

Abstract
The Hippo pathway effector YAP is implicated in the pathogenesis of cholangiocarcinoma (CCA). The Hippo pathway relies on signaling cross talk for its regulation. Given the importance of platelet derived growth factor receptor (PDGFR) signaling in CCA biology, our aim was to examine potential YAP regulation by PDGFR. We employed human and mouse CCA specimens and cell lines for these studies. Initially, we confirmed upregulation of PDGFRβ and PDGFR ligands in human and mouse CCA specimens and cell lines. YAP, a transcriptional co-activator, was localized to the nucleus in human CCA specimens and a cell line, as well as patient derived xenografts (PDX). PDGFR pharmacologic inhibition led to a redistribution of YAP from the nucleus to cytosol and downregulation of YAP target genes in a human CCA cell line. siRNA silencing of PDGFR-β similarly downregulated YAP target genes. YAP activation (nuclear localization and target gene expression) was regulated by Src family kinases (SFKs) downstream of PDGFR. SFK activity resulted in phosphorylation of YAP on tyrosine357 (YAPY357). The importance of YAPY357 phosphorylation in regulating YAP activation was confirmed utilizing the SB-1 cell line, a mouse cell line expressing YAP S127A precluding canonical serine phosphorylation. PDGFR inhibition decreased cellular abundance of the survival protein Mcl-1, a known YAP target gene, and accordingly increased cell death in CCA cells in vitro and in vivo. These preclinical data demonstrate that a PDGFR-SFK cascade regulates YAP activation via tyrosine phosphorylation in CCA. Inhibiting this cascade may provide a viable therapeutic strategy for this human malignancy.
Keywords: Cholangiocarcinoma, platelet derived growth factor, Src kinase family, hippo
Cholangiocarcinoma (CCA) is a devastating hepatobiliary malignancy with features of biliary epithelial differentiation, a rising incidence, and limited treatment options[1–3]. Currently, surgical extirpation offers a chance for cure, but only a small fraction of patients will present with tumors amenable to surgical therapy[4]. Available chemotherapeutic options only modestly increase survival[5]. Thus, a significant unmet need exists in this patient population for better therapeutic options and identification of patients suitable for those therapies. These therapeutic advances will require additional insights regarding the molecular mechanisms of biliary carcinogenesis and tumor progression.
The Hippo pathway has been implicated in CCA pathogenesis[6, 7]. The effectors of the Hippo pathway, YAP and its paralog TAZ, are transcriptional co-activators and are, in part, regulated by serine phosphorylation [8–10]. Specifically, phosphorylation of the serine127 residue of YAP by a kinase module consisting of MST1/2 and LATS1/2 sequesters YAP in the cytosol and limits transcriptional activity[8]. Hence, the Hippo kinase module restrains YAP signaling by limiting its access to the nucleus via serine127 phosphorylation. Based on the regulatory importance of this serine residue in CCA oncogenesis, we have previously demonstrated the ability to generate intrahepatic CCA in mice following biliary tract transduction with a YAP construct in which serine127 has been mutated to alanine (S127A)[11]. Although simultaneous transduction with activated Akt was also required to generate CCA in mice, these studies support a potent role for the Hippo pathway and its effector protein YAP in CCA pathogenesis. How YAP is activated in oncogenic pathways in CCA remains incompletely understood, as the Hippo pathway does not have a dedicated plasma membrane receptor nor extracellular ligand. Hence, cross-talk between other signaling pathways and the Hippo pathway are of considerable interest.
Our group has implicated the platelet derived growth factor receptor (PDGFR) signaling pathway in survival signals in CCA[12] and YAP is thought to co-activate transcription of potent survival mediators[13, 14]. How a receptor tyrosine kinase such as PDGFR may potentially activate YAP is unclear as YAP is thought to be largely regulated by serine phosphorylation. However, recent work has demonstrated that YAP tyrosine phosphorylation by a Src family kinase (SFK) mediates IL-6 inflammation associated carcinogenesis in the colon [13]. The IL6-GP130 complex phosphorylates SFK on tyrosine416, and, in turn, SFK phosphorylates YAP on tyrosine357. The tyrosine phosphorylated YAP may then potentially function as a co-transcriptional activator. YAP co-transcriptional activity is mediated by its binding to several different transcription factors. Classically, YAP associates with the TEAD transcription factors (TEAD1–4); however, evidence suggests multiple alternate binding partners are possible[15]. Thus, our knowledge regarding the specificity for YAP binding to transcription factors is incomplete.
Herein, we examine cross-talk between the PDGFR and Hippo signaling pathways. The results suggest PDGFR signaling regulates YAP nuclear localization and cognate gene expression via SFK-mediated YAP tyrosine phosphorylation. YAP tyrosine phosphorylation also appears to promote its association with the transcription factor TBX5, promoting CCA cell viability by inducing Mcl-1 expression. Pharmacologic inhibition of PDGFR signaling is cytotoxic in human cholangiocarcinoma cell lines and PDX models.
MATERIALS AND METHODS
Cell culture
The human cholangiocarcinoma cell line HUCCT-1, mouse cholangiocarcinoma cell line SB-1, and normal human cholangiocyte (NHC) cell line[16], were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum and 0.2% Primocin under standard conditions. The murine SB-1 cell line was isolated from our murine model of YAP associated CCA and will be described in detail in a separate publication. This cell line expresses Flag-tagged-YAPS127A and myr-AKT. Due to cell density and serum regulation of the Hippo pathway, cell culture experiments were performed at near confluence (~80%) in serum starved cells. Serum starved cells were cultured overnight in media without serum. The media was replaced again three hours prior to initiation of experimental conditions.
Antibodies and Reagent
Crenolanib (Selleck Chemicals) and Dasatinib (TSZ Chemicals) was added to cells at final concentrations from 0.1–10μM. The following primary antisera were used for immunoblot analysis: PDGF Receptor (28E1 Cell Signaling, Danvers, MA), actin (C-11 Santa Cruz, Dallas, TX), phospho YAPY357 (ab62751 abcam, Cambridge, MA), phospho YAPS127 (4911 Cell Signaling), total YAP (63.7 Santa Cruz), lamin B1 (D8P3U Cell Signaling), GAPDH (MAB374 Millipore, Temecula, CA), phospho SrcY416 (2101 Cell Signaling), Src (L4A1 Cell Signaling), and Mcl-1 (S-19 Santa Cruz). The following primary monoclonal antibodies were used for immunofluorescence: total YAP (63.7 Santa Cruz) and anti-FLAG M2 (F1804 Sigma Adrich). For ChIP immunoprecipitation assays we employed an anti-YAP monoclonal antibody (63.7 Santa Cruz). ProLong Antifade with 4′,6-diamidino-2-phenylindole (DAPI, Life Technologies) was used for nuclear staining. The following primers used are listed in Table 1.
Table 1.
PCR Primers
| Gene | Forward primer sequence (5′-3′) | Reverse primer sequence (5′-3′) |
|---|---|---|
| YAP | CCC TCG TTT TGC CAT GAA CC | TGC CGA AGC AGT TCT TGC TG |
| CTGF | GCA GCG GAG AGT CCTTCC AG | GGG CCA AAC GTG TCT TCC AG |
| CYR61 | GAG TGG GTC TGT GAC GAG GAT | GGT TGT ATA GGA TGC GAG GCT |
| PDGFRβ | TGA TGC CGA GGA ACT ATT CAT CT | TTT CTT CTC GTG CAG TGT CAC |
| PDGFB | CTG GCA TGC AAG TGT GAG AC | CGA ATG GTC ACC CGA GTT T |
| Fyn | TGT GAC CTC CAT CCC CAA CT | AAC TCA GGT CAT CTT CTG TCC GT |
| Lck | CAC GAA GGT GGC GGT GAA GA | GAA GGG GTC TTG AGA AAA TCC A |
| Lyn | GAG GCT CTA CGC TGT GGT CA | GAC TCG GAG ACC AGA ACA TTA GC |
| Src | CAG TGT CTG ACT TCG ACA ACG C | CCA TCG GCG TGT TTG GAG TA |
| Yes | GCT GCA CTG TAT GGT CGG TT | AGG GCA CGG CAT CCT GTA TC |
| 18S | CGC TTC CTT ACC TGG TTG AT | GAG CGA CCA AAG GAA CCA TA |
| GAPDH | GAT GTG CGA ACT GGA CAC AG | CAT AGG GGG CGT CAA ACA G |
| ChiP Primers | ||
| PDGFB Promoter | GGG GTG AGC CAC CGT GCC TG | GTG GCA GAA GAG GGC CC AGC A |
Immunoblot analyses
Whole-cell lysates were collected by adding ice cold lysis buffer (Cell Signaling Technologies) containing protease inhibitors (Sigma-Aldrich), phosphatase inhibitors (Roche Diagnostics) and 1 mM PMSF. Cells were then scraped to collect cells, vortexed and lysed on ice for approximately 20 minutes. The lysed cells were then centrifuged at 12,000g for 15 minutes to remove cellular debris. Supernatant was transferred to a clean tube and protein concentration determined by the Bradford (Sigma-Aldrich) protein assay. Proteins were then resolved by SDS-PAGE electrophoresis and transferred to nitrocellulose membranes. Membranes were blotted with primary antibodies at 4°C overnight in 5% BSA-TBS Tween. The primary antibody dilution was 1:1000 unless otherwise indicated. After overnight incubation membranes were washed for 30 minutes in TBS Tween and then horseradish peroxidase-conjugated secondary antibodies against mouse, rabbit and goat (Santa Cruz) were added to membrane at a concentration of 1:5000 and incubated for 1 hour at room temperature. Immunoblots were visualized with enhanced chemiluminescence (ECL) or ECL prime (GE Healthcare Life Sciences).
Immunofluorescence
Cells were grown on glass coverslips, once the cells reached desired confluency they were treated per experimental conditions. Cells were then fixed with 4% paraformaldehyde and permeabilized with 0.1% Triton X-100. The fixed cells were then incubated at room temperature with blocking buffer containing 5.0% bovine serum albumin (BSA) and 0.1% Glycine. After blocking the cells were incubated with primary antibody in blocking buffer over night at 4°C. After washing the cells were incubated with the corresponding secondary antibodies diluted in blocking buffer for 1 hour at room temperature in the dark. After secondary incubation the coverslips were again washed and mounted onto slides using ProLong Antifade (Invitrogen-Molecular Probes) containing DAPI. The slides were analyzed by fluorescent confocal microscopy (LSM 780, Zeiss)
Immunohistochemistry in Human Cholangiocarcinoma Specimens
Following IRB approval, resected tumors from patients who had given research authorization were fixed in 10% buffered formaldehyde and embedded in paraffin. Blocks were sectioned into 3.5 μM slices. Sections were deparaffinized, hydrated, and incubated with primary antibody overnight at 4° C. Sections were stained with antibody for YAP (1:50). Bound antibody was detected with biotin-conjugated secondary antibody and diaminobenzidine as a substrate.
Quantitative and qualitative
PCR-mRNA was isolated from cells using TRIzol Reagent (Ambion). Reverse transcription was performed using Moloney murine leukemia virus reverse transcriptase and random primers (Life Technologies). After reverse transcription, cDNA was analysed using real-time PCR (Light Cycler 480 II, Roche Diagnostics) for the quantitation of the target genes; SYBR Green (Roche Diagnostics) was used as the fluorophore. Expression was normalized to either 18 S or GAPDH and relative quantification performed according to the 2−ΔCT or 2−ΔΔCT method as previously described[17]. Data are reported as fold expression compared to calibrator as geometric mean and geometric standard deviation of expression relative to calibrator. Technical replicates were completed for each run and a minimum of three biologic replicates completed for each condition/cell line. The primers used are listed in Table 1.
RNA Interference
PDGFR-β was transiently knocked down in the HuCCT-1 cell line with a validated siRNA (Darmicon). Cells grown in 6 well plates and transfected with Lipofectamine RNAiMAX (Invitrogen) following manufacturer’s instructions. Following 48 hour transfection the cells were used for subsequent experiments.
Chromatin Immunoprecipitation
Cells were plated on 35 cm2 dishes corresponding to 1x107 – 5x107 per dish. Cross linking was performed with formaldehyde added to the media to a final concentration of 1.0% with gentle rocking at room temperature for 10 minutes. Glycine was then added to a final concentration of 125 mM and the cells incubated for an additional 5 minutes. Cells were then washed with PBS and collected in ice cold PBS. Cells were centrifuged for 5 minutes at 1,000g supernatant removed, and the cell pellet resuspended in lysis buffer (50 mM HEPES pH 7.5, 140 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.1% sodium deoxycholate, 0.1% SDS, protease inhibitors) with 750 μL per 1x107 cells. The resuspended pellet was sonicated to shear DNA to an average fragment size of 500 to 1,000 bp. Sonicated samples were centrifuged for 1 minute at 4°C at 8,000g and supernatant transferred to fresh tube. 40 μg of protein was used per IP sample at a 1:10 dilution with RIPA buffer (50 mM Tris-HCL pH 8, 150 mM MaCl, 2 mM EDTA, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, Protease inhibitors). Primary antibody was added at a 1:50 dilution to samples plus 40 μl of protein A/G beads (Santa Cruz). Next, the samples were incubated overnight at 4°C with rotation. The protein/bead complex was washed 3X by centrifuging the samples for 1 minute at 2,000 g. The supernatant was removed and the beads resuspended in wash buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 150 mM NaCl, 20 mM Tris-HCl pH 8). DNA was eluted by adding 150 μl elution buffer (1% SDS, 100 mM NaHCO3) to the A/G beads and incubating for 15 minutes at 30°C with rotation. Samples were cleared by centrifugation and the supernatant was transferred into fresh tubes. The DNA was purified using PCR purification kit (QIAquick PCR Purification Kit) and used for PCR analysis.
Quantification of Cell Death
Apoptosis was quantified by assessing the characteristic nuclear changes of apoptosis (chromatin condensation and nuclear fragmentation) using fluorescence microscopy after staining with 4′,6-diamidino-2-phenylindole (DAPI) (Sigma). Caspase-3/7 activity in cell cultures was assessed using the Apo-ONE homogenous caspase-3/7 kit (Promega) per the supplier’s protocol with a fluorescent microplate reader (Synergy H1, BioTek, Winooski, VT)..
PDX generation and treatment
Human CCA tumors were implanted in NOD/SCID mice as previously described by us[18]. Once the tumors had reached palpable size (~125mm3) size was recorded and treatment with crenolanib (15 mg/kg) or vehicle begun daily for 2 weeks via IP injection as previously described[19]. At the end of the treatment period all mice were sacrificed and tumor tissue obtained for further studies.
TUNEL staining
The fluorescent TUNEL assay (in situ cell death detection kit, Roche) was utilized on tissue sections. Sections were paraformaldehyde-fixed and hydrated. The TUNEL assay was then performed using the manufacturer’s protocol. Slides were mounted with ProLong Antifade (Invitrogen-Molecular Probes) containing DAPI. The slides were analyzed by fluorescent confocal microscopy (LSM 780, Zeiss). Dead cells were quantified by counting TUNEL-positive nuclei in 20 random microscopic fields (20X).
RNA sequencing
RNA was extracted from mouse cholangiocarcinoma samples utilizing TRIzol Reagent (Ambion) per manufacturer’s protocol. RNA quality was assessed on the Agilent TapeStation 4200 (High Sensitivity RNA ScreenTape®, Agilent) and all samples were of high quality with RIN values >8.6. Sequencing library was prepared using the TruSeq mRNA library prep kit v2 (Illumina, RS-122-2001). Whole transcriptome sequencing (paired end; 100bp) was performed on an Illumina HiSeq 4000 by the Genome Analysis Core of Mayo Clinic’s Medical Genome Facility, Rochester, MN. The application analytical tool MAP-Rseq (v 2.1, Mayo Clinic, Rochester, MN) was used to assess read quality based on percentage of mapped sequence reads, junction plots and base resolution expression profile. FASTQ reads were aligned to the Mus musculus genome, mm10, using the software TopHat2. Fusion events were evaluated using the TopHat-Fusion module (no gene fusions were identified in this data). Raw gene and exon counts were generated using the FeatureCounts module (Ref) using the gene definition files from ENSEMBL GRCm38.79 and normalized to RPKM expression values. Differential gene expression analysis was performed using the R-based tool from Bioconductor, edgeR (Robison et al., 2010).
RESULTS
The PDGF pathway is upregulated in CCA
Initially, we examined expression of PDGF pathway mediators in our mouse model of CCA (Table 2). Significant upregulation (log2 expression greater than 1.5) was observed in all four ligands (PDGF A–D). PDGF-B had the highest differential expression with a log2 expression of 4.4, representing a greater than 20-fold increase in expression in tumor tissue, with PDGF-D being the next highest expressed. Evaluation of PDGF receptors α and β demonstrated only upregulation of PDGFR-β with a log2 expression of 1.8, representing a greater than 3-fold increase; PDGFR-α was not differentially expressed. In the HuCCT-1 cells, the relative levels of PDGF receptor-beta (PDGFR-β) and the ligand PDGF-B were also found to be increased as compared to normal human cholangiocytes (NHC) (Fig. 1A). Upregulation of the PDGFR- β was confirmed by immunoblot (Fig. 1B). The activation status of the PDGFR-β was examined by phospho-blot analysis in the HuCCT-1 cell line; indeed, PDGFR-β was found to be phosphorylated in the absence of adding exogenous ligand, indicating constitutive activation (Fig. 1C). Hence, YAP associated mouse CCA, and the human HuCCT-1 CCA cell line are associated with PDGFR receptor expression and signaling, likely via an autocrine pathway given expression of both a receptor and ligands.
Table 2.
Differential Expression PDGF Pathway in Murine YAP-Driven CCA versus Adjacent Liver
| Gene | Fold Change(log2) |
|---|---|
| PDGF-B | 4.4 |
| PDGF-D | 3.7 |
| PDGF-A | 1.8 |
| PDGF-C | 1.7 |
| PDGFR-β | 1.8 |
| PDGFR-α | −0.1 |
Figure 1. Platelet Derived Growth Factor Signaling is Upregulated in CCA.

A, mRNA expression in the HuCCT-1 CCA cell line compared to NHC as a calibrator. Fold expression is depicted as geometric mean +/− geometric standard deviation (n=3). B, cell lysates from CCA cell line HuCCT-1 and NHC cell line were subjected to immunoblot for PDGFR-β, actin was used as a loading control. C, cell lysates from HuCCT-1 cell line were subjected to immunoblot for PDGFR- β and phospho-PDGFR-β.
The PDGFR pathway regulates YAP subcellular compartmentation and transcriptional activity
We next examined YAP subcellular compartmentation in human CCA specimens and 7 human patient derived xenografts, a human CCA cell line, and our mouse SB-1a CCA cell line. We observed YAP localized to the nucleus not only in the resected paraffin embedded human CCA specimens and all 7 of the PDX, but also in the human HuCCT-1 cells and the mouse SB-1a CCA line (Fig. 2A). Next we examined the role of PDGFR in regulating YAP nuclear localization utilizing an inhibitory paradigm. Pharmacologic inhibition was undertaken with crenolanib (Arog, Dallas, TX) a type 3 receptor tyrosine kinase inhibitor, including PDGFR-β. Incubation with crenolanib decreased expression of CTGF and Cyr61, cognate gene targets of YAP co-activation. In addition, crenolanib also decreased expression of PDGF-B suggesting it is also a YAP co-transcriptional gene target (Fig. 2B). The specificity of this response to crenolanib was confirmed utilizing an siRNA approach (Fig. 2C). siRNA to PDGFR-β similarly decreased mRNA expression of CTGF, Cyr61 and PDGF-B (Fig. 2D). These data suggest that PDGFR receptor signaling modulates YAP target gene levels including PDGF-B. As a next step in interrogating the relationship between PDGF signaling and YAP activation, we examined the nuclear localization of YAP by immunofluorescence under basal conditions and following incubation with crenolanib. Under basal conditions, HuCCT-1 cells displayed nuclear localization of YAP. In contrast, incubation with crenolanib resulted in a redistribution of YAP from the nucleus to the cytoplasm (Fig. 2E). These data suggest that PDGFR, a receptor tyrosine kinase, modulates the Hippo pathway in CCA cells.
Figure 2. Platelet Derived Growth Factor signaling modulates YAP localization and transcriptional activity.

A, YAP immunohistochemistry and immunofluorescence images in human tumor, CCA cell lines (HuCCT-1 and SB-1a), and patient-derived xenografts. Scale bar = 10 microns. B, mRNA expression in HuCCT-1 CCA cell line +/− PDGFR inhibitor crenolanib (1 μM). Fold expression treated/control is depicted as geometric mean +/− geometric standard deviation (n=3). C, cell lysates from HuCCT-1 cell lines transfected with targeting and non-targeting siRNA were subjected to immunoblot for PDGFR-β and mRNA expression depicted as geometric mean +/− geometric standard deviation (n=5) D, mRNA expression in HuCCT-1 cell line lines transfected with targeting and non-targeting siRNA. Fold expression targeting/non-targeting is depicted as geometric mean +/− geometric standard deviation (n=5). E, YAP immunofluorescence in HuCCT-1 cell line +/− PDGFR inhibitor crenolanib (10 μM) with quantification of subcellular localization (right panel). Scale bar = 10 microns.
YAP nuclear localization is regulated by PDGF-mediated tyrosine phosphorylation
To further explore how a receptor kinase may modulate YAP subcellular localization, we first examined phospho-YAPS127 and -YAPY357 in cells treated with crenolanib. Interestingly, no increase was observed for phospho-YAPS127 following incubation with crenolanib despite the relocation of YAP from the nucleus to the cytoplasm; indeed, a small decrease was actually noted (Fig. 3A). In contrast, a marked decrease of phospho-YAPY357 was observed during PDGFR inhibition with crenolanib. Next, we performed subcellular fractionation experiments in HuCCT-1 cells separating nuclei from the cytoplasm. Phospho immunoblot analysis identified abundant phospho-YAPY357 in the nuclear fraction with minimal tyrosine phosphorylated protein in the cytoplasm, while the opposite was observed for phospho-YAPS127 (Fig. 3B). The importance of phospho-YAPY357 in regulating subcellular localization was further confirmed by determining the subcellular localization of YAP in the mouse murine CCA cell line SB-1; a cell line in which flag tagged YAPS127A expression is enforced. The S127A mutation renders YAP insensitive to serine phosphorylation by LATS1/2, and as such permits examination of YAP subcellular distribution independent of S127 phosphorylation. Employing these cells, we performed immunofluorescence for the flag-tagged epitope of the mutant YAP. When SB-1 CCA cells were incubated with the crenolanib, flag-tagged YAP still redistributed from the nucleus to the cytoplasm (Fig. 3C). These data suggest that in CCA cells, tyrosine phosphorylation of YAP at Y357 may determine its subcellular localization independent of S127 phosphorylation.
Figure 3. Tyrosine phosphorylation regulates YAP localization in CCA.

A, cell lysates from the HuCCT-1 cell line were subjected to immunoblot for phosphorylated YAP (S127 and Y357) and YAP. Actin was used as a loading control. B, cell lysates from HuCCT-1 cell line were fractionated and subjected to immunoblot for phosphorylated YAP (S127 and Y357) and YAP. Lamin B1 and GAPDH were used as loading controls. C, immunofluorescence images from the SB-1a CCA cell line ± PDGFR inhibitor crenolanib (10 μM) with quantification of subcellular localization (right panel). Scale bar = 10 microns.
YAP tyrosine phosphorylation is via SFK
The SFK are capable of phosphorylating the Y357 residue of YAP. Therefore, their role in regulating YAPY357 phosphorylation and subcellular localization in CCA cells was examined. SFK were profiled in the CCA cell line HuCCT-1. Five of the eight SFK members, Src, Fyn, Yes, Lyn, and Lck, are expressed in HuCCT-1 cells (Fig. 4A). However, Yes, a protein known to bind YAP[20], was most abundant. To explore the prospect that PDGF may be result in SFK activation as a mechanism of YAPY357 phosphorylation, we examined the activating phosphorylation status of SFK. The SFK members are regulated by a common autophosphorylation event on tyrosine416 (Y416) and are of similar molecular weight precluding identification of the individual family members by immunoblot analysis. Under basal conditions, phospho-SFKY416 was readily identified; after incubation with crenolanib, a significant decrease of phospho-SFKY416 abundance was observed, which paralleled the decrease in phospho-YAPY357 (Fig. 4B). Next, the role of SFK inhibition in modulating YAP tyrosine phosphorylation was explored using dasatinib (developed as a Bcr-Abl tyrosine kinase inhibitor, but is also a potent SFK inhibitor especially at nM concentrations)[21]. Incubation of HuCCT-1 cells with dasatinib similarly lead to a decrease in phospho-SFKY416 and phospho-YAPY357 abundance, and a rapid redistribution of YAP from the nucleus to the cytosol (Figs. 4 C,D). These observations are consistent with a PDGFR-SFK-YAP tyrosine phosphorylation cascade in HuCCT-1 cells.
Figure 4. PDGF mediated YAP tyrosine phosphorylation is via Src Family Kinases.

A, mRNA expression in HuCCT-1 cell lines. Relative expression compared to 18S internal control is depicted as geometric mean (n=3). B,C cell lysates from the HuCCT-1 cell line ± PDGFR inhibitor crenolanib (10 μM) (B) or the SFK inhibitor dasatinib (1 μM) (C) were subjected to immunoblot for phosphorylated Src (Y416) and Src and phosphorylated YAP (Y357) and YAP. Actin was used as a loading control. D, immunofluorescence images from the HuCCT-1 CCA cell line ± SFK inhibitor dasatinib (1 μM) with quantification of subcellular localization (right panel). Scale bar = 10 microns.
PDGFR mediated SFK activation promotes PDGF-B expression via a YAP-TBX5 complex
We sought to determine the role of SFK activation on the transcriptional regulation of PDGF-B. Similar to the results with crenolanib, incubation of HuCCT-1 cells with dasatinib reduced the level of PDGF-B mRNA, consistent with SFK regulation of PDGF-B expression (Fig. 5A). In silico analysis of the PDGF-B promoter demonstrated a single TEAD binding site. However, chromatin immunoprecipitation assays targeting this site did not demonstrate YAP binding (data not shown). We have previously demonstrated YAP binding to the transcription factor TBX5 in CCA cells and in silico analysis demonstrated multiple potential TBX5 binding sites within the PDGF-B promoter. Chromatin immunoprecipitation studies identified binding of YAP to these sites by a mechanism disrupted by treating the cells with dasatinib (Fig. 5B). Taken together, these findings suggest that SFK activation is a mechanism for YAP-mediated regulation of PDGF-B expression consistent with a PDGF-YAP feed forward, autocrine signaling process in CCA cells.
Figure 5. SFKs mediate PDGF feed-forward signaling.

A, mRNA expression in HuCCT-1 cca cell line ± SFK inhibitor dasatinib (1 μM). Fold expression treated/control is depicted as geometric mean +/− geometric standard deviation (n=3). B, chromatin immunoprecipitation for TBX5 sites (top panel) in the PDGF-B promoter region (bottom panel) ± SFK inhibitor dasatinib (1 μM).
PDGFR inhibition is therapeutic in vitro and in vivo
Incubation of HuCCT-1 cells with crenolanib results in cell death by apoptosis as assessed by morphology and biochemically as measured by caspase 3/7 activation (Fig. 6A–C). Mcl-1, a prosurvival member of the Bcl-2 family, promotes CCA survival and its expression is regulated by YAP [18]. Therefore, we examined Mcl-1 protein abundance following crenolanib incubation, and, indeed, observed a time-dependent decrease in cellular Mcl-1 protein (Fig. 6D) and mRNA (Fig. 6E); these observations are consistent with Mcl-1 cellular depletion as a mechanism of apoptosis following PDGFR inhibition. Based on these in vitro findings, we next examined cell death in vivo utilizing PDX. Intrahepatic PDX from three separate patients were expanded into the bilateral flanks of NOD/SCOD mice. Once palpable tumors had developed (~125mm3) the mice were dosed daily with crenolanib for 14 days, sacrificed, and tumors removed. TUNEL staining demonstrated a significant increase in cell death cells within the tumors of mice treated with crenolanib as compared to vehicle treated controls (Fig. 6F). Immunohistochemistry for p-YAPY357 was attempted, but failure of the antibody to reliably stain control and treated tissues in our model limited this evaluation. However based on the TUNEL staining, crenolanib is tumor suppressive in PDX models likely by restraining PDGFR-YAP signaling.
Figure 6. PDGF inhibition is therapeutic in vitro and in vivo in CCA.

A–C the CCA cell line HuCCT-1 was incubated with the PDGFR inhibitor crenolanib (1 μM) and apoptosis evaluated by DAPI nuclear staining(A) and caspase 3/7 assay (C). *p<0.01 Morphologic changes are demonstrated in merged images (B) with arrows representing nuclei of cells undergoing apoptosis. Scale bar = 10 microns. D,E cell lysates from the HuCCT-1 cell line incubated with crenolanib (1 μM) were subjected to immunoblot for Mcl-1 and RT-PCR. Actin was used as loading control and mRNA expression is depicted as fold expression treated/control geometric mean +/− geometric standard deviation (n=3). F, in tissue from patient-derived xenograft bearing mice treated with crenolanib, apoptotic cells were quantified (right panel) by counting TUNEL-positive nuclei in 20 random microscopic fields (20x) using a fluorescent microscope *p<0.01, H and E staining demonstrated areas of cell death (right panel). Scale bar = 20 microns.
DISCUSSION
This study describes cross-talk between the PDGFR and Hippo signaling pathways in cholangiocarcinoma cells (Fig 7). These data indicate that: (1) PDGFR signaling can drive YAP co-transcriptional activity; (2) YAP nuclear localization appears to be regulated by SFK-mediated tyrosine phosphorylation; and (3) PDGF inhibition leads to cell death in vitro and in vivo. These findings are discussed in greater detail below.
Figure 7. PDGF signaling regulates YAP transcriptional activity via SFK-mediated tyrosine phosphorylation.
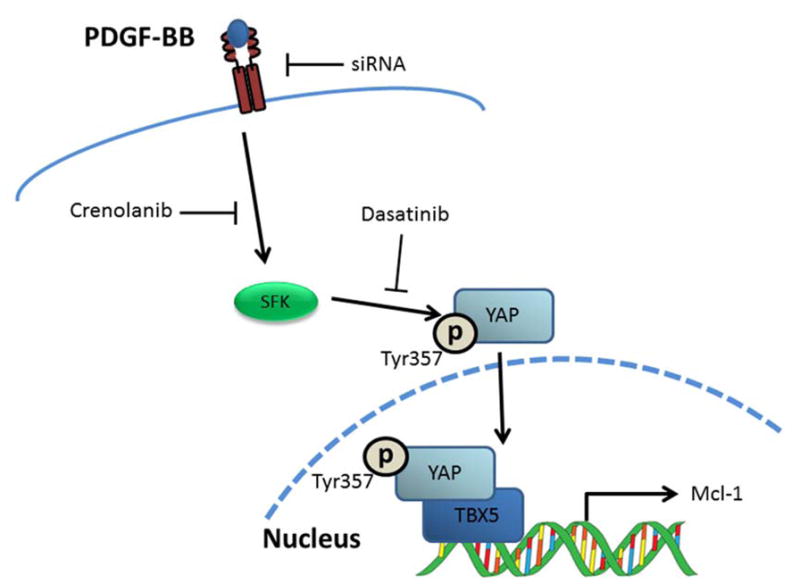
Proposed model of PDGF/Hippo pathway interaction and inhibitors evaluated in current study.
The role of various cell surface receptors in regulating Hippo signaling remains an active area of interest, based on the absence of a dedicated cell surface receptor and extracellular ligand for the Hippo pathway and the prevalence of disordered Hippo signaling in various human tumors. In the current study, the role of the PDGF signaling pathway in regulating YAP subcellular localization and transcriptional activity was examined given that multiple components of the pathway were differentially expressed in human and mouse CCA. We were able to demonstrate downregulation of multiple YAP co-transcriptional targets following PDGFR inhibition. Interestingly we also observed that PDGF-B was a YAP transcriptional target, suggesting an autocrine signaling loop whereby PDGFR signaling drives YAP mediated expression of a cognate ligand, as has been described for FGF, IL-6, and PGE2 signaling[13, 18, 22–25].
The regulation of YAP subcellular localization has focused on the serine/threonine kinase relay module involving MST1/2 and LATS1/2. YAP phosphorylation on residue S127 by serine/threonine kinases blocks its co-transcriptional activity by promoting its binding to protein 14-4-4 in the cytoplasm as well as targeting it for proteasomal degradation[10]. However, PDGFR inhibition of YAP co-transcriptional activity did not modulate phospho-YAPS127 abundance. Indeed, we observed that the mutant S127A YAP, which cannot undergo phosphorylation on this residue, still redistributed to the cytoplasm following PDGFE inhibition in our mouse CCA cells. Our study implicates YAP tyrosine phosphorylation also in the subcellular compartmentation of this transcriptional co-activator; an observation which has been suggested by others [13]. More detailed studies will be required to address the interplay between tyrosine and serine phosphorylation in YAP subcellular compartmentation.
Src-family kinases have been previously implicated in tyrosine phosphorylation and regulation of transcriptional activity of YAP in the setting of inflammation and colon cancer[14]; additionally SFKs have been associated with receptor tyrosine kinases via their Src-homology domains[26]. Given these known associations we evaluated the potential that SFKs could play a role in signal transduction from the activated (phosphorylated) PDGFR to YAP. In our models we did observe significant inhibition of the activating auto-phosphorylation of SFKs (and YAP Tyr357 phosphorylation) when cells were incubated with the PDGFR inhibitor crenolanib. These results were confirmed utilizing the SFK inhibitor dasatinib, a drug that has recently been shown to be especially effective in models of CCA bearing mutations in isocitrate dehydrogenase[27]. The specific SFK(s) responsible for YAP phosphorylation in CCA remain to be defined and the potential for redundancy and/or tissue specificity are intriguing questions yet to be answered.
In our current study, we observed SFK-dependent association of YAP to a cognate TBX5 binding site within the PDGF-B promoter, similar to our previous observation of YAP association with a TBX5 binding site in the Mcl-1 promoter[18]. This observation raises the possibility that in CCA biology, YAP association with TBX5 culminates in expression of prosurvival factors. This paradigm has also been observed in colorectal cancers in which YAP-TBX5 complexes have been demonstrated to regulate expression of the prosurvival protein Bcl-XL[14]. The potency of this survival function was manifest when cytotoxicity was observed following PDGFR pharmacologic inhibition both in a cell line and PDX models. Therapeutic targeting of pathways that culminate in an activating phosphorylation of YAP, as suggested by our data, is attractive as the classic regulatory Hippo kinase complex is inhibitory and thus is not a viable target.
Overall, our study contributes to and supports the accumulating data that receptor tyrosine kinases may modulate YAP signaling by autocrine, feed-forward loops. Our observations extend our understanding of YAP regulation by tyrosine phosphorylation, and provide additional possible therapeutic targets by identifying a role for SFK in YAP mediated gene expression and prosurvival function. Potent SFK inhibitors may prove to be therapeutically beneficial for human CCA, a therapeutic avenue which merits further attention.
Acknowledgments
Grant Support: This work was supported by National Institutes of Health Grants R01DK59427 (to G.J.G), Optical Microscopy Core services and a Pilot and Feasibility Award (to R.L.S) provided through the Mayo Clinic Center for Cell Signaling in Gastroenterology (P30DK084567), and by the Mayo Clinic.
References
- 1.Everhart JE, Ruhl CE. Burden of digestive diseases in the United States Part III: Liver, biliary tract, and pancreas. Gastroenterology. 2009;136(4):1134–44. doi: 10.1053/j.gastro.2009.02.038. [DOI] [PubMed] [Google Scholar]
- 2.Khan SA, et al. Guidelines for the diagnosis and treatment of cholangiocarcinoma: an update. Gut. 2012;61(12):1657–69. doi: 10.1136/gutjnl-2011-301748. [DOI] [PubMed] [Google Scholar]
- 3.Rizvi S, Gores GJ. Pathogenesis, diagnosis, and management of cholangiocarcinoma. Gastroenterology. 2013;145(6):1215–29. doi: 10.1053/j.gastro.2013.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jarnagin WR, et al. Staging, resectability, and outcome in 225 patients with hilar cholangiocarcinoma. Ann Surg. 2001;234(4):507–17. doi: 10.1097/00000658-200110000-00010. discussion 517–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Valle J, et al. Cisplatin plus gemcitabine versus gemcitabine for biliary tract cancer. N Engl J Med. 2010;362(14):1273–81. doi: 10.1056/NEJMoa0908721. [DOI] [PubMed] [Google Scholar]
- 6.Li H, et al. Deregulation of Hippo kinase signalling in human hepatic malignancies. Liver Int. 2012;32(1):38–47. doi: 10.1111/j.1478-3231.2011.02646.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pei T, et al. YAP is a critical oncogene in human cholangiocarcinoma. Oncotarget. 2015;6(19):17206–20. doi: 10.18632/oncotarget.4043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Piccolo S, Dupont S, Cordenonsi M. The biology of YAP/TAZ: hippo signaling and beyond. Physiol Rev. 2014;94(4):1287–312. doi: 10.1152/physrev.00005.2014. [DOI] [PubMed] [Google Scholar]
- 9.Moroishi T, Hansen CG, Guan KL. The emerging roles of YAP and TAZ in cancer. Nat Rev Cancer. 2015;15(2):73–9. doi: 10.1038/nrc3876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Johnson R, Halder G. The two faces of Hippo: targeting the Hippo pathway for regenerative medicine and cancer treatment. Nat Rev Drug Discov. 2014;13(1):63–79. doi: 10.1038/nrd4161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yamada D, et al. IL-33 facilitates oncogene-induced cholangiocarcinoma in mice by an interleukin-6-sensitive mechanism. Hepatology. 2015;61(5):1627–42. doi: 10.1002/hep.27687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fingas CD, et al. Myofibroblast-derived PDGF-BB promotes Hedgehog survival signaling in cholangiocarcinoma cells. Hepatology. 2011;54(6):2076–88. doi: 10.1002/hep.24588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Taniguchi K, et al. A gp130-Src-YAP module links inflammation to epithelial regeneration. Nature. 2015;519(7541):57–62. doi: 10.1038/nature14228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rosenbluh J, et al. beta-Catenin-driven cancers require a YAP1 transcriptional complex for survival and tumorigenesis. Cell. 2012;151(7):1457–73. doi: 10.1016/j.cell.2012.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ehmer U, Sage J. Control of Proliferation and Cancer Growth by the Hippo Signaling Pathway. Mol Cancer Res. 2016;14(2):127–40. doi: 10.1158/1541-7786.MCR-15-0305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.O’Hara SP, et al. ETS Proto-oncogene 1 Transcriptionally Up-regulates the Cholangiocyte Senescence-associated Protein Cyclin-dependent Kinase Inhibitor 2A. The Journal of biological chemistry. 2017;292(12):4833–4846. doi: 10.1074/jbc.M117.777409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nature protocols. 2008;3(6):1101–8. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- 18.Rizvi S, et al. A Hippo and Fibroblast Growth Factor Receptor Autocrine Pathway in Cholangiocarcinoma. J Biol Chem. 2016;291(15):8031–47. doi: 10.1074/jbc.M115.698472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zimmerman EI, et al. Crenolanib is active against models of drug-resistant FLT3-ITD-positive acute myeloid leukemia. Blood. 2013;122(22):3607–15. doi: 10.1182/blood-2013-07-513044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sudol M. Yes-associated protein (YAP65) is a proline-rich phosphoprotein that binds to the SH3 domain of the Yes proto-oncogene product. Oncogene. 1994;9(8):2145–52. [PubMed] [Google Scholar]
- 21.Davis MI, et al. Comprehensive analysis of kinase inhibitor selectivity. Nature biotechnology. 2011;29(11):1046–51. doi: 10.1038/nbt.1990. [DOI] [PubMed] [Google Scholar]
- 22.Fan R, Kim NG, Gumbiner BM. Regulation of Hippo pathway by mitogenic growth factors via phosphoinositide 3-kinase and phosphoinositide-dependent kinase-1. Proc Natl Acad Sci U S A. 2013;110(7):2569–74. doi: 10.1073/pnas.1216462110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Farrell J, et al. HGF induces epithelial-to-mesenchymal transition by modulating the mammalian hippo/MST2 and ISG15 pathways. J Proteome Res. 2014;13(6):2874–86. doi: 10.1021/pr5000285. [DOI] [PubMed] [Google Scholar]
- 24.Kim HB, et al. Prostaglandin E2 Activates YAP and a Positive-Signaling Loop to Promote Colon Regeneration After Colitis but Also Carcinogenesis in Mice. Gastroenterology. 2017;152(3):616–630. doi: 10.1053/j.gastro.2016.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim NG, Gumbiner BM. Adhesion to fibronectin regulates Hippo signaling via the FAK-Src-PI3K pathway. J Cell Biol. 2015;210(3):503–15. doi: 10.1083/jcb.201501025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bromann PA, Korkaya H, Courtneidge SA. The interplay between Src family kinases and receptor tyrosine kinases. Oncogene. 2004;23(48):7957–68. doi: 10.1038/sj.onc.1208079. [DOI] [PubMed] [Google Scholar]
- 27.Saha SK, et al. Isocitrate Dehydrogenase Mutations Confer Dasatinib Hypersensitivity and SRC Dependence in Intrahepatic Cholangiocarcinoma. Cancer Discov. 2016;6(7):727–39. doi: 10.1158/2159-8290.CD-15-1442. [DOI] [PMC free article] [PubMed] [Google Scholar]