

Abstract
Objectives
Interleukin-6 (IL-6) is implicated in rheumatoid arthritis (RA) pathophysiology. Unlike IL-6 receptor inhibitors, sirukumab is a human monoclonal antibody that selectively binds to the IL-6 cytokine. The phase III, multicentre, randomised, double-blind, placebo-controlled, parallel-group SIRROUND-D study (ClinicalTrials.gov identifier NCT01604343) evaluated the efficacy and safety of sirukumab in patients with active RA refractory to disease-modifying antirheumatic drugs.
Methods
Patients were randomised 1:1:1 to treatment with sirukumab 100 mg every 2 weeks, 50 mg every 4 weeks or placebo every 2 weeks subcutaneously. Results through week 52 are reported.
Results
Of 1670 randomised patients, significantly more patients achieved American College of Rheumatology 20% (ACR20) response at week 16 (coprimary endpoint) with sirukumab 100 mg every 2 weeks (53.5%) or 50 mg every 4 weeks (54.8%) versus placebo (26.4%; both p<0.001). Mean (SD) change from baseline in modified Sharp/van der Heijde score at week 52 (coprimary endpoint) was significantly lower with sirukumab (100 mg every 2 weeks: 0.46 (3.26); 50 mg every 4 weeks: 0.50 (2.96)) versus placebo (3.69 (9.25); both p<0.001). All major secondary endpoints (week 24 Health Assessment Questionnaire–Disability Index change from baseline, ACR50 response, 28-joint Disease Activity Score based on C reactive protein and major clinical response (ACR70 for six continuous months by week 52)) were met. The most common adverse events with sirukumab were elevated liver enzymes, upper respiratory tract infection, injection site erythema and nasopharyngitis.
Conclusions
Sirukumab 100 mg every 2 weeks and 50 mg every 4 weeks led to significant reductions in RA symptoms, inhibition of structural damage progression and physical function and quality of life improvements, with an expected safety profile.
Trial registration number
NCT01604343; Results.
Keywords: rheumatoid arthritis, cytokines, DMARDs (biologic), DMARDs (synthetic), treatment
Introduction
Patients with rheumatoid arthritis (RA) often have increased levels of interleukin (IL)-6 in serum and the synovial compartment where its levels are correlated to local disease activity.1–3 In the RA synovium, both tumour necrosis factor (TNF) and IL-1 can stimulate IL-6 production by multiple cell types.4 Local concentrations of IL-6 may stimulate leucocyte recruitment to the joint, promote osteoclast maturation and activation, suppress chondrocytes and stimulate synovial proliferation, summarily contributing to joint damage.5 Systemically, elevated IL-6 levels in patients with RA may induce haepatic production of acute-phase proteins6 and likely increase hepcidin and the development of anaemia of chronic inflammation.7 Elevated IL-6 may also be responsible for autoimmune features in RA, such as autoreactive T cell activation and hypergammaglobulinaemia.8 Therefore, IL-6 is an attractive target for the treatment of RA.
In patients with active RA and inadequate response to disease-modifying antirheumatic drug (DMARD) therapy, inhibition of the IL-6 receptor with the monoclonal antibody (mAb) tocilizumab reduced joint swelling and tenderness, improved physical function and reduced the rate of radiographic progression.9–12 Another anti–IL-6 receptor mAb, sarilumab, demonstrated similar efficacy in patients with RA and inadequate response to methotrexate (MTX).13 Although the clinical relevance of a different mechanism of targeting the IL-6 pathway is not fully understood, sirukumab is a human mAb that selectively binds to the IL-6 cytokine with high affinity. Sirukumab was shown to significantly improve signs and symptoms (eg, American College of Rheumatology 20% (ACR20) response at week 16), functionality and quality of life versus placebo in a difficult-to-treat population of RA patients refractory to anti-TNF and other biologicals.14 Two other antibodies to IL-6, clazakizumab and olokizumab, have demonstrated activity in phase II studies of RA patients with an inadequate response to MTX or failure to anti-TNF therapy, respectively.15 16 The SIRROUND-D study (ClinicalTrials.gov identifier NCT01604343) was designed to assess efficacy and safety of subcutaneous (SC) sirukumab in patients with active RA despite DMARD therapy over 52 weeks.
Methods
Patients
Patients from 18 countries (USA, Canada, Mexico, Colombia, Chile, South Africa, Lithuania, Poland, Russia, Ukraine, Serbia, Croatia, Bulgaria, Romania, Japan, South Korea, Taiwan and Malaysia) were enrolled and monitored between July 2012 and September 2015. Eligible patients were aged ≥18 years, had moderately to severely active RA and were refractory to single-agent or combination DMARD therapy including MTX or sulfasalazine, based on lack of benefit after ≥12 weeks. Patients needed ≥6/68 tender joints and ≥6/66 swollen joints at screening and baseline; C reactive protein (CRP) ≥8.0 mg/L; and ≥1 of the following three criteria to be met prior to treatment: (A) anticitrullinated peptide antibody-positive (measured by anticyclic citrullinated peptide antibody test) at screening; (B) rheumatoid factor positive at screening; or (C) documented history of radiographic evidence of erosive RA in the hands and/or feet. Patients using non-biological DMARDs must have been on a stable dose for ≥4 weeks prior to receiving study drug. Patients not currently using DMARDs must not have received DMARDs for ≥4 weeks prior to receiving study drug. Patients who previously were treated with biologicals were permitted, as long as they had not failed anti-TNF or tocilizumab for safety or efficacy reasons and had not received biologicals within the past 3 months (6 weeks for etanercept or yisaipu and 4 weeks for anakinra). Patients with a history of or current serious infection (including tuberculosis) were excluded.
Study design
This global, phase III, multicentre, randomised, double-blind, placebo-controlled, parallel-group study randomised patients 1:1:1 at week 0 to sirukumab 100 mg SC every 2 weeks, sirukumab 50 mg SC every 4 weeks or placebo SC every 2 weeks (see online supplementary figure S1). These doses were previously studied in a phase IIb dose-ranging study.17 Patients were stratified by baseline MTX use (none, up to 12.5 mg/week or ≥12.5 mg/week). Patients on placebo demonstrating <20% improvement from baseline in both swollen and tender joint counts at week 18 (early escape (EE)) or week 40 (late escape (LE)), or still on study treatment at week 52 (crossover), were rerandomised 1:1 to receive blinded treatment with one of the two sirukumab doses through week 104. A 16-week safety follow-up phase occurred after the final dose, making the total study duration 120 weeks excluding the screening period of up to 6 weeks. The study protocol was approved by the relevant institutional review boards or ethics committees, and all patients gave written informed consent. Data were collected by the investigators and analysed by the study sponsor.
annrheumdis-2017-211328supp002.jpg (240.9KB, jpg)
{kind=link}
Assessments
All analyses were prespecified unless otherwise noted; all randomised patients were included in population summaries and efficacy analyses, and all patients treated with ≥1 dose of study agent were included in safety analyses. The coprimary efficacy endpoints were proportion of patients who achieved an ACR20 response at week 16 and change from baseline in modified Sharp/van der Heijde score (SHS) at week 52. Radiographs of the hands and feet were taken at baseline, week 18 (for patients meeting EE criteria), week 24 (for patients who did not meet EE criteria) and week 52. Major secondary endpoints included change from baseline in Health Assessment Questionnaire–Disability Index (HAQ-DI) score at week 24, proportion of patients achieving an ACR50 response at week 24, proportion of patients with the 28-joint Disease Activity Score based on CRP (DAS28 (CRP)) <2.6 at week 24 and proportion of patients achieving major clinical response (defined as ACR70 response for six continuous months) by week 52. Additional endpoints included physical component summary (PCS) and mental component summary (MCS) of the patient-reported 36-item Short Form survey (SF-36) and proportions of patients achieving clinical disease activity index (CDAI) low disease activity (≤10.0) at week 24 and CDAI remission (≤2.8) at weeks 24 and 52 (analyses of CDAI low disease activity and remission were post hoc). Efficacy endpoints were also assessed over time. Safety assessments included monitoring of adverse events (AEs), standard clinical laboratory tests, vital signs evaluations and physical examinations. Serum samples were analysed for antibodies to sirukumab using a validated drug-tolerant electrochemiluminescent immunoassay method on the Meso Scale Discovery platform.
Statistical methods
A sample size of 550 patients per treatment group would provide approximately 98%–99% power to detect a treatment difference of 11%–17% in the proportion of patients who achieved ACR20 response at week 16 and approximately 98% power to detect a treatment difference of 1.0 in the mean change from baseline SHS at week 52.
The coprimary efficacy endpoints were tested in the following predefined order: (1) sirukumab 100 mg every 2 weeks versus placebo in week 16 ACR20 response; (2) sirukumab 100 mg every 2 weeks versus placebo in week 52 SHS change from baseline; (3) sirukumab 50 mg every 4 weeks versus placebo in week 16 ACR20 response; (4) sirukumab 50 mg every 4 weeks versus placebo in week 52 SHS change from baseline. If a given comparison was not significant at α=0.05 (two sided), the remaining treatment group comparisons were to be considered as supportive analyses. For week 16 ACR20, Cochran-Mantel-Haenszel tests stratified by baseline MTX use (none, up to 12.5 or ≥12.5 mg/week) were used for treatment comparisons. The last observation carried forward method was used for imputing missing ACR components if a patient had data for ≥1 ACR component at week 16. Patients were considered ACR20 non-responders if they did not have data for any ACR component at week 16 or if they met any of the following treatment failure criteria prior to week 16: initiated treatment with DMARDs, systemic immunosuppressives or biologicals for RA; increased their dose of MTX; initiated or increased oral corticosteroid treatment or received intravenous or intramuscular corticosteroids for RA; or discontinued study agent. For week 52 SHS change from baseline analyses, analysis of variance tests stratified by baseline MTX use on the van der Waerden normal scores were used for treatment comparisons. Missing SHS values at week 52 were imputed by a linear extrapolation of non-missing values before week 52. For patients who met EE criteria in the placebo treatment group, SHS value at week 52 was replaced by the imputed value from a linear extrapolation of non-missing values prior to escape.
Results
Patients
Overall, 1670 patients across 185 sites were randomised, administered ≥1 dose of study agent and included in efficacy (non-radiographic) and safety analyses (figure 1). Radiographic efficacy analyses included 1654 patients with non-missing baseline SHS. Demographic and baseline characteristics were generally well-balanced across all treatment groups (table 1). Of note, 583 (34.9%) of the enrolled patients had previously received ≥1 biological therapy (see online supplementary table S1).
Figure 1.

Patient disposition through week 52. EE, early escape; LE, late escape; q2w, every 2 weeks; q4w, every 4 weeks. aIncludes placebo patients who escaped (EE/LE) to sirukumab 50 mg q4w or 100 mg q2w.
Table 1.
Demographic and baseline characteristics
| Characteristic | Placebo (n=556) |
Sirukumab | Total (n=1670) |
|
| 50 mg q4w (n=557) |
100 mg q2w (n=557) |
|||
| Female sex, n (%) | 436 (78.4) | 447 (80.3) | 452 (81.1) | 1335 (79.9) |
| Age, years | 52.9 (11.9) | 52.9 (11.8) | 53.0 (11.3) | 52.9 (11.7) |
| Race, n (%) | ||||
| White | 403 (72.5) | 397 (71.3) | 408 (73.2) | 1208 (72.3) |
| Asian | 88 (15.8) | 89 (16.0) | 95 (17.1) | 272 (16.3) |
| Black or African-American | 16 (2.9) | 15 (2.7) | 10 (1.8) | 41 (2.5) |
| American Indian or Alaska Native | 6 (1.1) | 4 (0.7) | 4 (0.7) | 14 (0.8) |
| Other* | 39 (7.0) | 49 (8.8) | 38 (6.8) | 126 (7.5) |
| Not reported/unknown | 4 (0.7) | 3 (0.5) | 2 (0.4) | 9 (0.5) |
| Region, n (%) | ||||
| Eastern Europe | 271 (48.7) | 263 (47.2) | 273 (49.0) | 807 (48.3) |
| North America | 85 (15.3) | 96 (17.2) | 91 (16.3) | 272 (16.3) |
| Asia-Pacific | 87 (15.6) | 89 (16.0) | 91 (16.3) | 267 (16.0) |
| Latin America | 73 (13.1) | 75 (13.5) | 76 (13.6) | 224 (13.4) |
| South Africa | 40 (7.2) | 34 (6.1) | 26 (4.7) | 100 (6.0) |
| Weight, kg | 72.7 (17.4) | 72.3 (18.6) | 71.6 (17.1) | 72.2 (17.7) |
| Disease duration, years | 8.3 (7.0) | 8.7 (7.5) | 8.8 (7.6) | 8.6 (7.4) |
| BMI, kg/m2 | 27.4 (6.0) | 27.3 (6.4) | 27.2 (6.0) | 27.3 (6.2) |
| CRP, mg/L | 25 (34) | 24 (26) | 24 (26) | 24 (29) |
| RF positive, n (%) | 444 (79.9) | 433 (77.7) | 468 (84.0)† | 1345 (80.5) |
| Anti-CCP positive, n (%) | 467 (84.0) | 476 (85.5) | 484 (86.9) | 1427 (85.4) |
| SHS | 41.9 (46.7) | 41.8 (45.4) | 42.5 (49.3) | 42.1 (47.1) |
| HAQ-DI score, range: 0–3 | 1.6 (0.7) | 1.5 (0.6) | 1.5 (0.7) | 1.5 (0.6) |
| DAS28 (CRP) | 5.9 (0.9) | 5.9 (0.9) | 5.8 (0.9)‡ | 5.9 (0.9) |
| Prior medication use§ | ||||
| 1 DMARD | 183 (32.9) | 179 (32.1) | 173 (31.1) | 535 (32.0) |
| ≥2 DMARDs | 373 (67.1) | 378 (67.9) | 384 (68.9) | 1135 (68.0) |
| MTX | 547 (98.4) | 550 (98.7) | 548 (98.4) | 1645 (98.5) |
| Sulfasalazine | 174 (31.4) | 167 (30.0) | 152 (27.3) | 493 (29.5) |
| Systemic corticosteroids | 422 (75.9) | 407 (73.1) | 418 (75.0) | 1247 (74.7) |
| Baseline medication use, n (%) | ||||
| DMARDs | 508 (91.4) | 517 (92.8) | 511 (91.7) | 1536 (92.0) |
| NSAIDs | 434 (78.1) | 420 (75.4) | 454 (81.5)¶ | 1308 (78.3) |
| Corticosteroids | 341 (61.3) | 331 (59.4) | 360 (64.6) | 1032 (61.8) |
Values are mean (SD) unless otherwise indicated.
Differences in demographic and baseline characteristics among groups were not significant, except where noted.
*No Native Hawaiian or other Pacific Islanders were reported in any treatment group.
†p=0.01 versus sirukumab 50 mg q4w based on χ2 test.
‡p=0.02 versus placebo based on t-test.
§All randomised patients took ≥1 DMARD.
¶p=0.01 versus sirukumab 50 mg q4w based on χ2 test.
q2w, every 2 weeks; q4w, every 4 weeks; BMI, body mass index; CCP, cyclic citrullinated peptide; CRP, C reactive protein; DAS28 (CRP), 28-joint Disease Activity Score based on C reactive protein; DMARD, disease-modifying antirheumatic drug; HAQ-DI, Health Assessment Questionnaire–Disability Index; MTX, methotrexate; NSAID, nonsteroidal anti-inflammatory drug; RF, rheumatoid factor; SHS, Sharp/van der Heijde score.
annrheumdis-2017-211328supp001.docx (288.8KB, docx)
Efficacy
Both coprimary endpoints were met. The proportion of patients with ACR20 responses was significantly greater for both sirukumab doses compared with placebo at week 16 (both p<0.001; figure 2A). Differences in proportions of patients achieving ACR20 were observed as early as week 2 and sustained through week 52 (both doses p<0.001 vs placebo at weeks 24 and 52). ACR20 response rates in both sirukumab groups were higher compared with placebo, regardless of baseline MTX use (see online supplementary table S2). A summary of percent improvement in ACR components at week 16 is provided (see online supplementary table S3). Significant inhibition of radiographic progression (SHS mean change from baseline) was achieved at week 52 (coprimary endpoint), with differences observed as early as week 24 for sirukumab versus placebo (both doses p<0.001 vs placebo at both timepoints; figure 2B). Significantly higher proportions of patients treated with sirukumab did not show radiographic progression (score change of ≤0 from baseline in SHS total, erosion and joint space narrowing scores) compared with placebo at week 52 (both doses p<0.001 vs placebo; figure 2C). Smaller week 52 SHS mean changes from baseline were observed in both sirukumab groups compared with placebo, regardless of baseline MTX use. The probability plot of the SHS change from baseline at week 52 clearly shows separation between both sirukumab groups and the placebo group and no separation between the sirukumab groups (see online supplementary figure S2).
Figure 2.
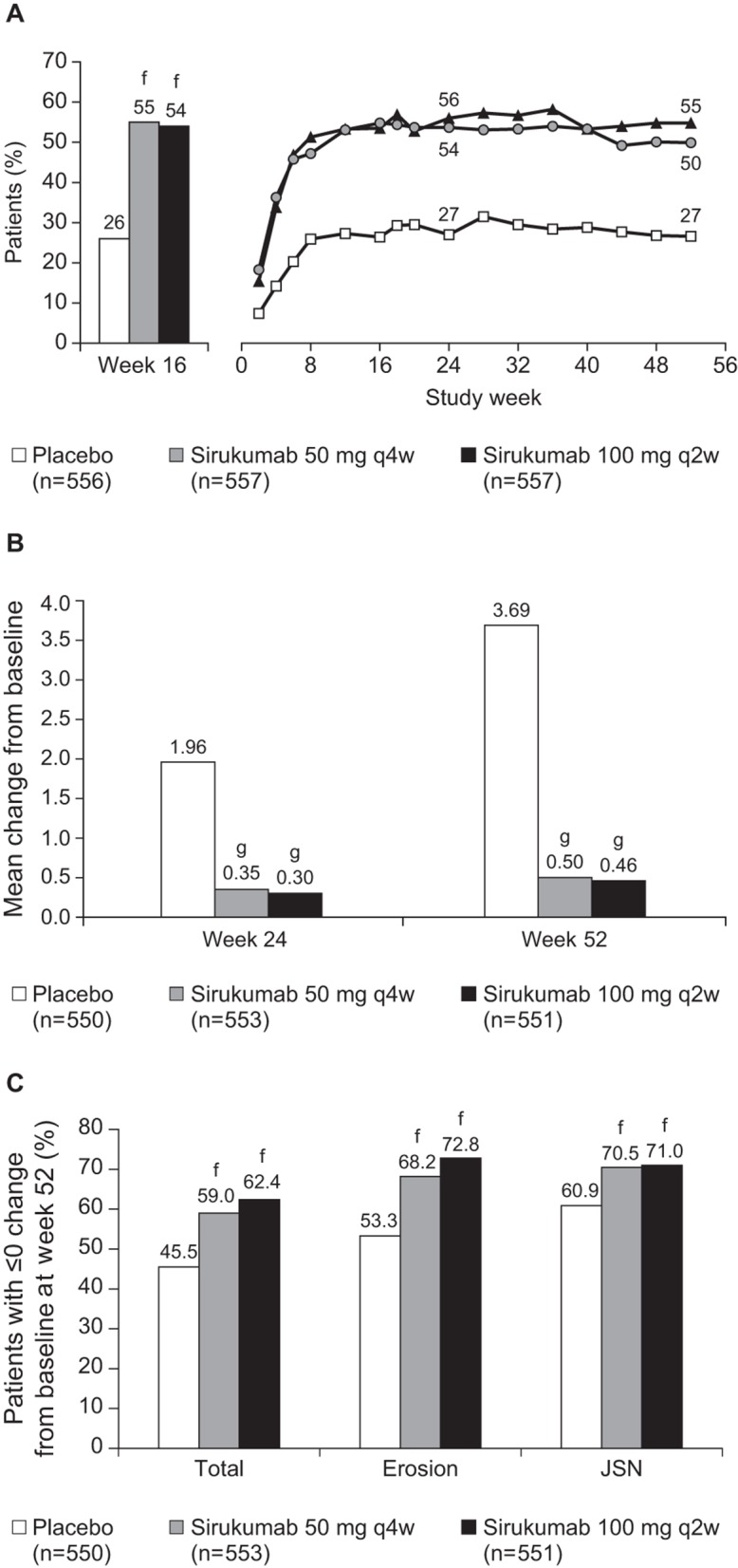
(A) Proportions of patients with an ACR20 response at week 16a and ACR20 response over time.b,c,d (B) Change from baseline in SHS results at weeks 24 and 52.e (C) Proportions of patients with no radiographic progression from baseline to week 52.e EE, early escape; JSN, joint space narrowing; LE, late escape; NR, non-responder; q2w, every 2 weeks; q4w, every 4 weeks; SHS, Sharp/van der Heijde score; TF, treatment failure. aBased on imputed values by missing data (NR)/TF(NR). bBased on imputed values by missing data (NR)/TF(NR)/EE(NR)/LE(NR). cp<0.001 for both doses of sirukumab versus placebo across all timepoints based on Cochran-Mantel-Haenszel test. dNot significant for sirukumab 50 mg q4w versus sirukumab 100 mg q2w across all timepoints based on Cochran-Mantel-Haenszel test. eBased on imputed values by EE rules and then missing data rules. fp<0.001 versus placebo based on Cochran-Mantel-Haenszel test. gp<0.001 versus placebo based on van der Waerden analysis of variance.
annrheumdis-2017-211328supp003.jpg (212.8KB, jpg)
{kind=link}
All major secondary efficacy endpoints demonstrated significant improvements for both sirukumab doses versus placebo (all p≤0.001; table 2). In addition, more patients on sirukumab achieved ACR70 as early as week 8, with treatment differences maintained through week 52 (both doses p<0.001 vs placebo at weeks 16, 24 and 52; table 2). ACR90 responses were achieved by a significantly greater proportion of patients on both sirukumab doses versus placebo at weeks 16, 24 and 52 (all p<0.05; not shown). The proportions of patients achieving CDAI low disease activity (≤10.0) for sirukumab 100 mg every 2 weeks and 50 mg every 4 weeks at week 24 were 30.2% and 29.4%, respectively, compared with 15.5% for placebo (both p<0.001 vs placebo); at week 52, proportions were 32.0% and 32.5%, respectively, compared with 15.3% for placebo (both p<0.001 vs placebo). The proportions of patients in CDAI remission (≤2.8) at week 24 for sirukumab 100 mg every 2 weeks and 50 mg every 4 weeks were 8.4% and 7.0%, respectively, compared with 3.1% for placebo (both p≤0.003 vs placebo). At week 52, the proportions of patients in CDAI remission for sirukumab 100 mg every 2 weeks and 50 mg every 4 weeks were 8.3% and 10.1%, respectively, compared with 3.8% for placebo (both p≤0.002 vs placebo).
Table 2.
Results of major and other key secondary endpoints
| Endpoint | Placebo (n=556) |
Sirukumab | |
| 50 mg q4w (n=557) |
100 mg q2w (n=557) |
||
| HAQ-DI change from baseline at week 24, mean (SD)* | –0.22 (0.53) | –0.43 (0.58)† | –0.46 (0.57)† |
| ACR50 at week 16, n (%)‡ | 60 (10.8) | 167 (30.0)§ | 146 (26.2)§ |
| ACR50 at week 24, n (%)¶ | 69 (12.4) | 168 (30.2)§ | 185 (33.2)§ |
| ACR50 at week 52, n (%)** | 77 (13.8) | 169 (30.3)§ | 198 (35.5)§ |
| ACR70 at week 16, n (%)‡ | 22 (4.0%) | 75 (13.5%)§ | 75 (13.5%)§ |
| ACR70 at week 24, n (%)¶ | 19 (3.4%) | 83 (14.9%)§ | 91 (16.3%)§ |
| ACR70 at week 52, n (%)** | 30 (5.4%) | 92 (16.5%)§ | 103 (18.5%)§ |
| DAS28 (CRP) <2.6 at week 24, n (%)¶ | 31 (5.6) | 145 (26.0)§ | 142 (25.5)§ |
| Major clinical response by week 52, n (%)** | 10 (1.8) | 30 (5.4)§ | 50 (9.0)§ |
| SF-36 PCS change from baseline at week 52, mean (SD)†† | 2.42 (6.81) | 5.66 (7.74)‡‡ | 6.16 (7.23)‡‡ |
| SF-36 MCS change from baseline at week 52, mean (SD)†† | 2.69 (9.57) | 5.35 (9.64)‡‡ | 4.77 (9.80)‡‡ |
*Based on imputed values by missing data (LOCF)/EE(LOCF).
†p≤0.001 versus placebo based on analysis of covariance.
‡Based on imputed values by missing data (NR)/TF(NR).
§p≤0.01 versus placebo based on Cochran-Mantel-Haenszel test.
¶Based on imputed values by missing data (NR)/TF(NR)/EE(NR).
**Based on imputed values by missing data (NR)/TF(NR)/EE(NR)/LE(NR).
††Based on imputed values by missing data (LOCF)/EE(LOCF)/LE(LOCF).
‡‡p≤0.001 versus placebo based on analysis of variance.
ACR50/70, American College of Rheumatology 50%/70%; DAS28 (CRP), 28-joint Disease Activity Score based on C reactive protein; EE, early escape; HAQ-DI, Health Assessment Questionnaire–Disability Index; LE, late escape; LOCF, last observation carried forward; MCS, mental component summary; NR, non-responder; PCS, physical component summary; q2w, every 2 weeks; q4w, every 4 weeks; SF-36, Short Form-36; TF, treatment failure.
Significantly greater improvements from baseline in health-related physical and emotional well-being were observed with sirukumab on the patient-reported SF-36 PCS and MCS scores at week 52 (p<0.001 for PCS and MCS, both sirukumab doses vs placebo; table 2). At weeks 24 and 52, greater improvements in all eight individual SF-36 domain scores were achieved with sirukumab compared with placebo (all p≤0.006), and significantly more sirukumab-treated patients achieved clinically meaningful improvements (≥5-point increase) from baseline in PCS and MCS scores compared with placebo (all p≤0.009).
Safety
Safety results were summarised in the ‘pure’ placebo-controlled period prior to EE (to week 18; see online supplementary table S4) and at the end of the placebo-controlled period (to week 52) for all AEs (table 3) and for specific AEs of interest (see online supplementary table S5). Through week 52, no disproportional increase from week 18 was observed in AE rates, and the overall AE profile was similar to that observed through week 18. The most common AEs (≥5%) through week 52 with sirukumab were elevated liver enzymes, upper respiratory tract infection, bronchitis, nasopharyngitis, injection site erythema and pruritus, leucopaenia, neutropaenia, headache, and hypertension (table 3). No dose relationship was apparent between sirukumab doses and the types or frequency of AEs other than injection site reactions (ISRs) and elevated liver enzymes, which were more frequent with sirukumab 100 mg every 2 weeks than 50 mg every 4 weeks. No ISRs were considered severe in intensity, and four patients (two in each sirukumab dose group) discontinued the study due to mild or moderate ISRs.
Table 3.
Summary of overall safety through week 52
| Variable | Placebo (n=556) |
Sirukumab*† | ||
| 50 mg q4w (n=663) |
100 mg q2w (n=662) |
Combined (n=1325) |
||
| Mean duration of follow-up, weeks | 36.18 | 45.76 | 45.12 | 45.44 |
| Mean number of study agent administrations | 17.45 | 21.88 | 21.56 | 21.72 |
| Patients with ≥1 AE, n (%) p Value versus placebo |
364 (65.5) | 528 (79.6) <0.001 |
531 (80.2) <0.001 |
1059 (79.9) <0.001 |
| Patients with ≥1 SAE, n (%) p Value versus placebo |
38 (6.8) | 73 (11.0) 0.012 |
65 (9.8) NS |
138 (10.4) 0.015 |
| Patients with ≥1 AE that caused study agent discontinuation, n (%) p Value versus placebo |
18 (3.2) | 53 (8.0) <0.001 |
51 (7.7) <0.001 |
104 (7.8) <0.001 |
| Patients with ≥1 serious infection, n (%) p Value versus placebo |
10 (1.8) | 27 (4.1) 0.021 |
22 (3.3) NS |
49 (3.7) 0.031 |
| Patients with ≥1 injection site reaction, n (%) p Value versus placebo |
14 (2.5) | 71 (10.7) <0.001 |
108 (16.3) <0.001 |
179 (13.5) <0.001 |
| Patients with ≥1 MACE, n (%) p Value versus placebo‡ |
2 (0.4) | 8 (1.2) NS |
3 (0.5) NS |
11 (0.8) NS |
| Patients with ≥1 malignancy, n (%) p Value versus placebo‡ |
2 (0.4) | 2 (0.3) NS |
5 (0.8) NS |
7 (0.5) NS |
| Patients with ≥1 GI perforation, n (%) p Value versus placebo‡ |
1 (0.2) | 1 (0.2) NS |
0 NS |
1 (0.1) NS |
| Death, n (%) p Value versus placebo‡ |
1 (0.2) | 7 (1.1) NS |
3 (0.5) NS |
10 (0.8) NS |
| Events of ≥5% frequency in any sirukumab group, n (%) | ||||
| Alanine aminotransferase increased p Value versus placebo |
25 (4.5) | 102 (15.4) <0.001 |
124 (18.7) <0.001 |
226 (17.1) <0.001 |
| Aspartate aminotransferase increased p Value versus placebo |
19 (3.4) | 58 (8.7) <0.001 |
82 (12.4) <0.001 |
140 (10.6) <0.001 |
| Upper respiratory tract infection p Value versus placebo |
63 (11.3) | 65 (9.8) NS |
66 (10.0) NS |
131 (9.9) NS |
| Injection site erythema p Value versus placebo |
6 (1.1) | 50 (7.5) <0.001 |
80 (12.1) <0.001 |
130 (9.8) <0.001 |
| Nasopharyngitis p Value versus placebo |
57 (10.3) | 62 (9.4) NS |
56 (8.5) NS |
118 (8.9) NS |
| Leucopaenia p Value versus placebo |
7 (1.3) | 37 (5.6) <0.001 |
37 (5.6) <0.001 |
74 (5.6) <0.001 |
| Bronchitis p Value versus placebo |
27 (4.9) | 39 (5.9) NS |
31 (4.7) NS |
70 (5.3) NS |
| Neutropaenia p Value versus placebo |
5 (0.9) | 38 (5.7) <0.001 |
29 (4.4) <0.001 |
67 (5.1) <0.001 |
| Hypertension p Value versus placebo |
21 (3.8) | 28 (4.2) NS |
33 (5.0) NS |
61 (4.6) NS |
| Headache p Value versus placebo |
22 (4.0) | 33 (5.0) NS |
26 (3.9) NS |
59 (4.5) NS |
| Injection site pruritus p Value versus placebo |
1 (0.2) | 11 (1.7) 0.009 |
41 (6.2) <0.001 |
52 (3.9) <0.001 |
*Includes patients from the placebo group rerandomised to treatment with sirukumab; thus, patients may be represented in >1 treatment group.
†p Values are nominal and from χ2 tests, unless otherwise noted.
‡p Values are nominal and from Fisher’s exact tests.
AE, adverse event; GI, gastrointestinal; MACE, major adverse cardiovascular event; NS, not significant; q2w, every 2 weeks; q4w, every 4 weeks; SAE, serious adverse event.
Through week 18, serious AEs (SAEs) were reported in 4.7%, 2.9% and 3.1% of patients in the sirukumab 100 mg every 2 weeks, 50 mg every 4 weeks and placebo groups, respectively; through week 52, SAEs were reported in 9.8%, 11.0% and 6.8% of patients, respectively. Serious infections were reported in 0.9%, 0.7% and 0.9% of patients, respectively, through week 18, and were numerically greater in the sirukumab 100 mg every 2 weeks combined (3.3%) and 50 mg every 4 weeks combined (4.1%) groups (including EE patients) compared with the placebo group (1.8%) through week 52. Two gastrointestinal (GI) perforations were reported: one upper GI (gastric) perforation in the placebo group and one lower GI perforation (perforated appendicitis) in a patient randomised to placebo with EE to sirukumab 50 mg every 4 weeks. During the 18-week period prior to EE, mortality rates were the same across the treatment groups, with one death each in the three groups. Through 52 weeks, there was a numerical imbalance in exposure-adjusted mortality rates (supplementary table S5); however, the interpretation of these results is confounded by the loss of randomisation as patients in the placebo group switched to sirukumab at EE and LE timepoints.
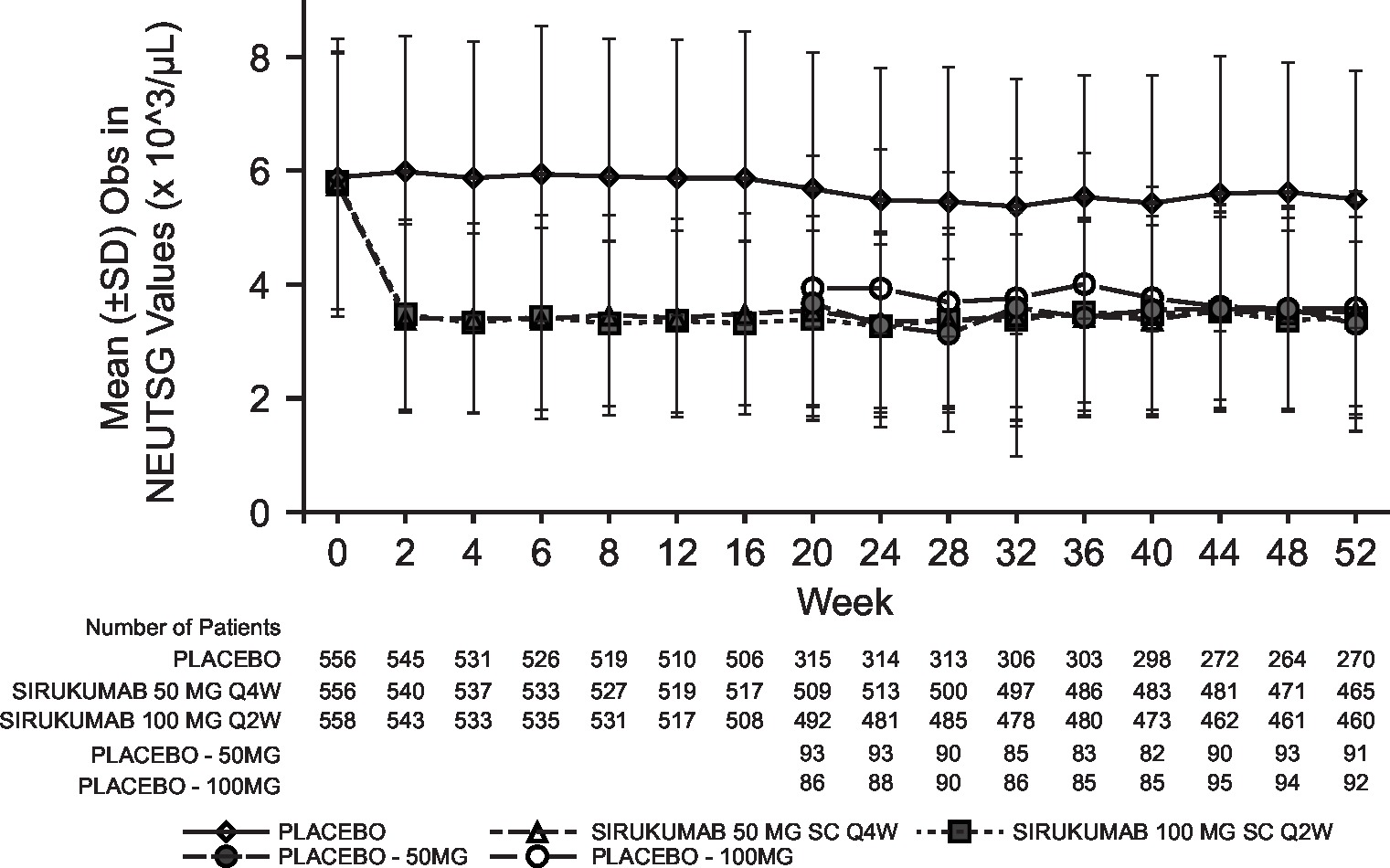
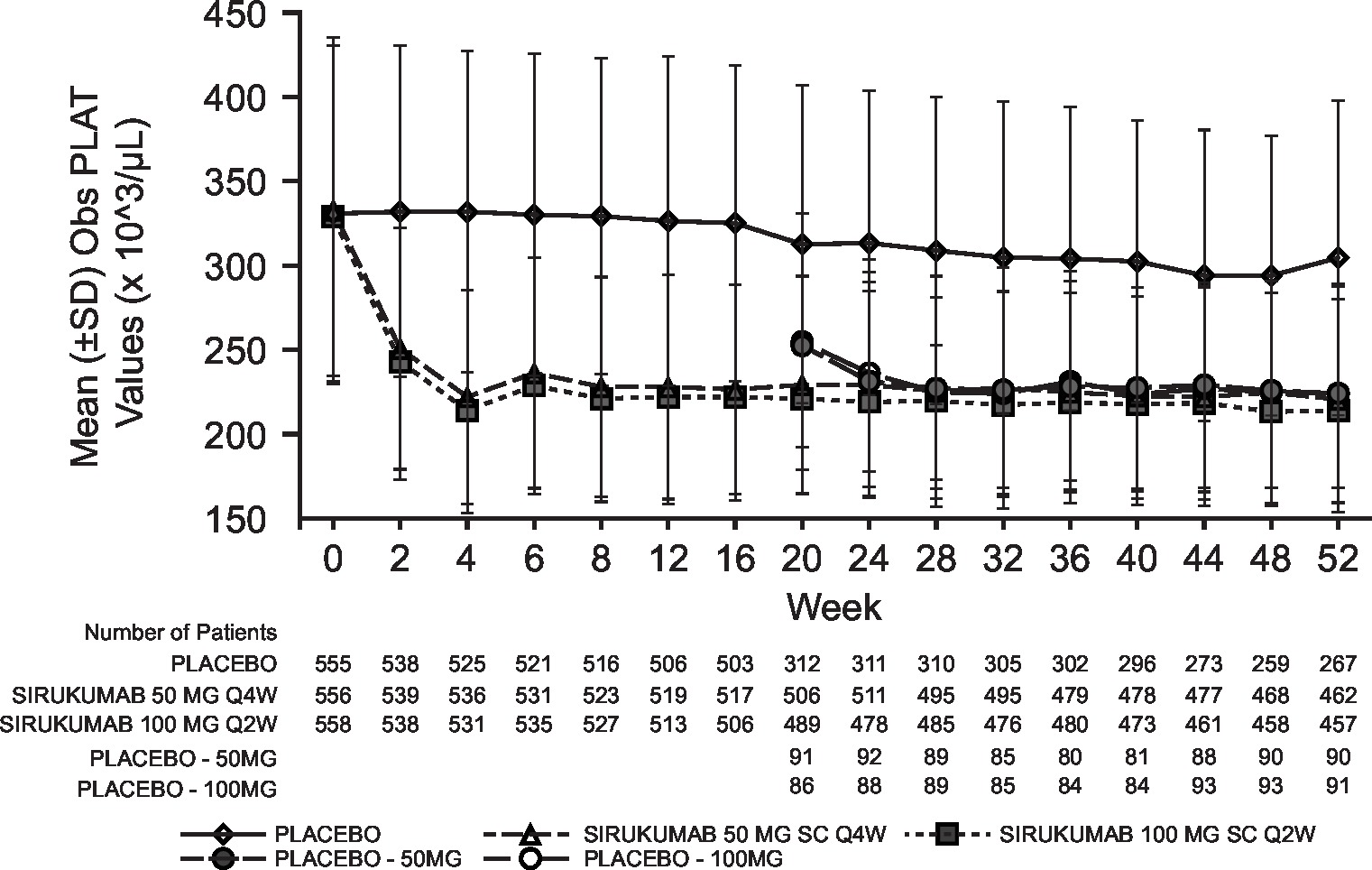
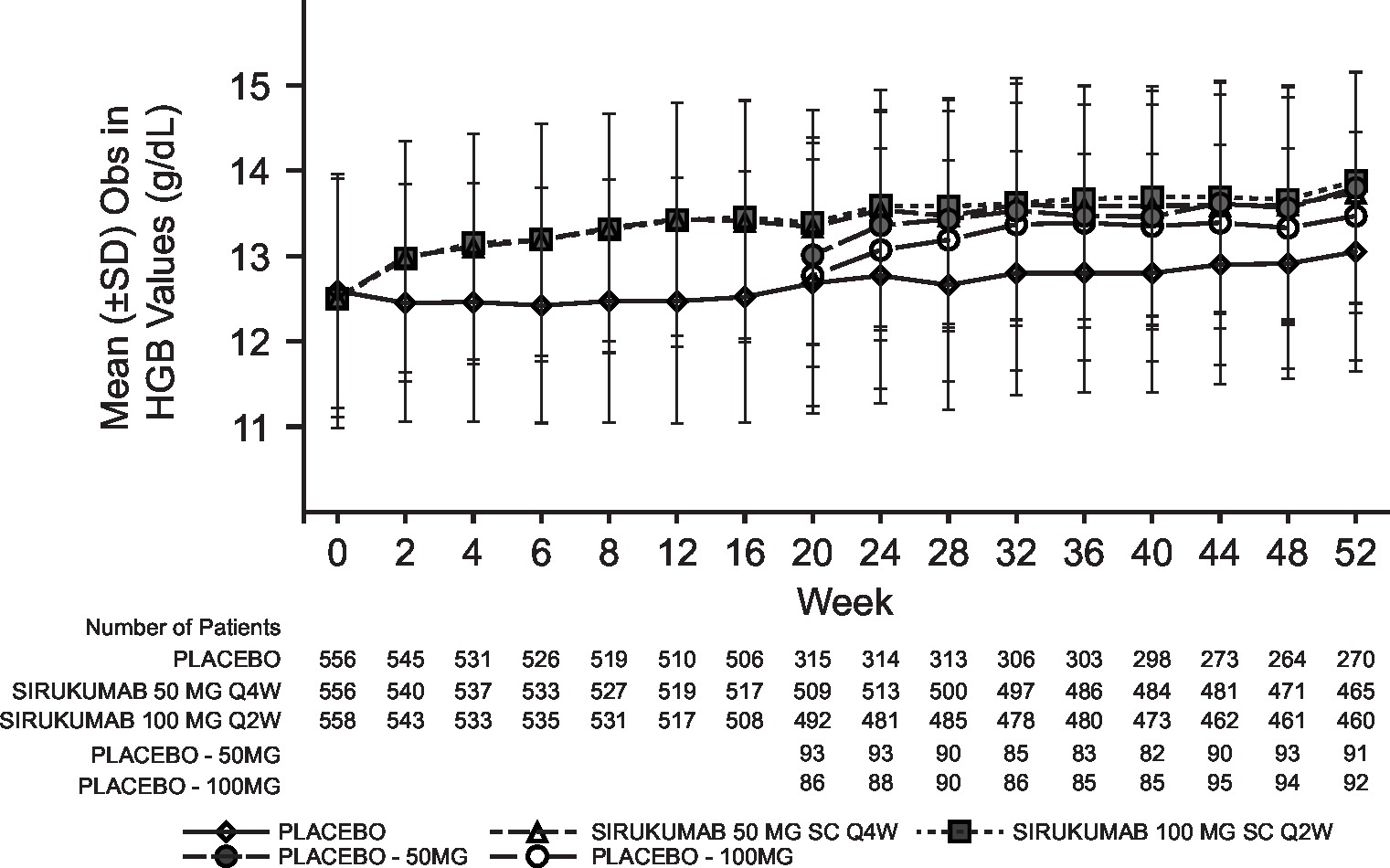
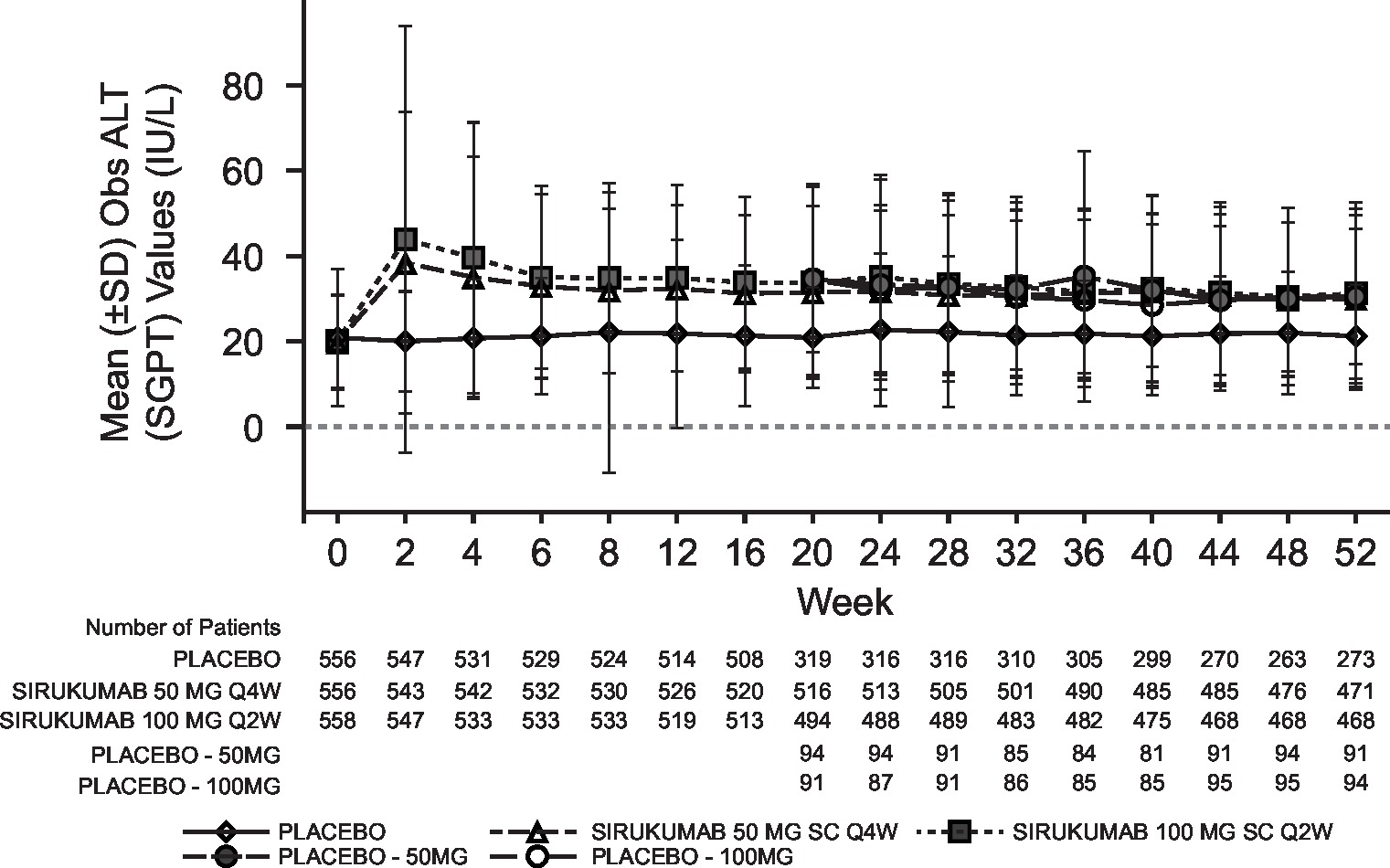
Laboratory abnormalities were similar for both sirukumab doses and were numerically higher than placebo; the incidence of grade 3/4 laboratory abnormalities with sirukumab was low for decreased platelets (0.2% grade 3; 0% grade 4), decreased neutrophils (4.1% grade 3; 0.2% grade 4), increased alanine aminotransferase (ALT; 3.2% grade 3, 0% grade 4) and increased aspartate aminotransferase (AST; 0.7% grade 3; 0% grade 4). Decreased neutrophil and platelet counts and increased haemoglobin, ALT and AST began at week 2 of sirukumab treatment and were sustained through week 52 (see online supplementary figure S3). Total and low-density lipoprotein cholesterol increased with both doses of sirukumab relative to placebo; however, the total cholesterol:high-density lipoprotein ratio remained below 4.0 for all treatment groups at week 52.
annrheumdis-2017-211328supp004.jpg (254.6KB, jpg)
{kind=link}
The overall incidence of antibodies to sirukumab through week 52 was 2.4% (16/654), occurring in 1.2% (4/328) of patients receiving sirukumab 100 mg every 2 weeks and 3.7% (12/326) of patients receiving sirukumab 50 mg every 4 weeks. Only one of these 16 patients (in the 50 mg every 4 weeks group) was positive for neutralising antibodies to sirukumab. In patients who were positive for antidrug antibodies, there was no apparent relationship between antibodies to sirukumab and clinical response or ISRs.
Discussion
This phase III, double-blind, randomised clinical trial evaluated the safety and efficacy of sirukumab, an IL-6 cytokine antibody, administered as 100 mg every 2 weeks or 50 mg every 4 weeks to patients with moderate-to-severe active RA refractory to conventional DMARDs, including MTX. Approximately one-third of enrolled patients in this large, global study were previously treated with biological DMARD therapy (noting that these patients could not have failed for safety or efficacy reasons) and over two-thirds had prior treatment with ≥2 conventional DMARDs.
All clinical efficacy endpoints demonstrated that sirukumab was effective at reducing signs and symptoms of active RA in a robust and rapid manner through 52 weeks. Improvements occurred as early as 2 weeks in patients treated with sirukumab who demonstrated an ACR20 response; responses plateaued at week 12 and were maintained through week 52. The clinical findings were supported by robust effects on structural damage inhibition at week 52. Significant inhibition of radiographic progression was observed with sirukumab as early as week 24 (the first timepoint assessed), and a significantly greater proportion of patients treated with sirukumab showed no progression compared with placebo at week 52. Positive clinical and radiographic effects were consistently associated with significant patient-reported improvements in physical and emotional health and functional status. In a phase IIb trial17 and in the current phase III study, clinical efficacy was largely similar between the 100 mg every 2 weeks and 50 mg every 4 weeks sirukumab doses, suggesting that the two doses do not differ in their effectiveness.
The safety profile of sirukumab did not raise any new concerns and was consistent with those reported for agents targeting the IL-6 receptor, such as tocilizumab18 19 and sarilumab.20 The proportions of patients experiencing AEs and SAEs were relatively similar between treatment groups, and the types of AEs and SAEs were similar through the 52-week study period. The most common AEs were elevated liver enzymes and injection site erythema, which was the only AE that was more frequent with sirukumab 100 mg every 2 weeks compared with 50 mg every 4 weeks. ISRs were all considered mild or moderate in severity and led to few discontinuations. No serious opportunistic infections were reported. Laboratory abnormalities included neutropaenia, thrombocytopaenia and increased levels of liver transaminases and lipids, all of which have been reported as class effects of anti–IL-6 therapies. There was no evidence of a dose response for sirukumab in these laboratory abnormalities, except for liver transaminases.
This study included a population of patients with RA who were refractory to DMARDs and who may or may not have received prior biological therapy. The results of this study are therefore not applicable to the full spectrum of patients with RA, but provide important information on the use of anti–IL-6 therapy as a possible first-line or alternate biological therapy in patients who are no longer responding to conventional DMARDs. Use of sirukumab in patients who cannot tolerate or are no longer responding to biological DMARDs was demonstrated in the SIRROUND-T study.14 The current study design led to loss of randomisation after the 18-week pure placebo-controlled period and, therefore, longer total exposure in patient-years to sirukumab relative to placebo, which confounded interpretation of safety comparisons between sirukumab-treated and placebo-only patients beyond week 18. The safety of sirukumab continues to be assessed in the long-term extension study.
In conclusion, in patients with active RA refractory to DMARDs, sirukumab 100 mg every 2 weeks and 50 mg every 4 weeks led to significant reductions in signs and symptoms of RA, improvement of physical function, inhibition of structural damage progression and improvement of quality of life. Both sirukumab dose regimens were similarly efficacious, and sirukumab also demonstrated an acceptable safety profile.
annrheumdis-2017-211328supp005.jpg (262.5KB, jpg)
{kind=link}
annrheumdis-2017-211328supp006.jpg (254.9KB, jpg)
{kind=link}
annrheumdis-2017-211328supp007.jpg (248.6KB, jpg)
{kind=link}
annrheumdis-2017-211328supp008.jpg (255.4KB, jpg)
{kind=link}
Footnotes
Handling editor: Tore K Kvien
Contributors: TT, CT, GK, SS, WX, RR, BH and PPT contributed to the study design. TT and KF contributed to patient recruitment and treatment. TT, CT, GK, WX, RR, KF and BH contributed to the conduct of the study. SS and KF collected the data. CT, GK, SS, WX, RR, KF, BH and PPT analysed the data. All authors interpreted the data, contributed to drafting the work or revising it critically for important intellectual content, provided final approval of the version published and agreed to be accountable for all aspects of the work.
Funding: This study was sponsored by Janssen Research & Development, LLC, in collaboration with GlaxoSmithKline. Writing and editorial support were provided by Allison Michaelis, PhD, of MedErgy, and were funded by Janssen Global Services, LLC and GlaxoSmithKline.
Competing interests: TT: grant/research support from Astellas Pharma, Bristol-Myers K.K., Chugai Pharmaceutical Co., Ltd., Daiichi Sankyo Co., Ltd., Eisai Co., Ltd., Mitsubishi Tanabe Pharma Co., Pfizer Japan Inc., Santen Pharmaceutical Co., Ltd., Takeda Pharmaceutical Co., Ltd., Teijin Pharma Ltd., AbbVie GK, Asahi Kasei Pharma Corp., Taisho Toyama Pharmaceutical Co., Ltd. and SymBio Pharmaceuticals Ltd; consultant for AstraZeneca K.K., Eli Lilly Japan K.K., Novartis Pharma K.K., Mitsubishi Tanabe Pharma Co., Asahi Kasei Medical K.K., AbbVie GK, Daiichi Sankyo Co., Ltd., Bristol-Myers K.K. and Nipponkayaku Co. Ltd.; speakers bureau for AbbVie GK, Bristol-Myers K.K., Chugai Pharmaceutical Co., Ltd., Eisai Co., Ltd., Janssen Pharmaceutical K.K., Mitsubishi Tanabe Pharma Co., Pfizer Japan Inc., Takeda Pharmaceutical Co., Ltd., Astellas Pharma, Daiichi Sankyo Co., Ltd., Celtrion and Nipponkayaku Co. Ltd. CT: grant/research support from Celgene, Novartis and Pfizer; advisory board and consultant for AbbVie, Amgen, Celgene, Hospira, Lilly, Novartis and Pfizer; steering committee for Janssen/Centocor/Johnson & Johnson. GK: grant/research support from Pfizer; consultant for Bristol-Myers Squibb, Genentech, GlaxoSmithKline, Janssen, Pfizer, Regeneron, Roche and Sanofi; speakers bureau for Bristol-Myers Squibb, Genentech, GlaxoSmithKline, Janssen, Pfizer, Regeneron, Roche and Sanofi. SS, WX, KF and BH: employees and shareholders of Janssen Research & Development, LLC. RR and PPT: employees and shareholders of GlaxoSmithKline.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1.Kishimoto T. Interleukin-6: from basic science to medicine-40 years in immunology. Annu Rev Immunol 2005;23:1–21. 10.1146/annurev.immunol.23.021704.115806 [DOI] [PubMed] [Google Scholar]
- 2.Tak PP, Smeets TJ, Daha MR, et al. . Analysis of the synovial cell infiltrate in early rheumatoid synovial tissue in relation to local disease activity. Arthritis Rheum 1997;40:217–25. 10.1002/art.1780400206 [DOI] [PubMed] [Google Scholar]
- 3.Arend WP, Dayer JM. Cytokines and cytokine inhibitors or antagonists in rheumatoid arthritis. Arthritis Rheum 1990;33:305–15. 10.1002/art.1780330302 [DOI] [PubMed] [Google Scholar]
- 4.Guerne PA, Zuraw BL, Vaughan JH, et al. . Synovium as a source of interleukin 6 in vitro. Contribution to local and systemic manifestations of arthritis. J Clin Invest 1989;83:585–92. 10.1172/JCI113921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Flannery CR, Little CB, Hughes CE, et al. . IL-6 and its soluble receptor augment aggrecanase-mediated proteoglycan catabolism in articular cartilage. Matrix Biol 2000;19:549–53. 10.1016/S0945-053X(00)00111-6 [DOI] [PubMed] [Google Scholar]
- 6.Xu Z, Bouman-Thio E, Comisar C, et al. . Pharmacokinetics, pharmacodynamics and safety of a human anti-IL-6 monoclonal antibody (sirukumab) in healthy subjects in a first-in-human study. Br J Clin Pharmacol 2011;72:270–81. 10.1111/j.1365-2125.2011.03964.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nemeth E, Rivera S, Gabayan V, et al. . IL-6 mediates hypoferremia of inflammation by inducing the synthesis of the iron regulatory hormone hepcidin. J Clin Invest 2004;113:1271–6. 10.1172/JCI200420945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ishihara K, Hirano T. IL-6 in autoimmune disease and chronic inflammatory proliferative disease. Cytokine Growth Factor Rev 2002;13:357–68. 10.1016/S1359-6101(02)00027-8 [DOI] [PubMed] [Google Scholar]
- 9.Emery P, Keystone E, Tony HP, et al. . IL-6 receptor inhibition with tocilizumab improves treatment outcomes in patients with rheumatoid arthritis refractory to anti-tumour necrosis factor biologicals: results from a 24-week multicentre randomised placebo-controlled trial. Ann Rheum Dis 2008;67:1516–23. 10.1136/ard.2008.092932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jones G, Sebba A, Gu J, et al. . Comparison of tocilizumab monotherapy versus methotrexate monotherapy in patients with moderate to severe rheumatoid arthritis: the AMBITION study. Ann Rheum Dis 2010;69:88–96. 10.1136/ard.2008.105197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nishimoto N, Hashimoto J, Miyasaka N, et al. . Study of active controlled monotherapy used for rheumatoid arthritis, an IL-6 inhibitor (SAMURAI): evidence of clinical and radiographic benefit from an x ray reader-blinded randomised controlled trial of tocilizumab. Ann Rheum Dis 2007;66:1162–7. 10.1136/ard.2006.068064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Smolen JS, Beaulieu A, Rubbert-Roth A, et al. . Effect of interleukin-6 receptor inhibition with tocilizumab in patients with rheumatoid arthritis (OPTION study): a double-blind, placebo-controlled, randomised trial. Lancet 2008;371:987–97. 10.1016/S0140-6736(08)60453-5 [DOI] [PubMed] [Google Scholar]
- 13.Genovese MC, Fleischmann R, Kivitz AJ, et al. . Sarilumab plus methotrexate in patients with active rheumatoid arthritis and inadequate response to methotrexate: results of a phase III study. Arthritis Rheumatol 2015;67:1424–37. 10.1002/art.39093 [DOI] [PubMed] [Google Scholar]
- 14.Aletaha D, Bingham CO, Tanaka Y, et al. . Efficacy and safety of sirukumab in patients with active rheumatoid arthritis refractory to anti-TNF therapy (SIRROUND-T): a randomised, double-blind, placebo-controlled, parallel-group, multinational, phase 3 study. Lancet 2017;389:1206–17. 10.1016/S0140-6736(17)30401-4 [DOI] [PubMed] [Google Scholar]
- 15.Takeuchi T, Tanaka Y, Yamanaka H, et al. . Efficacy and safety of olokizumab in Asian patients with moderate-to-severe rheumatoid arthritis, previously exposed to anti-TNF therapy: Results from a randomized phase II trial. Mod Rheumatol 2016;26:15–23. 10.3109/14397595.2015.1074648 [DOI] [PubMed] [Google Scholar]
- 16.Weinblatt ME, Mease P, Mysler E, et al. . The efficacy and safety of subcutaneous clazakizumab in patients with moderate-to-severe rheumatoid arthritis and an inadequate response to methotrexate: results from a multinational, phase IIb, randomized, double-blind, placebo/active-controlled, dose-ranging study. Arthritis Rheumatol 2015;67:2591–600. 10.1002/art.39249 [DOI] [PubMed] [Google Scholar]
- 17.Smolen JS, Weinblatt ME, Sheng S, et al. . Sirukumab, a human anti-interleukin-6 monoclonal antibody: a randomised, 2-part (proof-of-concept and dose-finding), phase II study in patients with active rheumatoid arthritis despite methotrexate therapy. Ann Rheum Dis 2014;73:1616–25. 10.1136/annrheumdis-2013-205137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fleischmann RM, Halland AM, Brzosko M, et al. . Tocilizumab inhibits structural joint damage and improves physical function in patients with rheumatoid arthritis and inadequate responses to methotrexate: LITHE study 2-year results. J Rheumatol 2013;40:113–26. 10.3899/jrheum.120447 [DOI] [PubMed] [Google Scholar]
- 19.Kivitz A, Olech E, Borofsky M, et al. . Subcutaneous tocilizumab versus placebo in combination with disease-modifying antirheumatic drugs in patients with rheumatoid arthritis. Arthritis Care Res 2014;66:1653–61. 10.1002/acr.22384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huizinga TW, Fleischmann RM, Jasson M, et al. . Sarilumab, a fully human monoclonal antibody against IL-6Rα in patients with rheumatoid arthritis and an inadequate response to methotrexate: efficacy and safety results from the randomised SARIL-RA-MOBILITY Part A trial. Ann Rheum Dis 2014;73:1626–34. 10.1136/annrheumdis-2013-204405 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
annrheumdis-2017-211328supp002.jpg (240.9KB, jpg)
annrheumdis-2017-211328supp001.docx (288.8KB, docx)
annrheumdis-2017-211328supp003.jpg (212.8KB, jpg)
annrheumdis-2017-211328supp004.jpg (254.6KB, jpg)
annrheumdis-2017-211328supp005.jpg (262.5KB, jpg)
annrheumdis-2017-211328supp006.jpg (254.9KB, jpg)
annrheumdis-2017-211328supp007.jpg (248.6KB, jpg)
annrheumdis-2017-211328supp008.jpg (255.4KB, jpg)