

Abstract
Modification of essential bacterial peptidoglycan (PG)-containing cell walls can lead to antibiotic resistance; for example, β-lactam resistance by L,D-transpeptidase activities. Predatory Bdellovibrio bacteriovorus are naturally antibacterial and combat infections by traversing, modifying and finally destroying walls of Gram-negative prey bacteria, modifying their own PG as they grow inside prey. Historically, these multi-enzymatic processes on two similar PG walls have proved challenging to elucidate. Here, with a PG-labelling approach utilizing timed pulses of multiple fluorescent D-amino acids, we illuminate dynamic changes that predator and prey walls go through during the different phases of bacteria:bacteria invasion. We show formation of a reinforced circular port-hole in the prey wall, L,D-transpeptidaseBd-mediated D-amino acid modifications strengthening prey PG during Bdellovibrio invasion, and a zonal mode of predator elongation. This process is followed by unconventional, multi-point and synchronous septation of the intracellular Bdellovibrio, accommodating odd- and even-numbered progeny formation by non-binary division.
Peptidoglycan (PG) is a shape-determining macromolecule common to the bacterial domain. The mature PG wall of bacteria is made by glycan polymerization and peptide crosslinking of a D-amino acid-rich muramyl pentapeptide subunit (Fig. 1a). These crosslinks give the PG wall its essential load-bearing properties against the bacterial cell’s turgor pressure and are made in two basic ways; either 3–4 crosslinks catalysed by normally essential and common penicillin-binding proteins or 3–3 crosslinks catalysed by normally disposable, variable, L,D-transpeptidases (Ldt) (Fig. 1b)1.
Fig. 1. Background and introduction to experimental procedures.
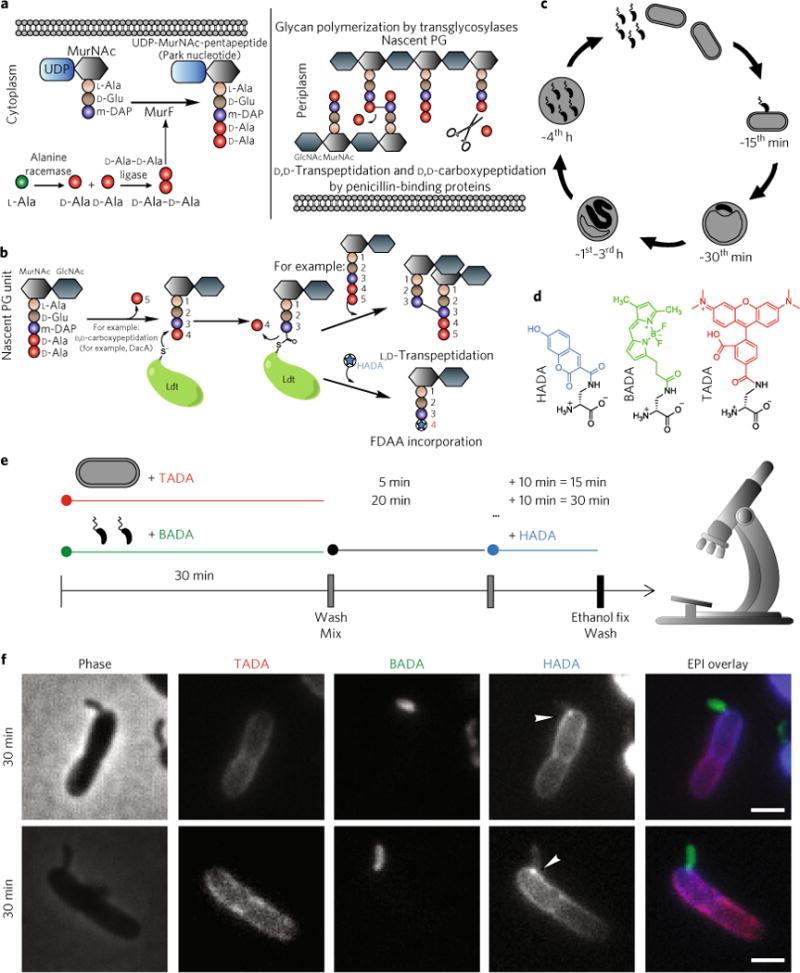
a, Biosynthesis of PG starts in the cytoplasm by sequential addition of L-alanine, D-glutamic acid, a diamino acid and a dipeptide of D-alanine-D-alanine to disaccharide units. This subunit is then incorporated into the murein sacculus by glycan polymerization via transglycosylases. The D-alanine at position 5 can also be cleaved by the actions of D,D-carboxypeptidases. b, L,D-transpeptidases cleave the D-alanine from position 4 and utilize the energy from cleaving this bond to form a 3–3 crosslink with another acyl-acceptor stem peptide or replace the D-alanine with a free D-amino acid such as a fluorescent D-amino acid (FDAA). c, Timed stages of the predatory cycle of B. bacteriovorus (black) bacteria invading E. coli prey (grey). At 0–15 min post-mixing of B. bacteriovorus and prey, B. bacteriovorus attach and begin to enter the outer layers of the prey. At 30 min, most of the B. bacteriovorus have entered the prey periplasm, modifying the prey cell to form a rounded ‘bdelloplast’. At 1–3 h, B. bacteriovorus growth occurs at the expense of the prey cell contents in the form of elongation as a filament. At 4h, this filament fragments into smaller attack-phase cells that break out from the bdelloplast. d, FDAAs used in this study; colours are representative of emission maxima. e, Multi-coloured-FDAA labelling scheme with time points observed by wide-field epifluorescence microscopy. Predator and prey cells were pre-labelled separately with BADA and TADA, respectively, before being washed and then mixed. Samples of this mixed infection were then pulse-labelled with HADA for 10 min before each time point before being fixed, washed, and then microscopically observed. f, Phase contrast and epi-fluorescent microscopy images of the early stages of B. bacteriovorus predation The B. bacteriovorus are false-coloured in green, the E. coli prey cells are false-coloured in red and the pulsed HADA signal is false-coloured in blue. Each channel is displayed independently in white and with all three fluorescence channels merged in an epifluorescence (EPI) overlay. HADA fluorescence signal on the prey wall has an intense focus at each point of B. bacteriovorus contact and spreads from this point across the rest of the wall. Scale bars, 1 μm. The two images are representative of between 321 and 10,546 cells for each time point, detailed in Supplementary Table 1.
Although penicillin-binding proteins and Ldts are evolutionary and structurally distinct transpeptidases, research in diverse bacteria showed that both enzyme types can exchange a range of naturally occurring D-amino acids (DAAs) with the fifth- and fourth-position D-alanines in the peptide stems of PG subunits, respectively2–4 (Fig. 1b). Such exchanges are associated with changes in a variety of biophysical properties of the wall5, 6, in particular the strength (as determined by osmolarity challenge2,7) in some bacteria. Substrate promiscuity of these transpeptidases toward a diverse set of DAAs8 has allowed the development of fluorescent D-amino acids (FDAAs) and their implementation as a means to visualize PG dynamics in situ9–12
Bdellovibrio bacteriovorus (approximately 1.0 × 0.3 μm) prey on (larger) Gram-negative bacterial species by breaching the prey outer membrane, residing in the modified prey periplasm (forming the ‘bdelloplast’), resealing and growing within13,14, before finally bursting out to invade more prey (Fig. 1c). The prey are killed some 20 min into predation when electron transport ceases as predator molecules pass across the prey inner membrane15; however, the prey bdelloplast is kept intact for 4 h to allow ‘private dining’ and consumption of prey contents by the predator. Early electron microscopic work16,17 led to the assumptions that the invading B. bacteriovorus would squeeze through the outer layers of the prey bacterium, degrading some type of entry pore in the prey PG-containing cell wall, re-sealing this, and modifying the rest of the prey PG. However, as the biochemically similar walls were obscured at the points of contact between the two bacterial cells, this bicellular multi-enzymatic process has, until now, been difficult to analyse. Therefore, other than recent work showing the mechanisms of prey cell rounding18, self-protection from auto-rounding19 and marking of the wall for later destruction20, B. bacteriovorus wall-invasion dynamics and enzymology have remained a subject of conjecture.
Here, we combine three differently coloured FDAAs9 in a timed series (Fig. 1d,e) to illuminate dynamic PG modifications during bacterial predation, simultaneously, in two bacterial species. Three-dimensional structured illumination microscopy (3D-SIM), resolved the B. bacteriovorus processes of: breaching the prey PG; constructing a reinforced port-hole in the prey cell wall; resealing the port-hole after entry; modifying the prey PG with L,D-transpeptidases; and eventually achieving filamentous, intra-bacterial zonal cell growth and synchronous, multi-site septation.
Results
Multi-colour-FDAA microscopy reveals prey versus predator cell wall modifications during invasion
A synchronous predatory invasion co-culture of Escherichia coli prey cells pre-labelled with a red FDAA, TADA, and B. bacteriovorus predator cells pre-labelled with a green FDAA, BADA, was established, and this invasive culture was further pulse-labelled with a blue FDAA, HADA, for 10 min at key points during the predation process. The cells were then fixed, washed and imaged (Fig. 1e).
Total cell wall fluorescence of now-dead prey cells (TADA) showed no appreciable change through the invasive process (Supplementary Fig. 1); however, both labelling patterns and signal intensities of pulsed HADA fluorescence showed dramatic differences depending on the stage of predation.
HADA pulses early in the infection, 15 or 30 min post-mixing of predators with prey, resulted in labelling of various subcellular features. In particular, intense, localized, focal HADA marks on the prey PG (and a gradient of blue HADA signal from that focal point) were seen associated with attached B. bacteriovorus cells, revealing the entry point of the B. bacteriovorus during the earliest predator-prey interaction (Fig. 1f).
To further characterize these subcellular features in early predation, we imaged these labelled cells with high-resolution 3D-SIM. 3D-SIM resolved most of these focal marks of HADA labelling as annular ring structures (~25% of all HADA-bright prey cells investigated at the earliest predation point, Fig. 2, Supplementary Table 2 and Supplementary Movie 1) having a width (~0.24 μm; Supplementary Table 2) slightly less than that of a B. bacteriovorus cell (~ 0.33 μm) at the point of predator invasive cell pole-prey contact. This is consistent with the B. bacteriovorus ‘squeezing through the entry pore’ idea suggested by electron micrographs in earlier work16,21,22. Therefore, these HADA foci probably indicate the specific modification of the prey cell wall by the predator during entry (Fig. 2a). The ring of HADA modification was on the prey PG rather than the predator PG, as it was always observed at the point of the prey PG, whether the predator was on the outside, inside, or partially entering the prey cell (Supplementary Fig. 2a–c). Furthermore, rare instances were observed where the predator had become detached from the prey but the HADA foci were still visible, confirming that these foci were indeed on the prey PG (Supplementary Fig. 2d).
Fig. 2. Three-dimensional structured illumination microscopy images of early predation by B. bacteriovorus (pre-labelled with BADA, false-coloured red) on prey E. coli cells after pulse labelling for 10 min with HADA (false-coloured cyan) to show early modification of cell walls.
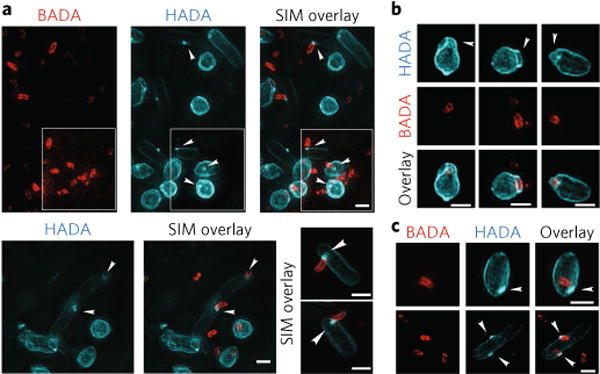
a, Predation 15min post-mixing reveals a ring of HADA-labelled prey cell wall modification at the point of B. bacteriovorus contact (arrowheads) and of similar width to the B. bacteriovorus cell (see Supplementary Table 2). Central pores in the labelled PG material can be seen where the B. bacteriovorus image is artificially removed from the overlay of the two channels. Such annuli may represent a thickened ring of PG modification. In the white inset, the lookup table for the BADA channel has been separately adjusted until all the BADA labelled predators are clearly visible. Three representative examples are displayed. b, Prey PG is deformed around the site of B. bacteriovorus invasion (arrowheads). c, The cells show HADA fluorescence at the end of the internal B. bacteriovorus cell (arrowheads), which probably represents transpeptidase activity re-sealing the hole in the prey PG after the B. bacteriovorus cell has entered. Images are representative of >100 3D-reconstructed cells in two independent experiments (Supplementary Table 2 for details of numbers analysed). Scale bars, 1 μm.
To establish that the dark channel in the HADA focal mark was indeed an entry pore in the prey PG, we needed to detect the reduction of prey-PG material at the HADA channel centre. Using a more outer-membrane-permeable E. coli imp4213− mutant strain as an alternative prey allowed us to label the prey PG uniformly and more completely with otherwise poorly outer-membrane-permeable TADA9. In these cells, dark pores in the TADA signal (arrowheads, TADA channel, Fig. 3a) were present, coincident with, and central within, the HADA ring (Fig. 3a and Supplementary Table 3). These results represent a direct observation of B. bacteriovorus generating a ringed pore in the prey PG; a process that had previously been only inferred from indirect evidence16,22,23.
Fig. 3. Three-dimensional structured illumination microscopy images of early predation by B. bacteriovorus (pre-labelled with BADA, false-coloured green) on prey E. coli imp4213 cells (which are more permeable and thus susceptible to the TADA pre-labelling, false coloured in red) after pulse labelling for 10min with HADA (false-coloured cyan) to show early modification of cell walls.
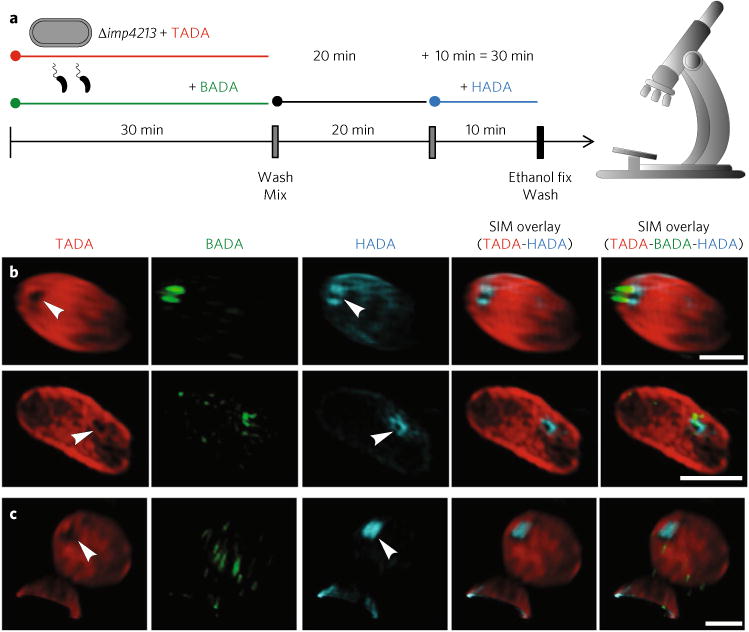
a, FDAA labelling scheme (using excess B. bacteriovorus to promote synchronous invasion of E. coli Δimp4213 mutant prey) with time points observed by 3D-SIM fluorescence microscopy. Predator and prey cells were pre-labelled separately with BADA and TADA, respectively, before being washed and then mixed. Samples of this mixed infection were then pulse-labelled with HADA for 10min before the time points of 15 or 30min. The cells were then fixed, washed and microscopically observed. b, Predation 30 min post-mixing with this prey strain reveals a pore in the TADA signal coincident with the ring of HADA-labelled prey cell wall modification at the point of B. bacteriovorus contact (arrowheads) and of similar width to the B. bacteriovorus cell (Supplementary Table 3). c, In several cases (Supplementary Table 3) where the B. bacteriovorus cell had entered into the prey cell and established itself in the periplasm of the bdelloplast, the pore in the TADA was coincident with a patch of HADA labelling and thus is likely to represent the sealing of the pore through which the B. bacteriovorus had entered. Images are representative of two independent experimental repeats. Scale bars, 1 μm.
Our approach also allowed us to distinguish clear deformations of the prey cell wall at the point where the B. bacteriovorus cell had entered (arrowheads, Fig. 2b; arrowheads, HADA channel, Supplementary Fig. 3; and Supplementary Table 2), clarifying visually previous suggestions that B. bacteriovorus enzymatic modifications of prey cell walls may act to soften them19,24.
To investigate dynamic changes in pores after invasion, we analysed (Supplementary Table 2, Fig. 2c and Supplementary Fig. 2e), ~400 HADA-labelled E. coli S17-1 bdelloplasts. In 27% of these containing internalized B. bacteriovorus, there was a HADA ring similar to the entry pore on bdelloplasts, located at the prey-predator contact point on the prey wall-proximal pole of the internalized B. bacteriovorus cells (red arrowheads, Supplementary Fig. 2e and Supplementary Table 2). In some cases (4%), the HADA patches were filled discs (white arrowheads, Fig. 2c and yellow arrowheads, Supplementary Fig. 2e). Such discs were also coincident with dark pores in the TADA label of E. coli imp4213 mutant bdelloplasts (Fig. 3c and Supplementary Table 3) suggesting that they are sealing discs made by internalized B. bacteriovorus to close the prey, keeping the bdelloplast intact for predator consumption of contents.
B. bacteriovorus establishment inside prey is accompanied by an L,D-transpeptidase-mediated prey wall modification
As the B. bacteriovorus cells enter the prey periplasm, the prey cells become rounded (Fig. 2a), forming a bdelloplast13. During this period, the extent of HADA incorporation to the whole rounding wall of the (now dead) prey substantially increased and peaked around 45 min post-mixing, with ~2 to 4 times more HADA signal intensity (blue line, Fig. 4a, see Methods for details) than the mean HADA labelling at later 2, 3 and 4 h predation time points.
Fig. 4. Quantitative and qualitative effects of two L,D-transpeptidases on prey cell wall modifications by FDAAs and their expression profiles.

a, Plot of mean HADA fluorescent signal of cells against time throughout the predation cycle. Measurements are total mean background-corrected fluorescent signal from wild-type B. bacteriovorus cells (grey line), Δ2ldt mutant (yellow line) or invaded prey bdelloplast. Mean fluorescent signal was significantly lower in the bdelloplasts invaded by the Δ2ldt mutant (orange line) compared with those invaded by the wild type (blue line). Time is in minutes postmixing of predator and prey and fluorescence is in relative fluorescent units (RFU). Data are from at least two independent repeats (see Supplementary Table 1 for details of n). Error bars are s.e.m. The HADA signal differences between E. coli preyed on by WT or Δ2ldt mutant were significant in each of the time points (****p< 0.0001 for all time points except 240 min, for which *P= 0.016 by the Mann-Whitney test). b, RT-PCR showing the expression of the predicted L,D-transpeptidase genes bd0886 and bd1176 or the control gene dnaK, over the predatory cycle of B. bacteriovorus. L: 100 bp DNA ladder; AP: attack-phase cells; 15–45, 1h–4h: minutes or hours respectively since mixing of B. bacteriovorus and prey. Ec: E. coli S17-1 RNA (negative control: no B. bacteriovorus); NT, no-RNA control; Gen: B. bacteriovorus HD100 genomic DNA (positive control). The schematic diagram above represents the different stages of predation. Expression of both genes peaked at 15–30 min post-mixing predator and prey. Two independent repeats were carried out and showed the same transcription pattern. c,d, FDAA labelling of B. bacteriovorus wild-type HD100 (c) and Δ2ldt mutant (d) predation and bdelloplast establishment. The white arrowheads point to HADA modification of the bdelloplast and HADA polar foci visible on the mutant predators inside the bdelloplast. The B. bacteriovorus are false-coloured green, the E. coli prey cells are false-coloured red and the HADA pulse-labelling is false-coloured blue. HADA fluorescence of the prey cell during predation with the L,D-transpeptidase mutant is less than for predation by the wild type. Scale bars, 1 μm. Images are representative of five independent replicates for the wild-type and two independent replicates for the Δ2ldt mutant (see Supplementary Table 1 for details of n).
Previous global transcriptomic work had shown that the predicted B. bacteriovorus L,D-transpeptidase (Ldt) genes, bd0886 and bd1176, are transcriptionally upregulated at 30 min from the start of predation about fivefold and sixfold, respectively25. These predicted L,D-transpeptidases, therefore, are good candidates for prey wall modification enzymes during bdelloplast establishment. Reverse transcription-PCR analysis confirmed that the expression of both genes peaked at 15–30 min into predation (Fig. 4b); time points at which HADA incorporation to the prey walls begins (blue line, Fig. 4a). Deletion of both of these ldt genes (leaving 17 ldtBd genes intact) resulted in a Δbd0886Δbd1176 predator (named A2ldt) that caused ~2–4 times less prey HADA incorporation activity than the wild type (blue line versus orange line, Fig. 4a and representative images in Fig. 4c versus Fig. 4d). This significant difference suggests that these two B. bacteriovorus ldt gene products are responsible for the majority of the overall HADA pulse incorporation into prey wall within the first 2 h of predation. A carboxy-terminal fusion of mCherry to one of these two Ldts (Bd1176) localized to the prey bdelloplast, suggesting that this transpeptidase was exported from predator to bdelloplast and so was acting on the prey PG (Supplementary Fig. 4).
Bdelloplast wall modification is largely by the action of B. bacteriovorus enzymes that act on uncrosslinked tetrapeptides of the prey PG
To test the nature of the bdelloplast wall modification, we quantified HADA incorporation in bdelloplasts formed by B. bacteriovorus predation on different E. coli prey lacking different PG modification functionalities. The prey strain E. coli BW25113 Δ6LDT lacks all of the 6 E. coli L,D-transpeptidases (and therefore any L,D-transpeptidation activity). It lacks tripeptides, 3–3 crosslinks and PG-attached Lpp, and is rich in tetrapeptides26,27. The prey strain E. coli BW25113 ΔdacA lacks the major E. coli D,D-carboxy-peptidase DacA and so contains more pentapeptides in its PG. The prey strain E. coli BW25113 Δ6LDTΔdacA lacks all 6 L,D-transpeptidases and the D,D-carboxypeptidase DacA and so contains mainly tetrapeptides, some pentapeptides, and lacks the modifications introduced by L,D-transpeptidases. Compared with the wild-type prey strain E. coli BW25113 WT, predation of these strains by B. bacteriovorus and pulse labelling with HADA at 35–45 min post-mixing of predator and prey resulted in significantly more HADA incorporation for both prey strains lacking the l,D-transpeptidase activity (Δ6LDT and Δ6LDT ΔdacA, Fig. 5a), but with no significant difference for prey lacking DacA alone (Fig. 5a). In the absence of B. bacteriovorus predation, prey cells in Ca/HEPES buffer pulsed with HADA showed a fraction of the HADA incorporation when compared with the prey strains subjected to B. bacteriovorus predation (~1.5–14.6% of HADA incorporation, controls versus + Bds, Fig. 5a). The majority of the E. coli self-labelling (in controls in the absence of B. bacteriovorus, Fig. 5a) was absent in the E. coli BW25113 Δ6LDT, showing the LdtEC to be responsible for this small amount of labelling. That predation of this strain actually resulted in more HADA incorporation further supports the notion that this incorporation is by Bdellovibrio-encoded enzymes rather than those of the prey. Altogether, these results suggest that a significant proportion of the strong HADA incorporation observed on the prey PG during predation involves predator l,D-transpeptidase activity on tetrapeptides of the prey bdelloplast PG (and not D,D-transpeptidase activity on pentapeptides). These data, along with Bd1176-mCherry and Δ2ldt data above, show that this activity comes from l,D-transpeptidases secreted by the B. bacteriovorus and not due to lingering activities of prey Ldt enzymes.
Fig. 5. Plots showing HADA incorporation in the PG of prey E. coli mutants upon B. bacteriovorus predation and showing the damage by osmotic shock to bdelloplasts formed by B. bacteriovorus Ldt mutants.

a, Chart of mean HADA fluorescent signal of prey strains preyed on by B. bacteriovorus (+Bd), and pulsed with HADA at 35–45 min post-mixing (the time point of maximal HADA incorporation for E. coli S17-1). Controls were in Ca/HEPES buffer without B. bacteriovorus predation, but pulsed with HADA at the same time point. Measurements are total mean background-corrected fluorescent signal of prey cells, reported in relative fluorescent units measured by MicrobeJ. Prey cells lacking all six L,D-transpeptidases (A6LDT) accumulated more HADA fluorescence following predation by B. bacteriovorus. Control samples without B. bacteriovorus predation accumulated considerably less HADA fluorescence. Controls of A6LDT prey cells without Bdellovibrio predation accumulated negligible HADA fluorescence. Data are from two (for the controls) or three independent repeats. Error bars are s.e.m. WT: E. coli BW25113 wild-type strain YB7421; A6LDT: E. coli BW25113 A6LDT strain deficient in all six L,D-transpeptidases; ΔdacA: E. coli BW25113 strain YB7423 deficient in dacΔ; Δ6LDTΔdacA: E. coli BW25113 Δ6LDTΔdacA strain YB7439 deficient in all six L,D-transpeptidases and dacA. NS, not significant; all other comparisons were significant P<0.0001, with the one exception shown, by the Mann-Whitney test. b, CPRG β-galactosidase assay measuring cytoplasmic leakage of shocked E. coli bdelloplasts formed by wild-type (E. coli + Bd WT) or bdelloplasts formed by Δ2ldt mutant B. bacteriovorus (E. coli + Bd Δ2ldt) with controls of uninvaded E. coli prey cells (E. coli alone) or B. bacteriovorus cells alone (Bd WT alone). Red colour from positive CPRG reaction was measured by spectrophotometry at 574nm and readings were normalized to each experiment. Bdelloplasts were harvested by centrifugation and shocked by resuspension in Ca/HEPES buffer for no shock, except centrifugation alone (Buffer), Ca/HEPES buffer supplemented with 750 mM NaCl (Upshock) or upshock followed by further centrifugation and resuspension in water (Downshock). Error bars are s.e.m. Statistical significance was determined by Student’s t-test (two-tailed) *P<0.05, **P< 0.01, ***P< 0.001. Data are the mean of seven independent repeats.
L,D-TranspeptidaseBd-mediated prey wall modification confers bdelloplast physical robustness
To determine the role of the L,D-transpeptidase activity, we assayed the stability of bdelloplasts produced by wild-type B. bacteriovorus or by Δ2ldt mutant predator under osmotic challenge using the β-galatosidase substrate chlorophenyl red-β-D-galactopyranoside (CPRG) method to screen for damage to bacterial cell walls28.
Bdelloplasts, at the point where peak Ldt FDAA transfer was observed (1 h post-synchronous infection of E. coli S17-1 lac+ prey) were subjected to osmotic upshock or downshock29. We observed increased β-galactosidase activity (Fig. 5b) in the supernatant from shocked bdelloplasts formed by Δ2ldt mutant predators relative to wild type in all conditions tested, including a small (but significant) increase in levels from bdelloplasts formed by Δ2ldt predators, subjected only to the stress of centrifugation and resuspension in buffer (Fig. 5b). These data suggest that Bd0886 and Bd1176 L,D-transpeptidase activities strengthen the bdelloplast wall to resist bursting during periods of B. bacteriovorus predatory intrabacterial growth, after prey entry.
To investigate whether this Ldt modification had any effect on the bdelloplast morphology, we measured the sizes and shapes of the prey and bdelloplasts. Early bdelloplasts (45–60 min) formed by the Ldt mutant B. bacteriovorus were slightly, but significantly (p < 0.0001), less round than those formed by the wild type (Supplementary Fig. 5). We hypothesize that the less robust bdelloplasts formed by the Ldt mutant result in more flexible walls that warped more by the invading B. bacteriovorus cell, visible at the earlier stage of invasion after the B. bacteriovorus cell squeezed into the full prey cell. At later stages of invasion (2–4 h), degradation of prey cell content may be why the differences between bdelloplasts formed by the mutant or the wild type are no longer significant.
Multi-coloured-FDAA labelling provides direct evidence for the zonal mode of elongation and synchronous division of B. bacteriovorus growing inside prey
B. bacteriovorus grow without binary fission, as a single multi-nucleoid filament inside prey30. At later time points, after 2 h post-mixing, we observed filamentous cell elongation of the B. bacteriovorus within bdelloplasts (Fig. 6a)30. Attack-phase B. bacteriovorus were added in excess to ensure efficient predation in our experiments and attack-phase predator cells that did not enter prey can be seen to retain substantial initial BADA labelling (Fig. 6a and yellow arrowheads, Fig. 6b), because they do not replicate outside prey. On the other hand, after 2–3 h post-mixing, we observe some green BADA transfer into the prey bdelloplast structure (BADA signal on bdelloplasts, Fig. 6a), which may represent a predator-to-prey DAA turnover and transfer event as the growing B. bacteriovorus make new PG during elongation. While potentially fascinating, quantifying this inter-wall transfer proved impossible to resolve with current reagents. The high level of BADA accumulation in these bdelloplast walls appears to be more than could have been accrued from just one invading Bdellovibrio. This may be a slow accumulation into the prey PG of free BADA present in the medium. This BADA may have been released from excess non-invading Bdellovibrio due to their self-PG turnover, and/or releasing of BADA transiently accumulated in their cell envelopes. This pool of free BADA would be present throughout the 4 h predatory cycle and so could incorporate into prey over a longer time compared with the 10 min pulses of HADA availability.
Fig. 6. Epifluorescence and 3D-SIM images of the later stages of predation to show PG modification of the growing internal B. bacteriovorus.
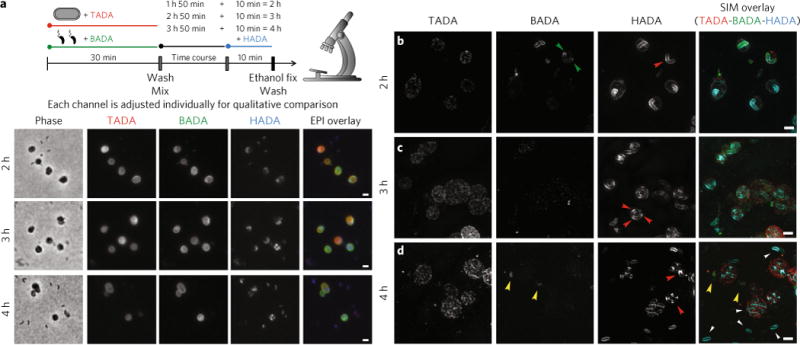
a–d, Phase-contrast (a) and epi-fluorescent microscopy and 3D-SIM (b–d) images of the later stages of B. bacteriovorus predation (after the peak of bdelloplast HADA labelling, by wild-type predator, has ended). The B. bacteriovorus were pre-labelled with BADA and are false-coloured in green; the E. coli prey cells were pre-labelled with TADA and are false-coloured in red. The cells were pulse-labelled for 10 min before each acquisition time point with HADA, which is false-coloured in cyan. Each channel is displayed independently and with all three fluorescence channels merged. The HADA fluorescence indicates synthesis of the B. bacteriovorus PG, which initiates at many points along the growing predator (2 h, b; red arrowhead) except the poles (2 h; b; green arrowheads), before developing into foci (3 h; c; red arrowheads), which become septa (4h; d; red arrowheads). After division, newly released B. bacteriovorus can be seen to modify their whole PG (4 h; d; white arrowheads). B. bacteriovorus that did not invade (there was an excess of B. bacteriovorus to ensure efficient predation) can be seen to have a strong BADA signal and low HADA signal (4h; d; yellow arrowheads). Images are representative examples from thousands of cells from five independent experiments (a) and of >100 3D-reconstructed cells in two independent experiments (b–d). See Supplementary Table 1 for numbers of cells analysed. Scale bars, 1 μm.
3D-SIM imaging showed that B. bacteriovorus cells elongate along the filament with numerous, focused zones of growth (labelled with HADA, red arrowhead, Fig. 6b) covering the entire cell surface except the apparently inert poles (preserving the original BADA signal, green arrowheads, Fig. 6b). Later, around 3 h post-mixing, new HADA incorporation appears as defined narrow foci along the filament (Fig. 6a and red arrowheads, Fig. 6c), at points in B. bacteriovorus where new division septa would be expected to form synchronously30. After 4 h post-mixing, these foci become the points of septum formation (Fig. 6a and yellow arrowheads, Fig. 6d). Finally, newly released, attack-phase B. bacteriovorus daughter cells (white arrowheads, Fig. 6d) incorporate pulsed HADA all over the cell and can therefore be distinguished from excess BADA-labelled predators that did not enter prey cells by the presence of a strong HADA fluorescent signal, but low BADA fluorescent signal.
Discussion
Here, using multi-coloured-FDAA labelling and super-resolution imaging, we directly visualize subcellular modifications by B. bacteriovorus on E. coli PG cell walls and their effects during predation. Our data define an entry port structure by which a B. bacteriovorus cell accesses the cytoplasmic membrane face of the prey cell wall and seals itself in. We also show the sites of PG growth in the nonbinary fission mode of predator growth. In addition, we show that L,D-transpeptidase enzymes from the B. bacteriovorus modify the PG of prey during residency of the predator to establish a stable intracellular niche.
Pioneering enzymology of prey bdelloplast extracts in the 1970s had detected bulk enzyme activities suggestive of extensive predator modification of prey PG. These included solubilization of 25% of the meso-diaminopimelic acid (m-DAP) residues on the PG23 and the addition of free m-DAP back to the bdelloplast31. m-DAP is a residue native to PG that has both L- and D-amino acid properties. Therefore, we see FDAAs in our studies acting as visible substrates for these enzymatic, fresco-like changes to the walls of invaded prey caused by B. bacteriovorus enzymes. Indeed, we show the B. bacteriovorus-facilitated, localized breakdown of the prey wall to form a pore, and its re-sealing while also rounding the prey cell wall to form an osmotically stable bdelloplast.
The initial ring of intense FDAA incorporation matches with the gap on the prey cell wall at the contact point with the B. bacteriovorus pole (Supplementary Tables 2 and 3, and Figs. 2a and 3a). Such a remodelling of the prey PG probably strengthens the predator entry point. We also show here (Figs. 2c and 3b) that such entry ports have accumulated centralized FDAA signal after B. bacteriovorus entry, which might represent a gradual ring-to-disc re-sealing activity of this pore; a process that had previously been only inferred by indirect evidence of ‘scars’ left behind on the prey cell wall at the point of entry32.
The most extensive prey cell wall modification occurs 30–45 min after mixing B. bacteriovorus with the prey; involving the L,D- transpeptidases with major contributions from 2 of the 19 LdtBd enzymes encoded by the genes bd0886 and bd1176 (Fig. 4a). These observations may be due to pulsed FDAAs mimicking the incorporation of previously solubilized m-DAP reported in early B. bacteriovorus studies23,31, but this is beyond the scope of our present work. While we were able to isolate fluorescent FDAA-labelled sacculi, the amounts were not sufficient for mass spectrometry-based identification of sites of D-amino acid incorporation in Bdellovibrio or E. coli (Supplementary Fig. 7). Incorporation of non-canonical D-amino acids into the cell wall is a stress response in Vibrio cholerae, which is shown to stabilize the PG integrity of the cells in stationary phase2. The incorporation of native m-DAP31 and/or D-amino acids into the prey cell wall by B. bacteriovorus Ldts early in the predation (15 min–1 h) could represent an analogous means of forming a stabilized and stress-resistant bdelloplast. The susceptibility of bdelloplasts formed by the Δ2ldt mutant predator to bursting during osmotic stress (Fig. 5b) supports this hypothesis.
FDAA labelling also elucidated the growth of the intraperiplasmic B. bacteriovorus predator directly (Fig. 6). Growth starts in patches along the length of the B. bacteriovorus cell, but not at the poles (Figs. 6a,b). After B. bacteriovorus septation, final predator self-PG-modification produces attack-phase B. bacteriovorus (Fig. 6d) that each emerge with one flagellated and one piliated pole21,33. These experiments provide evidence that both predator poles can carry out bilateral growth, along the length of the cell, rather than one ‘old’ pole remaining attached to the membrane and growth emanating solely from specific regions30,34. Synchronous septum construction (that results in odd or even progeny numbers) is seen along the length of the filamentous B. bacteriovorus growing within the bdelloplast (Fig. 6a,c,d), confirming earlier movies of this synchronous division30.
In conclusion, the ability to distinctly label the PG-containing cell walls of two different genera of interacting bacteria with different coloured FDAAs has illuminated a series of dynamic molecular modifications that predatory B. bacteriovorus make to prey cell walls and self cell walls during their intraperiplasmic lifestyle. These modifications (pore formation and resealing without bacterial bursting and PG remodelling with free small molecules, that is, DAAs, in dual-cell systems) are previously uncharacterized in bacteria, and are key mechanisms of B. bacteriovorus predation. Given the inherent promiscuity of virtually all PG-containing bacteria to incorporate FDAAs in situ9,35, we expect this general approach to be helpful for visualizing interactions of other complex bacterial communities, such as microbiota. Accordingly, we would not be surprised if this and similar approaches illuminate other examples of inter-generic PG modifications with novel functions.
Methods
RNA isolation from predatory cycle and RT-PCR analysis
Synchronous predatory infections of B. bacteriovorus HD100 on E. coli S17-1 in Ca/HEPES buffer (2 mM CaCl2, 25 mM HEPES, pH 7.6), or strain S17-1 suspended in Ca/HEPES alone, were set up as previously described36 with samples throughout the time course being taken and total RNA isolated from them. This semiquantitative PCR allows the evaluation of specific predator transcripts in the presence of fluctuating levels of prey RNA as the predator degrades it. RNA was isolated from the samples using a Promega SV total RNA isolation kit with the RNA quality being verified by an Agilent Bioanalyzer using the RNA Nano kit. RT-PCR was performed with the Qiagen One-step RT-PCR kit with the following reaction conditions: one cycle 50 °C for 30 min, 95 °C for 15 min, then 25 cycles of 94 °C for 1 min, 50 °C for 1 min, 72 °C for 1 min, a 10 min extension at 72 °C after the 30 cycles, and finally a 4 °C hold. Two independent repeats were carried out. Primers to anneal to bd0886 were 5′-AGCCTCTACATGGGTGCAAG-3′ and 5′-AACTTGGCTGCATACCAACC-3′. Primers to anneal to bd1176 were 5′-GCCAACGCCAGCGTGAATGC-3′ and 5′-GGCCGTCGTTGAGTTGCTGC-3′.
Generating gene deletion mutants in B. bacteriovorus
Markerless deletion of both the bd0886 and bd1176 genes from B. bacteriovorus HD100 was achieved sequentially as described previously19,37. Primers designed to amplify to the upstream region of bd0886 were: Bd0886F 5′-ACGGGGTACCCACGATCCCATCTTATAAGC -3′ and Delbd0886F 5′-GGAGATTATATGAAAGCTTTCTAGAATGGACTCTGTTCCTGCGC-3′. Primers designed to amplify to the downstream region of bd0886 were: Delbd0886R 5′-GCGCAGGAACAGAGTCCATTCTAGAAAGCTTTCATATA ATCTCC-3′ and Bd0886R 5′-CTGTAGCATGCTTCAGATCCTCGCTGAAACC-3′. Primers designed to amplify to the upstream region of bd1176 were: Bd1176-F 5′-GCGCAAAAGCTTTCGCAAGCTGGGTGTTCAGC-3′ and Delbd1176F 5′-GATTGCCAGCTCCCCTATGTCTAGAAATCCTCCGAAG ATCGTTT-3. Primers designed to amplify to the downstream region of bd1176 were: Delbd1176R 5′-AAACGATCTTCGGAGGATTTCTAGACATAGGGGA GCTGGCAATC-3′ and Bd1176-R 5′-ACGGGGTACCGGATGTGATTCATACCAGCC-3′.
Construction of an E. coli strain lacking all six L,D-transpeptidases
E. coli BW25113Δ6LDT lacks all five previously published L,D-transpeptidase genes (erfK, ybiS, ycfS, ynhG, ycbB)27,38 plus a sixth gene encoding a putative L,D-transpeptidase, yafK. Gene deletions were generated and combined by transferring kan-marked alleles from the Keio E. coli single-gene knockout library39 into relevant background strains using P1 phage transduction40. The Keio pKD13-derived kan cassette is flanked by FRT sites, allowing removal of the kan marker via expression of FLP recombinase from plasmid pCP20 to generate unmarked deletions with a FRT-site scar sequence39,41. The gene deletions present in BW25113Δ6LDT were verified by PCR, and the analysis of the PG composition showed that muropeptides generated by the activities of L,D-transpeptidases were below the limit of detection.
Fluorescent tagging of Bd1176
The bd1176 gene lacking its stop codon was cloned into the conjugable vector pK18mobsacB in such a way as to fuse the gene at the C terminus with the mCherry gene. This fusion was introduced into B. bacteriovorus by conjugation as described previously42. Cloning was carried out using the NEB Gibson cloning assembly kit and the primers used (5′–3′) were: cgttgtaaaacgacggccagtgccaATGACAAAGATTAATACGCGCC, ccttgctcaccatGT TGTTGCCGCCTCTTCTTG, aggcggcaacaacATGGTGAGCAAGGGCGAG and cagctatgaccatgattacgTTACTTGTACAGCTCGTCCATGCC. Epi-fluorescence microscopy was undertaken using a Nikon Eclipse E600 through a 100× objective (NA 1.25) and acquired using a Hammamatsu Orca ER Camera. Images were captured using Simple PCI software (version 6.6). An hcRED filter block (excitation: 550–600 nm; emission: 610–665 nm) was used for visualization of mCherry tags.
Labelling of cells with FDAAs and imaging
Bdellovibrio bacteriovorus HD100 cells were grown predatorily for 16 h at 30 °C on stationary-phase E. coli S17-1 prey, until these were lysed. The B. bacteriovorus were then filtered through a 0.45 μm filter (yielding ~2 × 108 pfu ml−1) and concentrated 30× by centrifugation at 12,000g for 5 min. The resulting pellet was resuspended in Ca/HEPES buffer, (2 mM CaCl2, 25 mM, HEPES pH 7.6) and then pre-labelled with a final concentration of 500 μM BADA (by addition of 5 μl of a 50 mM stock in dimethylsulfoxide (DMSO)) for 30 min at 30 °C. The cells were then washed twice in Ca/HEPES buffer before being resuspended in an equal volume of Ca/HEPES buffer. E. coli S17-1 or E. coli imp4213 cells were grown for 16 h in LB at 37 °C with shaking at 100 r.p.m. and were back diluted to OD600 nm 1.0 in fresh LB, (yielding ~1 × 109 cfu ml−1) and labelled with a final concentration of 500 μM TADA (by addition of 5 μl of a 50 mM stock in DMSO) for 30 min at 30 °C, before being washed twice in Ca/HEPES buffer and then resuspended in an equal volume of Ca/HEPES buffer. E. coli BW25113 strains were grown as for strain S17-1, except strains YB7423, YB7424 and YB7439 were supplemented with 50 μg ml−1 kanamycin sulfate for incubation and washed of this by centrifugation at 5,000g for 5 min, resuspension in an equal volume of LB broth and further centrifugation at 12,000g for 5 min before back-dilution to OD600 nm 1.0 in Ca/HEPES buffer. This resulted in similar numbers of cells for each strain; E. coli BW25113 Δ6LDT 5.1 × 108 ± 3.6 × 107, YB7423 5.2 × 108 ± 1.8 × 108, YB7424 4.9 ×108 ± 2 × 107, YB74394.3 × 108 ± 1.6 × 108 as determined by colony-forming units.
Defined ratios of approximately five B. bacteriovorus predators to one E. coli prey were then prepared for semi-synchronous predation experiments to allow FDAA labelling of dynamic PG changes as the predators were invading and replicating within the prey. Five hundred microlitres of the pre-labelled B. bacteriovorus were mixed with 400 μl of the pre-labelled E. coli and 300 μl of Ca/HEPES buffer and incubated at 30 °C. For HADA pulse-labelling, 120 μl samples of these predatory cultures were added to 1.2 μl of a 50 mM stock of HADA in DMSO 10 min before each sampling time point for microscopy and returned to 30 °C incubation. These experimental timescales are consistent and are shown in diagrams above the figures (for example 30 min predation time point: 20 min of predator mixed with prey, plus 10 min of subsequent HADA labelling, followed by immediate fixation and then washing). At each time point, all of the 120 μl predator-prey sample was transferred to 175 μl ice-cold ethanol and incubated at −20 °C for at least 15 min to fix the cells. The cells were pelleted by centrifugation at 12,000g for 5 min, washed with 500 μl PBS and resuspended in 5 μl Slowfade (Molecular Probes) and stored at −20 °C before imaging. Samples of 2 μl volume were imaged using a Nikon Ti-E inverted fluorescence microscope equipped with a Plan Apo 60×/1.40 Oil Ph3 DM objective with ×1.5 intermediate magnification, or a Plan Apo 100×/1.45 Ph3 objective, a CFP/YFP filter cube and an Andor DU885 EMCCD or an Andor Neo sCMOS camera using CFP settings for detection of HADA (emission maximum 450 nm), a FITC filter cube for detection of BADA (emission maximum 512 nm) and others (acquisition and image processing details are in the Equipment and settings section in the Supplementary Information). Later time points were prepared with similar HADA pulses carried out on further samples of the continuing predator-prey culture, which extended to 4 h of incubation at 30 °C; the point at which new B. bacteriovorus predators emerge from lysed E. coli prey.
Super-resolution microscopy
3D structured illumination microscopy was performed using a Delta Vision OMX Imaging System equipped with an Olympus UPlanSApo 100×/1.40 Oil PSF objective and a Photometrics Cascade II EMCCD camera. The samples were excited with lasers at 405 nm, 488 nm and 561 nm, and the emission was detected through 419 nm-465 nm, 500 nm-550 nm and 609 nm-654 nm emission filters. The image processing was conducted by SoftWorx imaging software. Further image analysis and processing was conducted via ImageJ or Icy (http://www.bioimageanalysis.org/). Acquisition and image processing details are in the Equipment and settings section in the Supplementary Information.
Quantification of fluorescent signal
For quantification of fluorescent signal, images were acquired as above, but with unvarying exposure and gain settings. The exposures were chosen to give values that did not exceed the maximum, so that saturation was not reached for any of the fluorescent channels. Images were analysed using the MicrobeJ plugin for the ImageJ (FIJI distribution) software (http://www.indiana.edu/~microbej/index.html)43, which automates detection of bacteria within an image. The E. coli prey cells and Bdellovibrio cells were detected using the resulting binary mask from both the phase contrast and either the TADA or the BADA channels respectively. The E. coli prey cells and B. bacteriovorus cells were differentiated by defining two cell types based on size: Cell Type 1 (for E. coli) were defined by area 0.9–6 μm2, length 1.5–7 μm, width 0.4–3 μm and all other parameters as default; Cell Type 2 (for the smaller B. bacteriovorus cells) were defined by area 0–1 μm2, length 0.5–1.5 μm, width 0.2–0.8 μm and all other parameters as default. Manual inspection of the analysed images confirmed that the vast majority of cells were correctly assigned. Bdellovibrio cells were linked hierarchically with the E. coli prey cells, in order to distinguish between internalized, attached and unattached predator cells. The shape measurements including the angularity, area, aspect ratio, circularity, curvature, length, roundness, sinuosity, solidity and width were measured for each type of cell. Background-corrected mean fluorescent intensity was measured for each cell and then the mean of these measurements was determined for each cell type, for each independent experiment. Typically, 500–5,000 cells were measured at each time point for each independent experiment (details of n for each sample in each experiment are presented in Supplementary Table 1).
Code availability
The images and the data were analysed by MicrobeJ (5.11 v), a freely available and open-source software. The code source is available upon request from A.Du.
CPRG assay of leakage of osmotically shocked bdelloplasts derived from predation by Ldt mutant versus wild-type B. bacteriovorus
To evaluate whether DAA transfer to prey bdelloplast cell walls altered the physical stability of those walls to osmotic changes, an assay for leakage of cytoplasmic contents, including β-galactosidase, was used, with the CPRG as a detection reagent.
E. coli S17-1 (lac+) prey cells were grown for 16 h in YT broth at 37 °C with 200 r.p.m. shaking, before being supplemented with 200 μgml−1 IPTG for 2 h to induce expression of lacZ. These prey cells were then centrifuged at 5,100g for 5 min and resuspended in Ca/HEPES buffer (2 mM CaCl2, 25 mM HEPES, pH 7.6) and then diluted to OD600 nm 1.0 in Ca/HEPES buffer. Bdellovibrio bacteriovorus HD100 or Δ2ldt strains were grown predatorily for 16 h at 29 °C on stationary-phase E. coli S17-1 prey until these were fully lysed, and then B. bacteriovorus were filtered through a 0.45 μm filter, concentrated 50× by centrifugation at 5,100g for 20 min and resuspended in Ca/HEPES buffer. Total protein concentration of these concentrated suspensions was determined by Lowry assay, and matched amounts of 50 μg of each strain were used for semi-synchronous infections (between 115 and 284 μl of concentrated suspension made up to a total of 800 μl in Ca/HEPES buffer) with 400 μl of diluted E. coli S17-1 prey cells. This resulted in a multiplicity of infection (MOI of B. bacteriovorus cells:E. coli cells) of 1.4 to 10.5 for the wildtype strain HD100 as determined by plaque assay. The excess of predators resulted in > 99.4% of E.coli prey cells rounded by invasion of strain HD100 and > 99.6% of prey cells rounded by invasion of Δ2ldt mutant after incubation at 29 °C for 1 h with shaking at 200 r.p.m.
A control of prey alone (400 μl diluted prey cells with 800 μl Ca/HEPES buffer) resulted in no rounded prey cells and a control of wild-type B. bacteriovorus HD100 cells alone (50 μg in a total of 1,200 μl Ca/HEPES buffer) was included. After incubation, bdelloplasts (or cells in the controls) were harvested by centrifugation at 17,000g for 2 min and supernatant was removed. The pellets were resuspended in: Ca/HEPES buffer supplemented with 20 μg ml−1 CPRG (Sigma) for centrifugation shock alone; Ca/HEPES buffer supplemented with 750 mM NaCl and 20 μg ml−1 CPRG for upshock; Ca/HEPES buffer supplemented with 750 mM NaCl, incubated for 30 min at 29 °C followed by centrifugation at 17,000g for 2 min and supernatant removed, then the pellet resuspended in water supplemented with 20 μg ml−1 CPRG for downshock. These were then incubated for 30 min at 29 °C before purifying the supernatant, containing any bdelloplast leakage products, for β-galactosidase assay by removing cells by centrifugation at 17,000g for 2 min followed by filtration through a 0.2 μm filter. The β-galactosidase assay was carried out by incubation at 29 °C for 26 h and colour change was monitored by spectrophotometry at 574 nm. Data were normalized for each experiment.
Extra experimental considerations
The Δ2ldt mutant strain exhibited a plaquing phenotype, forming mostly very small plaques with ~1% forming larger plaques similar to the wild-type HD100 strain (see Supplementary Fig. 6) and as such an accurate MOI could not be measured by plaques for this strain. To confirm that matching the input cells by Lowry assay resulted in similar numbers of B. bacteriovorus, and therefore a similar MOI, images of the mixed prey and predators were analysed. After the 1 h incubation at 29 °C, 40 μl samples were mixed with 2 μl of 0.3 μm polystyrene beads (Sigma; diluted 500 times and washed 5 times with water). Samples of 10 μl volume were dropped onto microscope slides with a 1% agarose pad made with Ca/HEPES buffer and 20 fields of view were imaged at × 1,000 phase contrast with a Nikon Ti-E inverted microscope. Images were analysed with the MicrobeJ plugin as described above, but including a third cell type definition for quantifying the beads defined by area 0–1, length 0.1–0.8, width 0.1–0.6 and all other parameters 0-max. This confirmed that there were not significantly different ratios of beads to B. bacteriovorus cells in the two strains (6.1 ± 3.9 for HD100, 6.9 ±0.7 for Δ2ldt mutant) and that all visible prey cells were rounded up after 1 h of incubation, indicating that an MOI of > 1 was achieved (which was required for semisynchronous infection). To confirm that the defective plaquing phenotype of the Δ2ldt mutant was not a result of low yield in liquid culture, images were analysed at the start and end of predatory growth in liquid. The average result of five Lowry assays was taken to match the starting amounts of B. bacteriovorus: 245 μl of strain HD100 and 337 pl of the Δ2ldt mutant strain (after filtration through a 0.45 μm filter, but not concentrated) were made up to 800 μl in Ca/HEPES buffer and added to 400 μl prey E. coli diluted to OD600 nm 1.0 in Ca/HEPES buffer. This mix was imaged with beads as described above at time 0 and 24 h (after incubation at 29 °C with 200 r.p.m. shaking) and analysed using the MicrobeJ plugin as described above. The increase in numbers of B. bacteriovorus cells per bead was not significantly different between the 2 strains (1.9 ± 0.5 for HD100 and 2.1 ± 0.8 for the Δ2ldt mutant). In both cases, the prey cells were almost eradicated after 24 h with only 8–13 cells detected by MicrobeJ in the 20 fields of view for each experiment (reduced to 1.0 ± 0.4 % of starting values for HD100 and 3.3 ± 0.8 % for the Δ2ldt mutant).
Data availability
The raw data that support the findings of this study are available from the corresponding author upon request.
Supplementary Material
Acknowledgments
We thank D. Kearns and his laboratory (Indiana University, USA) for facilities and hospitality to culture B. bacteriovorus, A. Lovering (University of Birmingham, UK) for insights and assistance with the alignment of L,D-transpeptidase protein sequences in B. bacteriovorus, T. Pilizota (University of Edinburgh, UK) for advice on osmotic stress conditions, and R. Lowry (University of Nottingham, UK) for assistance in image acquisition. This work was supported by BBSRC grant [BB/M010325/1] to C.L., a Leverhulme Trust (UK) Research Leave Fellowship RF-2013-348 to R.E.S., NIH GM113172 grant to M.VN. and Y.V.B. and R35GM122556 and GM51986 to Y.V.B. A.De. was supported by an EMBO long-term fellowship, and W.V. was supported by funds from the Wellcome Trust (101824/Z/13/Z).
Footnotes
Author contributions
E.K. and R.E.S. conceived the study and carried out the experiments along with C.L. using reagents constructed by M.V.N. and J.R., and bacterial strains constructed by R.T. and A. De. J.G. and J.B. performed muropeptide analysis in the laboratory of W.V. A. Du. wrote code and aided C.L. and E.K. with image analysis. Y.V.B. provided microscopy facilities and with M.V.N. and W.V. provided helpful comments. E.K., C.L. and R.E.S. wrote the manuscript with input and comments from the other authors.
Competing interests
The authors declare no competing financial interests.
Supplementary information is available for this paper at doi:10.1038/s41564-017-0029-y.
References
- 1.Mainardi JL, et al. A novel peptidoglycan cross-linking enzyme for a β-lactam-resistant transpeptidation pathway. J Biol Chem. 2005;280:38146–38152. doi: 10.1074/jbc.M507384200. [DOI] [PubMed] [Google Scholar]
- 2.Cava F, de Pedro MA, Lam H, Davis BM, Waldor MK. Distinct pathways for modification of the bacterial cell wall by non-canonical D-amino acids. EMBO J. 2011;30:3442–3453. doi: 10.1038/emboj.2011.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Magnet S, et al. Specificity of L,D-transpeptidases from Gram-positive bacteria producing different peptidoglycan chemotypes. J Biol Chem. 2007;282:13151–13159. doi: 10.1074/jbc.M610911200. [DOI] [PubMed] [Google Scholar]
- 4.Fura JM, Kearns D, Pires MM. D-amino acid probes for penicillin binding protein-based bacterial surface labeling. J Biol Chem. 2015;290:30540–30550. doi: 10.1074/jbc.M115.683342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gupta R, et al. The Mycobacterium tuberculosis protein LdtMt2 is a nonclassical transpeptidase required for virulence and resistance to amoxicillin. Nat Med. 2010;16:466–469. doi: 10.1038/nm.2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Peltier J, et al. Clostridium difficile has an original peptidoglycan structure with a high level of N-acetylglucosamine deacetylation and mainly 3-3 cross-links. J Biol Chem. 2011;286:29053–29062. doi: 10.1074/jbc.M111.259150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lam H, et al. D-amino acids govern stationary phase cell wall remodeling in bacteria. Science. 2009;325:1552–1555. doi: 10.1126/science.1178123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Radkov AD, Moe LA. Bacterial synthesis of D-amino acids. Appl Microbiol Biotechnol. 2014;98:5363–5374. doi: 10.1007/s00253-014-5726-3. [DOI] [PubMed] [Google Scholar]
- 9.Kuru E, Tekkam S, Hall E, Brun YV, Van Nieuwenhze MS. Synthesis of fluorescent D-amino acids and their use for probing peptidoglycan synthesis and bacterial growth in situ. Nat Protoc. 2015;10:33–52. doi: 10.1038/nprot.2014.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fleurie A, et al. MapZ marks the division sites and positions FtsZ rings in Streptococcus pneumoniae. Nature. 2014;516:259–262. doi: 10.1038/nature13966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tsui HC, et al. Pbp2x localizes separately from Pbp2b and other peptidoglycan synthesis proteins during later stages of cell division of D39. Mol Microbiol. 2014;94:21–40. doi: 10.1111/mmi.12745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pilhofer M, et al. Discovery of chlamydial peptidoglycan reveals bacteria with murein sacculi but without FtsZ. Nat Commun. 2013;4:2856. doi: 10.1038/ncomms3856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stolp H, Starr M. P Bdellovibrio bacteriovorus gen et sp n., a predatory, ectoparasitic, and bacteriolytic microorganism. Antonie van Leeuwenhoek. 1963;29:217–248. doi: 10.1007/BF02046064. [DOI] [PubMed] [Google Scholar]
- 14.Sockett RE. Predatory lifestyle of Bdellovibrio bacteriovorus. Annu Rev Microbiol. 2009;63:523–539. doi: 10.1146/annurev.micro.091208.073346. [DOI] [PubMed] [Google Scholar]
- 15.Rittenberg SC, Shilo M. Early host damage in the infection cycle of Bdellovibrio bacteriovorus. J Bacteriol. 1970;102:149–160. doi: 10.1128/jb.102.1.149-160.1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Abram D, Castro e Melo J, Chou D. Penetration of Bdellovibrio bacteriovorus into host cells. J Bacteriol. 1974;118:663–680. doi: 10.1128/jb.118.2.663-680.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Abram D, Davis BK. Structural properties and features of parasitic Bdellovibrio bacteriovorus. J Bacteriol. 1970;104:948–965. doi: 10.1128/jb.104.2.948-965.1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lerner TR, et al. Specialized peptidoglycan hydrolases sculpt the intrabacterial niche of predatory Bdellovibrio and increase population fitness. PLoS Pathog. 2012;8:e1002524. doi: 10.1371/journal.ppat.1002524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lambert C, et al. Ankyrin-mediated self-protection during cell invasion by the bacterial predator Bdellovibrio bacteriovorus. Nat Commun. 2015;6:8884. doi: 10.1038/ncomms9884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lambert C, et al. Interrupting peptidoglycan deacetylation during Bdellovibrio predator-prey interaction prevents ultimate destruction of prey wall, liberating bacterial-ghosts. Sci Rep. 2016;6:26010. doi: 10.1038/srep26010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Evans KJ, Lambert C, Sockett RE. Predation by Bdellovibrio bacteriovorus HD100 requires type IV pili. J Bacteriol. 2007;189:4850–4859. doi: 10.1128/JB.01942-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Koval SF, et al. Bdellovibrio exovorus sp. nov., a novel predator of Caulobacter crescentus. Int J Syst Evol Microbiol. 2013;63:146–151. doi: 10.1099/ijs.0.039701-0. [DOI] [PubMed] [Google Scholar]
- 23.Thomashow MF, Rittenberg SC. Intraperiplasmic growth of Bdellovibrio bacteriovorus 109J: solubilization of Escherichia coli peptidoglycan. J Bacteriol. 1978;135:998–1007. doi: 10.1128/jb.135.3.998-1007.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Volle CB, Ferguson MA, Aidala KE, Spain EM, Nunez ME. Quantitative changes in the elasticity and adhesive properties of Escherichia coli ZK1056 prey cells during predation by Bdellovibrio bacteriovorus 109J. Langmuir. 2008;24:8102–8110. doi: 10.1021/la8009354. [DOI] [PubMed] [Google Scholar]
- 25.Lambert C, Chang CY, Capeness MJ, Sockett RE. The first bite-profiling the predatosome in the bacterial pathogen Bdellovibrio. PLoS ONE. 2010;5:e8599. doi: 10.1371/journal.pone.0008599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sanders AN, Pavelka MS. Phenotypic analysis of Escherichia coli mutants lacking L,D-transpeptidases. Microbiology. 2013;159:1842–1852. doi: 10.1099/mic.0.069211-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Magnet S, et al. Identification of the L,D-transpeptidases responsible for attachment of the Braun lipoprotein to Escherichia coli peptidoglycan. J Bacteriol. 2007;189:3927–3931. doi: 10.1128/JB.00084-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pilizota T, Shaevitz JW. Origins of Escherichia coli growth rate and cell shape changes at high external osmolality. Biophys J. 2014;107:1962–1969. doi: 10.1016/j.bpj.2014.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pilizota T, Shaevitz JW. Plasmolysis and cell shape depend on solute outer-membrane permeability during hyperosmotic shock in E. coli. Biophys J. 2013;104:2733–2742. doi: 10.1016/j.bpj.2013.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fenton AK, Kanna M, Woods RD, Aizawa SI, Sockett RE. Shadowing the actions of a predator: backlit fluorescent microscopy reveals synchronous nonbinary septation of predatory Bdellovibrio inside prey and exit through discrete bdelloplast pores. J Bacteriol. 2010;192:6329–6335. doi: 10.1128/JB.00914-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Araki Y, Ruby EG. A soluble enzyme activity that attaches free diaminopimelic acid to bdelloplast peptidoglycan. Biochemistry. 1988;27:2624–2629. doi: 10.1021/bi00407a053. [DOI] [PubMed] [Google Scholar]
- 32.Shilo M. Morphological and physiological aspects of the interaction of bdellovibrio with host bacteria. Curr Top Microbiol Immunol. 1969;50:174–204. doi: 10.1007/978-3-642-46169-9_6. [DOI] [PubMed] [Google Scholar]
- 33.Iida Y, et al. Roles of multiple flagellins in flagellar formation and flagellar growth post bdelloplast lysis in Bdellovibrio bacteriovorus. J Mol Biol. 2009;394:1011–1021. doi: 10.1016/j.jmb.2009.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Eksztejn M, Varon M. Elongation and cell division in Bdellovibrio bacteriovorus. Arch Microbiol. 1977;114:175–181. doi: 10.1007/BF00410781. [DOI] [PubMed] [Google Scholar]
- 35.Kuru E, et al. In situ probing of newly synthesized peptidoglycan in live bacteria with fluorescent D-amino acids. Angew Chem. 2012;51:12519–12523. doi: 10.1002/anie.201206749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lambert C, et al. Characterizing the flagellar filament and the role of motility in bacterial prey-penetration by Bdellovibrio bacteriovorus. Molec Microbiol. 2006;60:274–286. doi: 10.1111/j.1365-2958.2006.05081.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lambert C, Sockett R. Nucleases in Bdellovibrio bacteriovorus contribute towards efficient self-biofilm formation and eradication of pre-formed prey biofilms. FEMS Microbiol Lett. 2013;320:109–116. doi: 10.1111/1574-6968.12075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Magnet S, Dubost L, Marie A, Arthur M, Gutmann L. Identification of the L,D-transpeptidases for peptidoglycan cross-linking in Escherichia coli. J Bacteriol. 2008;190:4782–4785. doi: 10.1128/JB.00025-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Baba T, et al. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol Syst Biol. 2006;2(2006):0008. doi: 10.1038/msb4100050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Thomason LC, Sawitzke JA, Li X, Constantino N, Court DL. Recombineering: genetic engineering in bacteria using homologous recombination. Curr Protoc Mol Biol. 2014;14:1–39. doi: 10.1002/0471142727.mb0116s106. [DOI] [PubMed] [Google Scholar]
- 41.Datsenko KA, Wanner BL. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci USA. 2000;97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fenton AK, Lambert C, Wagstaff PC, Sockett RE. Manipulating each MreB of Bdellovibrio bacteriovorus gives diverse morphological and predatory phenotypes. J Bacteriol. 2010;192:1299–1311. doi: 10.1128/JB.01157-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ducret A, Quardokus EM, Brun YV. MicrobeJ, a tool for high throughput bacterial cell detection and quantitative analysis. Nat Microbiol. 2016;1:16077. doi: 10.1038/nmicrobiol.2016.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw data that support the findings of this study are available from the corresponding author upon request.