

Abstract
Restrictive cardiomyopathy (RCM) is the least common among cardiomyopathies. It can be idiopathic, familial, or secondary to systematic disorders. Marked increase in left and/or right ventricular filling pressures causes symptoms and signs of congestive heart failure. Electrocardiographic findings are nonspecific and include atrioventricular conduction and QRS complex abnormalities and supraventricular and ventricular arrhythmias. Echocardiography and cardiac magnetic resonance (CMR) play a major role in diagnosis. Echocardiography reveals normal or hypertrophied ventricles, preserved systolic function, marked biatrial enlargement, and impaired diastolic function, often with restrictive filling pattern. CMR offering a higher spatial resolution than echocardiography can provide detailed information about anatomic structures, perfusion, ventricular function, and tissue characterization. CMR with late gadolinium enhancement (LGE) and novel approaches (myocardial mapping) can direct the diagnosis to specific subtypes of RCM, depending on the pattern of scar formation. When noninvasive studies have failed, endomyocardial biopsy is required. Differentiation between RCM and constrictive pericarditis (CP), nowadays by echocardiography, is important since both present as heart failure with normal-sized ventricles and preserved ejection fraction but CP can be treated by means of anti-inflammatory and surgical treatment, while the treatment options of RCM are dictated by the underlying condition. Prognosis is generally poor despite optimal medical treatment.
1. Introduction
According to a recent position statement of the European Society of Cardiology (ESC) working group on myocardial and pericardial diseases, cardiomyopathy is defined as a myocardial disorder in which there is abnormality in the structure and function of the myocytes, in the absence of coronary artery disease, hypertension, valvular disease, and congenital heart disease that could justify this abnormality. In the past, cardiomyopathies were also separated from systematic disorders that could involve the myocardium. ESC classification proposes a shift from diagnosing by exclusion criteria and focusing on the morphology and function of the heart [1].
Restrictive cardiomyopathy (RCM) is the least common of cardiomyopathies. It is characterized by impaired diastolic function with restrictive filling and reduced diastolic volume of either or both ventricles, preserved systolic function, and invariably normal or mildly increased wall thickness [2]. Due to increased myocardial stiffness, a small increase in volume leads to a precipitous rise of pressure within the ventricle with an accentuated filling in early diastole, which ceases abruptly at the end of the rapid filling phase showing the characteristic “dip-and-plateau” pattern [3] during cardiac catheterization.
There are many classification criteria for the causes of RCM. According to the ESC Working group in myocardial and pericardial disease RCM can be idiopathic, familial (autosomal dominant, autosomal recessive or X-linked inheritance), or secondary to systematic disorders [1] (Table 1). For the purposes of this review, we will refer to the most common of them.
Table 1.
Causes of restrictive cardiomyopathy.
| (1) Amyloidosis (AL, ATTR, SSA) |
| (2) Sarcoidosis |
| (3) Hemochromatosis |
| (4) Eosinophilic myocardial disease |
| (5) Idiopathic RCM |
| (6) Progressive systemic sclerosis (scleroderma) |
| (7) Postradiation therapy (Hodgkin's lymphoma, breast cancer etc) |
| (8) Anderson Fabry disease |
| (9) Danon's disease |
| (10) Friedreich's ataxia |
| (11) Diabetic cardiomyopathy (restrictive phenotype) |
| (12) Drug induced (anthracycline toxicity, methysergide, ergotamine, mercurial agents, etc.) |
| (13) Mucopolysaccharidoses (Hurler's cardiomyopathy) |
| (14) Myocardial oxalosis |
| (15) Wegener's granulomatosis |
| (16) Metastatic malignancies |
2. Clinical Presentation and Physical Examination
Patients commonly present with dyspnea, fatigue and limited exercise capacity, palpitations, and syncope, while angina may also occur. Others may present with thromboembolic complications. Physical examination may reveal signs of congestive heart failure (distended jugular veins occasionally with Kussmaul sign, peripheral bilateral edema, respiratory rales, hepatomegaly, ascites, and S3 and S4 gallops). The ECG may reveal sinus rhythm with signs of left or biatrial enlargement or atrial fibrillation. There may be nonspecific ST and T wave abnormalities. QRS voltage may be low in the precordial leads and conductive disturbances may also occur, particularly in infiltrative diseases. Specific ECG findings may occur depending on the underlying cause and aid in the diagnostic work-up.
3. Diagnosis
3.1. Laboratory Tests
A complete evaluation should be performed as restrictive cardiomyopathy may originate from systematic disorders or can in turn affect other organs as heart failure develops. Hematocrit (HCT), serum electrolytes, blood urea nitrogen (BUN), creatinine, 24 hr urine total protein, and liver function should be assessed. Arterial blood gas (ABG) should also be obtained to monitor hypoxia. Serum brain natriuretic peptide (BNP) or N-terminal pro-b-type natriuretic peptide (NT-proBNP) and troponin T are indicative of the heart failure and in many cases predictors of survival [5–7]. Specific disorders will require more sophisticated exams (angiotensin converting enzyme (ACE) in sarcoidosis [8], complete blood count (CBC) with peripheral smear helping to establish eosinophilia [9] in hypereosinophilic syndromes, serum iron concentrations, total iron-binding capacity and ferritin levels in hemocromatosis, immunoglobulin free light κ, λ chain testing, and serum and urine immunofixation in amyloidosis, etc.).
3.2. Echocardiography
Echocardiography is the first imaging modality for the assessment of patients with dyspnea and/or heart failure. In patients with RCM, it usually discloses nonspecific findings such as normal (not dilated) ventricles with normal or increased wall thickness (Figures 1(a) and 1(b)) although it can occasionally give specific clues to the diagnosis. Transmitral spectral Doppler often shows restrictive filling pattern (accentuated early diastolic velocity with low or absent late filling velocity with E/A ratio > 2, E wave deceleration time < 150 ms, and short isovolumic relaxation time < 60 ms, Figure 1(c)) although this is commonly a sign of advanced myocardial involvement. There is marked left or biatrial dilatation usually as a consequence of chronically elevated filling pressures (Figures 1(a) and 1(b)). Although classically systolic function as expressed by means of ejection fraction (EF) is normal or near normal, novel techniques like tissue Doppler and speckle tracking echocardiography (STE) reveal the presence of latent systolic myocardial impairment which may be specific to the disease state. Tissue Doppler of the mitral annulus shows reduced systolic waves (s wave), blunted early diastolic waves, and preserved or blunted late diastolic waves, depending on the degree of atrial involvement in the myopathic process [10, 11] (Figures 1(d) and 1(e)).
Figure 1.

Echocardiographic images from a patient with restrictive cardiomyopathy. Two-dimensional 4-chamber view in diastole (a) and systole (b), showing normal left ventricular volume, wall thickness, and systolic function (EF 62%). There is marked biatrial enlargement. Pulse wave Doppler from the left ventricular inflow (c) showing restrictive filling pattern with an E wave velocity of 1 m/sec, an A wave velocity of 0.4 m/sec, and an E wave deceleration time of 145 msec. Spectral tissue Doppler from the lateral (d) and septal (e) mitral annulus. There is marked reduction in systolic annular velocities indicative of latent systolic dysfunction. E/e′ (e′ measured as the average between the two annular e′ velocities) is 16, indicative of increased left ventricular filling pressure. Note the marked reduction in lateral a′ velocity (<4 cm/sec) indicative of left atrial systolic dysfunction.
One of the main utilities of echocardiography is the differential diagnosis between constrictive pericarditis (CP) and RCM. Both present as heart failure with normal-sized ventricles and preserved EF, dilated atria, and Doppler findings of increased filling pressure (often restrictive filling pattern). Differential diagnosis is important since CP can be treated by means of anti-inflammatory drugs or surgery while the treatment options of RCM are dictated by the underlying condition. For the main differences between those disease states, the reader can refer to Table 2 and recent research findings [12, 13].
Table 2.
Differential diagnosis between restrictive cardiomyopathy and constrictive pericarditis.
| Clinical and investigation features | Restrictive cardiomyopathy | Constrictive pericarditis |
|---|---|---|
| History | Systemic disease (e.g., sarcoidosis, hemochromatosis). | Prior history of pericarditis or conditions affecting the pericardium. |
|
| ||
| Physical examination | ± Kussmaul sign, S3 and S4 gallop, murmurs of mitral and tricuspid regurgitation | Pericardial knock |
|
| ||
| Chest X-ray | Atrial dilatation | Pericardial calcification |
|
| ||
| ECG | Low QRS voltages (mainly amyloidosis), conduction disturbances, nonspecific ST abnormalities | Nonspecific ST and T abnormalities, low QRS voltage (<50%) |
|
| ||
| 2D echocardiography | ± Wall and valvular thickening, sparkling myocardium | ± Pericardial thickening, respiratory ventricular septal shift. |
|
| ||
| Doppler echocardiography | Decreased variation in mitral and/or tricuspid inflow E velocity, increased hepatic vein inspiratory diastolic flow reversal, presence of mitral and tricuspid regurgitation | Increased variation in mitral and/or tricuspid inflow E velocity, hepatic vein expiratory diastolic reversal ratio ≥ 0.79 medial e′/lateral e′ ≥ 0.91 (Annulus Reversus) [4] |
|
| ||
| Catheterization hemodynamics | LVEDP – RVEDP ≥ 5 mmHg RVSP ≥ 55 mmHg RVEDP/RVSP ≤ 0.33 |
LVEDP – RVEDP < 5 mmHg RVSP < 55 mmHg RVEDP/RVSP > 0.33 Inspiratory decrease in RAP < 5 mmHg Systolic area index > 1.1 (Ref CP in the modern era) Left ventricular height of rapid filling wave > 7 mmHg |
|
| ||
| CT | Normal pericardium | Thickened/calcified pericardium |
|
| ||
| MRI | Measurement of iron overload, various types of LGE (late gadolinium enhancement) | Thickened pericardium |
|
| ||
| Biopsy | May reveal underlying cause. | Normal myocardium |
3.3. Cardiac Magnetic Resonance
Echocardiography is unable to establish the definite diagnosis of specific RCM subtypes due to its inherent poor tissue characterization and limited assessment of the ventricular apex and the right ventricle. Cardiac Magnetic Resonance (CMR) with a higher spatial resolution and late gadolinium enhancement (LGE) can provide detailed information about anatomic structures, perfusion, ventricular function, and tissue characterization [14]. Late gadolinium enhancement (LGE) depending on the pattern of scar formation can direct the diagnosis to specific subtypes of RCM. Finally it can accurately measure pericardial thickness [15, 16] and visualize pericardial inflammation [17], aiding in the diagnosis of CP.
3.4. Cardiac CT
As of this writing, Cardiac CT has no specific role in the work-up of patients with RCM. It can accurately exclude the presence of coronary obstructive artery disease in patients with angina symptom. Its main role is in the differential diagnosis between RCM and CP where it can give invaluable information about the thickness and composition of the pericardium [18].
3.5. Invasive Catheterization
In contemporary practice, invasive catheterization and coronary angiography are rarely needed for the establishment of diagnosis. In cases where chest pain is the predominant symptom, it can reveal the presence or absence of obstructive coronary artery disease. Left and right heart catheterization can be used when findings of noninvasive tests, including the differential diagnosis between CP and RCM, are nonconclusive [19].
3.6. Cardiac Biopsy
Histological examination can be valuable in setting a definite diagnosis when noninvasive studies have failed, although there are periprocedural risks of endomyocardial biopsy (EMB) [20]. Moreover, in diseases where there is focal involvement of the myocardium (e.g., sarcoidosis), biopsy may miss the area of lesion, giving false negative findings.
4. Prognosis
Prognosis varies among the specific types of RCM but is generally poor with progressive deterioration, particularly in children, despite optimal medical treatment.
5. Specific Types of Restrictive Cardiomyopathy
5.1. Cardiac Amyloidosis
This is the most common cause of RCM. It can be either systemic or localized to the heart muscle. It is characterized by deposition of amyloid fibrils in the extracellular space. These fibrils are culprit proteins misfold into β-pleated sheets antiparallel to one another, resistant to proteolysis, and they cause oxidative stress, disruption of the myocardium, and myocardial damage [21, 22]. There are many proteins that can form amyloid but not all of them affect the heart. The main difference among them is the origin of the protein formation, the percentage of heart involvement, and the rate of progress. Amyloid stains pink with hematoxylin and eosin and demonstrates apple-green birefringence when viewed under polarized light.
The deposition begins in the subendocardium and extends within the myocardium between the muscle fibres. Rarely, the pericardium may be the primary site of amyloid deposition without any endomyocardial involvement [23]. Increased wall thickness is therefore the result of interstitial deposition and not of hypertrophied myocardial cells, in contrast to hypertensive heart disease or hypertrophic cardiomyopathy (HCM). Small intramyocardial vessels may also be infiltrated leading to anginal symptoms while epicardial arteries are angiographically normal.
The conductive system is also affected causing several degrees of atrioventricular (AV) block. Other characteristic ECG abnormalities are low voltage QRS complexes which, in combination with the finding of increased wall thickness in echocardiography, can differentiate this disease from HCM. These findings are more commonly seen in light chain amyloid (AL) [24] than in familial transthyretin-related amyloidosis (ATTR) [25] subtype. Other ECG findings include a pseudoinfarct pattern (poor R wave progression in precordial leads), intraventricular conduction delay, and atrial arrhythmias while ventricular arrhythmias are rare [26–30] (Figure 2).
Figure 2.

ECG from a patient with cardiac amyloidosis. There are low voltage QRS complexes with left axis deviation and marked 1st-degree atrioventricular block.
Echocardiographic amyloidosis may present with biventricular hypertrophy, although up to 1/3 of cases may have normal wall thickness. The constellation of low voltage QRS complexes with hypertrophy, often with a restrictive filling pattern, is a classic feature but it indicates advanced disease. Left ventricular EF is commonly preserved in the early stages of the disease process as it is primary calculated from the contraction of the heart along its short axis, while the subendocardial myocytes that are prone to damage are longitudinally oriented and therefore the longitudinal contraction will be impaired [31]. The endocardium may also be infiltrated and there may be thrombi. A granular, speckled appearance of the ventricular myocardium, (Figure 3) while nonspecific (also seen in glycogen storage disease, HCM, Anderson-Fabry disease, hypertensive heart disease, and end-stage renal disease) [32], combined with clinical suspicion and other laboratory findings, is suggestive of cardiac amyloidosis.
Figure 3.

Parasternal long (a) and short axis (b) view from the patient whose ECG appears in Figure 2. There is marked LV hypertrophy. Also note the granular, speckled myocardial appearance.
Atrioventricular valves, if infiltrated, may be thickened with mild to moderate regurgitation. Mild or moderate pericardial effusion may be present. Thickening of the interatrial septum without sparing of the area of the fossa ovalis is characteristic of the disease but apparent only in late stages [33].
Speckle tracking echocardiography, with the ability of measuring novel indices of deformation, has expanded its role in the diagnosis. It has been found that in cardiac amyloidosis systolic longitudinal strain (LSsys) is impaired in the mid and basal intraventricular septum compared to the apex (Figure 4). In a recent study, septal apical to basal LSsys ratio > 2.1 combined with deceleration time of early filling < 200 msec differentiated amyloidosis from other forms of LV hypertrophy with a sensitivity of 88%, specificity of 85%, positive predictive value of 67%, and negative predictive value of 96% [34] (Figure 4).
Figure 4.
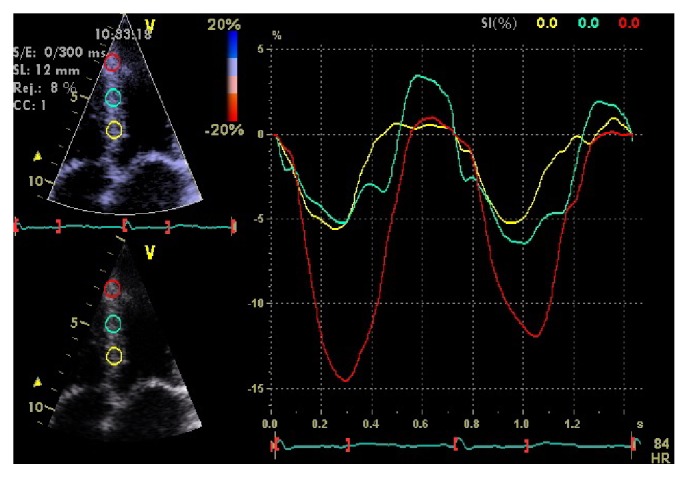
Strain curves from the interventricular septum of the same patient appearing in Figures 2 and 3. In this example, strain curves were constructed with the Doppler Myocardial Imaging technique. There is reduced peak systolic strain of the basal (yellow curve) and mid (green curve) septal segments, compared to the apical segment (red curve) with an apex to base ratio > 2.1.
CMR will show diffuse LGE throughout both ventricles, particularly the subendocardium with a characteristic zebra-stripe appearance of the subendocardial enhancement of the LV and RV endocardium, sparing the mid-wall of the interventricular septum [35]. Sometimes there is a patchy transmural pattern [36–38]. There is a faster contrast washout from myocardium and blood pool than normal. Enhancement of the atrial wall has also been reported [39] (Figure 5). The technique of T1 mapping may reveal cardiac involvement at an earlier stage compared to overt LGE images [40] and may also prove useful in patients with end-stage renal dysfunction where the administration of gadolinium based contrast is contraindicated due to the risk of nephrogenic systemic sclerosis. Recent studies have shown high sensitivity of the technique in the diagnosis of AL [41] and its ability to differentiate between AL and ATTR [42]. Moreover the combination of T1 mapping with LGE may have prognostic implications [43].
Figure 5.

CMR short axis (a) and four-chamber view (b). LGE images showing diffuse, nonhomogenous myocardial enhancement involving both ventricles and atria. The pattern of enhancement is consistent with cardiac amyloidosis. Note the presence of a right atrial thrombus. Images courtesy of Professor Dr. Jan Bogaert, University Hospital Leuven.
Radionuclide imaging using single-photon emission computed tomography (SPECT), or positron emission tomography (PET), has been recently introduced in the diagnostic work-up. An intense myocardial uptake of imaging agents Tc-99m pyrophosphate (PYP, available in the USA) or Tc-99m 3,3-diphosphono-1,2-propanodicarboxylic acid (DPD, available in Europe) identifies patients with ATTR and can differentiate them from those with AL [44, 45]. A recent study has shown that Tc-99m DPD uptake is an independent predictor of adverse events [46].
Histological confirmation is mandatory for the diagnosis. Renal, myocardial, or liver biopsy is invasive, expensive with high periprocedural risks [47, 48]. Preferably biopsy can be obtained by the iliac crest bone marrow [49] combined with subcutaneous abdominal fat aspiration [50] which will identify amyloidosis in 85% of cases [51]. If negative, biopsy directly from an involved organ should be obtained. A negative myocardial biopsy in suspected cases practically excludes cardiac amyloidosis, since myocardial involvement is usually widespread. In a positive examination though it is crucial to determine the type of amyloid as this in turn defines treatment and prognosis.
5.1.1. Light Chain (AL) Amyloidosis
AL amyloidosis (formerly called primary) is the commonest and most aggressive form. Amyloid is derived from monoclonal light chains (or chain fragments) secondary to a plasma cell disorder (produced at the bone marrow). The most common plasma cell disorder is multiple myeloma; however only a minority of these patients will develop amyloidosis and vice versa. Cardiac deposition occurs in almost every case of biopsy or autopsy while clinical involvement is found in about half of the cases and has a major prognostic implication [30] with a median survival as low as 4 months [52].
The clinical presentation is that described generally for RCM above. Extracardiac organs infiltrated are the kidneys, skin, liver, peripheral, and autonomic nervous system and symptoms may rise, respectively.
Treatment must combine the underlying plasma dyscrasia and the subsequent heart failure, though patients with amyloidosis respond minimally to conventional heart failure treatment. There is no evidence of beneficial effect of beta blockers, except in atrial fibrillation for relative rate control; angiotensin converting enzyme (ACE) inhibitors and angiotensin receptor blockers (ARBs) are difficultly tolerated because of hypotension which is almost always present (in grounds of low cardiac output and autonomic neuropathy). Calcium channel blockers are contraindicated as they may produce a negative inotropic effect. Cautious titration to diuretics is the most effective treatment. Digoxin should only be used for rate control in atrial fibrillation with extreme caution since the risk of toxicity is high due to possible binding of the drug to amyloid fibrils. Melphalan plus dexamethasone supported with autologous stem-cell transplantation [31, 51] is considered a standard treatment which can increase the median survival. Novel agents such as thalidomide, lenalidomide, and bortezomib are also promising but most of the trials are still ongoing. Recently the results of a phase 1 trial involving the use of a drug called (R)-1-[6-[(R)-2-carboxy-pyrrolidin-1-yl]-6-oxo-hexanoyl]pyrrolidine-2-carboxylic acid (CPHPC) have been reported. The drug succeeded in triggering clearance of amyloid deposits in patients with systemic amyloidosis. Future trials will test its effectiveness in cardiac amyloidosis [53]. The ideal treatment would be a heart transplantation (though most of the patients are unsuitable by precise criteria suggested) with autologous stem-cell transplantation since the underlying systemic disorder still exists.
5.1.2. Familial (ATTR) Amyloidosis
ATTR amyloidosis originates from a number of DNA mutations (autosomal dominant inheritance, more than 100 mutations) of the transthyretin (TTR) protein which is synthesized in the liver. The cardiac deposits vary depending on the mutation and the phenotype may be from exclusive neuropathy to cardiomyopathy or combination of the two [54]. Carpal tunnel syndrome may be an early indicator. Comparatively to AL amyloidosis ATTR has a more favorable prognosis of 3–5 years as it has a slower clinical course. Heart failure symptoms are treated with beta blockers and ACE inhibitors which are better tolerated than in AL amyloidosis. Liver transplantation is effective in reversing clinical disease in patients with TTR Val30Met mutation but it can accelerate progression of heart disease in the rest. Similar to AL amyloidosis the ideal treatment would be heart transplantation (neuropathy is a major contraindication) along with liver transplantation preferably at an early stage.
5.1.3. Senile Systemic Amyloidosis (SSA)
SSA amyloidosis, formerly called wild type amyloidosis, is an exclusive cardiac disease with carpal tunnel syndrome being the only extracardiac manifestation. SSA has a more indolent course than AL with a median survival of 4–6 years after diagnosis [55] and is seen almost always in elderly males. Treatment is currently palliative, with diuretics being the cornerstone of therapy and as the clinical expression is mild, ACE inhibitors or ARBs, beta blockers, or nitrates are generally well tolerated. If atrial fibrillation occurs oral anticoagulants are indicated. Amiodarone is preferred over other antiarrhythmic agents to maintain sinus rhythm. If permanent pacemaker is needed because of high degree AV block, biventricular pacing should be considered as right ventricular pacing may worsen an already affected cardiac output.
Less common forms of amyloidosis are the AA (formerly called secondary) which is a result of chronic inflammatory conditions affecting mostly the liver and kidneys with less than 2% of the cases involving the heart and the isolated atrial amyloid where atrial natriuretic peptide (ANP) is synthesized by atrial myocytes and can be deposited as amyloid and is nowadays considered part of the ATTR or SSA subtypes.
5.2. Sarcoidosis
Sarcoidosis is a systematic noncaseating granulomatous disorder affecting several tissues. Most commonly the respiratory system is affected and patients typically present with hilar lymphadenopathy on chest X-ray. Skin, gastrointestinal, ocular, and nervous system involvement follows in frequency. Cardiac sarcoidosis (CS) is clinically present in about 5% of patients, although an autopsy study showed a prevalence of cardiac involvement in approximately 25% of cases [56] which portends worse prognosis [57].
Sarcoid granulomas consist of macrophages and epithelioid histiocytes and lymphocytes [58]. There are three histological stages: edema, granulomatous infiltration, and fibrosis but the latest is the most severe, and its presence is an independent predictor of mortality [59]. Cardiac granulomas are usually found in the basal septum, AV node, bundle of His, the ventricular free walls, and papillary muscles [60].
Blood test abnormalities are nonspecific but may reveal raised levels of ACE because it is produced by noncaseating granulomas. ECG findings include conduction abnormalities, ventricular arrhythmias (including ventricular tachycardia), and atypical infarction patterns.
There is a wide spectrum of echocardiographic abnormalities that vary from wall thickening due to edema and infiltration, to wall thinning due to fibrosis. Ventricular chambers may be normal or dilated with regional wall motion abnormalities or globally impaired systolic function. Scar retraction may lead to aneurysm formation, particularly after corticosteroid treatment. Compared to idiopathic dilated and ischemic cardiomyopathy, in CS, normokinetic segments may alternate with affected ones [58] and typically do not show a coronary distribution [61].
CMR may display all three stages of edema, inflammation, and scar formation [62]. LGE is patchy and typically found in the myocardium and epicardium of the basal and lateral LV walls [63] (Figure 6), though subendocardial or transmural hyperenhancement has also been observed, resembling an ischemic pattern [35]. The presence, extent, and location of LGE are predictive for the development of ventricular arrhythmias or death and patient response to corticosteroid treatment [59, 64, 65].
Figure 6.

LGE images of a patient with pulmonary sarcoidosis with cardiac involvement. Four-chamber view (a) showing mid-wall focal enhancement in the lateral wall of the left ventricle and in the apex. Note also the enhancement of the mediastinal lymph nodes. Short axis view (b) demonstrating enhancement of the LVOT and possibly of the RVOT. Images courtesy of Professor Dr. Jan Bogaert, University Hospital Leuven.
Radionuclide imaging with 18fluorodeoxyglucose- (FDG-) PET has also been used in the diagnostic work-up [66]. Perfusion imaging is provided with the tracer rubidium and metabolic activity is assessed according to the degree of 18FDG uptake. Decreased myocardial perfusion with enhanced 18FDG uptake is consistent with inflammation, while decreased perfusion with reduced 18FDG uptake is consistent with scarring. As PET identifies active inflammation earlier than CMR [67], hybrid techniques, combining PET and CMR scanners enables simultaneous acquisition of structural and functional data, therefore increasing the diagnostic accuracy [68].
The first international guidelines for the diagnosis of CS [69] incorporated the two modalities, PET and CMR in the diagnostic algorithm. When clinical presentation along with imaging findings set the suspicion, biopsy from an affected extracardiac tissue should be obtained to make the diagnosis probable, with the exception of Lofgren's syndrome (bilateral hilar lymphadenopathy, arthritis, and erythema nodosum with an incidence of 30% of cases) where biopsy is not needed [60].
Definite diagnosis is posed only by EMB but it is not preferred because of its potential risks and low sensitivity (20–30%) [20] due to the patchy distribution of granulomas. PET or CMR directed cardiac biopsy has an increased sensitivity, but still a negative EMB cannot definitely exclude CS.
Treatment of CS incorporates the guidelines directed medical therapy (GDMT) of heart failure with the addition of corticosteroids to reduce the inflammatory process (except asymptomatic hilar adenopathy where no therapy is required). FDG-PET is suggested in the treatment algorithm for the follow-up [69], defining whether satisfactory response to steroids is achieved or a second-line agent (i.e., methotrexate) should be added. If sustained VT or VF occurs, particularly in grounds of myocardial scarring, implantable cardiac defibrillator (ICD) is indicated.
5.3. Hemochromatosis
Hemochromatosis is a group of disorders which leads to excessive accumulation of iron within the cells of liver, pancreas, heart, and several endocrine glands leading to cirrhosis, diabetes, heart failure, skin pigmentation, and so forth. It may be hereditary or secondary. In the former, properly called hemochromatosis [70], transferrin iron-binding capacity (TIBC) falls short of plasma iron content while erythropoiesis is normal. Secondary, properly called hemosiderosis, is due to increased catabolism of erythrocytes and occurs in patients undergoing frequent blood transfusions (e.g., in thalassemia major). Cardiac involvement develops later in comparison to other organs and defines prognosis. In the early stages, ECG is normal while with advanced disease low voltage QRS complexes with nonspecific ST-T segment abnormalities and supraventricular arrhythmias. Echocardiographic features may be those of dilated or less often RCM and are not specific [71]. Liver biopsy is the gold standard to establish diagnosis in hemochromatosis. Serum ferritin concentration which has been traditionally used for establishing diagnosis is a rough estimation of total body iron load.
Recently CMR with the use of T2 star (T2∗) technique has been used to quantify myocardial iron overload [72]. T2∗ values smaller than 20 msec are predictive of adverse cardiac events [73]. It can also accurately predict the development of heart failure and arrhythmias in patients undergoing repeated transfusions [67]. Some authors advocate its use for the assessment of excess cardiac iron from the early age of 5 years in patients with thalassemia major and suboptimal chelation therapy [74]. Consequently it is regarded the gold standard for follow-up of iron chelation therapy and therapeutic guidance [75, 76].
Treatment by limitation of transfusions, phlebotomy, and chelation can reverse cardiomyopathy in hemosiderosis but liver and/or heart transplantation may be needed in advanced disease and in patients with hemochromatosis [77].
5.4. Eosinophilic Endomyocardial Disease
Eosinophilic infiltration of the heart may be caused by a heterogeneous group of disorders, referred to as hypereosinophilic syndromes (HES) and is an uncommon cause of restrictive cardiomyopathy. HES are either primary, first described by Löffler [78], or secondary (Table 3). Eosinophilia is defined as an eosinophilic count greater than 500 eos/mm3, while the heart is most commonly affected with a count greater than 5000 eos/mm3 [79]. Independent of the underlying causative factor, eosinophilic degranulation causes endocardial damage (Löffler endocarditis) [80]. Primary HES is defined as an eosinophilic count greater than 1500 eos/mm3 for more than 6 months in the absence of a secondary cause without other organ involvement [81, 82].
Table 3.
Causes of eosinophilia.
| Infectious (helminths, HIV, tuberculosis) | Allergic reactions |
| Inflammatory (Churg-Strauss, Crohn's, Wegener, rheumatoid arthritis) | Drug hypersensitivity (NSAIDS, sulfonamides, antimicrobial) |
| Malignancies (Hodgkin lymphoma, Non-Hodgkin lymphoma, acute leukemia) | Idiopathic hypereosinophilic syndrome |
Cardiac infiltration consists of three stages: (a) acute necrotic stage, (b) thrombotic stage, and (c) fibroting stage. The first stage is characterized by inflammation where eosinophilic degranulation releases toxic proteins that induce endocardial necrosis and apoptosis. Rarely, acute necrotizing myocarditis may ensue with high early mortality without appropriate treatment. If left untreated, the first stage evolves into the thrombotic stage due to continuous eosinophilic activation. It consists of thrombus formation over the affected endocardium. Typically there is thrombotic obliteration of left and/or right ventricular apex which may extend to the ventricular outflow tracts, the basal ventricular segments, or even the atria. Thromboembolic complications dominate the clinical picture at this stage. The final stage is due to scarring and irreversible fibrosis of the endocardium (endomyocardial fibrosis, EMF [1]) which may affect the subvalvular apparatus of both atrioventricular valves. Patients at this stage present with advanced heart failure and prognosis is poor.
ECG findings are nonspecific and include sinus tachycardia with conduction disturbances or nonspecific ST-T segment abnormalities. At the first stage, echocardiography may reveal increased LV wall thickness due to interstitial edema, impaired systolic function with abnormal wall kinetics, although most often ventricular systolic function remains preserved. CMR with LGE may additionally reveal endomyocardial involvement, often with patchy distribution [83]. At the second stage, echocardiography shows the presence and location of thrombi while CMR may be more sensitive for thrombus detection [84] and localization. At the third stage, Doppler echocardiography may show echo-dense regions suggestive of myocardial scarring with restrictive filling pattern. CMR, again, is more sensitive and specific for the detection and localization of fibrosis.
The goals of initial therapy are twofold: first to decrease the eosinophil count and attenuate the inflammatory process and second to treat the underlying cause (if any). Corticosteroids are the mainstay of treatment combined with disease specific interventions, for example, antihelminthic therapy for parasitic infections. Anticoagulation should be initiated at the second stage to prevent thromboembolic complications. Once severe EMF has developed, surgical resection with extended endocardial dissection is the treatment of choice but with poor long-term prognosis. Atrioventricular valve replacement should be undertaken at the time of operation if the valves are affected.
5.5. Anderson-Fabry Disease
Anderson-Fabry disease is a rare X-linked recessive lysosomal storage disorder, caused by mutations of the GLA gene that encodes α-galactosidase A which breaks down neutral glycosphingolipids [85]. This results in intracellular accumulation of glycosphingolipids causing cardiac, renal, and cerebrovascular disease. Males are more commonly affected and diagnosed early in childhood (boys at an average age of 5-6 years and girls approximately 3-4 years older) [86–88] while cardiac involvement occurs in adulthood with a median survival of 50 years. Chronic neuropathic pain, typically affecting hands and feet, can be the first symptom. Children can present with poor growth and difficulties following school activities. Main symptoms are gastrointestinal like postprandial diarrhea and abdominal pain, angiokeratomas, lymphedema, and hypohydrosis. Microalbuminuria, proteinuria, and renal failure present late in the disease process and are a major source of morbidity and mortality [89]. Transient ischemic attacks are also likely to occur. Cardiac disease is caused by accumulation of globotriaosylceramide in all cellular components of the heart. The classic phenotype is that of HCM and less often of the restrictive one. ECG may reveal LV hypertrophy, with preexcitation [90] or prolonged [91] PR interval. Arrhythmias may occur since childhood and include bradycardia, supraventricular tachycardia, atrial fibrillation, or atrial flutter. Echocardiographic concentric LV hypertrophy is the most frequent finding with asymmetric septal hypertrophy developing in approximately 5% of patients [92] (Figures 7(a), 7(b), and 7(c)). RV hypertrophy is a common finding (Figure 7(d)). Systolic anterior motion (SAM) of the anterior mitral valve leaflet, with obstruction of the LV outflow tract, may also occur. Systolic function is preserved and diastolic function is impaired but restrictive physiology is rare [93]. CMR reveals a LGE pattern of the basal inferolateral LV segment and typically spares the subendocardium [35]. Diagnosis in males can be set by measuring α-galactosidase A activity or by GLA gene sequencing, while female carriers have a normal enzyme activity and gene sequencing is needed [86].
Figure 7.

Echocardiographic images from a patient with advanced Anderson-Fabry disease and end-stage renal failure. Parasternal long axis (a), short axis (b), and apical four-chamber view (c). There is left ventricular hypertrophy, more pronounced at the interventricular septum. There is also thickening of the right ventricular free wall apparent at the modified apical four-chamber view (d).
Treatment includes GDMT of heart failure (with careful beta blocker use) and enzyme replacement therapy which is effective in myocardial remodeling and improves peak systolic strain and strain rate [94] but its use is limited in advanced cases [95]. Patients should receive genetic counseling. Offsprings of affected families may be screened by means of α-galactosidase A activity or by GLA gene sequencing for males and gene sequencing for females. This can aid in early diagnosis with early implementation of enzyme replacement therapy before overt end-organ damage occurs.
5.6. Diabetic Cardiomyopathy
Diabetic cardiomyopathy (DMCMP) was firstly described as a dilated phenotype with reduced ejection fraction (HFrEF). Recent studies [96, 97] showed a restrictive phenotype as well with preserved systolic function (HFpEF). These distinct patterns have common pathophysiologic mechanisms (autoimmunity, coronary microvascular rarefaction, hyperglycaemia, lipotoxicity, etc.) [98] which seem to be of variable relevance. This distinction is of therapeutic importance as the standard heart failure treatment is of uncertain value for the restrictive DMCMP. Diagnosis of DMCMP with restrictive phenotype (HFpEF) requires exclusion of CAD, valvular, congenital, or hypertensive heart disease, as well as exclusion of infiltrative cardiomyopathies which may require endomyocardial biopsy.
5.7. Idiopathic Restrictive Cardiomyopathy
This is the primary restrictive cardiomyopathy, more commonly affecting women, where hemodynamic abnormalities occur without any specific histological changes and in the absence of ischemic, valvular, and congenital heart disease or hypertension. Patients usually present with overt heart failure. Echocardiographic findings are nonspecific as well, with marked dilation of the atria and normal-sized ventricles with preserved systolic function. Doppler demonstrates elevated LV filling pressures [99] more often with restrictive filling pattern.
5.8. Progressive Systemic Sclerosis (Scleroderma)
Systemic sclerosis is an autoimmune disorder that practically affects almost any organ system. Myocardial fibrosis results in heart failure symptoms, while fibrosis of the conduction system may lead to ventricular arrhythmias and sudden cardiac death. Physical examination depends on the organs involved with cardiac manifestations mainly caused by right heart failure (peripheral bilateral edema and raised jugular venous pressure). Echocardiographic findings are consistent with restrictive physiology.
5.9. Postradiotherapy Cardiomyopathy
Radiotherapy used against thoracic malignancies (Hodgkin's lymphoma, breast cancer, etc.) may provoke several cardiovascular disorders such as coronary artery disease, myocardial fibrosis, valvular heart disease, pericardial disease, and ECG conduction abnormalities. Diffuse interstitial fibrosis after relatively low doses of radiation [100] ultimately leads to decrease in tissue elasticity and distensibility [101] which causes restrictive cardiomyopathy.
5.10. Danon Disease
Danon disease is an X-linked disorder, due to primary deficiency of lysosome-associated membrane protein 2 (LAMP2). Symptoms occur in adolescence and include heart failure, mental retardation, and skeletal myopathy. Most patients die of heart failure in the third decade of life [102]. Laboratory tests reveal creatine kinase (CK) elevation two to three times the normal value in males, liver enzyme (aspartate transaminase (AST), alanine aminotransferase (ALT), and lactate dehydrogenase (LDH)) and serum aldolase elevation in at least one-half of patients. As in Anderson-Fabry disease ECG may reveal normal or increased QRS voltage often with extreme hypertrophy (Sokolov score ≥ 50) [103] and/or preexcitation. Other ECG abnormalities include nonspecific intraventricular conduction delay, variable degrees of atrioventricular block, sinus bradycardia, reentrant atrial, and ventricular tachycardia.
Echocardiography reveals severe hypertrophy of both ventricles, resembling HCM but the electrophysiological abnormalities, particularly ventricular preexcitation, may give a clue for differentiating the two conditions [104]. In Danon disease, CMR may show subendocardial LGE [105]. Definite diagnosis is set by biopsy of skeletal muscle. There is no specific treatment.
5.11. Friedreich Ataxia
It is an autosomal recessive disorder caused by repeated Guanine-Adenine-Adenine triplets in the frataxin gene on chromosome 9 [106]. Manifestations include neuromuscular impairment of the respiratory muscles with frequent respiratory infections and heart failure. Echocardiography reveals increased wall thickness of the intraventricular septum or posterior wall with preserved systolic function [107].
6. Conclusions
Restrictive cardiomyopathies are a group of diseases with various genetic or acquired etiologies. They are characterized by marked increase in ventricular filling pressures which cause symptoms and signs of congestive heart failure. Electrocardiogram, echocardiography, CMR with LGE, or novel radionuclide imaging can direct the diagnosis to specific subtypes of RCM which is important as treatment and prognosis differ significantly, but endomyocardial biopsy is the definite means of establishing the diagnosis. Genetic counseling and familial screening are necessary in hereditary forms.
Acknowledgments
Dr. Georgios Vovas has been supported by a grant from the Hellenic Cardiological Society.
Conflicts of Interest
The authors declare that they have no conflicts of interest.
References
- 1.Elliott P., Andersson B., Arbustini E., et al. Classification of the cardiomyopathies: a position statement from the european society of cardiology working group on myocardial and pericardial diseases. European Heart Journal. 2008;29(2):270–276. doi: 10.1093/eurheartj/ehm342. [DOI] [PubMed] [Google Scholar]
- 2.Ammash N. M., Seward J. B., Bailey K. R., Edwards W. D., Tajik A. J. Clinical profile and outcome of idiopathic restrictive cardiomyopathy. Circulation. 2000;101(21):2490–2496. doi: 10.1161/01.CIR.101.21.2490. [DOI] [PubMed] [Google Scholar]
- 3.Higano S. T., Azrak E., Tahirkheli N. K., Kern M. J. Hemodynamic rounds series II: Hemodynamics of constrictive physiology: Influence of respiratory dynamics on ventricular pressures. Catheterization and Cardiovascular Interventions. 1999;46(4):473–486. doi: 10.1002/(SICI)1522-726X(199904)46:4<473::AID-CCD21>3.0.CO;2-W. doi: 10.1002/(SICI)1522-726X(199904)46:4<473::AID-CCD21>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 4.Welch T. D., Ling L. H., Espinosa R. E., et al. Echocardiographic diagnosis of constrictive pericarditis Mayo Clinic criteria. Circulation: Cardiovascular Imaging. 2014;7(3):526–534. doi: 10.1161/CIRCIMAGING.113.001613. [DOI] [PubMed] [Google Scholar]
- 5.Leya F. S., Arab D., Joyal D., et al. The efficacy of brain natriuretic peptide levels in differentiating constrictive pericarditis from restrictive cardiomyopathy. Journal of the American College of Cardiology. 2005;45(11):1900–1902. doi: 10.1016/j.jacc.2005.03.050. [DOI] [PubMed] [Google Scholar]
- 6.de Lemos J. A., McGuire D. K., Drazner M. H. B-type natriuretic peptide in cardiovascular disease. The Lancet. 2003;362(9380):316–322. doi: 10.1016/s0140-6736(03)13976-1. [DOI] [PubMed] [Google Scholar]
- 7.Dispenzieri A., Gertz M. A., Kyle R. A., et al. Serum cardiac troponins and N-terminal pro-brain natriuretic peptide: a staging system for primary systemic amyloidosis. Journal of Clinical Oncology. 2004;22(18):3751–3757. doi: 10.1200/JCO.2004.03.029. [DOI] [PubMed] [Google Scholar]
- 8.Ainslie G. M., Benatar S. R. Serum angiotensin converting enzyme in sarcoidosis: Sensitivity and specificity in diagnosis: correlations with disease activity, duration, extra-thoracic involvement, radiographic type and therapy. QJM: An International Journal of Medicine. 1985;55(3-4):253–270. doi: 10.1093/oxfordjournals.qjmed.a067873. [DOI] [PubMed] [Google Scholar]
- 9.Rothenberg M. E. Eosinophilia. The New England Journal of Medicine. 1998;338(22):1592–1600. doi: 10.1056/NEJM199805283382206. [DOI] [PubMed] [Google Scholar]
- 10.Liu S., Ma C., Ren W., et al. Regional left atrial function differentiation in patients with constrictive pericarditis and restrictive cardiomyopathy: a study using speckle tracking echocardiography. The International Journal of Cardiovascular Imaging. 2015;31(8):1529–1536. doi: 10.1007/s10554-015-0726-7. [DOI] [PubMed] [Google Scholar]
- 11.Pislaru C., Abraham T. P., Belohlavek M. Strain and strain rate echocardiography. Current Opinion in Cardiology. 2002;17(5):443–454. doi: 10.1097/00001573-200209000-00002. [DOI] [PubMed] [Google Scholar]
- 12.Talreja D. R., Nishimura R. A., Oh J. K., Holmes D. R. Constrictive pericarditis in the modern era: novel criteria for diagnosis in the cardiac catheterization laboratory. Journal of the American College of Cardiology. 2008;51(3):315–319. doi: 10.1016/j.jacc.2007.09.039. [DOI] [PubMed] [Google Scholar]
- 13.Jaber W. A., Sorajja P., Borlaug B. A., Nishimura R. A. Differentiation of tricuspid regurgitation from constrictive pericarditis: Novel criteria for diagnosis in the cardiac catheterisation laboratory. Heart. 2009;95(17):1449–1454. doi: 10.1136/hrt.2008.155523. [DOI] [PubMed] [Google Scholar]
- 14.Quarta G., Sado D. M., Moon J. C. Cardiomyopathies: Focus on cardiovascular magnetic resonance. British Journal of Radiology. 2011;84(3):S296–S305. doi: 10.1259/bjr/67212179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bogaert J., Dymarkowski S., Taylor A. M. Clinical Cardiac MRI. 1st. Berlin, Germany: Springer; 2005. [Google Scholar]
- 16.Misselt A. J., Harris S. R., Glockner J., Feng D., Syed I. S., Araoz P. A. MR Imaging of the Pericardium. Magnetic Resonance Imaging Clinics of North America. 2008;16(2):185–199. doi: 10.1016/j.mric.2008.02.011. [DOI] [PubMed] [Google Scholar]
- 17.Taylor A. M., Dymarkowski S., Verbeken E. K., Bogaert J. Detection of pericardial inflammation with late-enhancement cardiac magnetic resonance imaging: Initial results. European Radiology. 2006;16(3):569–574. doi: 10.1007/s00330-005-0025-0. [DOI] [PubMed] [Google Scholar]
- 18.Bogaert J., Francone M. Pericardial disease: Value of CT and MR imaging. Radiology. 2013;267(2):340–356. doi: 10.1148/radiol.13121059. [DOI] [PubMed] [Google Scholar]
- 19.Sorajja P. Invasive hemodynamics of constrictive pericarditis, restrictive cardiomyopathy, and cardiac tamponade. Cardiology Clinics. 2011;29(2):191–199. doi: 10.1016/j.ccl.2011.01.003. [DOI] [PubMed] [Google Scholar]
- 20.Cooper L. T., Baughman K. L., Feldman A. M., et al. The role of endomyocardial biopsy in the management of cardiovascular disease: a scientific statement from the American Heart Association, the American College of Cardiology, and the European Society of Cardiology. Endorsed by the Heart Failure Society of America and the Heart Failure Association of the European Society of Cardiology. Journal of the American College of Cardiology. 2007;50(19):1914–1931. doi: 10.1016/j.jacc.2007.09.008. [DOI] [PubMed] [Google Scholar]
- 21.Merlini G., Bellotti V. Molecular mechanisms of amyloidosis. The New England Journal of Medicine. 2003;349(6):583–596. doi: 10.1056/nejmra023144. [DOI] [PubMed] [Google Scholar]
- 22.Shah K. B., Inoue Y., Mehra M. R. Amyloidosis and the heart: a comprehensive review. JAMA Internal Medicine. 2006;166(17):1805–1813. doi: 10.1001/archinte.166.17.1805. [DOI] [PubMed] [Google Scholar]
- 23.Singh V., Fishman J. E., Alfonso C. E. Primary systemic amyloidosis presenting as constrictive pericarditis. Cardiology. 2011;118(4):251–255. doi: 10.1159/000329062. [DOI] [PubMed] [Google Scholar]
- 24.Murtagh B., Hammill S. C., Gertz M. A., Kyle R. A., Tajik A. J., Grogan M. Electrocardiographic findings in primary systemic amyloidosis and biopsy-proven cardiac involvement. American Journal of Cardiology. 2005;95(4):535–537. doi: 10.1016/j.amjcard.2004.10.028. [DOI] [PubMed] [Google Scholar]
- 25.Sattianayagam P. T., Hahn A. F., Whelan C. J., et al. Cardiac phenotype and clinical outcome of familial amyloid polyneuropathy associated with transthyretin alanine 60 variant. European Heart Journal. 2012;33(9):1120–1127. doi: 10.1093/eurheartj/ehr383. [DOI] [PubMed] [Google Scholar]
- 26.Lee G. Y., Kim K., Choi J.-O., et al. Cardiac amyloidosis without increased left ventricular wall thickness. Mayo Clinic Proceedings. 2014;89(6):781–789. doi: 10.1016/j.mayocp.2014.01.013. [DOI] [PubMed] [Google Scholar]
- 27.Namdar M., Steffel J., Jetzer S., et al. Value of electrocardiogram in the differentiation of hypertensive heart disease, hypertrophic cardiomyopathy, aortic stenosis, amyloidosis, and fabry disease. American Journal of Cardiology. 2012;109(4):587–593. doi: 10.1016/j.amjcard.2011.09.052. [DOI] [PubMed] [Google Scholar]
- 28.Perlini S., Salinaro F., Cappelli F., et al. Prognostic value of fragmented QRS in cardiac AL amyloidosis. International Journal of Cardiology. 2013;167(5):2156–2161. doi: 10.1016/j.ijcard.2012.05.097. [DOI] [PubMed] [Google Scholar]
- 29.Granstam S.-O., Rosengren S., Vedin O., et al. Evaluation of patients with cardiac amyloidosis using echocardiography, ECG and right heart catheterization. Amyloid. 2013;20(1):27–33. doi: 10.3109/13506129.2012.761967. [DOI] [PubMed] [Google Scholar]
- 30.Reyners A. K. L., Hazenberg B. P. C., Reitsma W. D., Smit A. J. Heart rate variability as a predictor of mortality in patients with AA and AL amyloidosis. European Heart Journal. 2002;23(2):157–161. doi: 10.1053/euhj.2001.2972. [DOI] [PubMed] [Google Scholar]
- 31.Falk R. H., Dubrey S. W. Amyloid Heart Disease. Progress in Cardiovascular Diseases. 2010;52(4):347–361. doi: 10.1016/j.pcad.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 32.Aljaroudi W. A., Desai M. Y., Tang W. H. W., Phelan D., Cerqueira M. D., Jaber W. A. Role of imaging in the diagnosis and management of patients with cardiac amyloidosis: State of the art review and focus on emerging nuclear techniques. Journal of Nuclear Cardiology. 2014;21(2):271–283. doi: 10.1007/s12350-013-9800-5. [DOI] [PubMed] [Google Scholar]
- 33.Oh J. K., Seward J. B., Talik A. J. The Echo Manual. 3rd 2007. [Google Scholar]
- 34.Liu D., Hu K., Niemann M., et al. Effect of combined systolic and diastolic functional parameter assessment for differentiation of cardiac amyloidosis from other causes of concentric left ventricular hypertrophy. Circulation: Cardiovascular Imaging. 2013;6(6):1066–1072. doi: 10.1161/CIRCIMAGING.113.000683. [DOI] [PubMed] [Google Scholar]
- 35.Karamitsos T. D., Francis J. M., Myerson S., Selvanayagam J. B., Neubauer S. The Role of Cardiovascular Magnetic Resonance Imaging in Heart Failure. Journal of the American College of Cardiology. 2009;54(15):1407–1424. doi: 10.1016/j.jacc.2009.04.094. [DOI] [PubMed] [Google Scholar]
- 36.Selvanayagam J. B., Hawkins P. N., Paul B., Myerson S. G., Neubauer S. Evaluation and Management of the Cardiac Amyloidosis. Journal of the American College of Cardiology. 2007;50(22):2101–2110. doi: 10.1016/j.jacc.2007.08.028. [DOI] [PubMed] [Google Scholar]
- 37.Maceira A. M., Joshi J., Prasad S. K., et al. Cardiovascular magnetic resonance in cardiac amyloidosis. Circulation. 2005;111(2):186–193. doi: 10.1161/01.CIR.0000152819.97857.9D. [DOI] [PubMed] [Google Scholar]
- 38.Cheng A. S. H., Banning A. P., Mitchell A. R. J., Neubauer S., Selvanayagam J. B. Cardiac changes in systemic amyloidosis: Visualisation by magnetic resonance imaging. International Journal of Cardiology. 2006;113(1):E21–E23. doi: 10.1016/j.ijcard.2006.07.107. [DOI] [PubMed] [Google Scholar]
- 39.Lyne J. C., Petryka J., Pennell D. J. Atrial enhancement by cardiovascular magnetic resonance in cardiac amyloidosis. European Heart Journal. 2008;29(2):p. 212. doi: 10.1093/eurheartj/ehm351. [DOI] [PubMed] [Google Scholar]
- 40.Maceira A. M., Prasad S. K., Hawkins P. N., Roughton M., Pennell D. J. Cardiovascular magnetic resonance and prognosis in cardiac amyloidosis. Journal of Cardiovascular Magnetic Resonance. 2008;10(1, article no. 54) doi: 10.1186/1532-429X-10-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Karamitsos T. D., Piechnik S. K., Banypersad S. M., et al. Noncontrast T1 mapping for the diagnosis of cardiac amyloidosis. JACC: Cardiovascular Imaging. 2013;6(4):488–497. doi: 10.1016/j.jcmg.2012.11.013. [DOI] [PubMed] [Google Scholar]
- 42.Dungu J. N., Valencia O., Pinney J. H., et al. CMR-Based Differentiation of AL and ATTR Cardiac Amyloidosis. JACC: Cardiovascular Imaging. 2014;7(2):133–142. doi: 10.1016/j.jcmg.2013.08.015. [DOI] [PubMed] [Google Scholar]
- 43.Fontana M., Banypersad S. M., Treibel T. A., et al. Native T1 Mapping in Transthyretin Amyloidosis. JACC: Cardiovascular Imaging. 2014;7(2):157–165. doi: 10.1016/j.jcmg.2013.10.008. [DOI] [PubMed] [Google Scholar]
- 44.Bokhari S., Castaño A., Pozniakoff T., Deslisle S., Latif F., Maurer M. S. 99mTc-pyrophosphate scintigraphy for differentiating light-chain cardiac amyloidosis from the transthyretin-related familial and senile cardiac amyloidoses. Circulation: Cardiovascular Imaging. 2013;6(2):195–201. doi: 10.1161/CIRCIMAGING.112.000132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Perugini E., Guidalotti P. L., Salvi F., et al. Noninvasive etiologic diagnosis of cardiac amyloidosis using 99mTc-3,3-diphosphono-1,2-propanodicarboxylic acid scintigraphy. Journal of the American College of Cardiology. 2005;46(6):1076–1084. doi: 10.1016/j.jacc.2005.05.073. [DOI] [PubMed] [Google Scholar]
- 46.Rapezzi C., Quarta C. C., Guidalotti P. L., et al. Role of 99mTc-DPD scintigraphy in diagnosis and prognosis of hereditary transthyretin-related cardiac amyloidosis. JACC: Cardiovascular Imaging. 2011;4(6):659–670. doi: 10.1016/j.jcmg.2011.03.016. [DOI] [PubMed] [Google Scholar]
- 47.Von Hutten H., Mihatsch M., Lobeck H., Rudolph B., Eriksson M., Röcken C. Prevalence and origin of amyloid in kidney biopsies. The American Journal of Surgical Pathology. 2009;33(8):1198–1205. doi: 10.1097/PAS.0b013e3181abdfa7. [DOI] [PubMed] [Google Scholar]
- 48.Kieninger B., Eriksson M., Kandolf R., et al. Amyloid in endomyocardial biopsies. Virchows Archiv. 2010;456(5):523–532. doi: 10.1007/s00428-010-0909-5. [DOI] [PubMed] [Google Scholar]
- 49.Petruzziello F., Zeppa P., Catalano L., et al. Amyloid in bone marrow smears of patients affected by multiple myeloma. Annals of Hematology. 2010;89(5):469–474. doi: 10.1007/s00277-009-0857-9. [DOI] [PubMed] [Google Scholar]
- 50.Van Gameren I. I., Hazenberg B. P. C., Bijzet J., et al. Amyloid load in fat tissue reflects disease severity and predicts survival in amyloidosis. Arthritis Care & Research. 2010;62(3):296–301. doi: 10.1002/acr.20101. [DOI] [PubMed] [Google Scholar]
- 51.Gertz M. A. Immunoglobulin light chain amyloidosis: 2014 update on diagnosis, prognosis, and treatment. American Journal of Hematology. 2014;89(12):1133–1140. doi: 10.1002/ajh.23828. [DOI] [PubMed] [Google Scholar]
- 52.Kyle R. A., Gertz M. A. Primary systemic amyloidosis: clinical and laboratory features in 474 cases. Seminars in Hematology. 1995;32:45–59. [PubMed] [Google Scholar]
- 53.Richards D. B., Cookson L. M., Berges A. C., et al. Therapeutic clearance of amyloid by antibodies to serum amyloid P component. The New England Journal of Medicine. 2015;373(12):1106–1114. doi: 10.1056/NEJMoa1504942. [DOI] [PubMed] [Google Scholar]
- 54.Rapezzi C., Quarta C. C., Obici L., et al. Disease profile and differential diagnosis of hereditary transthyretin-related amyloidosis with exclusively cardiac phenotype: An Italian perspective. European Heart Journal. 2013;34(7):520–528. doi: 10.1093/eurheartj/ehs123. [DOI] [PubMed] [Google Scholar]
- 55.Ruberg F. L., Maurer M. S., Judge D. P., et al. Prospective evaluation of the morbidity and mortality of wild-type and V122I mutant transthyretin amyloid cardiomyopathy: The Transthyretin Amyloidosis Cardiac Study (TRACS) American Heart Journal. 2012;164(2):222–228.e1. doi: 10.1016/j.ahj.2012.04.015. [DOI] [PubMed] [Google Scholar]
- 56.Silverman K. J., Hutchins G. M., Bulkley B. H. Cardiac sarcoid: a clinicopathologic study of 84 unselected patients with systemic sarcoidosis. Circulation. 1978;58(6):1204–1211. doi: 10.1161/01.CIR.58.6.1204. [DOI] [PubMed] [Google Scholar]
- 57.Bejar D., Colombo P. C., Latif F., Yuzefpolskaya M. Infiltrative cardiomyopathies. Clinical Medicine Insights: Cardiology. 2015;9:29–38. doi: 10.4137/CMC.S19706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dubrey S. W., Falk R. H. Diagnosis and management of cardiac sarcoidosis. Progress in Cardiovascular Diseases. 2010;52(4):336–346. doi: 10.1016/j.pcad.2009.11.010. [DOI] [PubMed] [Google Scholar]
- 59.Greulich S., Deluigi C. C., Gloekler S., et al. CMR imaging predicts death and other adverse events in suspected cardiac sarcoidosis. JACC: Cardiovascular Imaging. 2013;6(4):501–511. doi: 10.1016/j.jcmg.2012.10.021. [DOI] [PubMed] [Google Scholar]
- 60.Iannuzi M. C., Rybicki B. A., Tierstein A. S. Sarcoidosis. The New England Journal of Medicine. 2007;357:2153–2165. doi: 10.1056/NEJMra071714. [DOI] [PubMed] [Google Scholar]
- 61.Yazaki Y., Isobe M., Hiramitsu S., et al. Comparison of clinical features and prognosis of cardiac sarcoidosis and idiopathic dilated cardiomyopathy. American Journal of Cardiology. 1998;82(4):537–540. doi: 10.1016/S0002-9149(98)00377-4. [DOI] [PubMed] [Google Scholar]
- 62.Gupta A., Singh Gulati G., Seth S., Sharma S. Cardiac MRI in restrictive cardiomyopathy. Clinical Radiology. 2012;67(2):95–105. doi: 10.1016/j.crad.2011.05.020. [DOI] [PubMed] [Google Scholar]
- 63.Smedema J., Snoep G., van Kroonenburgh M. P. G., et al. Evaluation of the accuracy of gadolinium-enhanced cardiovascular magnetic resonance in the diagnosis of cardiac sarcoidosis. Journal of the American College of Cardiology. 2005;45(10):1683–1690. doi: 10.1016/j.jacc.2005.01.047. [DOI] [PubMed] [Google Scholar]
- 64.Crawford T., Mueller G., Sarsam S., et al. Magnetic resonance imaging for identifying patients with cardiac sarcoidosis and preserved or mildly reduced left ventricular function at risk of ventricular arrhythmias. Circulation: Arrhythmia and Electrophysiology. 2014;7(6):1109–1115. doi: 10.1161/CIRCEP.113.000156. [DOI] [PubMed] [Google Scholar]
- 65.Ise T., Hasegawa T., Morita Y., et al. Extensive late gadolinium enhancement on cardiovascular magnetic resonance predicts adverse outcomes and lack of improvement in LV function after steroid therapy in cardiac sarcoidosis. Heart. 2014;100(15):1165–1172. doi: 10.1136/heartjnl-2013-305187. [DOI] [PubMed] [Google Scholar]
- 66.Britton K. A., Stevenson W. G., Levy B. D., Katz J. T., Loscalzo J. The beat goes on. The New England Journal of Medicine. 2010;362(18):1662–1726. doi: 10.1056/NEJMcps0801473. [DOI] [PubMed] [Google Scholar]
- 67.Erthal F., Juneau D., Lim S. P., et al. Imaging of cardiac sarcoidosis. The Quarterly Journal of Nuclear Medicine and Molecular Imaging. 2016;60(3):252–263. [PubMed] [Google Scholar]
- 68.White J. A., Rajchl M., Butler J., Thompson R. T., Prato F. S., Wisenberg G. Active cardiac sarcoidosis first clinical experience of simultaneous positron emission tomography-magnetic resonance imaging for the diagnosis of cardiac disease. Circulation. 2013;127(22):e639–e641. doi: 10.1161/CIRCULATIONAHA.112.001217. [DOI] [PubMed] [Google Scholar]
- 69.Birnie D. H., Nery P. B., Ha A. C., Beanlands R. S. B. Cardiac Sarcoidosis. Journal of the American College of Cardiology. 2016;68(4):411–421. doi: 10.1016/j.jacc.2016.03.605. [DOI] [PubMed] [Google Scholar]
- 70.Pietrangelo A. Hereditary hemochromatosis—a new look at an old disease. The New England Journal of Medicine. 2004;350(23):2383–2430. doi: 10.1056/NEJMra031573. [DOI] [PubMed] [Google Scholar]
- 71.Kremastinos D. T., Farmakis D., Aessopos A., et al. β-thalassemia cardiomyopathy: History, present considerations, and future perspectives. Circulation: Heart Failure. 2010;3(3):451–458. doi: 10.1161/CIRCHEARTFAILURE.109.913863. [DOI] [PubMed] [Google Scholar]
- 72.Anderson L. J., Holden S., Davis B., et al. Cardiovascular T2-star (T2*) magnetic resonance for the early diagnosis of myocardial iron overload. European Heart Journal. 2001;22(23):2171–2179. doi: 10.1053/euhj.2001.2822. [DOI] [PubMed] [Google Scholar]
- 73.Kirk P., Roughton M., Porter J. B., et al. Cardiac T2* magnetic resonance for prediction of cardiac complications in Thalassemia Major. Circulation. 2009;120(20):1961–1968. doi: 10.1161/circulationaha.109.874487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chen X., Zhang Z., Zhong J., et al. MRI assessment of excess cardiac iron in thalassemia major: When to initiate? Journal of Magnetic Resonance Imaging. 2016;42(3):737–745. doi: 10.1002/jmri.24781. [DOI] [PubMed] [Google Scholar]
- 75.Tanner M. A., Galanello R., Dessi C., et al. A randomized, placebo-controlled, double-blind trial of the effect of combined therapy with deferoxamine and deferiprone on myocardial iron in thalassemia major using cardiovascular magnetic resonance. Circulation. 2007;115(14):1876–1884. doi: 10.1161/circulationaha.106.648790. [DOI] [PubMed] [Google Scholar]
- 76.Tanner M. A., Galanello R., Dessi C., et al. Combined chelation therapy in thalassemia major for the treatment of severe myocardial siderosis with left ventricular dysfunction. Journal of Cardiovascular Magnetic Resonance. 2008;10, article 12 doi: 10.1186/1532-429x-10-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Nishio M., Endo T., Nakao S., Sato N., Koike T. Reversible cardiomyopathy due to secondary hemochromatosis with multitransfusions for severe aplastic anemia after successful non-myeloablative stem cell transplantation. International Journal of Cardiology. 2008;127(3):400–401. doi: 10.1016/j.ijcard.2007.04.066. [DOI] [PubMed] [Google Scholar]
- 78.Löffler W. L. Endocarditis parietal fibroplastica with eosinophilia: a strange diseas. Schweizerische Medizinische Wochenschrift. 1936;66:817–820. (Ger). [Google Scholar]
- 79.Chusid M. J. Eosinophilia in childhood. Immunology and Allergy Clinics of North America. 1999;19(2):327–346. doi: 10.1016/S0889-8561(05)70091-6. [DOI] [Google Scholar]
- 80.Séguéla P. E., Iriart X., Acar P., Montaudon M., Roudaut R., Thambo J. B. Eosinophilic cardiac disease: molecular, clinical and imaging aspects. Archives of Cardiovascular Diseases. 2015;108(4):258–268. doi: 10.1016/j.acvd.2015.01.006. [DOI] [PubMed] [Google Scholar]
- 81.Chusid M. J., Dale D. C., West B. C., Wolff S. M. The hypereosinophilic syndrome: analysis of fourteen cases with review of the literature. Medicine. 1975;54(1):1–27. doi: 10.1097/00005792-197501000-00001. [DOI] [PubMed] [Google Scholar]
- 82.Feske S. K., Goldberg M., Dudzinski D. M., Gonzalez R. G., Kovach A. E. Case 29-2015—A 38-year-old pregnant woman with headache and visual symptoms. The New England Journal of Medicine. 2015;373(12):1154–1164. doi: 10.1056/NEJMcpc1404335. [DOI] [PubMed] [Google Scholar]
- 83.Chun W., Grist T. M., Kamp T. J., Warner T. F., Christian T. F. Images in cardiovascular medicine. Infiltrative eosinophilic myocarditis diagnosed and localized by cardiac magnetic resonance imaging. Circulation. 2004;110(3):p. e19. doi: 10.1161/01.CIR.0000135586.94417.3C. [DOI] [PubMed] [Google Scholar]
- 84.Mollet N. R., Dymarkowski S., Volders W., et al. Visualization of ventricular thrombi with contrast-enhanced magnetic resonance imaging in patients with ischemic heart disease. Circulation. 2002;106(23):2873–2876. doi: 10.1161/01.CIR.0000044389.51236.91. [DOI] [PubMed] [Google Scholar]
- 85.O'Mahony C., Elliott P. Anderson-Fabry disease and the heart. Progress in Cardiovascular Diseases. 2010;52(4):326–335. doi: 10.1016/j.pcad.2009.11.002. [DOI] [PubMed] [Google Scholar]
- 86.Ramaswami U., Whybra C., Parini R., et al. Clinical manifestations of Fabry disease in children: Data from the Fabry Outcome Survey. Acta Paediatrica. 2006;95(1):86–92. doi: 10.1080/08035250500275022. [DOI] [PubMed] [Google Scholar]
- 87.MacDermot K. D., Holmes A., Miners A. H. Anderson-Fabry disease: clinical manifestations and impact of disease in a cohort of 98 hemizygous males. Journal of Medical Genetics. 2001;38(11):750–760. doi: 10.1136/jmg.38.11.750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.MacDermot K. D., Holmes A., Miners A. H. Anderson-Fabry disease: clinical manifestations and impact of disease in a cohort of 60 obligate carrier females. Journal of Medical Genetics. 2001;38(11):769–775. doi: 10.1136/jmg.38.11.769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zarate Y. A., Hopkin R. J. Fabry's disease. The Lancet. 2008;372(9647):1427–1435. doi: 10.1016/S0140-6736(08)61589-5. [DOI] [PubMed] [Google Scholar]
- 90.Linhart A., Elliott P. M. The heart in Anderson-Fabry disease and other lysosomal storage disorders. Heart. 2007;93(4):528–535. doi: 10.1136/hrt.2005.063818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Pochis W. T., Litzow J. T., King B. G., Kenny D. Electrophysiologic findings in Fabry's disease with a short PR interval. American Journal of Cardiology. 1994;74(2):203–204. doi: 10.1016/0002-9149(94)90106-6. [DOI] [PubMed] [Google Scholar]
- 92.Goldman M. E., Cantor R., Schwartz M. F., Baker M., Desnick R. J. Echocardiographic abnormalities and disease severity in Fabry's disease. Journal of the American College of Cardiology. 1986;7(5):1157–1161. doi: 10.1016/S0735-1097(86)80238-8. [DOI] [PubMed] [Google Scholar]
- 93.Linhart A., Lubanda J.-C., Palecek T., et al. Cardiac manifestations in Fabry disease. Journal of Inherited Metabolic Disease. 2001;24(2):75–83. doi: 10.1023/A:1012428009627. [DOI] [PubMed] [Google Scholar]
- 94.Weidemann F., Breunig F., Beer M., et al. Improvement of cardiac function during enzyme replacement therapy in patients with fabry disease: A prospective strain rate imaging study. Circulation. 2003;108(11):1299–1301. doi: 10.1161/01.CIR.0000091253.71282.04. [DOI] [PubMed] [Google Scholar]
- 95.Weidemann F., Niemann M., Breunig F., et al. Long-term effects of enzyme replacement therapy on fabry cardiomyopathy. Evidence for a better outcome with early treatment. Circulation. 2009;119(4):524–529. doi: 10.1161/CIRCULATIONAHA.108.794529. [DOI] [PubMed] [Google Scholar]
- 96.Marwick T. H. Diabetic cardiomyopathy. In: Crawford M. H., DiMarco J. P., Paulus W. J., editors. Cardiology. 3rd. Philadelphia, Pa, USA: Mosby-Elsevier; 2010. pp. 1107–1109. [Google Scholar]
- 97.Forbes J. M., Cooper M. E. Mechanisms of diabetic complications. Physiological Reviews. 2013;93(1):137–188. doi: 10.1152/physrev.00045.2011. [DOI] [PubMed] [Google Scholar]
- 98.Seferović P. M., Paulus W. J. Clinical diabetic cardiomyopathy: a two-faced disease with restrictive and dilated phenotypes. European Heart Journal. 2015;36(27):1718–1727. doi: 10.1093/eurheartj/ehv134. [DOI] [PubMed] [Google Scholar]
- 99.Nihoyannopoulos P., Dawson D. Restrictive cardiomyopathies. European Heart Journal - Cardiovascular Imaging. 2009;10(8):iii23–iii33. doi: 10.1093/ejechocard/jep156. [DOI] [PubMed] [Google Scholar]
- 100.Jaworski C., Mariani J. A., Wheeler G., Kaye D. M. Cardiac complications of thoracic irradiation. Journal of the American College of Cardiology. 2013;61(23):2319–2328. doi: 10.1016/j.jacc.2013.01.090. [DOI] [PubMed] [Google Scholar]
- 101.Taunk N. K., Haffty B. G., Kostis J. B., Goyal S. Radiation-induced heart disease: pathologic abnormalities and putative mechanisms. Frontiers in Oncology. 2015;5, article 39 doi: 10.3389/fonc.2015.00039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Charron P., Villard E., Sébillon P., et al. Dano's disease as a cause of hypertrophic cardiomyopathy: A systematic survey. Heart. 2004;90(8):842–846. doi: 10.1136/hrt.2003.029504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Elliott P. M., Anastasakis A., Borger M. A., et al. 2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy: the Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC) European Heart Journal. 2014;35(39):2733–2779. doi: 10.1093/eurheartj/ehu284. [DOI] [PubMed] [Google Scholar]
- 104.Arad M., Maron B. J., Gorham J. M., et al. Glycogen storage diseases presenting as hypertrophic cardiomyopathy. The New England Journal of Medicine. 2005;352(4):362–372. doi: 10.1056/NEJMoa033349. [DOI] [PubMed] [Google Scholar]
- 105.Piotrowska-Kownacka D., Kownacki L., Kuch M., et al. Cardiovascular magnetic resonance findings in a case of Danon disease. Journal of Cardiovascular Magnetic Resonance. 2009;11(1, article no. 12) doi: 10.1186/1532-429X-11-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Dürr A., Cossee M., Agid Y., et al. Clinical and genetic abnormalities in patients with Friedreich's ataxia. The New England Journal of Medicine. 1996;335(16):1169–1175. doi: 10.1056/NEJM199610173351601. [DOI] [PubMed] [Google Scholar]
- 107.Alboliras E. T., Shub C., Gomez M. R., et al. Spectrum of cardiac involvement in Friedreich's ataxia: Clinical, electrocardiographic and echocardiographic observations. American Journal of Cardiology. 1986;58(6):518–524. doi: 10.1016/0002-9149(86)90026-3. [DOI] [PubMed] [Google Scholar]