30694. doi: 10.1074/jbc.M109.040386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigetti D, Ori M, Viola M, Genasetti A, Karousou E, Rizzi M, Pallotti F, Nardi I, Hascall VC, De Luca G, Passi A. Molecular cloning and characterization of UDP-glucose dehydrogenase from the amphibian Xenopus laevis and its involvement in hyaluronan synthesis. J Biol Chem. 2006;281:8254–8263. doi: 10.1074/jbc.M508516200. [DOI] [PubMed] [Google Scholar]
- Vigetti D, Viola M, Karousou E, De Luca G, Passi A. Metabolic control of hyaluronan synthases. Matrix Biol. 2014;35:8–13. doi: 10.1016/j.matbio.2013.10.002. [DOI] [PubMed] [Google Scholar]
- Villa C, Combi R. Potassium Channels and Human Epileptic Phenotypes: An Updated Overview. Front Cell Neurosci. 2016;10:81. doi: 10.3389/fncel.2016.00081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walther H, Lambert JD, Jones RS, Heinemann U, Hamon B. Epileptiform activity in combined slices of the hippocampus, subiculum and entorhinal cortex during perfusion with low magnesium medium. Neurosci Lett. 1986;69:156–161. doi: 10.1016/0304-3940(86)90595-1. [DOI] [PubMed] [Google Scholar]
- Walz W. Role of astrocytes in the clearance of excess extracellular potassium. Neurochem Int. 2000;36:291–300. doi: 10.1016/s0197-0186(99)00137-0. [DOI] [PubMed] [Google Scholar]
- Wang L, Dufour S, Valiante TA, Carlen PL. Extracellular Potassium and Seizures: Excitation, Inhibition and the Role of Ih. Int J Neural Syst. 2016;26:1650044. doi: 10.1142/S0129065716500441. [DOI] [PubMed] [Google Scholar]
- Weigel PH. Hyaluronan Synthase: The Mechanism of Initiation at the Reducing End and a Pendulum Model for Polysaccharide Translocation to the Cell Exterior. Int J Cell Biol. 2015;2015:367579. doi: 10.1155/2015/367579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss SA, Faber DS. Field effects in the CNS play functional roles. Front Neural Circuits. 2010;4:15. doi: 10.3389/fncir.2010.00015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weissmann B, Meyer K. The structure of hyalobiuronic acid and of hyaluronic acid from umbilical cord. J Am Chem Soc. 1954;76:1753–57. [Google Scholar]
- Weissmann B, Meyer K, Sampson P, Linker A. Isolation of oligosaccharides enzymatically produced from hyaluronic acid. J Biol Chem. 1954;208:417–429. [PubMed] [Google Scholar]
- Xiao F, Nicholson C, Hrabe J, Hrabetova S. Diffusion of flexible random-coil dextran polymers measured in anisotropic brain extracellular space by integrative optical imaging. Biophys J. 2008;95:1382–1392. doi: 10.1529/biophysj.107.124743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie L, Kang H, Xu Q, Chen MJ, Liao Y, Thiyagarajan M, O’Donnell J, Christensen DJ, Nicholson C, Iliff JJ, Takano T, Deane R, Nedergaard M. Sleep drives metabolic clearance from the adult brain. Science. 2013;342:373–377. doi: 10.1126/science.1241224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaari Y, Konnerth A, Heinemann U. Spontaneous epileptiform activity of CA1 hippocampal neurons in low extracellular calcium solutions. Exp Brain Res. 1983;51:153–156. doi: 10.1007/BF00236813. [DOI] [PubMed] [Google Scholar]
- Yaari Y, Konnerth A, Heinemann U. Nonsynaptic epileptogenesis in the mammalian hippocampus in vitro. II. Role of extracellular potassium. J Neurophysiol. 1986;56:424–438. doi: 10.1152/jn.1986.56.2.424. [DOI] [PubMed] [Google Scholar]
- Yamaguchi Y. Lecticans: Organizers of the brain extracellular matrix. Cell Mol Life Sci. 2000;57:276–289. doi: 10.1007/PL00000690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto H, Tobisawa Y, Inubushi T, Irie F, Yamaguchi Y. A mammalian homolog of the zebrafish Transmembrane Protein 2 (TMEM2) is the long-sought-after cell surface hyaluronidase. J Biol Chem. 2017;292:7304–7313. doi: 10.1074/jbc.M116.770149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida H, Nagaoka A, Kusaka-Kikushima A, Tobiishi M, Kawabata K, Sayo T, Sakai S, Sugiyama Y, Enomoto H, Okada Y, Inoue S. KIAA1199, a deafness gene of unknown function, is a new hyaluronan binding protein involved in hyaluronan depolymerization. Proc Natl Acad Sci USA. 2013;110:5612–5617. doi: 10.1073/pnas.1215432110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young IJ. Reversible seizures produced by neuronal hyaluronic acid depletion. Exp Neurol. 1963;8:195–202. [PubMed] [Google Scholar]
- Zeuthen T, MacAulay N. Cotransport of water by Na+–K+–2Cl– cotransporters expressed in Xenopus oocytes: NKCC1 versus NKCC2. J Physiol. 2012;590:1139–1154. doi: 10.1113/jphysiol.2011.226316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Ladas TP, Qiu C, Shivacharan RS, Gonzalez-Reyes LE, Durand DM. Propagation of epileptiform activity can be independent of synaptic transmission, gap junctions, or diffusion and is consistent with electrical field transmission. J Neurosci. 2014;34:1409–1419. doi: 10.1523/JNEUROSCI.3877-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou B, Weigel JA, Saxena A, Weigel PH. Molecular cloning and functional expression of the rat 175-kDa hyaluronan receptor for endocytosis. Mol Biol Cell. 2002;13:2853–2868. doi: 10.1091/mbc.02-03-0048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziburkus J, Cressman JR, Barreto E, Schiff FJ. Interneuron and pyramidal cell interplay during in vitro seizure-like events. J Neurophysiol. 2006;95:3948–3954. doi: 10.1152/jn.01378.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuckermann EC, Glaser GH. Hippocampal epileptic activity induced by localized ventricular perfusion with high-potassium cerebrospinal fluid. Exp Neurol. 1968;20:87–110. doi: 10.1016/0014-4886(68)90126-x. [DOI] [PubMed] [Google Scholar]