

Abstract
A single molecular scaffold can be adapted to interact with diverse targets, either separately or simultaneously. Nucleosides and nucleotides in which ribose is substituted with bicyclo[3.1.0]hexane are an example of a versatile drug-like scaffold for increasing selectivity at their classical targets: kinases, polymerases, adenosine and P2 receptors. Also, by applying structure-based functional group manipulations, rigidified adenosine derivatives can be repurposed to satisfy pharmacophoric requirements of various GPCRs, ion channels, enzymes and transporters, initially detected as off-target activities. Recent examples include 5HT2B serotonin receptor antagonists and novel dopamine transporter allosteric modulators. This directable target diversity establishes rigid nucleosides as privileged scaffolds.
Graphical abstract
Introduction
It is now recognized that many pharmaceuticals on the market for central nervous system (CNS) diseases, cancer and other conditions hit multiple targets [1–3]. The pharmacological spectrum of a given compound can contribute to its net biological benefit in a disease state or detract from it through undesired side-effects [4]. Thus, it is important to assess and direct, if possible, the multiple actions of a compound or compound class. Here, we analyze in detail the polypharmacology of a class of nucleoside derivatives that were initially introduced as antiviral agents [5,6] and as selective ligands of purine receptors in the cell membrane [7]. Structural modification within this class can direct a given compound toward multitarget action or a single interaction, either at the original receptor or through previously undetected mechanisms.
The term ‘privileged scaffold’ was coined by Evans et al. [8,9] as a core structure that can be adapted to different protein targets by functionalization, early examples of which were benzodiazepines, indoles and 1,4-dihydropyridines [10]. This concept has been explored widely for chemically diverse scaffolds, especially flat heterocyclic systems. Benzodiazepines, otherwise known as allosteric enhancers at the γ-aminobutyric acid (GABA)A ionotropic receptor, can be functionalized to achieve high-affinity binding at various G-protein-coupled receptors (GPCRs) of interest such as cholecystokinin (CCK) receptors [8].
Biologically relevant chemical space is immense, and various attempts have been made to chart and categorize it [11–13]. Within that space, privileged structures, fragments or scaffolds have been identified by diverse screening and by design, including combinatorial design of bicyclic structures [14–16]. Purine nucleobases were previously identified as privileged structures for medicinal chemistry [14,15]. We and others have modified nucleoside derivatives to expand the range of their target proteins and to shift their selectivity. It is now apparent that nucleosides can serve as privileged scaffolds to bind to diverse proteins, and the many nucleoside drugs approved for therapy testify to the pharmacological versatility of this scaffold [17].
Conventional targets of nucleosides and small nucleotides
Nucleosides and nucleotides constitute a large class of drug-like molecules. Many nucleosides have proven their favorable physicochemical properties for use in humans in anticancer and antiviral therapies [17], which account for more than 30 pharmaceuticals currently on the market, mostly in those two categories. Complex nucleoside derivatives are also useful as antibiotics [18]. Therapies based on oligonucleotides and aptamers are also under development [19,20]. Various other nucleoside drugs, such as kinase inhibitors, can mimic a substrate and compete for a common binding site on their targets.
Naturally occurring extracellular nucleosides and nucleotides bind to and activate a variety of cell surface receptors that have diverse and important signaling roles in the body [21–23]. These receptors include family A (rhodopsin-like) GPCRs [i.e., adenosine receptors (ARs) and P2Y receptors (P2YRs)] and ligand-gated ion channels [i.e., P2X receptors (P2XRs)]. There are four AR subtypes and eight subtypes of P2YRs, which can function as monomeric GPCRs, as dimeric species in some cases or as higher order aggregates. The P2YRs have two subfamilies: P2Y1R-like Gq-coupled and P2Y12R-like Gi-coupled. In addition, there are seven P2XR subunits that form obligatory trimeric channels activated by ATP. The heterotrimeric or homotrimeric composition of each determines a characteristic pharmacology. Purine and pyrimidine nucleotides are important in the activation of P2YRs, whereas ARs and P2XRs are principally activated by adenine nucleosides and nucleotides, respectively. The concentrations of extracellular nucleosides and nucleotides, and consequently the levels of endogenous stimulation of these receptors, are controlled by enzymatic, transport and channel processes and by cell damage causing the release of these ligands from intracellular sources. The production and degradation of extracellular nucleosides and nucleotides along with their signaling functions through 19 receptors can be considered a ‘purinome’ [24]. Purinergic signaling through these receptors, transporters and enzymes has a role in most physiological processes and constitutes a major system for homeostatic control in the body. This has led to numerous therapeutic concepts, such as selective A3AR activation for cancer, inflammatory disease, chronic neuropathic pain and other conditions [25,26].
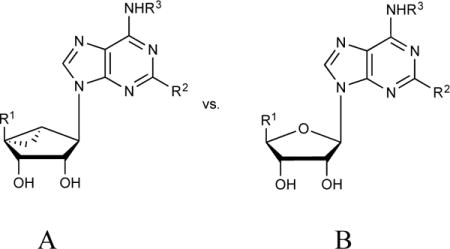
Our studies of nucleoside polypharmacology have focused recently on methanocarba nucleoside and nucleotide analogs, in which the tetrahydrofuryl core of the ribose ring system is replaced with a rigid bicyclo[3.1.0]hexane. This ring system was first introduced in nucleosides by Marquez and colleagues, and applied to conformational control of substrates of kinases and for transporters, aptamers and oligonucleotides [5,6]. There are two isomeric forms in the methanocarba series, depending on the fusion site of the cyclopropane ring: North (N)-methanocarba and South (S)-methanocarba (Fig. 1). These constrained [rings act as bioisosteres of ribose to pre-establish the conformation that is preferred by the target protein(s) or nucleic acids, thus lowering energy barriers for binding. The bicyclo[3.1.0] hexane ring lacks a furanose-type oxygen, which in some cases offers stabilizing effects. However, compared with the other locked nucleosides such as locked nucleic acids (LNAs) (containing an extra methylene bridge connecting the 4′ carbon and 2′ oxygen as an ether) [27], methanocarba rings feature the following advantages in addition to conformational rigidity: (i) availability of 2′ and 3′ hydroxyl groups for interaction with biomolecules, which are essential for ribose-like behavior; (ii) less sterically crowded than LNAs around the furanose plane.
FIGURE 1.
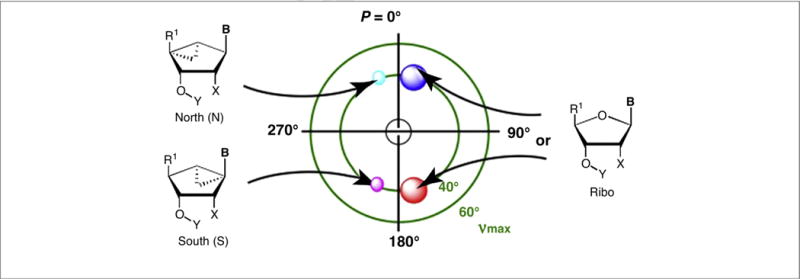
Relationship between ribose ring structure and favored conformations as depicted on the pseudorotational cycle, made by a mathematical formula to describe all twists of the ribose ring [5]. P = pseudorotational angle; ν = out of plane angle. On right side: red circle = region of North (N) conformation in nature; with cyan and link circles representing the conformations of the methanocarba rings (left side). Typically: B = nucleobase; Y = H; X = OH. R1 can be oxymethylene or carbonyl moieties.
The synthesis of these conformationally locked nucleosides requires long synthetic routes, the various stages of which are described in detail elsewhere [28–32]. Optimization of the synthetic approaches has made this nucleoside class more synthetically tractable and allowed stereochemical purity to be achieved. Common intermediates allow the introduction of diverse functional groups that can direct the polypharmacology of this compound class. The (S)-methanocarba nucleosides were initially prepared and tested as a mixture of enantiomers [39], but in 2008 the synthesis of the pure enantiomer series was reported [33], which enabled conformational studies.
Receptor targets
The methanocarba ring constraint was shown to be useful for designing ligands of ARs, P2YRs and P2XRs, as well as antiviral and anticancer compounds (Fig. 2). For example, a potent agonist of the A3AR, MRS5980 (1), contains a (N)-methanocarba ring [34]. MRS5980 and its congeners were demonstrated to have drug-like properties in ADMET tests, including oral bioavailability [34]. The high affinity (N)-methanocarba agonist MRS5980 (1) was twofold and 32-fold more-potent in binding to the human (h) and mouse (m)A3AR, respectively, than the corresponding ribose derivative MRS7294 (2) [35]. Similarly, the N6-propyl equivalents of 1 and 2 (not shown) displayed even more pronounced difference; the binding affinity of the (N)-methanocarba analog was sixfold higher than the ribose equivalent at the hA3AR and 88-fold in affinity higher at the mA3AR [35]. At the rat (r)A3AR, agonist affinity was better maintained with respect to hA3AR in the methanocarba series compared with the ribose series [7]. Selectivity for the A3AR over the other ARs, especially the A2AAR, was also increased by this ring modification. Thus, the (N)-methanocarba nucleosides, simple and hypermodified, either preserved or enhanced the affinity at the A3AR in multiple species.
FIGURE 2.
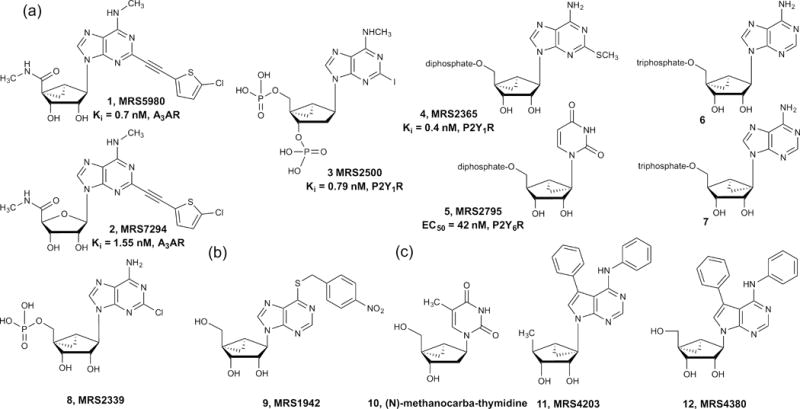
Structures of methanocarba nucleosides that interact with: (a) P2Y1R, (b) ENT1, (c) polymerases and kinases [35,45,62].
The (N)- and (S)-methanocarba rings were systematically incorporated in most of the native nucleotide ligands of the P2YR family. At the P2Y1R, the (N) conformer was highly favored, whereas at the P2Y6R, which is also coupled to Gq-protein, the (S) conformer was highly favored [28]. MRS2365 (4) was enhanced in affinity (Ki = 0.4 nM) as well as selectivity as an agonist of the P2Y1R, a platelet receptor that is important in aggregation, in comparison to its ribose analog 2-methylthio-adenosine 5′-diphosphate (2-MeSADP, Ki ~ 100 nM). Compound 4 activated only the P2Y1R, whereas 2-MeSADP, like the native agonist ADP, additionally activated the Gi-coupled P2Y12R and P2Y13R. In fact, neither the (N)- or (S)-methanocarba analogs of the native agonists of the Gi-coupled P2YR subfamily activated those receptors, perhaps because of steric hindrance of the cyclopropyl ring by a conserved Val3.30 residue in TM3 [36]. However, at the Gq-coupled P2Y6R, the (N) analog of native agonist UDP was inactive, whereas the (S) analog, MRS2795 (5, enantiomerically pure), was 7-fold more potent than UDP in a functional assay [37]. Thus, even within the same GPCR family that has a relatively high sequence identity (33% for hP2Y1R and hP2Y6R compared with 21% for hP2Y1R and hP2Y12R), the conformational preference of the ribose was variable and dramatic, leading to increased selectivity of the methanocarba nucleotide analogs.
A 2′-deoxy-methanocarba nucleotide, (1′R,2′S,4′S,5′S)-4-(2-iodo-6-methylamino-purin-9-yl)-1-[(phosphato)-methyl]-2-(phosphato)-bicyclo[3.1.0]hexane (MRS2500, 3) is the most potent known competitive antagonist for the ADP-activated P2Y1R and has antithrombotic activity [38]. The X-ray structure of the P2Y1R in complex with 3 confirmed the (N) conformation of this inhibitor (Fig. 3). The nucleotide binding site was located almost exclusively in the receptor’s extracellular region, which is unusual for an orthosteric ligand binding region (i.e., the same site at which a native agonist binds). The hydrophobic nucleobase of 3 was inserted in a hydrophobic pocket at the top of the binding site between the extracellular tips of transmembrane helix (TM)6 and TM7 and the N-terminal domain. The hydrophilic and charged, phosphorylated pseudoribose moiety was anchored at the bottom of the binding site, coordinated by residues in the second extracellular loop (EL2). The phosphate moieties at 3′ and 5′ positions contributed to ligand recognition by associating with positively charged and H-bonding amino acids of the receptor’s outer regions, which was confirmed by site-directed mutagenesis [38].
FIGURE 3.
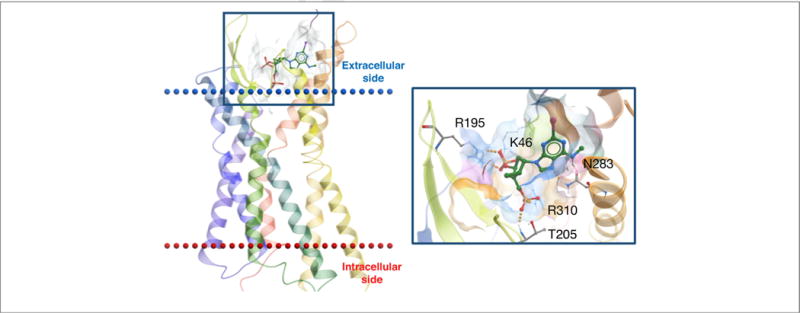
X-ray structure of the hP2Y1R showing the binding mode of orthosteric antagonist MRS2500 (3) [38]. In this inactive state, the 3′-phosphate is coordinated by K46 (EL1) and R195 (EL2), the 5′-phosphate deeper in the binding site by T205 (EL2) and R310 (7.39) and N6 by N283 (6.58).
Four subtypes of P2XRs in primary rat neurons (homotrimeric P2X1, P2X2 and P2X3Rs and a heteromeric P2X2/3R) also displayed a strong preference for the (N)-methanocarba analog of ATP (6) compared with the (S)-methanocarba analog 7 (racemic), which was weak or inactive [39]. An (N)-methanocarba equivalent (MRS2339, 8) of 2-chloro-AMP that activates a P2 × 4R on cardiac myocytes is an experimental drug for treating heart failure [40].
Kinase and RNA targets
Marquez and colleagues have extensively characterized rigidified nucleosides and nucleotides at diverse kinases and polymerases [5,6]. Although in general (S)-methanocarba isomers are the preferred substrates for nucleoside kinases and (N) isomers for nucleotide kinases, in many cases the nonpreferred isomers are metabolized at biologically significant rates, depending on the substrate and enzyme variant. For instance, h-deoxycytidine kinase and nucleoside-diphosphate kinases (NDPKs) readily phosphorylate (S)- and (N)-methanocarba-cytidine [41], but there is no human counterpart that metabolizes (N)-methanocarbathymidine (N-MCT, 10; Fig. 2). Nevertheless, N-MCT is a potent inhibitor of herpes simplex virus (HSV) [42] because of its species-dependent selectivity as a substrate for HSV thymidine kinases (HSV-TKs). It is also being evaluated for treatment of Kaposi’s-sarcoma-associated herpes virus [43], and a clinical trial of N-MCT was initiated for shingles [44]. Hence, in a diseased state the combination of properties of nucleoside/nucleotide-metabolizing enzymes (i.e., viral and cellular kinases and polymerases) was exploited to turn N-MCT into a potent HSV inhibitor [45,46]. Similarly, most polymerases [e.g., hDNA polymerase and HIV reverse transcriptase (HIV-RT)] incorporated only (N)-methanocarba nucleoside triphosphates [47–49]. A qualitative study of locked nucleotides as substrates of ectonucleotidases also suggested some degree of bias. The rNTPDase1 hydrolyzed (N)- and (S)-isomers at about half the rate of ATP, whereas rNTPDase2 did not hydrolyze (N)-methanocarba-ATP (6) [50]. The corresponding (S)-isomer 7 was hydrolyzed three-times slower than ATP by rNTPDase2. Also, both the isomers of methanocarba-AMP were relatively stable to 5′-ectonucleotidase (CD73) [50].
Recently, (N)- and (S)-methanocarba nucleosides MRS4203 11 and MRS4380 12 were also introduced as inhibitors of adenosine kinase (ADK), another conventional purine target [51]. Inhibitors of hADK have pronounced antiepileptic and antiepileptogenic effects [52]. Compounds 11 and 12 were comparable in potency to a standard ADK carbocyclic nucleoside inhibitor A-134974 [51]. A year after the first report of methanocarba nucleosides by Marquez and colleagues [53], Altmann et al. independently disclosed that the stability of oligodeoxynucleotide heteroduplexes involving (N)-methanocarba-T (TN) increased, and the (S)-methanocarba-T destabilized the heteroduplex [54,55]. Later, the stability of oligonucleotides having multiple TN was found to be additive in nature [5,56]. This led to a study on RNAi of siRNAs with TN modifications. Compared with LNAs, in addition to the increased thermal and serum stability of siRNA-duplexes in A-form, the North locked TN congeners were less subject to innate immunostimulation but with comparable gene-silencing activities [57].
Unconventional nucleoside targets
Recent findings suggest that the utility of nucleosides and nucleotides extends beyond the classical targets, especially among analogs that contain a sterically constrained substitution of ribose [58]. In some cases, a secondary activity, such as antioxidant, can be engineered into the nucleoside or nucleotide [59]. In other cases, the nucleobase alone displays a spectrum of activities (e.g., adenine derivatives that hit multiple targets). Therefore, methanocarba nucleosides and nucleotides are particularly suited for the exploration of polypharmacology, which needs to be considered in the biological characterization of any nucleoside or nucleotide. Furthermore, the rigidity of this scaffold facilitates the 3D exploration of ligand–protein interactions.
The methanocarba ring system is one of many constrained small ring systems that have been applied to conformational control in drug design. This is a general modification of nucleosides and nucleotides that can enhance high-affinity interactions with a variety of protein targets. The methanocarba modification has been shown to increase the affinity or selectivity, or both parameters, compared with ribose at a target such as a GPCR by enforcing a specific conformation that approximates the target-preferred conformation, such as at a GPCR [24]. Thus, drug-like methanocarba AR ligands have been repurposed to satisfy the pharmacophoric requirements of various GPCRs and other protein targets. The ability to introduce diverse chemical functionality at multiple sites in this privileged scaffold class provides the ability to adapt to different biological targets. By systematically identifying off-target sites as minor activities of nucleosides and then enhancing those activities by stepwise structural modification, this approach can be considered as scaffold repurposing.
Representative nucleosides, designed for interaction with ARs, were screened in radioligand binding assays at 53 diverse off-target activities performed by the Psychoactive Drug Screening Program (PDSP) at the University of North Carolina. The weak offtarget hits at non-nucleoside receptors and transporters included the α- and β-adrenergic receptors, serotonin receptors, δ-opioid receptor (DOR), sigma receptors, the translocator protein (TSPO), among others. A prototypical methanocarba nucleoside training set of ten congeneric compounds was used to probe the orientation of these ligands in selected off-target proteins (e.g., GPCRs) for which structural information is available. This led to SAR studies of nucleoside derivatives at the off-target GPCRs and other protein targets. For example, the potent agonist of h and mA3AR MRS5698 (13, Ki 3 nM) and its congeners additionally bound to the rat TSPO in the 200–300 nM range. Thus, this systematic effort revealed a modest number of unanticipated interactions of these rigidified and highly substituted nucleosides and their substructures with diverse off-target sites.
Subsequent studies focused on particular off-target hits to enhance the activity and/or selectivity at these sites as well as to minimize the activity at the previous on-target site (i.e., the Ars). Being conformationally constrained, (N)-methanocarba derivatives increase awareness of the spatial environment at diverse receptors of known structure. In some cases, we have arrived at self-consistent computational models for recognition of nucleosides at these unconventional targets. These nucleosides are not pan-assay interference compounds (PAINS) because each compound has no more than a few diverse interactions. However, the versatility of substitution extends the relevance of this chemical class to diverse targets. Thus, we have expanded the range of target proteins that interact with nucleoside derivatives.
Off-target activity of nucleosides at other (non-adenosine) GPCRs
A detailed examination of the polypharmacology of methanocarba nucleosides and their derivatives revealed moderate cross reactivity with other non-purine GPCRs, usually at higher concentrations. This cross reactivity could be modulated depending on the ligand functionalization. The SARs of the (N)-methanocarba nucleoside derivatives that were synthesized to achieve selective activation of a subtype of the ARs (e.g., A3AR) were analyzed with respect to off-target GPCR activities. The contributions to the new SAR of large hydrophobic N6 groups, adenine nitrogens, 5′-functionalization or extended C2-alkynyl groups were explored. Thus, functional group substitution on the methanocarba adenosine derivatives was customized to favor each class of new targets.
The ARs belong to the rhodopsin-like α-branch GPCRs, and other members include the biogenic amine receptors. In an initial study of polypharmacology, various off-target activities of AR agonists were found at the biogenic amine receptors, with Ki values as low as 61 nM. To design nucleosides that are selective for the biogenic amine receptors, it was necessary to incorporate the SAR features favoring these off-target receptors, to deselect the well-defined activity at the ARs. Some nucleoside derivatives synthesized in the context of AR activity interacted weakly with 5HT2B/5HT2C serotonin receptors (Table 1). This simplified table indicates the typical contributions of each structural feature to the overall affinity of these nucleosides at the indicated target. The SAR of these (N)-methanocarba adenosine derivatives was then probed to design related molecules that were even more potent in binding at 5HT2B and 5HT2C receptors than our original fortuitous hits. Using computational docking and molecular dynamics, putative interactions of these rigid nucleosides with 5HT2B and 5HT2C receptors were predicted. Functional assays demonstrated that the nucleosides were antagonists at serotonin receptors. N6-dicyclopropylmethyl 5′-methylamide (N)-methanocarba derivative MRS7185 (15;) was 170-fold selective in functional assays as an antagonist of the 5HT2BR compared with the 5HT2CR. The corresponding 5′-ethyl ester MRS7221 (14) bound with higher affinity but not selectivity at the 5HT2BR.
Table 1.
Interactions of representative adenosine derivatives with receptors and transporters. This simplified table indicates the typical contributions of each structural feature to the overall affinity of these nucleosides at the indicated target, with the beneficial gain in binding affinity indicated as +++ > ++ > + > −. Values were determined from several examples and are not necessarily general to all examples.
|
| ||||||
|---|---|---|---|---|---|---|
| Structural feature | hA1AR | hA3AR | mA3AR | h5HT2B/2C | hDAT | hNET |
| References: | 62, 35, 72 | 62, 35, 72 | 62 | 58, 62 | 35, 72 | 35, 72 |
| Ribose modifications (A vs. B), R1 at 5′ | ||||||
| A (N)-methanocarba | +++ | +++ | +++ | +++ | +++ | + |
| 5′-CONHCH3 | +++ | +++ | +++ | ++ | +++ | + |
| 5′-COOMe or 5′-COOEt | ++ | ++a | − | ++ | +++ | + |
| 5′-COOPr | ND | ++ | + | + | +++ | − |
| 5′-COO-i-Pr | ND | + | − | + (2C) | − | − |
| 5′-COOH | ND | + | − | + | + | − |
| 5′-CONH-(CH2)2-NH2 | − | − | − | + | + | − |
| 4′-truncated | + | ++a | + | + | − | ND |
| B 9-riboside (CH2OH) | +++ | ++ | + | ++ | ++ | + |
| Adenine modifications: R2 at C2; R3 at N6 | ||||||
| C2-Cl | +++ | +++ | +++ | ++ | − | − |
| C2-C≡C-(5-Cl-thien-2-yl) | − | +++ | ++ | + | +++ | + |
| N6-Me | ++ | +++ | ++ | + | +++ | + |
| N6-Pr | ++ | +++ | ++ | ND | ++ | − |
| N6-CH(cPr)2 | +++ | ++ | ND | +++ | − | − |
| N6-CH2Ph | ++ | +++ | +++ | ++ | − | − |
| N6-(Me)2 | − | + | + | ++ | − | − |
| 1-deaza | +++ | +++ | ++ | + | − | − |
ND, not determined.
reduces agonist efficacy.
The pharmacokinetics of 15 and the methyl ester homolog of 14 demonstrated prolonged exposure in vivo. The ester derivative was shown to be eliminated slowly in the rat, and therefore not rapidly hydrolyzed by esterases. Nucleoside derivatives typically do not readily cross the blood–brain barrier, and thus many of the (N)-methanocarba nucleosides described in this work are expected to attain much higher concentrations in the periphery than in the brain. Peripherally acting 5HT2BR antagonists might be useful for protection of liver and heart tissue because activation of the 5HT2BR causes fibrosis in these tissues.
The orientation of the methanocarba nucleosides in each target protein, such as a GPCR, can be analyzed by molecular modeling or in the best case by X-ray crystallography, which aids in their subsequent derivatization. In different GPCRs, the relative orientation of the pseudoribose and the nucleobase can be similar or even reversed. Docking and molecular dynamics (MD) analyses of the rigid adenosine derivative MRS7185 (15) at h5HT2BR suggested a binding mode with ligand inserted deeply in the TM bundle and lying almost parallel to the membrane plane (a). The adenine core of the ligand engaged in a π-π stacking interaction with Phe3406.51 (superscript refers to a numbering convention for GPCRs that identifies the TM and the relationship of the residue to the most conserved point in that helix), the N6-dicyclopropylmethyl moiety pointed toward TM5 and TM6 by establishing hydrophobic contacts with, among other residues, Met2185.39.
In modeling the interactions of nucleosides in diverse GPCRs it is important to consider the part played by water molecules. Following MD simulations of MRS7185 (15) in the 5HT2BR, the ribose moiety was anchored in the binding site by extended water-mediated H-bond interactions. Indeed, as depicted in b, several tightly bound water molecules connected the 2′- and 3′-OH groups to the conserved Asp1353.32, among other residues. A water molecule anchored the 5′-carbonyl group to the side chain of Gln3597.32 (acting as H-bond donor) and the Leu209EL2 backbone (acting as H-bond acceptor). This hypothetical binding mode enabled us to rationalize the greater affinity of this nucleoside series for the h5HT2BR. Indeed, the h5HT2CR features a Glu7.32 residue in place of Gln7.32 of 5HT2BR and a shorter EL2. The Glu7.32 side-chain could not act as a H-bond donor and therefore would not enable a H-bond network to occur in the h5HT2CR as described above for the h5HT2BR. The shorter EL2 in the h5HT2CR is expected to affect the 3D arrangement of the downstream region of the loop as well as of the extracellular tip of TM5, where the two key residues binding to the 5′-carbonyl group in h5HT2BR, namely Leu209EL2 (conserved) and Met5.39 (occurring as Val5.39 in h5HT2CR), are located. A direct comparison of h5HT2BR binding between (N)-methanocarba nucleosides and the corresponding ribosides indicated that the bicyclic ring system enhanced affinity at this nonpurine receptor. MRS5698 13 also bound to the δ-opioid receptor with a Ki value of 2.44 μM. Thus, the opioid receptor system is also a potential target family for (N)-methanocarba nucleosides. Other GPCRs that recognize various (N)-methanocarba nucleosides in the μM range are α2A, α2B, α2C and β3 adrenergic receptors.
Nucleoside off-target activity at other (non-nucleoside) transporters
Cell surface transporters for neurotransmitters, such as biogenic amines, ions, metabolites and nucleosides, belong to the large family of solute carrier (SLC) membrane transport proteins, which consists of >300 members. Two of the 52 families of SLC proteins are transporters for adenosine and other nucleosides [i.e., the equilibrative transport (ENT, SLC29) and sodium-coupled concentrative transport (CNT, SLC28) proteins]. The SAR of adenosine and its derivatives at the ENTs and, especially, at the CNTs is more restrictive than the corresponding SAR at ARs. ENT1 is potently inhibited by the (N)-methanocarba nucleoside equivalents of known inhibitors, such as the (N)-methanocarba derivative MRS1942 (9). (S) conformers either inhibit or are less potent substrates at ENTs and CNTs, and the permeability in CNTs is greater for (N) conformers than for (S) conformers.
Unexpectedly, a few of the nucleoside analogs interacted with the dopamine transporter (DAT, SLC6A3), a member of the same SLC membrane transporter family as ENTs. DAT is the protein target of cocaine and its blockade -the source of cocaine’s behavioral stimulant effects. However, a strange phenomenon was noted in the initial radioligand binding results at hDAT – (N)-methanocarba nucleosides dramatically increased radioligand binding (up to sevenfold the control value) rather than inhibiting it. In collaboration with the Janowsky laboratory, we characterized the nucleosides as novel allosteric DAT modulators, at a previously unaccessed site on the DAT protein – not the same (orthosteric) site where cocaine-like molecules (tropanes) bind. These nucleosides () increased the affinity of radioligands at DAT and inhibited DAT-mediated dopamine uptake. Thus, they have a complex mixture of positive allostery with respect to tropanes and negative allostery with respect to functional activity of DAT in the absence of tropanes. MRS5980 (1), a 5′-methylamide that also potently activates the A3AR, displayed an EC50 value of 35 nM in enhancing DAT binding. Some compounds were also found to interact with the norepinephrine transporter (NET, SLC6A2), which is an important target for treating pain and mood disorders. The mode of interaction at NET appears to be similar to DAT (i.e., radioligand binding enhancement at the orthosteric site and transport inhibition). The SAR of this chemical series was then explored to design related molecules that were even more potent in enhancing binding at DAT than the original hits. The corresponding 5′-methyl ester, MRS7292 (16), and 5′-ethyl ester, MRS7232 (17), were equipotent to the amide 1 at hDAT and even more potent than 1 in enhancing binding at hNET. The nucleosides also demonstrated probe dependence depending on which class of DAT radioligands was used – a characteristic of allosteric modulators. At mDAT, 16 and 17 were selective in comparison to mA3AR. A feature that was particularly important for transporter interaction was an extended C 2 group terminating in a 5-bromo- or 5-chlorothienyl group; an unsubstituted alkynylthienyl group was considerably less potent. Also, an N6-methyl group was strongly favored over larger groups. As with the h5HT2BR, the (N)-methanocarba modification enhanced DAT interaction in comparison to the corresponding riboside. The ribose equivalent of 16 (not shown) was weaker in DAT interaction (EC50 127 nM). Thus, new nucleoside analogs were synthesized that were especially potent and efficacious in their interaction with DAT, and they tended to be less potent than earlier compounds at the original target of ARs. Potential applications of peripherally acting allosteric inhibitors of DAT and NET might be similar to the current use of biogenic amines such as dopamine (i.e., as cardiac inotropic agents and to increase kidney/splanchnic circulation). Other non-GPCR targets to which various (N)-methanocarba nucleosides bind in the μM range are the σχ and σ2 receptors and an ion channel 53 serotonin receptor. However, these interactions have not been optimized in this chemical series.
Does endogenous adenosine interact with the 5HT2B and 5HT2CRs or with DAT? The affinity of adenosine has not been determined at these proteins; however, 2-chloroadenosine, often taken as a close mimic of adenosine, lacks significant binding affinity at these sites. In fact, no off-target actions of 2-chloroadenosine at 10 μM were found in the standard PDSP screen. Therefore, it is unlikely that endogenous adenosine also interacts with serotonin receptors or other off-target sites described here.
Relationship of nucleoside ligands to other privileged structures
How do nucleosides compare to other scaffolds that were deemed privileged? Most small molecules approved as pharmaceuticals as well as the contents of currently available chemical libraries contain flat heterocycles, and it is suggested that adding three-dimensionality could provide greater utility in hitting protein targets. With respect to the nucleobase, adenine adds hydrophobic character; adenine was reported to be the most frequently appearing bicyclic structure in approved drugs. An advantage of nucleosides over the nucleobases as privileged scaffolds is that they contain separate domains of predominately either sp2 or sp3 atoms. Therefore, methanocarba nucleosides combine flat and 3D characteristics and are desirable as privileged structures because they contain planar (nucleobase) and 3D (ribose or ribose-like) components. The concept of combining these features has already been noted in the literature. For example, Kombarov et al. described an approach to drug discovery using privileged structures termed ‘BioCores’, which, like nucleosides, contain pairs of saturated and aromatic heterocyclic moieties.
For ARs, these two domains (i.e., the adenine and ribose) have separate functions in AR recognition. The adenine moiety with its C2 and N6 substituents corresponds to the address portion of the ligands, and the ribose moiety is responsible for AR activation, which could be called the message portion (a). This amphiphilic feature and the high degree of rigidity, especially of the methanocarba nucleosides, can be useful in predicting an energetically favorable binding mode in the binding site of a given protein. Moreover, the divergent physicochemical properties of these two moieties can delineate preferred binding regions in the canonical GPCR binding site in the central cavities of rhodopsin-like GPCRs, which are often amphiphilic. This hybrid feature of nucleosides could be advantageous for interaction with a wide range of proteins in addition to the purine receptors.
Concluding remarks
Nucleosides are well represented as pharmaceuticals and have proven to be a generally well-tolerated drug class. We focus on a subcategory of nucleosides (and nucleotides) that introduces a significant steric constraint on the ribose moiety, which has the effect of enhancing pharmacological properties (e.g., potency and selectivity) and directing their activity at conventional and unconventional targets. The scope of action of these conformationally locked nucleosides has now been extended through embracing their potential usefulness for diverse targets that normally do not recognize ribonucleosides. Thus, the rigid methanocarba nucleoside scaffold that has been well explored at purine receptors, enzymes and transporters has been repurposed to satisfy the pharmacophoric requirements of unrelated GPCRs and transporters, such as biogenic amine carrier proteins. For example, this effort provided novel, selective 5HT2BR antagonists and the first allosteric modulators of the dopamine transporter, in some cases providing an array of structural information for interaction with a family of ligand substructures. These novel ligands might be useful as antifibrotic agents (5HT2BR antagonists) or inotropic agents (peripheral DAT modulators). The systematic correlation of protein interaction of the ligands with specific amino acid residues and regions of receptors, for example, could eventually lead to predicting multitarget interactions of new analogs within the same ligand family. Methanocarba nucleosides could be considered a privileged scaffold given their expansive applicability to many drug areas, but clearly they are not generally promiscuous compounds. The repurposing of this chemically well-explored scaffold to new and diverse biological targets could serve as an example for similar analyses for other drug and compound classes.
FIGURE 4.
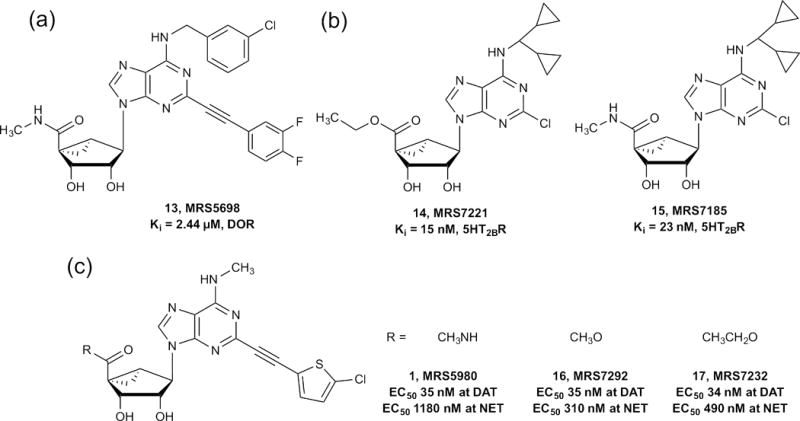
(a) Structures of (N)-methanocarba nucleosides that interact with conventionally non-purine sites. (a) DOR, (b) 5HT2Rs, (c) DAT.
FIGURE 5.
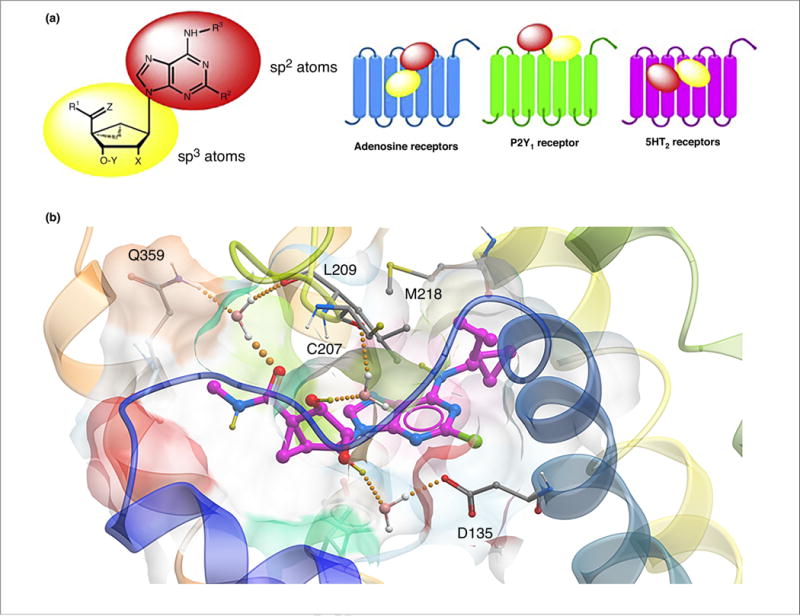
(a) Separate roles of the two moieties in rigid nucleosides in recognition at ARs and other sites, as illustrated for the general case of (N)-methanocarba adenosine derivatives. The relative orientations in the binding sites of ARs (based on X-ray structures of other nucleosides bound in the A2AAR), P2Y1R (from X-ray structure of the MRS2500 (3) complex) and in the 5HT2BR, as predicted following induced fit docking and molecular dynamics simulations, are contrasted. Typical substituents: X = H, phosphate; Y = H, OH; Z = H2, O; R1, R2, R3 = H, alkyl, O-alkyl; ethynyl, etc. (b) Hypothetical binding mode of antagonist MRS7185 (15) at the h5HT2BR. The H-bonding contacts (all through water bridges) are: 5′-carbonyl with L209 backbone (EL2) and Q359 (7.32), the 2′-hydroxyl to D135 (3.32) and the 3′-hydroxyl to C207 backbone (EL2). M218 (5.39) forms a hydrophobic contact with the N6 group.
Highlights.
Weak off-target activity of nucleosides at novel targets can be optimized.
Conformationally constrained nucleosides are privileged scaffolds.
The affinity and selectivity at conventional nucleoside targets can be enhanced.
Unconventional targets are the dopamine transporter and non-purine GPCRs.
Acknowledgments
We thank the Intramural Research Program of the NIH, National Institute of Diabetes and Digestive and Kidney Diseases for support.
Biographies

Kenneth A. Jacobson, PhD, is Chief of the Laboratory of Bioorganic Chemistry and the Molecular Recognition Section at the National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health in Bethesda, Maryland, USA. He is a medicinal chemist with interests in the structure and pharmacology of G-protein-coupled receptors (GPCRs), in particular receptors for nucleosides and nucleotides. He obtained his PhD in chemistry from the University of California, San Diego, and completed postdoctoral work at the Weizmann Institute. He was inducted into American Chemical Society’s Division of Medicinal Chemistry Hall of Fame in 2009.

Dilip K. Tosh, PhD, is a Staff Scientist at the Laboratory of Bioorganic Chemistry, National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health in Bethesda, Maryland, USA. He received his PhD in organic chemistry from the Indian Institute of Technology, Bombay, under the supervision of Prof. V.K. Singh. Subsequently, he joined as a postdoc in the laboratory of Prof. Lak Shin Jeong, at Ewha Womans University, Seoul, Korea, where he worked on medicinal chemistry and discovery employing nucleoside chemistry. Currently, he works on drug discovery based on G-protein-coupled receptors (GPCRs).

Kiran S. Toti, PhD, joined Dr Jacobson’s research group at NIH, USA, as a post-doctoral fellow in 2014. Here, he works on the synthesis of conformationally locked methanocarba nucleos(t)ides targeting GPCRs and kinases. Kiran received his PhD (2014) under the guidance of Prof. Serge Van Calenbergh, for the thesis Synthesis and Biological Evaluation of 4′-Hydroxymethyl Deleted, Transposed and Modified Nucleosides at Ghent University, Belgium. He obtained synthetic chemistry training at Aurigene Discovery Technologies, Bengaluru (2007–2009), and at NCL, Pune (2004–2007) after completing an MS in organic chemistry (2004) at Karnatak University, Dharwad, India

Antonella Ciancetta, PhD, received her Master’s Degree in pharmaceutical chemistry and technology and her PhD in drug sciences from the University of Chieti, Italy. After her PhD, she joined Prof. Stefano Moro’s group at the University of Padova, Italy, as a postdoc. She is currently a Visiting Fellow at the National Institutes of Health in Bethesda, Maryland, USA, in Dr Jacobson’s lab. Antonella’s research focuses on the application and development of structure-based modeling techniques for the design of GPCR ligands and the elucidation of ligand–GPCR recognition mechanisms.
References
- 1.Talevi A. Multi-target pharmacology: possibilities and limitations of the skeleton key approach from a medicinal chemist perspective. Front Pharmacol. 2015;6:205. doi: 10.3389/fphar.2015.00205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.de Lera A, Ganesan A. Epigenetic polypharmacology: from combination therapy to multitargeted drugs. Clin Epigenet. 2016;8:105. doi: 10.1186/s13148-016-0271-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nikolic K, et al. Drug design for CNS diseases: polypharmacological profiling of compounds using cheminformatic, 3D-QSAR and virtual screening methodologies. Front Neurosci. 2016;10:265. doi: 10.3389/fnins.2016.00265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Anighoro A, et al. Polypharmacology: challenges and opportunities in drug discovery. J Med Chem. 2014;57:7874–7887. doi: 10.1021/jm5006463. [DOI] [PubMed] [Google Scholar]
- 5.Marquez VE, et al. Nucleosides with a twist: can fixed forms of sugar ring pucker influence biological activity in nucleosides and oligonucleotides? J Med Chem. 1996;39:3739–3747. doi: 10.1021/jm960306+. [DOI] [PubMed] [Google Scholar]
- 6.Marquez VE. The properties of locked methanocarba nucleosides in biochemistry biotechnology and medicine. In: Herdewijn P, editor. Modified Nucleosides in Biochemistry Biotechnology and Medicine. Wiley-VCH; 2008. pp. 307–341. [Google Scholar]
- 7.Tchilibon S, et al. (N)-Methanocarba 2,N6-disubstituted adenine nucleosides ashighlypotentandselectiveAßadenosinereceptoragonists. J Med Chem. 2005;48:1745–1758. doi: 10.1021/jm049580r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Evans BE, et al. Methodsfordrugdiscovery: developmentofpotent, selective, orally effective cholecystokinin antagonists. J Med Chem. 1988;31:2235. doi: 10.1021/jm00120a002. [DOI] [PubMed] [Google Scholar]
- 9.Horton DA, et al. The combinatorial synthesis of bicyclic privileged structures or privileged substructures. Chem Rev. 2003;103:893–930. doi: 10.1021/cr020033s. [DOI] [PubMed] [Google Scholar]
- 10.Ioan P, et al. 1,4-Dihydropyridine scaffold in medicinal chemistry, the story so far and perspectives (part 1): action in ion channels and GPCRs. Cun Med Chem. 2011;18:4901–4922. doi: 10.2174/092986711797535173. [DOI] [PubMed] [Google Scholar]
- 11.Marcus A, et al. Charting biologically relevant chemical space: a structural classification of natural products (SCONP) Proc Natl Acad Sci U S A. 2008;102:17272–17277. doi: 10.1073/pnas.0503647102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Humbeck L, Koch O. What can we learn from bioactivity data? Chemoinformatics tools and applications in chemical biology research. ACS Chem Biol. 2017;12:23–35. doi: 10.1021/acschembio.6b00706. [DOI] [PubMed] [Google Scholar]
- 13.Reymond JL, Awale M. Exploring chemical space for drug discovery using the chemical universe database. ACS Chem Neurosci. 2012;3:649–657. doi: 10.1021/cn3000422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Costantino L, Daniela B. Privileged structures as leads in medicinal chemistry. Front Med Chem. 2010;5:381–422. [PubMed] [Google Scholar]
- 15.Welsch ME, et al. Privileged scaffolds for library design and drug discovery. Curr Opin Chem Biol. 2010;14:347–361. doi: 10.1016/j.cbpa.2010.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kombarov R, et al. Bio Cores: identification of a drug/natural product-based privileged structural motif for small-molecule lead discovery. Mol Divers. 2010;14:193–200. doi: 10.1007/s11030-009-9157-5. [DOI] [PubMed] [Google Scholar]
- 17.Jordheim LP, et al. Advances in the development of nucleoside and nucleotide analogues for cancer and viral diseases. Nat Rev Drug Discov. 2013;12:447–464. doi: 10.1038/nrd4010. [DOI] [PubMed] [Google Scholar]
- 18.Niu G, Tan H. Nucleoside antibiotics: biosynthesis, regulation, and biotechnology. Trends Microbiol. 2016;23:110–119. doi: 10.1016/j.tim.2014.10.007. [DOI] [PubMed] [Google Scholar]
- 19.Johnson ZL, et al. Structural basis of nucleoside and nucleoside drug selectivity by concentrative nucleoside transporters. eLife. 2014;3:e03604. doi: 10.7554/eLife.03604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sun H, et al. Oligonucleotide aptamers: new tools for targeted cancer therapy. Mol Ther Nucleic Acids. 2014;3:e182. doi: 10.1038/mtna.2014.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen JF, et al. Adenosine receptors as drug targets—what are the challenges? Nat Rev Drug Discov. 2013;12:265–286. doi: 10.1038/nrd3955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cekic C, Linden J. Purinergic regulation of the immune system. Nat Rev Immunol. 2016;16:177–192. doi: 10.1038/nri.2016.4. [DOI] [PubMed] [Google Scholar]
- 23.Antonioli L, et al. CD39 and CD73 in immunity and inflammation. Trends Mol Med. 2013;19:355–367. doi: 10.1016/j.molmed.2013.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jacobson KA, et al. John Daly Lecture: structure-guided drug design for adenosine and P2Y receptors. Comput Struct Biotechnol J. 2015;13:286–298. doi: 10.1016/j.csbj.2014.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fishman P, et al. Pharmacological and therapeutic effects of A3 adenosine receptor (A3AR) agonists. Drug Discov Today. 2012;17:359–366. doi: 10.1016/j.drudis.2011.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Janes K, et al. Identification of A3 adenosine receptor agonists as novel nonnarcotic analgesics. Br J Pharmacol. 2016;173:1253–1267. doi: 10.1111/bph.13446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Astakhova IK, Wengel J. Acc Chem Res. 2014;47:1768–1777. doi: 10.1021/ar500014g. [DOI] [PubMed] [Google Scholar]
- 28.Tosh DK, Jacobson KA. Methanocarba ring as a ribose modification in ligands of G protein-coupled purine and pyrimidine receptors: synthetic approaches. MedChemComm. 2013;4:619–630. doi: 10.1039/C2MD20348K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Choi WJ, et al. Preparative and stereoselective synthesis of the versatile intermediate for carbocyclic nucleosides: effects of the bulky protecting groups to enforce facial selectivity. J Org Chem. 2004;69:2634–2636. doi: 10.1021/jo0356762. [DOI] [PubMed] [Google Scholar]
- 30.Lee JA, et al. Stereoselective synthesis of 2′-C-methyl-cyclopropyl-fused carbanucleosides as potential anti-HCV agents. Org Lett. 2006;8:5081–5083. doi: 10.1021/ol061959f. [DOI] [PubMed] [Google Scholar]
- 31.Michel BY, Strazewski P. Total syntheses of a conformationally locked north-type methanocarba puromycin analogue and a dinucleotide derivative. Chem Eur J. 2009;15:6244–6257. doi: 10.1002/chem.200802629. [DOI] [PubMed] [Google Scholar]
- 32.Joshi BV, et al. Synthesis of ethyl (1S, 2R, 3S, 4S, 5S)-2, 3-O-(isopropylidene)-4-hydroxy-bicyclo[3.1.0]hexane-carboxylate from L-ribose: a versatile chiral synthon for preparation of adenosine and P2 receptor ligands. Nucleosides Nucleotides Nucleic Acids. 2008;27:279–291. doi: 10.1080/15257770701845253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Melman A, et al. Synthesis of enantiomerically pure (S)-methanocarbaribo uracil nucleoside derivatives for use as antiviral agents and P2Y receptor ligands. J Org Chem. 2008;73:8085–8088. doi: 10.1021/jo801224j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tosh DK, et al. In vivo phenotypic screening for treating chronic neuropathic pain: modification of C2-arylethynyl group of conformationally constrained A3 adenosine receptor agonists. J Med Chem. 2014;57:9901–9914. doi: 10.1021/jm501021n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tosh DK, et al. Scaffold repurposing of nucleosides (adenosine receptor agonists): enhanced activity at the human dopamine and norepinephrine sodium symporters. J Med Chem. 2017;60:3109–3123. doi: 10.1021/acs.jmedchem.7b00141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Trujillo K, et al. Molecular modeling of the human P2Y14 receptor: a template for structure-based design of selective agonist ligands. Bioorg Med Chem. 2015;23:4056–4064. doi: 10.1016/j.bmc.2015.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Maruoka H, et al. Pyrimidine ribonucleotides with enhanced selectivity as P2Y6 receptor agonists: novel 4-alkyloxyimino, (S)-methanocarba, and 5′-triphosphate γ-ester modifications. J Med Chem. 2010;53:4488–4501. doi: 10.1021/jm100287t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang D, et al. Two disparate ligand-binding sites in the human P2Y1 receptor. Nature. 2015;520:317–321. doi: 10.1038/nature14287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dunn PM, et al. Northern ring conformation of methanocarba-adenosine 5′-triphosphate required for activation of P2X receptors. Drug Dev Res. 2004;61:227–232. doi: 10.1002/ddr.10381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kumar TS, et al. 5′-Phosphate and 5′-phosphonate ester derivatives of (N)-methanocarba adenosine with in vivo cardioprotective activity. J Med Chem. 2013;56:902–914. doi: 10.1021/jm301372c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sjuvarsson E, et al. Selective phosphorylation of South and North-cytidine and adenosine methanocarba-nucleosides by human nucleoside and nucleotide kinases correlates with their growth inhibitory effects on cultured cells. Nucleosides Nucleotides Nucleic Acids. 2015;34:544–564. doi: 10.1080/15257770.2015.1031248. [DOI] [PubMed] [Google Scholar]
- 42.Bernstein DI, et al. N-methanocarbathymidine is more effective than acyclovir for treating neonatal herpes simplex virus infection in guinea pigs. Antiviral Res. 2011;92:386–388. doi: 10.1016/j.antiviral.2011.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Guerra KP, et al. Synthetic survey and activity of 2′-deoxy-methanocarba nucleosides. Curr Org Synth. 2013;10:210–240. [Google Scholar]
- 44.Evaluating the Safety and Plasma Levels of N-Methanocarbathymidine (N-MCT) in Normal Patients. ClinicalTrials.gov Identifier: NCT02778386. Available at: http://www.clinicaltrials.gov.
- 45.Marquez VE, et al. Experimental and structural evidence that herpes 1 kinase and cellular DNA polymerase(s) discriminate on the basis of sugar pucker. J Am Chem Soc. 2004;126:543–549. doi: 10.1021/ja037929e. [DOI] [PubMed] [Google Scholar]
- 46.Marquez VE, et al. The investigation of a conformational concept leads to the discovery of a potent and selective nucleoside antiviral agent. Antiviral Res. 2006;71:268–275. doi: 10.1016/j.antiviral.2006.04.012. [DOI] [PubMed] [Google Scholar]
- 47.Marquez VE, et al. HIV-1 reverse transcriptase can discriminate between two conformationally locked carbocyclic AZT triphosphate analogues. J Am Chem Soc. 1998;120:2780–2789. [Google Scholar]
- 48.Marquez VE, et al. Experimental and structural evidence that herpes 1 kinase and cellular DNA polymerase(s) discriminate on the basis of sugar pucker. J Am Chem Soc. 2004;126:543–549. doi: 10.1021/ja037929e. [DOI] [PubMed] [Google Scholar]
- 49.Boyer PL, et al. Fixed conformation nucleoside analogs effectively inhibit excision-proficient HIV-1 reverse transcriptases. J Mol Biol. 2005;345:441–450. doi: 10.1016/j.jmb.2004.10.021. [DOI] [PubMed] [Google Scholar]
- 50.Ravi RG, et al. Adenine nucleotide analogues locked in a Northern methanocarba conformation: enhanced stability and potency as P2Y1 receptor agonists. J Med Chem. 2002;45:2090–2100. doi: 10.1021/jm010538v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Toti KS, et al. South (S)- and North (N)-methanocarba-7-deazaadenosine analogues as inhibitors of human adenosine kinase. J Med Chem. 2016;59:6860–6877. doi: 10.1021/acs.jmedchem.6b00689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Boison D. Adenosine kinase: exploitation for therapeutic gain. Pharmacol Rev. 2013;65:906–943. doi: 10.1124/pr.112.006361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rodriguez JB, et al. Synthesis of cyclopropane-fused dideoxycarbocyclic nucleosides structurally related to neplanocin C. Tetrahedron Lett. 1993;34:6233–6236. [Google Scholar]
- 54.Altmann KH, et al. 4′,6′-Methano carbocyclic thymidine: a conformationally constrained building block for oligonucleotides. Tetrahedron Lett. 1994;35:2331–2334. [Google Scholar]
- 55.Altmann KH, et al. 1′,6′-Methano carbocyclic thymidine: synthesis and X-ray crystal structure, and effect on nucleic acid duplex stability. Tetrahedron Lett. 1994;35:7625–7628. [Google Scholar]
- 56.Jung ME, et al. Synthesis and duplex-stabilizing properties of fluorinated N-methanocarbathymidine analogues locked in the C3′-endo conformation. Angew Chem Int Ed. 2014;53:9893–9897. doi: 10.1002/anie.201405283. [DOI] [PubMed] [Google Scholar]
- 57.Terrazas M, et al. Effect of North bicyclo[3.1 0]hexane 2′-deoxypseudosugars on RNA interference: a novel class of siRNA modification. Chembiochem. 2011;12:1056–1065. doi: 10.1002/cbic.201000791. [DOI] [PubMed] [Google Scholar]
- 58.Paoletta S, et al. Structural probing of off-target G protein-coupled receptor activities within a series of adenosine/adenine congeners. PLoS One. 2014;9:e97858. doi: 10.1371/journal.pone.0097858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Azran S, et al. Identification of highly promising antioxidants/neuroprotectants based on nucleoside 5′-phosphorothioate scaffold. Synthesis, activity, and mechanisms of action. J Med Chem. 2015;58:8427–8443. doi: 10.1021/acs.jmedchem.5b00575. [DOI] [PubMed] [Google Scholar]
- 60.Vincetti P, et al. Discovery of multitarget antivirals acting on both the Dengue virus NS5-NS3 interaction and the host Src/Fyn kinases. J Med Chem. 2015;58:4964–4975. doi: 10.1021/acs.jmedchem.5b00108. [DOI] [PubMed] [Google Scholar]
- 61.Stockdale TP, Williams CM. Pharmaceuticals that contain polycyclic hydrocarbon scaffolds. Chem Soc Rev. 2015;44:7737–7763. doi: 10.1039/c4cs00477a. [DOI] [PubMed] [Google Scholar]
- 62.Tosh DK, et al. Structure-based scaffold repurposing for G protein-coupled receptors: transformation of adenosine derivatives into 5HT2B/5HT2C serotonin receptor antagonists. J Med Chem. 2016;59:11006–11026. doi: 10.1021/acs.jmedchem.6b01183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jacobson KA, et al. Computational studies to predict or explain GPCR polypharmacology. Trends Pharmacol Sci. 2014;35:658–663. doi: 10.1016/j.tips.2014.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gilberg E, et al. Highly promiscuous small molecules from biological screening assays include many pan-assay interference compounds but also candidates for polypharmacology. J Med Chem. 2016;59:10285–10290. doi: 10.1021/acs.jmedchem.6b01314. [DOI] [PubMed] [Google Scholar]
- 65.Baell JB, Holloway GA. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J Med Chem. 2010;53:2719–2740. doi: 10.1021/jm901137j. [DOI] [PubMed] [Google Scholar]
- 66.Schaddelee MP, et al. Brain penetration of synthetic adenosine A1 receptor agonists in situ: role of the rENT1 nucleoside transporter and binding to blood constituents. Eur J Pharm Sci. 2005;24:59–66. doi: 10.1016/j.ejps.2004.09.010. [DOI] [PubMed] [Google Scholar]
- 67.Carlin JL, et al. Hypothermia in mouse is caused by adenosine A1 and A3 receptor agonists and AMP via three distinct mechanisms. Neuropharmacology. 2017;114:101–113. doi: 10.1016/j.neuropharm.2016.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ballesteros JA, Weinstein H. Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. Meth Neurosci. 1995;25:366–428. [Google Scholar]
- 69.Lin L, et al. SLC transporters as therapeutic targets: emerging opportunities. Nat Rev Drug Discov. 2015;14:543–560. doi: 10.1038/nrd4626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lee K, et al. Synthesis using ring closure metathesis and effect on nucleoside transport of a (N)-methanocarba S-(4-nitrobenzyl)thioinosine derivative. Org Lett. 2001;3:597–599. doi: 10.1021/ol006999c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Damaraju VL, et al. Influence of sugar ring conformation on the transportability of nucleosidesbyhuman nucleoside transporters. Chembiochem. 2011;12:2774–2778. doi: 10.1002/cbic.201100567. [DOI] [PubMed] [Google Scholar]
- 72.Janowsky A, et al. Rigid adenine nucleoside derivatives as novel modulators of the human sodium symporters for dopamine and other neurotransmitters. J Pharmacol Exp Ther. 2016;357:24–35. doi: 10.1124/jpet.115.229666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lovering F, et al. Escape from flatland: increasing saturation as an approach to improving clinical success. J Med Chem. 2009;52:6752–6756. doi: 10.1021/jm901241e. [DOI] [PubMed] [Google Scholar]
- 74.Taylor RD, et al. Combining molecular scaffolds from FDA approved drugs: application to drug discovery. J Med Chem. 2017;60:1638–1647. doi: 10.1021/acs.jmedchem.6b01367. [DOI] [PubMed] [Google Scholar]
- 75.Venkatakrishnan AJ, et al. Molecular signatures of G-protein-coupled receptors. Nature. 2013;494:185–194. doi: 10.1038/nature11896. [DOI] [PubMed] [Google Scholar]
- 76.Xu F, et al. Structure of an agonist-bound human A2A adenosine receptor. Science. 2011;332:322–327. doi: 10.1126/science.1202793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lebon G, et al. Agonist-bound adenosine A2A receptor structures reveal common features of GPCR activation. Nature. 2011;474:521–525. doi: 10.1038/nature10136. [DOI] [PMC free article] [PubMed] [Google Scholar]