

Abstract
Adsorbate vibrational excitations are an important fingerprint of molecule/surface interactions, affecting temperature contributions to the free energy and impacting reaction rate and equilibrium constants. Furthermore, vibrational spectra aid in identifying species and adsorption sites present in experimental studies. Despite their importance, knowledge of how adsorbate frequencies scale across materials is lacking. Here, by combining previously reported experimental data and our own density-functional theory calculations, we reveal linear correlations between vibrational frequencies of adsorbates on transition metal surfaces. Through effective-medium theory, linear muffin-tin orbital theory, and the d-band model, we rationalize the squares of the frequencies to be fundamentally linear in their scaling across transition metal surfaces. We identify the adsorbate-binding energy as a descriptor for certain molecular vibrations and rigorously relate errors in frequencies to errors in adsorption energies. We also discuss the impact of scaling on surface thermochemistry and adsorbate coverage.
Vibrational excitations are a fingerprint of molecule–surface interactions, but knowing how they scale across materials is tricky. Here, the authors discover correlations between the vibrational frequencies of adsorbates on transition metals, developing a predictive theory to allow interpretation of complex experimental spectra.
Introduction
A breakthrough in computational catalysis was the introduction of linear scaling relations (LSRs), by Nørskov and co-workers1, which link the binding energy of a partially hydrogenated adsorbate AHX to the binding energy of its respective atomic adsorbate A (ΔE A) across transition metal surfaces,
| 1 |
where the slope, m E, depends on the valencies of A and AHX. LSRs provide a means to estimate thermodynamic properties of reaction intermediates on heterogeneous catalyst surfaces, enabling in silico catalyst screening2, 3. While LSRs can predict the scaling of binding energies, there is not yet a way to scale zero-point energies (ZPE) and temperature contributions to Gibbs free energy across surfaces. These temperature effects, which are composed of a molecule’s heat capacity and entropy, result entirely from vibrations for immobilized chemisorbed surface species4. As vibrations of adsorbates are often considered to be invariant with respect to the metal surface5, temperature effects and pre-exponentials are typically taken to be constant in microkinetic modeling of heterogeneously catalyzed systems6, 7.
There is evidence suggesting that frequencies should scale with electronic energy of the relevant chemical bonds. For example, both experimental gas-phase diatomic frequencies, and density functional theory (DFT)-determined force constants scale with bond dissociation energy8, 9. A DFT study of ammonia adsorbed on transition metals indicates that N–H vibrational stretching and bending frequencies experience slight trends with binding energy10. Furthermore, the C–O stretching vibration for chemisorbed carbon monoxide varies linearly with coverage below 300 K11. Coverage effects on frequencies can be separated into static and dynamic frequency shifts12–15. The former can be treated as a consequence of the metal’s d-band centre shift with coverage, similar to the effects on adsorption energy16, implying that frequencies should relate to energies, but the explicit dependence is lacking17. On a more fundamental level, the vibrational force constants can be linked analytically to the electron density18, and DFT is based on the knowledge that the electronic density completely specifies a systems energy19.
Here we develop vibrational scaling relations (VSRs) for CHX, NHX, and OHX species, as well as for CH2CH3, adsorbed on transition metal surfaces. Using effective-medium theory (EMT)20–24 and linear muffin-tin orbital (LMTO)21, 25, 26 theory, we derive an expression that fundamentally relates the slopes of the LSRs and the VSRs. On the basis of this theory, we successfully predict the VSRs from the corresponding LSRs, adsorbate geometry, and reduced mass on any single transition metal surface. We show the influence of the VSRs and vibrational frequencies on thermodynamic and kinetic model predictions and rationalize their accuracy.
Results
Vibrational scaling theory and connection to energy scaling
Here we provide the essential theory and equations for understanding vibrational scaling. A more detailed derivation with discussion is provided in Supplementary Note 1. According to tight-binding theory and the d-band model27, the adsorption energy (ΔE ads) can be separated into sp-band (ΔE sp) and d-band (ΔE d) contributions:
| 2 |
Furthermore, every bulk transition metal has its outer s-orbital half-filled, and the coupling of the adsorbate to the metal sp-band is constant across transition metals27. The slope of the LSR in Eq. (1) is then attributed to the coupling of the adsorbate’s s and p orbitals with the transition metal’s d-band1. Consequently, we can reformulate Eq. (1) as follows:
| 3 |
Within the harmonic approximation, the frequency (v) of a normal mode is given according to
| 4 |
with reduced mass (μ) and force constant (k), where k is the second derivative of the potential energy surface (PES) at equilibrium with respect to the direction of a normal mode displacement.
Due to the additive nature of adsorption energy according to Eq. (2), k can be taken as a sum of contributions from adsorbate coupling to the metal sp-band (k sp) and d-band (k d):
| 5 |
From EMT, ΔE sp is exponential with respect to separation between the adsorbate and metal surface. We show in Supplementary Fig. 1 that a Morse potential adequately describes this interaction. The second derivative of any sum of exponential functions results in k sp being proportional to an adsorbate-dependent constant (η) and the sp contribution to the adsorbate-binding energy at equilibrium (ΔE sp).
According to Anderson’s LMTO theory, the d-band contribution to binding energy (ΔE d) is proportional to adsorbate- and metal-dependent constants M A and M M, and is related to the distance (r) between the nuclei of the adsorbate and the nearest metal atoms. Formulating Anderson’s expression to account for the existence of both adsorbate s and p states results in the following:
| 6 |
Here, f s and f p represent the fractional contributions of the adsorbate’s s and p orbitals to the adsorbate-metal bond, respectively, such that f s + f p = 1 and
| 7 |
Constants n s and n p depend on the angular momenta of the adsorbate (l a) and metal d orbitals (l d)21, 25. Inserting second derivatives of ΔE sp and ΔE d into Eq. (4) allows Eq. (8) to describe any frequency driven by the adsorbate-metal interaction:
| 8 |
where μ is the reduced mass of the corresponding normal mode, g(θ) relates the displacement of the normal mode to a change in r, and R n is expressed in terms of previously defined quantities:
| 9 |
We note that η is negative and R n is positive. The importance of their signs will be discussed later.
In this work, f s and f p were estimated from the total number of adsorbate’s s and p electrons in the atom bound to the metal. Thus, for all OHX, NHX, and CHX species, f s was equal to 1/3, 2/5, and 1/2, respectively, resulting in the following equality:
| 10 |
A projected density of states analysis suggests that 2/5 may be a more appropriate value for CHX species. We also find that the value of f s does not affect the predicted scaling significantly when Eq. (10) holds (Supplementary Fig. 2). Here we shall address the case where g(α) = 1, which is strictly true only for the perpendicular frequency (ν ⊥) at an atop site.
By representing ν AH X with Eq. (8), we can find its scaling with respect to ν A. First, because is described according to Eq. (3), we can replace in 8 with its functional dependence on ν A. Then a series of substitutions and taking the derivative of ν AH X with respect to ν A results in
| 11 |
where m E is the LSR slope, μ is assumed to be the total mass of the adsorbate, and R n is determined from Eq. (9). For AHX species, the light mass of hydrogen allows the reduced mass of the species to be approximated as the total mass.
Upon substitution of Eq. (8) into Eq. (11) and subsequent simplifications, as outlined in Supplementary Note 1, we get the expression for the first derivative of (ν AH X)2 with respect to (ν A)2:
| 12 |
where the terms in R n can be determined from the equilibrium geometry and electronic structure of an adsorbate on a reference metal. In this work, the reference metal is taken to be Pt. Upon integration, we obtain a linear scaling relationship between the squared frequencies of AHX and A:
| 13 |
with slope (m v) given by Eq. (12), such that . The intercept (b v) is derived in Supplementary Note 2 by setting k d = −k sp and, like the LSR intercept1, is independent of the d-band contribution to binding energy:
| 14 |
The slope and intercept are specific to any A/AHX pair of adsorbates. We call the scaling relations of Eq. (13) vibrational scaling relationships (VSRs).
These VSRs should hold for any vibrational mode primarily driven by interaction of the adsorbate with the metal. We test the predictive power of the derived relations in the following section by calculating DFT normal mode frequencies for AHX adsorbates and investigating trends across transition metals.
Scaling of normal mode vibrations calculated using DFT
In Fig. 1, we report DFT-computed frequencies of adsorbed species based on the harmonic approximation with normal mode analysis. The data support the existence of linear correlations between squared vibrational frequencies of AHX and atomic A species across transition metal surfaces, for A=C, N, and O, and X = 1 up to one less than the valency of the atomic species. Due to the relatively narrow range of frequencies, the error in predicting frequencies is not significantly higher when a linear model is used. The average root mean square error in predicting frequencies of these models is 22 ± 5 cm−1 (square model) and 23 ± 5 cm−1 (linear model), at the atop site for all facets. The standard errors in the regressed VSR slopes for O/OH, NH/NH2, and CH2/CH3, all with valency ratios of 1/2, are 0.062, 0.047, and 0.051, respectively, on the (100) surface when the scaling is with respect to frequency squared and 0.089, 0.055, and 0.067, respectively, for a linear fit. While a linear model fits the data reasonably well, the squared relation arises naturally from theory and, as discussed later, can be used to predict the VSR slopes over a wide range of values.
Fig. 1.

Scaling of the ν ⊥ modes for AHX adsorbates with their atomic counterparts from DFT. Species are a CHX, b NHX, and c OHX. Adsorption is on the atop site of (100) transition metal surfaces. For a and b, respectively: green triangles = CH and NH, blue circles = CH2 and NH2, and red squares = CH3. For OHX species c, ν ⊥ frequencies on the (110) and (111) facets are also included and green triangles = (100), blue circles = (111) and red squares = (100). The slopes of the regressed lines in a are 0.14, 0.42, and 0.86 for the red, blue, and green lines, respectively. In b, the slopes of the regressed lines are 0.26 and 0.64 for the blue and green lines, respectively. In c the slopes of the regressed lines are 0.43, 0.42, and 0.48 for the red, blue and green lines, respectively. All intercepts are positive
Comparison of ν⊥ (Fig. 1) and ν|| (Supplementary Note 3 and Supplementary Figs. 3–5) AHX frequency trends reveal universal linear scaling behaviour, as frequencies of species adsorbed on top sites, three and four-fold hollow sites, and bridge sites all correlate. Interestingly, the VSR intercepts are positive, as opposed to negative for their respective LSRs. A negative intercept for the LSR is a result of its slope being less than one, as the adsorption energy on pure transition metals is more negative for atomic adsorbates than their hydrogenated counterparts. The positive intercept for the VSR is then expected from Eq. (14), as m E < 1 for all A/AHX pairs, and is usually less than one as well. As already mentioned, for the AHX adsorbates considered here, the fraction of the adsorbate s and p orbitals interacting with the metal d-band was taken to be constant. Thus, R n is primarily a function of the distance between the adsorbate and the metal atom. These distances are provided in Supplementary Table 1.
In Fig. 2, the theoretical slopes of the VSRs predicted by Eq. (12) are compared to those found by fitting the DFT-calculated normal mode frequencies shown in Fig. 1, according to Eq. (13). When the LSR and VSR fittings exhibit small standard errors, such as for CHX species, the slopes match well. To explain deviations in O/OH, we note that the value of / is roughly constant across metals, so that the uncertainty in the O/OH VSR is primarily associated with the variance in its LSR (Supplementary Table 1). For comparison, LSR slopes for O/OH, NH/NH2, and CH2/CH3, all with valency ratios of 1/2, have standard errors of 0.12, 0.04, and 0.05, respectively, on the (100) surface at the atop site. The standard errors for other NHX and CHX LSR slopes are about 0.05. Our model successfully predicts VSR slopes that range from 0.14 (C/CH3) to 0.86 (C/CH).
Fig. 2.

Predicted VSR slopes for AHX species. Values are predicted based on Eq. (12). Labels indicate adsorbate A/B for CHX (blue), NHX (green), and OHX (red). The black line indicates parity. The fitted slopes to DFT data are from the data in Fig. 1 and fit according to Eq. (13). Included are the perpendicular frequencies of the adsorbates on the atop sites of the (100) (circles), (110) (triangles), and (111) (squares) facets
Our model is based on an assumption that f s and f p are invariant within the same class of AHX species. While this is generally true28, a projected density of states analysis (Supplementary Table 2) onto atom A shows that the fraction of electron density attributed to p-electrons monotonically increases by a small amount with increasing X for all adsorbates. Such observations have already been reported for CHX species on Pd29. If the p character of the adsorbate-bonding orbital increases in a similar manner, this would explain the tendency of our model to slightly under-predict the VSR slope between two species AHX1/AHX2 when X 2 > X 1.
The results for frequency scaling in this work are valid for normal modes primarily driven by interaction of the adsorbate with the metal surface atoms, such as metal-adsorbate ν ⊥ and ν || stretches, as well as δ⊥ rotations. For molecular adsorbates, the ν || mode is often dependent on surface geometry. See Supplementary Note 3 and Supplementary Figs. 3–5 for scaling of ν || frequencies and the section on extension to complex molecules for δ⊥ rotations.
A/B vibrational scaling
To this point, we have only covered scaling of A/AHX species. Calle-Vellejo et al. showed that linear scaling holds for binding energies of AHX and BHX, where A and B have the same number of valence electrons, such as for CHX and SiHX species30. They also showed that for the adsorption energy of O (ΔE O) and N (ΔE N) on bimetallic alloys of Pt, a general scaling relation exists:
| 15 |
In Eq. (15), γ is a constant with respect to E N, and g is a function of the number of valence electrons ( and ) in the transition metals. It is straightforward to show that if is small and is constant, the squares of the frequencies should scale with a slope that is approximately equal to γ. The DFT data plotted in Fig. 3 and the experimental data in Fig. 4 highlight that frequencies between species A/B do indeed scale, and that this scaling is universal across various sites and facets, as all frequencies lie on the same line.
Fig. 3.
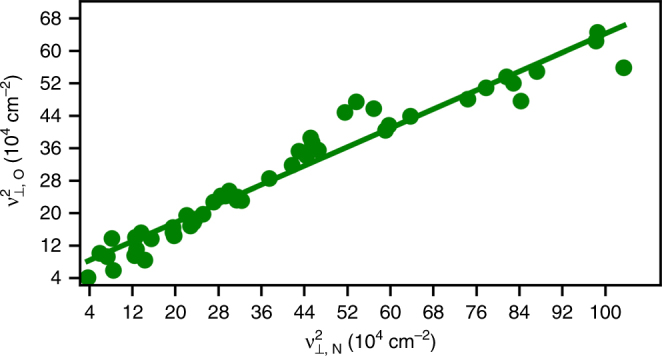
DFT ν ⊥ normal mode frequencies for atomic O vs. N. The data include hollow, bridge, and atop site adsorption on the (111), (110), and (100) facets of Ag, Au, Cu, Ir, Pd, Pt, and Rh. The slope of the regressed line is 0.58 and the intercept is 59,800 cm−2
Fig. 4.

Experimental ν ⊥ frequencies for atomic O vs. N. Label Cu(100)* indicates ordered adsorbate layers at high coverage. Atomic O adsorption is typically at low coverage. Atomic N was often co-adsorbed with other species. The slope of the regressed line is 1.37 and the intercept is −64,500 cm−2
Figure 3 shows perpendicular frequency scaling of atomic N vs. atomic O on several sites and facets of fcc metals from DFT. The best fit value of γ used in scaling adsorption energy on Pt-bimetallic alloys was 0.6730. Our value of 0.58 for the VSR slope indicates that either is not zero or is not constant, but the minor deviation from 0.67 suggests such assumptions are a good approximation. Accounting for g(θ), as we do in Supplementary Note 1 for AHX frequencies at hollow and bridge sites, results in a slope of 0.63.
In Fig. 4, we plot previously reported low-coverage experimental vibrational frequencies of atomic O, and N on ideal metal surfaces, obtained by High-Resolution electron Energy Loss Spectroscopy (HREEL) at temperatures between 30 and 500 K31–49. Figure 4 is not expected to match Fig. 3 for several reasons: (1) the infinite metal mass approximation in DFT calculations, (2) the possibility of different experimental binding sites for atomic O and N, and (3) presence of co-adsorbed species during experimental measurement of atomic N frequencies.
The discovered universality of vibrational scaling for chemically different adsorbates and metals evidently stems from the universal electronic structure description of bonding on transition metal surfaces26, 50. Therefore, we expect similar correlations should exist between vibrational frequencies of more complex species as well.
Extension to more complex molecules
To determine whether the VSR theory is applicable to large molecules, we calculated frequencies for CH2CH3 adsorbed on atop sites. Figure 5 indicates that ν⊥(Metal-CH2CH3) frequencies correlate with the corresponding frequencies of CH3 at the atop site. While all atoms in CH2CH3 are displaced in this normal mode, the frequency is primarily attributed to the stretching of the metal-carbon bond. Because the reduced mass can only be calculated exactly for a two-body system, the theoretical scaling relation for the AHX species, Eq. (12), provides bounds. Generally, an adsorbate’s reduced mass for a ν ⊥ mode will lie between the mass of the adsorbate atom nearest to the surface and the total mass of the adsorbed molecule. The predicted frequency slope for ν ⊥ scaling of CH2CH3 vs. CH3 is between 0.57 and 1.10, when the reduced mass is set to the total mass of the adsorbate or the mass of the bonding carbon atom, respectively. The actual slope is between 0.82 and 0.90 depending on facet. Furthermore, while scaling on more complex surfaces is expected to exist51, geometric effects on the reduced mass that are surface dependent are likely to be important.
Fig. 5.
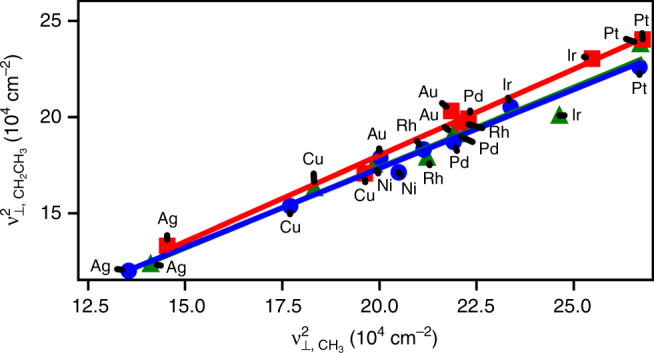
Scaling of ν ⊥ for CH2CH3 vs. CH3. The data is calculated from DFT at the atop site. The data include vibrations on (100) (green triangles), (111) (blue circles), and (110) (red squares) transition metal surfaces. The slopes of the regressed lines are 0.90, 0.82, and 0.83 for the red, blue and green lines, respectively
Scaling of frequencies with adsorption energy
The correlation between frequencies and adsorption energies is fairly complex, due to the force constant involving contributions from both the metal sp-band and d-band (ηΔE sp and ΔE d R n, respectively) according to Eq. (8). As the magnitude of ΔE d R n increases relative to ηΔE sp, the scaling takes a square root dependence on energy. In contrast, when ηΔE sp is dominant, the frequency becomes independent of adsorption energy. The order of the scaling therefore lies between 0 and 1/2. Substituting Eq. (2) into Eq. (8) results in the following frequency dependence on adsorption energy,
| 16 |
Because R n is positive and approximately constant (see Supplementary Note 1), and frequencies increase with increasing binding energy, η must increase in magnitude as binding energy increases. Allowing η to be a linear function of ΔE d gives the following empirical relation for the dependence of frequency on adsorption energy:
| 17 |
where α and β are constants.
Figure 6 depicts scaling of ν ⊥ DFT-calculated frequencies of CHX species, for X = 0 to 3, on fcc hollow (green squares) and atop (blue circles) sites for close-packed surfaces of fcc metals against the electronic adsorption energies. Scaling of ν ⊥ with adsorption energy for OHX and NHX, as well as ν || for CHX, NHX, and OHX, are shown in Supplementary Note 4 and Supplementary Figs. 6 and 7. The data reveal that (1) adsorbates with larger frequency and adsorption energy ranges have less variance, and (2) smaller equilibrium distances between the adsorbate and metal atoms, result in greater d-band contributions (ΔE d R n) to frequency scaling. These characteristics are expected from Eqs. (8), (9) and (16). Frequencies, which can be measured spectroscopically, provide a means of estimating and comparing adsorption energies at different sites and on different surfaces.
Fig. 6.

Scaling of frequencies with adsorption energy. Frequencies are for the ν ⊥ mode and calculated from DFT for CHX species, where X ≥ 0. Included are modes and adsorption energies at atop sites (blue circles) and fcc hollow sites (green squares). Fits to Eq. (17) are shown by solid blue and dashed green lines, respectively. The slopes of the regressed lines are −192,000 and −63,100 for the blue and green lines, respectively. The intercepts are also both negative
Interpreting complex experimental vibrational spectra
Based on selection rules and our own scaling studies of experimental O2 and O frequencies in the Supporting Information, we identify frequencies associated with molecular oxygen in the range of 500 and 800 cm−1 as frustrated rotations (δ ⊥). Scaling frequencies provide additional information to single metal frequency studies. Conclusions drawn from previous experimental studies on a single metal are discussed in Supplementary Note 5. Because the δ ⊥(M–O2) motion is primarily perpendicular to the surface, its corresponding HREELS peak should be much more intense than any ν(O–O) stretch where the intra-oxygen bond is parallel to the surface52. Partially because of these selection rules, we suggest frequencies identified experimentally between 600 and 1000 cm−1 are a combination of O–O stretches at the bridge site and metal-oxide stretches on stronger binding metals. The frequency of both of these experimentally observed modes increases as the atomic oxygen frequency increases, as seen in Supplementary Fig. 8, which is consistent with modes driven by interaction of the oxygen species with the surface, not a pure O–O stretch.
To support the mode assignments based on scaling of literature experimental frequencies, we performed our own DFT studies for molecular oxygen and atomic oxygen on the (111) and (100) facets of transition metal surfaces at their most stable site (Fig. 7) As expected, the δ ⊥(M–O2) mode (Fig. 7; green triangles) correlates positively with the ν ⊥(M–O) frequency. While the slopes determined from experimental data in Supplementary Fig. 8 are slightly lower than from DFT, the differences are well within uncertainty, lack of precise knowledge regarding the experimental adsorption site, and the infinite metal mass approximation. Of significance is that the experimental and computational 500–800 cm−1 frequencies scale positively with the ν ⊥(M–O) frequency.
Fig. 7.

Scaling of O2 frequencies vs. atomic O frequencies calculated from DFT. Modes shown include the ν ⊥ stretch mode of O2 (blue circles), δ ⊥ rotation of O2 (green triangles), and the O–O stretch (red squares). Frequencies of atomic oxygen are for the ν ⊥ mode. DFT calculations are for adsorption on (100) and (111) fcc transition metal surfaces. Atomic O is at the most stable site in each case and O2 is at the bridge site parallel to the surface. Only the points corresponding to the ν ⊥(M–O2) modes are labeled, as all DFT frequencies are for these metals. The slopes of the regressed lines are −2.19, 0.87, and 0.63 for the red, green, and blue lines, respectively. The intercepts are all positive
Our DFT-calculated ν(O–O) stretch (Fig. 7; red squares) shows a negative slope when scaled with the ν ⊥(M–O) stretch, in direct contrast to the experimentally observed modes shown in Supplementary Fig. 8. As the DFT-calculated ν(O–O) frequency matches experiment for O2 on some metals, such as Pt(111), the deviations are most likely due to the presence of oxides in experiment on stronger binding metals52. Bond-order conservation53 rules and back donation of metal’s d-electrons to antibonding O2 orbitals54 both suggest that the ν(O–O) stretch should decrease with increasing ν ⊥(M–O2) stretch8, 27. Thus, our DFT calculations and scaling experimental data support that the 500–800 cm−1 frequencies belong to frustrated rotations and that 600–1000 cm−1 frequencies observed experimentally are not purely due to an O–O stretching mode.
Vibrational scaling relations can also provide quantitative insight from complex surface spectra where the exact surface structure is unknown, making it difficult or impossible to replicate the spectra from DFT alone. Already, machine learning has turned FTIR and Raman spectroscopy into quantitative tools for detecting bulk concentrations and distinguishing between different strains of bacteria55. As an example from heterogeneous catalysis, previous DFT and experimental studies have shown that Ni-Pt-Pt (Ni on top of subsurface Pt, followed by core Pt) is very active for ammonia decomposition3. Experimental HREELS studies assigned the 494 cm−1 peak to both surface N and NH2 by comparing to known peaks on single crystal monometallics. Our scaling relations in Fig. 1b reveals that if the 494 cm−1 peak is assigned to chemisorbed atomic N, the frequency of NH2 should be 424 cm−1 and that of NH should be 469 cm−1. These NHX scaling relations suggest that the 494 cm−1 peak should be attributed to a combination of N and NH whereas the 440 cm−1 peak cited in the paper should include contributions from NH2, in addition to the background CO. Given the vibrational peak of atomic N, scaling relations developed in this work can be used to determine the concentration of each species on the surface through spectral deconvolution56.
Finally, in combination with machine learning, the scalings developed herein may be valuable to characterize the surfaces of complex materials, like those of bimetallics. Such surfaces are non-ideal. Rather, they contain different terminations, e.g., patches of Ni on Pt adjacent to Pt patches of varying size, steps, and single point defects. We envision that the proposed scaling relations could, in conjunction with machine learning, be used to unravel atomistic structural detail from the spectra to complement imaging techniques whose operando use is still under development. Future work will be needed to fully explore this opportunity.
Effect of VSRs on predicted thermochemistry
VSRs can be used to discern whether scaling vibrational contributions to Gibbs free energy (Gvib) is important. In Supplementary Note 6 and Supplementary Figs. 9 and 10 we show that, for the range of normal modes found for chemisorbed CO, Gvib can be approximated with a first order Taylor expansion of the nonlinear terms resulting in a linear contribution to Gvib from individual vibrational modes. Because different normal modes within the same molecule scale both with each other and with adsorption energy, Gvib often correlates linearly with adsorption energy. For certain molecules, such as for CO, scaling of Gvib is significant. For example, at 400 K, Gvib for CO on Pd is over 0.20 eV higher than on Ag, impacting microkinetic model predictions by over three orders of magnitude.
In Fig. 8 we report errors in carbon monoxide surface coverage for competitive adsorption of CO and O2, caused by neglect of Gvib scaling. The error in coverage is plotted for different binding energies of CO and O, as well as the positions with respect to the close-packed facet for Ir, Ni, and Rh on the binding energy map. The region where both CO and O2 are co-adsorbed in appreciable amounts is also the region where neglecting vibrational scaling effects produces the greatest error. This region is important because it is the catalytically active region for forming CO2 from CO and O2 57. While coverage effects have not been included, the effects of scaling are expected to hold in the presence of lateral effects at higher coverages58. Clearly, accounting for Gvib scaling with frequencies is necessary to predict accurate coverages.
Fig. 8.
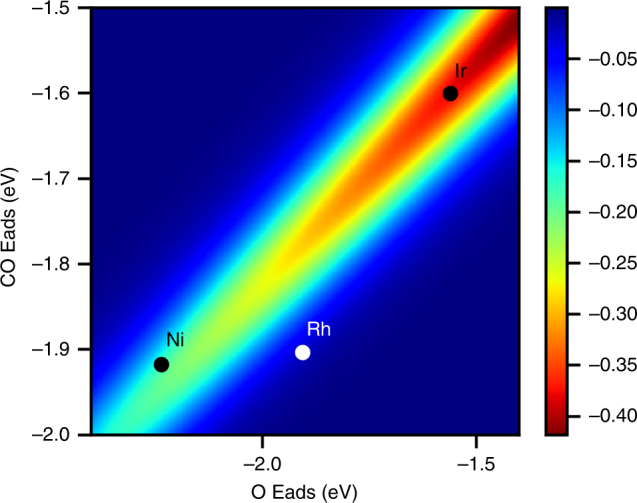
Error in predicted CO coverage. Error is without scaling of Gvib at 400 K, 1 bar and 100 ppm CO in O2 based on vibrational contributions for CO and O on Pd(111) at the fcc hollow site. x and y axes correspond to electronic adsorption energy calculated using the RPBE functional with D3 dispersion corrections. The color indicates the error in coverage, with the highest possible coverage of 1. Lack of vibrational scaling lowers the CO coverage
Discussion
Surfaces contain an ensemble of chemical conformations, adsorption sites, and reaction pathways. Fortunately, vibrational spectra are mode specific and can achieve precision to within 1 cm−1. This precision often corresponds to less than 0.1 and 1% uncertainties for the gas and surface species, respectively54. Experimentally, frequencies can shift by a few percent over large temperature changes59. These shifts, however, result from changes in surface structure60 or co-adoption of other species. Parameter fitting to experimental gas-phase vibrational frequencies, rather than strictly using energies, has already improved DFT functional performance for predicting geometries beyond those achieved by CCSD(T) calculations61. While our calculated adsorbate frequencies were usually accurate to within 5%, the ν ⊥ frequencies of adsorbed atomic nitrogen on Ni(111)42 and Cu(111)38 were higher than experiment by 15%. Spectroscopic studies on these surfaces therefore warrant more attention. Still, uncertainties in binding energies are often much larger than this62.
Equation (8) implies that the relative error in ν is proportional to the square root of the sum of errors for ηΔE sp and R nΔE d. From standard error propagation, this is equivalent to half the relative error. Furthermore, if errors in ΔE sp and ΔE d are in the same direction (e.g., due to DFT-inherent overbinding)63, their contribution to the error in frequency partially cancel, since η is negative and R n is positive, where R n is given in Eq. (9). As a result, the error in frequency can be small, even when the error in adsorption energy is large. This cancellation of errors explains why DFT is usually able to predict vibrational frequencies accurately, despite errors in binding energies. A schematic depicting the competing nature of the sp-band and d-band contributions to binding energy, based on atomic oxygen on Pd(111)64, is shown in Fig. 9. The contribution from the d-band (ΔE d) and sp-band (ΔE sp) to adsorption energy (ΔE ads) is given in Eq. (2). The dependence of ΔE d and ΔE sp on distance were determined from LMTO and EMT, respectively.
Fig. 9.

Interaction energy of metal orbitals with an adsorbate. The contributions from the d-band (ΔE d; red) and sp-band (ΔE sp; blue) combine to make the total adsorption energy (ΔE ads; green). The illustration is representative of interaction energies for atomic oxygen with the Pd(111) surface
Insights into the errors for vibrations driven by the adsorbate-metal interaction are important for several reasons. First, DFT frequencies can reliably be compared to experiment in order to determine binding sites. Not only are frequencies more accurate than adsorption energies, but they are sensitive to binding site as well. Second, the contribution to Gibbs energy from frequencies that scale across transition metals, as shown in the previous section, can confidently be included in microkinetic models. Accurate free energy scaling and microkinetic modeling calculations are essential in rapid screening of novel materials for catalyst discovery.
Both experimental data from literature and new DFT calculations reveal that vibrational frequencies of adsorbates scale both with each other and with adsorption energy on transition metal surfaces. Using effective-medium theory and linear muffin-tin orbital theory for adsorbate coupling to the transition metal sp-band and d-band, respectively, we derived vibrational scaling relations for modes driven by interaction between the adsorbate and metal surface. These VSRs result in correlations between the vibrational Gibbs energy of a species with the electronic adsorption energy, making them straightforward to include in kinetic and thermodynamic models and necessary for accurate thermochemistry predictions. We also showed that the competing nature of the sp-band and d-band can result in cancellation of errors, allowing accurate frequencies irrespective of the errors in adsorption energies. VSR-based contributions to thermochemistry should prove especially important for large molecules that contain a greater number of vibrational modes driven by interaction between the adsorbate and metal surface.
Methods
DFT calculations
We calculated binding energies and vibrational frequencies using the Vienna ab initio Simulation Package (VASP) version 5.4 with the projector augmented wave method (PAWs)65. We employ the RPBE density functional66 with D3 dispersion corrections67. Simulation methods were similar to those of our previous study68, including use of spin-polarized calculations for gas-phase species and ferromagnetic metals, a 3 × 3 × 1 Monkhorst-Pack k-point sampling grid for all slab calculations69, and a 400 eV plane-wave cutoff. The initial magnetic moments for each atom in spin-polarized calculations were set to VASP default values.
Unit cell setup
For gas calculations, the supercell size was 10 × 10 × 10 Å. A Brillouin zone was sampled at the gamma point; a 0.005 eV/Å force cut-off was used in geometry optimizations. For slab calculations, the force cut-off was set to 0.02 eV/Å with 20 Å of vacuum space. The periodic cell consisted of four layers with 16 metal atoms in each layer; the bottom two layers were fixed at their bulk values, determined using a 15 × 15 × 15 k-point grid using the tetrahedron method with Blöchl corrections. Bulk metal lattice constants were pre-optimized with DFT using the Birch–Murnaghan equation of state70. All input files were created using the Atomistic Simulation Environment (ASE).
Adsorption sites and reference
We study adsorption at the fcc hollow, bridge, and atop sites of (100), (110), and (111) transition metal facets. The adsorption energy is calculated according to
| 18 |
where the adsorption energy (ΔE ads) is determined from the energies of the combined adsorbate-slab system (Energy(slab/adsorbate)), the slab (E slab), and the gaseous adsorbate (E gas).
Perpendicular (ν ⊥) and parallel (ν ||) vibrations (see Supplementary Fig. 12) are identified based on the criteria developed in the Supplementary Methods.
Data availability
The authors declare that all data supporting the findings of this study are available within the Article and its Supplementary Information files. An excel file is also provided that contains frequencies, energies, and other values calculated from DFT used in generating the figures contained in this work. Any other data will be provided by the corresponding author upon request.
Electronic supplementary material
Description of Additional Supplementary Files
Acknowledgements
Financial support from the Defense Advanced Research Project Agency under grant W911NF-15-2-0122 for Lansford and Vlachos is gratefully acknowledged. Support for Mironenko was from the Catalysis Center for Energy Innovation, an Energy Frontier Research Center funded by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Award no. DE-SC0001004. We also appreciate helpful discussions with Dr. Glen Jenness.
Author contributions
Lansford performed all DFT calculations, experimental data collection, and derivations. Mironenko advised on theory. Vlachos contributed the idea. All authors contributed to writing.
Competing interests
The authors declare no competing financial interests.
Footnotes
Electronic supplementary material
Supplementary Information accompanies this paper at doi:10.1038/s41467-017-01983-6.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Abild-Pedersen F, et al. Scaling properties of adsorption energies for hydrogen-containing molecules on transition-metal surfaces. Phys. Rev. Lett. 2007;99:016105. doi: 10.1103/PhysRevLett.99.016105. [DOI] [PubMed] [Google Scholar]
- 2.Andersson MP, et al. Toward computational screening in heterogeneous catalysis: Pareto-optimal methanation catalysts. J. Catal. 2006;239:501–506. doi: 10.1016/j.jcat.2006.02.016. [DOI] [Google Scholar]
- 3.Hansgen DA, Vlachos DG, Chen JG. Using first principles to predict bimetallic catalysts for the ammonia decomposition reaction. Nat. Chem. 2010;2:484–489. doi: 10.1038/nchem.626. [DOI] [PubMed] [Google Scholar]
- 4.Vannice MA, Hyun SH, Kalpakci B, Liauh WC. Entropies of adsorption in heterogeneous catalytic reactions. J. Catal. 1979;56:358–362. doi: 10.1016/0021-9517(79)90128-3. [DOI] [Google Scholar]
- 5.Rankin RB, Greeley J. Trends in selective hydrogen peroxide production on transition metal surfaces from first principles. ACS Catal. 2012;2:2664–2672. doi: 10.1021/cs3003337. [DOI] [Google Scholar]
- 6.Chen D, Lødeng R, Svendsen H, Holmen A. Hierarchical multiscale modeling of methane steam reforming reactions. Ind. Eng. Chem. Res. 2011;50:2600–2612. doi: 10.1021/ie1006504. [DOI] [Google Scholar]
- 7.Calle-Vallejo F, et al. Finding optimal surface sites on heterogeneous catalysts by counting nearest neighbors. Science. 2015;350:185–189. doi: 10.1126/science.aab3501. [DOI] [PubMed] [Google Scholar]
- 8.Ibach H, Hopster H, Sexton B. Analysis of surface reactions by spectroscopy of surface vibrations. Appl. Surf. Sci. 1977;1:1–24. doi: 10.1016/0378-5963(77)90003-4. [DOI] [Google Scholar]
- 9.Kraka E, Cremer D. Characterization of CF Bonds with multiple-bond character: bond lengths, stretching force constants, and bond dissociation energies. ChemPhysChem. 2009;10:686–698. doi: 10.1002/cphc.200800699. [DOI] [PubMed] [Google Scholar]
- 10.Sälli E, Martiskainen S, Halonen L. Computational study of the vibrational structure of the ammonia molecule adsorbed on the fcc (111) transition metal surfaces. J. Phys. Chem. C. 2012;116:14960–14969. doi: 10.1021/jp303955z. [DOI] [Google Scholar]
- 11.Ortega A, Huffman FM, Bradshaw AM. The adsorption of CO on Pd(100) studied by IR reflection absorption spectroscopy. Surf. Sci. 1982;119:79–94. doi: 10.1016/0039-6028(82)90189-3. [DOI] [Google Scholar]
- 12.Woodruff DP, Hayden BE, Prince K, Bradshaw AM. Dipole coupling and chemical shifts in IRAS of CO adsorbed on Cu(110) Surf. Sci. 1982;123:397–412. doi: 10.1016/0039-6028(82)90336-3. [DOI] [Google Scholar]
- 13.Bertolo M, Jacobi K. NO adsorption on Pd(111) in the temperature range between 20 and 300 K. Surf. Sci. 1990;226:207–220. doi: 10.1016/0039-6028(90)90486-R. [DOI] [Google Scholar]
- 14.Efrima S, Metiu H. The role of the electrostatic interaction in shifting the vibrational frequencies for two adsorbed molecules. Surf. Sci. 1981;109:109–126. doi: 10.1016/0039-6028(81)90515-X. [DOI] [Google Scholar]
- 15.Ueba H. Chemical effects on vibrational properties of adsorbed molecules on metal surfaces: Coverage dependence. Surf. Sci. 1987;188:421–455. doi: 10.1016/S0039-6028(87)80198-X. [DOI] [Google Scholar]
- 16.Hammer B. Coverage dependence of N2 dissociation at an N, O, or H precovered Ru(0001) surface investigated with density functional theory. Phys. Rev. B. 2001;63:205423. doi: 10.1103/PhysRevB.63.205423. [DOI] [Google Scholar]
- 17.Loffreda D, Simon D, Sautet P. Dependence of stretching frequency on surface coverage and adsorbate–adsorbate interactions: a density-functional theory approach of CO on Pd (111) Surf. Sci. 1999;425:68–80. doi: 10.1016/S0039-6028(99)00186-7. [DOI] [Google Scholar]
- 18.Anderson AB, Parr RG. Vibrational force constants from electron densities. J. Chem. Phys. 1970;53:3375–3376. doi: 10.1063/1.1674492. [DOI] [Google Scholar]
- 19.Kohn W. Nobel lecture: electronic structure of matter-wave functions and density functionals. Rev. Modern Phys. 1999;71:1253–1266. doi: 10.1103/RevModPhys.71.1253. [DOI] [Google Scholar]
- 20.Jacobsen KW, Norskov JK, Puska MJ. Interatomic interactions in the effective-medium theory. Phys. Rev. B. 1987;35:7423–7442. doi: 10.1103/PhysRevB.35.7423. [DOI] [PubMed] [Google Scholar]
- 21.Norskov JK. Effective medium potentials for molecule–surface interactions: H2 on Cu and Ni surfaces. J. Chem. Phys. 1989;90:7461–7471. doi: 10.1063/1.456679. [DOI] [Google Scholar]
- 22.Smith JR, Ying SC, Kohn W. Charge densities and binding energies in hydrogen chemisorption. Phys. Rev. Lett. 1973;30:610–613. doi: 10.1103/PhysRevLett.30.610. [DOI] [Google Scholar]
- 23.Nørskov JK, Lang ND. Effective-medium theory of chemical binding: Application to chemisorption. Phys. Rev. B. 1980;21:2131–2136. doi: 10.1103/PhysRevB.21.2131. [DOI] [Google Scholar]
- 24.Esbjerg N, Nørskov JK. Dependence of the He-scattering potential at surfaces on the surface-electron-density profile. Phys. Rev. Lett. 1980;45:807–810. doi: 10.1103/PhysRevLett.45.807. [DOI] [Google Scholar]
- 25.Andersen OK, Jepsen O. Explicit, first-principles tight-binding theory. Phys. Rev. Lett. 1984;53:2571–2574. doi: 10.1103/PhysRevLett.53.2571. [DOI] [Google Scholar]
- 26.Hammer B, Nørskov JK. Electronic factors determining the reactivity of metal surfaces. Surf. Sci. 1995;343:211–220. doi: 10.1016/0039-6028(96)80007-0. [DOI] [Google Scholar]
- 27.Hammer B, Norskov JK. Theoretical surface science and catalysis—calculations and concepts. Adv. Catal. 2000;45:71–129. [Google Scholar]
- 28.Bent HA. An appraisal of valence-bond structures and hybridization in compounds of the first-row elements. Chem. Rev. 1961;61:275–311. doi: 10.1021/cr60211a005. [DOI] [Google Scholar]
- 29.Paul JF, Sautet P. Chemisorption and transformation of CHx fragments (x = 0−3) on a Pd(111) surface: a periodic density functional study. J. Phys. Chem. B. 1998;102:1578–1585. doi: 10.1021/jp9733227. [DOI] [Google Scholar]
- 30.Calle-Vallejo F, Martínez JI, García-Lastra JM, Rossmeisl J, Koper MTM. Physical and chemical nature of the scaling relations between adsorption energies of atoms on metal surfaces. Phys. Rev. Lett. 2012;108:116103. doi: 10.1103/PhysRevLett.108.116103. [DOI] [PubMed] [Google Scholar]
- 31.de Mongeot FB, Valbusa U, Rocca M. Oxygen adsorption on Ag(111) Surf. Sci. 1995;339:291–296. doi: 10.1016/0039-6028(95)00594-3. [DOI] [Google Scholar]
- 32.So SK, Franchy R, Ho W. The adsorption and reactions of NO on Ag(111) at 80 K. J. Chem. Phys. 1989;91:5701. doi: 10.1063/1.457524. [DOI] [Google Scholar]
- 33.Baca AG, Klebanoff LE, Schulz MA, Paparazzo E, Shirley DA. Dissociative adsorption of CO and O2 on Cr(100, Cr(110), and Cr(111) in the temperature range 300–1175 K. Surf. Sci. 1986;173:215–233. doi: 10.1016/0039-6028(86)90117-2. [DOI] [Google Scholar]
- 34.Fukuda Y, Masayasu N. Molecular nitrogen with extremely low N-N stretching frequency on Cr(111) studied by HREELS and XPS. Surf. Sci. 1988;203:L651–L655. doi: 10.1016/0039-6028(88)90185-9. [DOI] [Google Scholar]
- 35.Baddorf AP, Wendelken JF. High coverages of oxygenon Cu(110) investigated with XPS, LEED, and HREELS. Surf. Sci. 1991;256:264–271. doi: 10.1016/0039-6028(91)90869-T. [DOI] [Google Scholar]
- 36.Asfin B, Davies PR, Pashusky A, Roberts MW, Vincent D. Reaction pathways in the oxydehydrogenation of ammonia. Surf. Sci. 1993;284:109–120. doi: 10.1016/0039-6028(93)90529-S. [DOI] [Google Scholar]
- 37.Sueyoshi T, Sasaki T, Iwasawa Y. Molecular and atomic adsorption states of oxygen on Cu(111) at 100–300 K. Surf. Sci. 1996;365:310–318. doi: 10.1016/0039-6028(96)00738-8. [DOI] [Google Scholar]
- 38.Higgs V, Hollins P, Pemble ME, Pritchard J. Formation of a surface nitride on copper(111) and its influence on carbon monoxide adsorption: investigation by leed, rairs and eels. J. Electron Spectrosc. Relat. Phenom. 1986;39:137–144. doi: 10.1016/0368-2048(86)85041-1. [DOI] [Google Scholar]
- 39.Erley W, Ibach H. Vibrational excitations and structure of oxygen on Fe(110) Solid State Commun. 1981;37:937–942. doi: 10.1016/0038-1098(81)91190-X. [DOI] [Google Scholar]
- 40.Erley W, Ibach H. Vibrational spectra of ammonia adsorbed on Fe(110) Surf. Sci. 1982;119:L357–L362. doi: 10.1016/0039-6028(82)90180-7. [DOI] [Google Scholar]
- 41.Zion BD, Hanibiki AT, Sibener SJ. Kinetic energy effects on the oxidation of Ni(111) using O2 molecular beams. Surf. Sci. 1998;417:L1154–L1159. doi: 10.1016/S0039-6028(98)00737-7. [DOI] [Google Scholar]
- 42.Gland JL, Fisher GB, Mitchell GE. Vibrational characterization of adsorbed NH on the Ni(111) surface. Chem. Phys. Lett. 1985;119:89–92. doi: 10.1016/0009-2614(85)85426-9. [DOI] [Google Scholar]
- 43.Gorodetskii VV, Sametova AA, Matveev AV, Tapilin VM. From single crystals to supported nanoparticles in experimental and theoretical studies of H2 oxidation over platinum metals (Pt, Pd): Intermediates, surface waves and spillover. Catal. Today. 2009;144:219–234. doi: 10.1016/j.cattod.2008.12.014. [DOI] [Google Scholar]
- 44.Masuaki matsumoto kF, et al. Study of the adsorption structure of NO on Pt(111) by scanning tunneling microsopy and high-resolution electron energy-loss spectroscopy. Surf. Sci. 2000;454-456:101–105. doi: 10.1016/S0039-6028(00)00266-1. [DOI] [Google Scholar]
- 45.Dietrich H, Jacobi K, Ertl G. Coverage, lateral order, and vibrations of atomic nitrogen on Ru(0001) J. Chem. Phys. 1996;105:8944. doi: 10.1063/1.472624. [DOI] [Google Scholar]
- 46.Froitzheim H, Ibach H, Lehwald S. Surface vibrations of oxygen on W(100) Phys. Rev. B. 1976;14:1362–1369. doi: 10.1103/PhysRevB.14.1362. [DOI] [Google Scholar]
- 47.Sellidj A, Erskine JL. Electron energy loss spectroscopy studies of nitrogen adsorption on W (100) Surf. Sci. 1989;220:253–267. doi: 10.1016/0039-6028(89)90231-8. [DOI] [Google Scholar]
- 48.Mohamed MH, Kesmodel LL. Vibrational HREELS spectra of atomic nitrogen and oxygen on Cu(100) Surf. Sci. 1987;185:L467–L474. doi: 10.1016/S0039-6028(87)80602-7. [DOI] [Google Scholar]
- 49.Schick M, Xie J, Mitchell WJ, Weinberg WH. Interaction of gas‐phase atomic deuterium with the Ru(001)–p(1 × 2)–O surface: kinetics of hydroxyl and water formation. J. Chem. Phys. 1996;104:7713–7718. doi: 10.1063/1.471452. [DOI] [Google Scholar]
- 50.Newns DM. Self-consistent model of hydrogen chemisorption. Phys. Rev. 1969;178:1123–1135. doi: 10.1103/PhysRev.178.1123. [DOI] [Google Scholar]
- 51.Fernández EM, et al. Scaling relationships for adsorption energies on transition metal oxide, sulfide, and nitride surfaces. Angew. Chem. 2008;120:4761–4764. doi: 10.1002/ange.200705739. [DOI] [PubMed] [Google Scholar]
- 52.Sebastian KL. The selection rule in electron energy loss spectroscopy of adsorbed molecules. J. Phys. C: Solid State Phys. 1980;13:L115. doi: 10.1088/0022-3719/13/6/004. [DOI] [Google Scholar]
- 53.Somayajulu GR. Dependence of force constant on electronegativity, bond strength, and bond order. VII. J. Chem. Phys. 1958;28:814. doi: 10.1063/1.1744276. [DOI] [Google Scholar]
- 54.Hoffmann FM. Infrared reflection—absorption spectroscopy of adsorbed molecules. Surf. Sci. 1983;3:107–192. doi: 10.1016/0167-5729(83)90001-8. [DOI] [Google Scholar]
- 55.Goodacre R. Explanatory analysis of spectroscopic data using machine learning of simple, interpretable rules. Vib. Spectrosc. 2003;32:33–45. doi: 10.1016/S0924-2031(03)00045-6. [DOI] [Google Scholar]
- 56.Kauppinen JK, Moffatt DJ, Mantsch HH, Cameron DG. Fourier self-deconvolution: a method for resolving intrinsically overlapped bands. Appl. Spectmsc. 1981;35:271–276. doi: 10.1366/0003702814732634. [DOI] [Google Scholar]
- 57.Reuter K, Scheffler M. First-principles kinetic Monte Carlo simulations for heterogeneous catalysis: Application to the CO oxidation at RuO2(110) Phys. Rev. B. 2006;73:045433. doi: 10.1103/PhysRevB.73.045433. [DOI] [Google Scholar]
- 58.Grabow LC, Hvolbæk B, Nørskov JK. Understanding trends in catalytic activity: the effect of adsorbate–adsorbate interactions for CO oxidation over transition metals. Top Catal. 2010;53:298–310. doi: 10.1007/s11244-010-9455-2. [DOI] [Google Scholar]
- 59.Skoplyak O, Menning CA, Barteau MA, Chen JG. Experimental and theoretical study of reactivity trends for methanol on Co∕Pt(111) and Ni∕Pt(111) bimetallic surfaces. J. Chem. Phys. 2007;127:114707. doi: 10.1063/1.2768520. [DOI] [PubMed] [Google Scholar]
- 60.Saldivar-Garcia AJ, Lopez HF. Temperature effects on the lattice constants and crystal structure of a Co-27Cr-5Mo low-carbon alloy. Metal. Mater. Trans. A. 2004;35:2517–2523. doi: 10.1007/s11661-006-0232-6. [DOI] [Google Scholar]
- 61.Lin CY, George MW, Gill PMW. EDF2: a density functional for predicting molecular vibrational frequencies. Aust. J. Chem. 2004;57:365–370. doi: 10.1071/CH03263. [DOI] [Google Scholar]
- 62.Wellendorff J, et al. A benchmark database for adsorption bond energies to transition metal surfaces and comparison to selected DFT functionals. Surf. Sci. 2015;640:36–44. doi: 10.1016/j.susc.2015.03.023. [DOI] [Google Scholar]
- 63.Scott AP, Random L. Harmonic vibrational frequencies: an evaluation of hartree-fock, moller-plesset, quadratic configuratino interaction,density functional theory, and semiempirical scale factors. J. Phys. Chem. 1996;100:16502–16513. doi: 10.1021/jp960976r. [DOI] [Google Scholar]
- 64.Hammer B. Reactivity of a stepped surface NO dissociation on Pd(211) Faraday Discuss. 1998;110:323–333. doi: 10.1039/a801126e. [DOI] [Google Scholar]
- 65.Kresse G, Furthmüller J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B. 1996;54:11169–11186. doi: 10.1103/PhysRevB.54.11169. [DOI] [PubMed] [Google Scholar]
- 66.Hammer B, Hansen LB, Norskov JK. Improved adsorption energetics within density-functional theory using revised Perdew-Burke-Ernzerhof functionals. Phys. Rev. B. 1999;59:7413–7421. doi: 10.1103/PhysRevB.59.7413. [DOI] [Google Scholar]
- 67.Grimme S, Antony J, Ehrlich S, Krieg H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010;132:154104. doi: 10.1063/1.3382344. [DOI] [PubMed] [Google Scholar]
- 68.Mironenko AV, et al. Ring activation of furanic compounds on ruthenium-based catalysts. J. Phys. Chem. C. 2015;119:6075–6085. doi: 10.1021/jp512649b. [DOI] [Google Scholar]
- 69.Monkhorst HJ, Pack JD. Special points for Brillouin-zone integrations. Phys. Rev. B. 1976;13:5188–5192. doi: 10.1103/PhysRevB.13.5188. [DOI] [Google Scholar]
- 70.Murnaghan F. The compressibility of media under extreme pressures. Proc. Natl Acad. Sci. 1944;30:244–247. doi: 10.1073/pnas.30.9.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Description of Additional Supplementary Files
Data Availability Statement
The authors declare that all data supporting the findings of this study are available within the Article and its Supplementary Information files. An excel file is also provided that contains frequencies, energies, and other values calculated from DFT used in generating the figures contained in this work. Any other data will be provided by the corresponding author upon request.