

Abstract
The current integrative pathobiological hypothesis states that pancreatic cancer (PDAC) develops and progresses in response to an interaction between known oncogenes and downstream epigenomic regulators. Congruently, this study tests a new combinatorial therapy based on the inhibition of the Aurora kinase A (AURKA) oncogene and one of its targets, the H3K9 methylation-based epigenetic pathway. This therapeutic combination is effective at inhibiting the in vitro growth of PDAC cells both, in monolayer culture systems, and in 3D spheroids and organoids. The combination also reduces the growth of PDAC xenografts in vivo. Mechanistically, it was found that inhibiting methyltransferases of the H3K9 pathway in cells, which are arrested in G2/M after targeting Aurora kinase A, decreases H3K9 methylation at centromeres, induces mitotic aberrations, triggers an aberrant mitotic check point response, and ultimately leads to mitotic catastrophe. Combined, this data describes for the first time a hypothesis-driven design of an efficient combinatorial treatment that targets a dual oncogenic-epigenomic pathway to inhibit PDAC cell growth via a cytotoxic mechanism that involves perturbation of normal mitotic progression to end in mitotic catastrophe. Therefore, this new knowledge has significant mechanistic value as it relates to the development of new therapies, as well as biomedical relevance.
Keywords: pancreatic cancer, epigenetic therapy, Aurora A, mitotic catastrophe, checkpoint
Introduction
Pancreatic cancer is the 4th leading cause of cancer-related deaths in the US, with a median survival of 6 months [1]. PDAC presents with aggressive biology, rapid dissemination, and late diagnosis, leading to an incurable stage for which therapy is a challenge. Surgery remains the only curative modality, but is only applicable to 10% of patients, with 5-year survival barely 20% [2]. Notably, PDAC is highly resistant to chemoradiation [3]. Thus, there remains a crucial need for the development and evaluation of novel targeted therapies.
Current evidence suggests that PDAC results from a crosstalk between oncogenes and epigenetic regulators, which together are responsible of the malignant phenotype. Thus, combinatorial therapies targeting both components may provide a useful approach to ameliorate PDAC initiation and progression. Regarding pharmacologically targetable oncogenes, recent studies have examined the effects of inhibiting Aurora kinase A (AurkA) in PDAC [4, 5]. AurkA is a G2/M kinase that plays crucial roles in cell division [6]. AurkA expression is aberrant in many malignancies, including PDAC [7]. Altered AurkA activity contributes to tumor progression by inducing abnormal G2/M transition, centrosome amplification and chromosome instability [8]. Both genetic and pharmaceutical AurkA inhibition has cytostatic effects through G2/M arrest [9, 10] and senescence [10]. Since these combined cellular effects lead to growth inhibition in PDAC cells and xenografts [9], AurkA has become a molecular target for PDAC treatment. MLN8237 (Alisertib) selectively inhibits AurkA [11] and is currently under Phase I and II clinical trials for solid tumors and hematologic malignancies. However, its underlying mechanisms are not fully understood deserving further investigations into combinatorial therapies that may improve its effects.
Interestingly, AurkA phosphorylates proteins associated with mitotic regulation through spatially and temporally controlled mechanisms [6], which includes epigenetic regulators. We have shown that a member of the methyl lysine 9 histone 3 (H3K9me) pathway, HP1γ (CBX3), is phosphorylated by AurkA to support normal mitotic progression [12]. The K9H3me histone mark to which HP1 binds is established by the H3K9 histone methyltransferases (HMTs) G9a, GLP, SUV39H1 and SUV39H2. HP1, in conjunction with these proteins, forms a multimeric complex that in a timely manner, deposits and reads the H3K9me mark, genome wide. Here, we investigate the effect of targeting both the AurkA and H3K9me pathways on PDAC growth. We find that treatment of PDAC cells with individual drugs against AurkA or H3K9me HMTs inhibits cell growth. However, the combination of these agents has an improved effect of reducing cell growth through a cytotoxic effect. Enhanced inhibition of cell growth with the combination is also observed in spheroid and organoid models. In vivo treatment of PDAC orthotopic xenografts with the H3K9me HMT inhibitor alone reduces PDAC growth, and increased efficacy in PDAC growth reduction is observed in combination with the AurkA inhibitor over individual treatments. Confocal and electron microscopy, along with cell cycle analysis, demonstrate that the cytotoxic effect of this combination is due to induction of mitotic cell death. The cellular response to combined treatment was also observed at the RNA-seq level, reflected by expression changes of genes involved in drug metabolism, mitotic chromosome-associated non-coding regulatory RNAs (mCARs), and genes associated to chromosome structure. Moreover, we observed a dramatic reduction in centromeric H3K9 methylation in the combination compared to individual inhibitors. Biochemical alterations of centromeres are known to activate cell cycle checkpoints and induce chromosomal instability [13]. Congruently, at the molecular level, we find that while AurkA inhibition alone triggers a Chk1-Cdc25c-Cdk1-dependent G2/M arrest, the combination with H3K9me HMT inhibition regulates checkpoint proteins in a manner that allows the bypass of this G2/M arrest with down-regulation of Chk1-p53 pathway to ultimately culminate in mitotic catastrophe. Therefore, our data demonstrate that combined targeting of AurkA and the epigenetic H3K9me HMT pathway decreases cytostatic effects to increase their cytotoxicity. Furthermore, our experiments unravel cellular and molecular mechanisms on how this combinatorial therapy works to bring about mitotic catastrophe, which is a desirable therapeutic outcome.
Materials and Methods
Cell lines and reagents
Human PDAC cell lines were obtained from the American Type Culture Collection (ATCC, Rockville, MD) and maintained according to their recommendations. L3.6, originally isolated by Bruns et al. [14], was cultured in appropriate media. The mouse PDAC cell line Pan02, syngeneic to C57BL/6, was obtained from DTP, DCTD Tumor Repository (National Cancer Institute) and maintained according to supplier’s recommendations. All cell lines were cultured at 37°C in a humidified incubator supplemented with 5% CO2 and passaged a maximum of 30 times prior to restarting cultures from cryogenically preserved stocks. Cell lines tested negative for mycoplasma within the last 6 months, and due to low passage, were not authenticated by our laboratory. For orthotopic tumors, Pan02 cells were incubated 72hrs with lentivirus to induce stable expression of luciferase. For in vitro studies, compounds were dissolved in DMSO and stored as 100uM aliquots at −80°C (MLN8237, Selleck Chemicals, ApexBio) or −20°C (chaetocin, Sigma-Aldrich).
Cell growth and synergy assays
Cell lines were treated with various concentrations of MLN8237 or chaetocin, as indicated. Relative cell viability was determined by colorimetric MTS assay (CellTiter 96® AQueous One Solution Cell Proliferation Assay; Promega) according to the manufacturer’s suggestions. For drug combinations, cells were treated with 8 independent concentrations at a fixed ratio (1:2) or 5 independent concentrations at fixed ratios (1:3 or 1:5) of chaetocin:MLN8237. The fraction affected (Fa) was calculated based on MTS measurements of treated samples normalized to untreated controls and the maximum biological effect of each cell line. A combination index (CI) was calculated by the median-drug effect analysis method developed by Chou and Talalay [15] using the CompuSyn software (ComboSyn, Inc, Paramus, NJ), where CI < 1, 1, or > 1 indicates synergistic, additive and antagonistic effect respectively.
Clonogenic assays
PDAC cells were plated on 60mm dishes at 10% confluence (1.5E4). After drug exposure, cells were fixed for 10min with 10% methanol/10% acetic acid and subsequently stained with 0.1% crystal violet (Sigma-Aldrich). Cell density was quantified by Image J [16].
Spheroid and organoid assays
PANC-1 cells were allowed to form spheroids in a 3D methylcellulose matrix [17]. After 4 days, media was replaced and drugs added. After 72hrs of treatment, viability was measured by APH assay [17] on the BioRad iMark Microplate reader at 415nM. Organoid cultures were established following the protocol from Dr. Tuveson’s lab [18]. Briefly, male Ela-Kras mice [19] were euthanized, pancreas aseptically removed, digested, and ducts were isolated and grown in matrigel (Corning) domes. One day prior to treatment, organoids were split into 96-well plates with matrigel. Viability was assessed with the CellTiter-Glo 3D assay (G9682, Promega) on the Promega’s GloMax. Bright field images were captured by a Canon EOS Rebel Xsi camera mounted on a Nikon Eclipse TS100 microscope at 10× magnification.
Orthotopic Xenografts
Animal experiments were reviewed by Mayo Clinic and followed the guidelines outlined in the Guide for Care and Use of Laboratory Animals (NIH) under IACUC approved protocol (A24815-15). C57Bl/6 mice (Jackson labs, aged 6–8wks) were anesthetized, subjected to a small incision to expose the tail of the pancreas, and injected with 5E5 Pan02+luciferase cells in 25% matrigel (BD bioscience). Mice were observed daily and recovered 1 week before treatment initiation. MLN8237 was resuspended in 10% 2-hydroxypropyl-β-cyclodextrin (2OHpβCD; Sigma-Aldrich)/1% sodium bicarbonate solution at 3mg/mL. Chaetocin was resuspended at 1mM in DMSO and diluted to 100uM in saline. Mice were randomly divided into four groups and treated daily with vehicle, MLN8237 (30mg/kg, gavage), chaetocin (0.279mg/kg, intraperiotenal, i.p.), or combination (N=5/group). For tumor monitoring, mice were anesthetized with 0.5% inhaled isoflourane, injected with D-luciferin (112.5mg/kg) and total flux (p/s) was quantified by Xenogen’s IVIS-200 luminescence imaging system. Animals were euthanized after 10 treatments and pancreatic tissues were formalin fixed and paraffin embedded for histology.
Cell Viability, Apoptosis, and Cell Cycle FACS Analysis
Cells were plated at 5.0E3 cells/well, treated, and cell viability and apoptosis were measured with the ApoTox-Glo™ Triplex Assay (G6321, Promega) on Promega’s GloMax at indicated times. For FACS, cells were prepared as previously described [20]. DNA content was analyzed on a Becton Dickinson LSR II FACS instrument at the Mayo Flow Cytometry Core Facility. 20,000–40,000 events were collected and analyzed with appropriate gating in FlowJo software (FlowJo, LLC, Asland, OR).
Immunofluorescence and confocal microscopy
Immunofluorescence and confocal microscopy were performed as previously described [21]. For mitotic aberrations, cells were stained with α/γ-tubulin primary antibodies (Supplementary Table 1) and counterstained with 4′,6-diamidino-2-phenylindole (DAPI) mounting media (Vector Laboratories). Images were obtained on a Zeiss AxioObserver LSM780 with objective alpha plan-apochromat 100×/1.46 oil iris.
Electron microscopy (EM)
PANC-1 cells were treated and harvested for fixation in Trump’s solution (4% formaldehyde + 1% glutaraldehyde in 0.1M phosphate buffer) overnight at room temperature. Subsequently, cells were fixed in 1% osmium tetroxide and 1% aqueous uranyl acetate, dehydrated in a graded series of ethanol, and embedded in Embed 812/Araldite (EMS, Hatfield, PA). Thin sections (0.1μm) were collected on copper grids and post-stained with lead citrate. Images were acquired in the Mayo Clinic Microscopy and Cell Analysis Core facility using a JEOL1400 TEM operating at 80kV (Jeol USA, Inc) equipped with a Gatan Orius832 camera (Gatan Inc., Pleasanton, CA).
Immunohistochemistry
IHC was performed on pancreatic tissue as previously described [22]. Slides were incubated overnight with primary antibody (Supplementary Table 1) and developed with Nova Red (Vector Labs) and hematoxylin counterstain. Five random fields (40× NA 0.65 A-Plan objective using Mikroscan D2 Digital Slide Scanner with Q-Skan) per pancreas were imaged and counted [23]. Each field contained at least 1000 cells.
RNA-sequencing and real time qPCR
RNA was isolated using RNeasy mini kit with optional on column DNA digestion (Qiagen). Libraries were prepared (Illumina TruSeq mRNA v2) and run on the Illumina High Seq-2000 (101 bp paired end reads) in the Mayo Clinic Sequencing Core. RNA-Seq data was analyzed using MAP-RSeq v.1.2.1 [24]. MAP-RSeq consists of alignment with TopHat 2.0.6 [25] against the hg19 genome build and gene counts with the HTSeq software 0.5.3p9 (http://www.huber.embl.de/users/anders/HTSeq/doc/overview.html) using gene annotation files obtained from Illumina (http://cufflinks.cbcb.umd.edu/igenomes.html). Principal component analysis and un-supervised hierarchical clustering were performed on the samples using Partek® Genomics Suite® software, version 6.6©; 2016, Partek Inc., St. Louis, MO, USA. Normalization and differential expression analysis were performed using edgeR2.6.2 [26]. Data is available through the GEO repository under accession GSE97215. Real time qPCR was performed using RT2 SYBR® Green qPCR Mastermix (Qiagen) and RT2 qPCR Primers (Qiagen) on the Bio-Rad CFX96 system. Fold changes (ΔΔCt) and p-values were calculated using Bio-Rad CFX manager and SABioscience’s RT2 Profiler PCR Array Data Analysis software. Treatment samples were compared to vehicle and normalized to a panel of housekeeping genes.
Western blot analysis
Western blots were performed as previously described [12]. Membranes were blocked in 3% BSA/TBST and incubated overnight at 4°C with primary antibody (Supplementary Table 1). Blots were developed by Pierce ECL chemiluminescent substrate (Thermo Scientific).
Results
Similar to the Aurora A inhibitor, MLN8237, the pan-H3K9me-HMT inhibitor, chaetocin, is effective at reducing PDAC cell growth in vitro
Although MLN8237 has been actively tested for controlling PDAC growth with initial promising results [27, 28], its efficacy remains to be improved. Thus, searching for novel pathways to serve as additional targets could potentially enhance the effects of MLN8237. The H3K9me mark is critical for maintaining the proper structure and function of centromeres [29], which if disrupted causes several types of mitotic stress. We hypothesized that chaetocin, an inhibitor of all known H3K9me HMTs [30], will generate an added stress to mitotically-arrested cells by MLN8237 and improve its beneficial effects. As an initial step toward defining the consequences of this combination, we first tested the effects of both drugs alone in PDAC cell lines. Figure 1A shows that MLN8237 (200nM) reduced cell viability in 5 different human PDAC cells after 72 hours of treatment: BxPC-3 (76.0±2.4% of control), Capan-2 (63.0±4.6%), L3.6 (70.6±1.6%), MIA PaCa-2 (78.9± 3.0%), and PANC-1 (89.5±2.0%), as well as the mouse-derived PDAC line Pan02 (77.2±4.5%). Similarly, treatment with the H3K9me HMT inhibitor, chaetocin (CH), demonstrated a dose-dependent inhibition of cell growth in the same cell lines (Figure 1B–G). At 48 hours, the percentage of viability compared to no treatment control cells with 25nM, 50nM, 75nM and 100nM chaetocin was 89.1±5.0%, 65.6±8.3%, 55.0±9.2% and 37.2±5.7%, respectively for BxPC-3 (of control), 88.3±4.0%, 71.7±3.5%, 60.0±2.4%, and 58.2±1.2%, for Capan-2, 118.5±6.5%, 92.5±4.0%, 91.1±10.5%, and 47.2±2.2%, for L3.6, 81.7±5.5%, 59.2±4.9%, 54.4±3.6%, and 42.5±2.6%, for MIA PaCa-2, 124.5±6.1%, 114.1±6.4%, 81.9±6.7%, and 68.0±4.2%, for PANC-1, and 99.1±6.8%, 61.9±4.3%, 49.1±7.7%, and 27.3±2.9%, for Pan02. Combined, these results demonstrate for the first time inhibition of PDAC cell growth in response to treatment with the pan-H3K9me HMT inhibitor, chaetocin, and also confirm that the AurkA inhibitor is similarly effective in this process. These baseline experiments guided the subsequent combination of these two inhibitors.
Figure 1. MLN8237 and Chaetocin (CH) demonstrate a dose-dependent inhibition of PDAC cell growth.
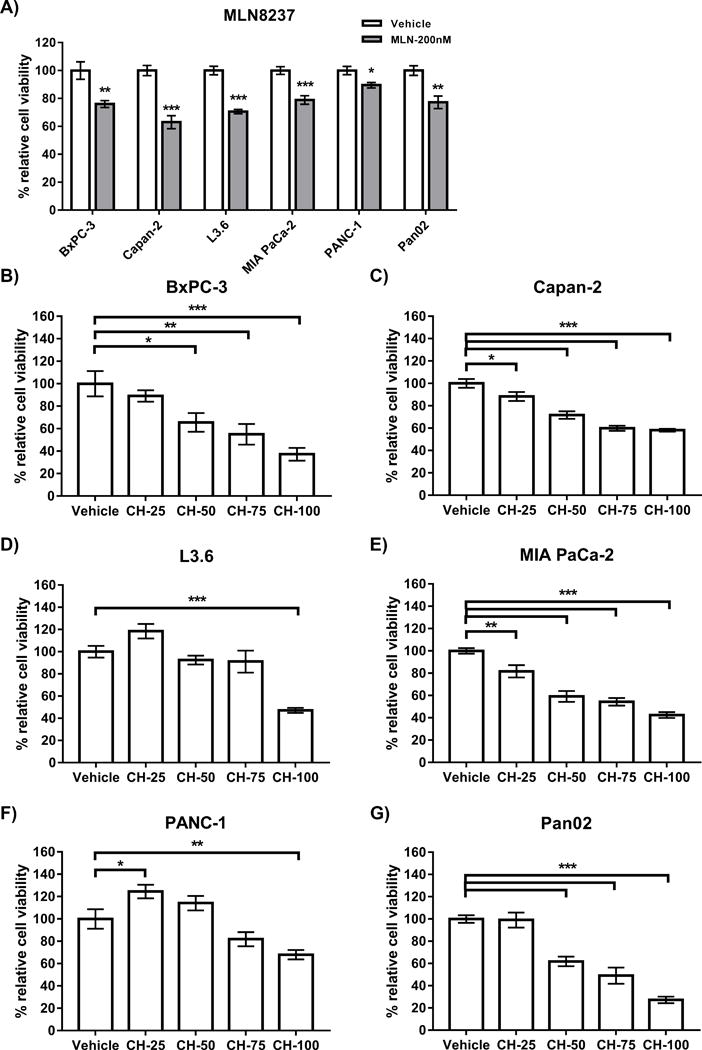
A) PDAC cell lines (5E4 per well of 96 well) were plated and treated with MLN8237 for 72hrs at a dose of 200nM. Treatment of B) BxPC-3, C) Capan-2, D) L3.6, E) MIA PaCa-2, F) PANC-1 and G) Pan02 PDAC cell lines (5E4 per well of 96 well) with escalating doses of chaetocin (25nM, 50nM, 75nM and 100nM) for 48hrs, demonstrated a dose-dependent inhibition of cell growth. Cell viability was measured by MTS, normalized to a vehicle control and data (technical triplicate with biological n=3) are represented as mean±SEM. For MLN8237, treated cells were compared to control by Student’s t-test. For Chaetocin treated cell lines, statistical significance was calculated by 1-way ANOVA with multiple comparisons. * indicates p-value ≤0.05, ** indicates p-value ≤0.005, *** indicates p-value ≤0.0005
Chaetocin enhances the inhibitory effects of MLN8237 on PDAC cell growth in culture and in vivo
We combined MLN8237 and chaetocin at various concentrations to determine their combination index (CI), using CompuSyn software, which is based on the theory of Chou-Talalay Method to define additive effect (CI = 1), synergism (CI < 1), or antagonism (CI > 1) in drug combinations [15]. We performed a 8×8 matrix format using MTS assays, which investigated concentration ranges of 60–320nM of MLN8237 and 5–50nM of chaetocin on the same mouse (Pan02) and human (BxPC-3, Capan-2, L3.6, MIA PaCa-2, and PANC-1) PDAC cells tested with individual drugs. Based on initial concentrations (Figure 1), MLN8237 or chaetocin were added to cells in an expanded matrix format to determine IC50 values of each drug alone. For MLN8237, the IC50 values ranged between 81 and 144 nM, and for chaetocin, values varied from 22 to 66 nM (Supplementary Table 2). Subsequently, the drugs were combined at a fixed concentration ratio of chaetocin:MLN8237 at 1:2 based on their average IC50 values (48 nM and 110nM, respectively), as well as in a 5×5 matrix format at fixed ratios of 1:3 and 1:5. The CI values, which provide a quantitative measure of the degree of drug interaction, were calculated at Fraction Affected (Fa) values of 0.50, 0.75, 0.90, and 0.95 after 72 hours of combined treatment. Figure 2A–G shows that the majority of PDAC cells evaluated with combination treatment had CIs < 1, indicating a synergism between these two inhibitors. Each individual cell line is shown with its corresponding CI values at Fa50, 75, 90 and 95 from each of the fixed ratios, 1:2, 1:3, and 1:5. Overall, the 1:3 fixed ratio resulted in the most effective inhibition with the combination, having mean CI values among all tested cell lines of 0.725, 0.565, 0.504, 0.482 at Fa50, 75, 90 and 95, respectively (Figure 2A). With the exception of MIA Paca-2 (Figure 2E), CI values improved from Fa50 to Fa95 at the 1:3 ratio, with the strongest CIs at higher Fa values. This bears relevance as CI values at higher Fa values are more appropriate than the lower Fa when considering cancer therapy [15]. At a ratio of 1:2, mean CI values were 0.881 (Fa50), 0.683 (Fa75), 0.557 (Fa90), and 0.493 (Fa95) (Figure 2A), with enhanced CI values from Fa50 to Fa95 in all cell lines (Figure 2B–G). Mean CI values for the 1:5 ratio were 0.835 (Fa50), 0.604 (Fa75), 0.486 (Fa90), and 0.429 (Fa95) with better CIs at higher Fa values (Figure 2A). In summary, the Fa-CI plots demonstrate that the MLN8237-chaetocin combination exhibited the highest synergy at high Fa values for the tested ratios.
Figure 2. Dual targeting of Pan-H3K9 HMTs and AurkA synergizes to inhibit PDAC cell growth.
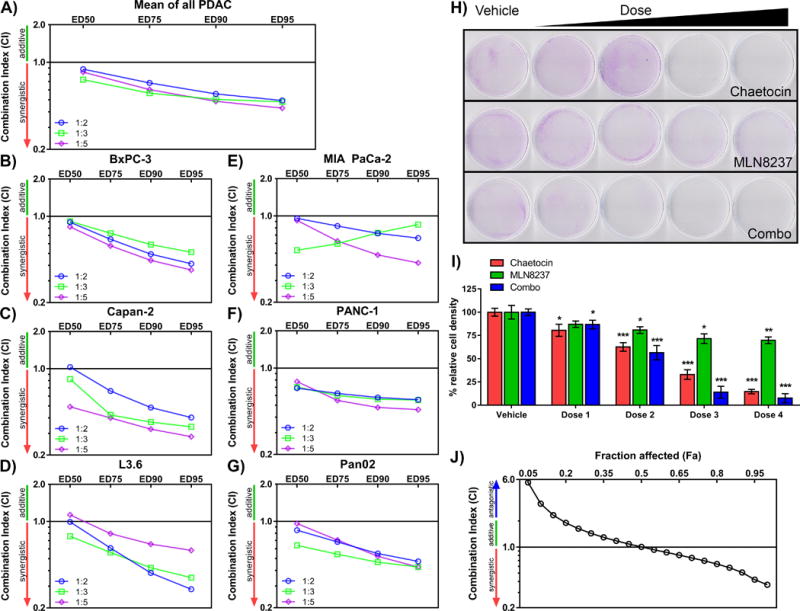
The CI of chaetocin and MLN8237 was calculated for 1:2, 1:3 and 1:5 ratios as an A) average and in the individual human B) BxPC-3, C) Capan-2, D) L3.6, E) MIA PaCa-2, F) PANC-1 and G) mouse Pan02 PDAC cell lines. The CIs from at least three independent experiments were averaged to determine CI values at Fa 0.50, 0.75, 0.90, and 0.95 for each cell line. H) L3.6 cells were plated for a clonogenic cell survival assay and stained for cell density after 7 days of treatment with chaetocin or MLN8237 alone or in combination (Dose 1: 7.5nM chaetocin, 22.5nM MLN8237; Dose 2: 15nM chaetocin, 45nM MLN8237; Dose 3: 22.5nM chaetocin, 67.5nM MLN8237; Dose 4: 30nM chaetocin, 90nM MLN8237). Representative images of clonogenic staining for the vehicle (DMSO) treated cells and dose escalation (left to right) are presented. I) Average density of the clonogenic assay (mean±SEM, n=3), normalized to vehicle for each treatment, and statistical significance calculated by Student’s t-tests for each dose compared to vehicle (* indicates p-value ≤0.05, ** indicates p-value ≤0.005, *** indicates p-value ≤0.0005). J) The CI for the clonogenic assay indicated that the drugs act synergist
We performed clonogenic survival assays on L3.6, as one of the cell lines that displayed a strong synergistic effect with better CIs at higher Fa values (Figure 2D). Cells were exposed to concentration ranges of 22.5–90nM for MLN8237 and 7.5–30nM for chaetocin or a combination at a fixed ratio of chaetocin:MLN8237 at 1:3 for 7 days. MLN8237 alone did not substantially reduce surviving colonies, whereas the pan-H3K9me HMT inhibitor, chaetocin, induced a moderate reduction in clonogenic survival. However, as shown in Figure 2H–I, in particular, the combination of MLN8237 at a concentration of 67.5nM and chaetocin at 22.5nM significantly reduced clonogenic survival with over 85% inhibition (86.1±6.3%) compared to MLN8237 alone (28.4±5.2%; 67.5nM) and 67.1±5.2% inhibition with chaetocin alone (22.5nM). The CI values at Fa 75, 90 and 95 were 0.695, 0.510, and 0.421, respectively (Figure 2J). Similar experiments were performed with PANC-1 cells (Supplementary Figure 1A–C), which demonstrated CI values Fa 75, 90 and 95 at 1.016, 0.944, and 0.912. Therefore, the combination of MLN8237 with chaetocin demonstrated synergy for reduction of clonogenic survival.
3D cultures more closely simulate in vivo conditions and often demonstrate increased resistance to drug treatments. Therefore, we repeated treatments on PDAC cell lines grown as spheroids, which are solid, 3D aggregates of cancer cells [17]. Formed spheroids were grown for 4 days before adding chaetocin, MLN8237 or their combination (Figure 3A–C). Similar to 2D cultures, the drug combination significantly reduced growth of PANC-1 spheroids than either drug alone as measured by acidic phosphatase (APH) assay (Figure 3A; 41.9±2.4% reduction of growth with combination treatment vs 15.9±1.6% with 900nM MLN8237 alone or 25.2±1.5% with 300nM chaetocin alone). Images of the 3D spheroids confirm that the dense core of cells was reduced following combination treatment (yellow circles, Figure 3C). Performing a 5×5 matrix dose curve, spheroids were exposed to concentration ranges of 150–1800nM for MLN8237 and 50–600nM for chaetocin or a combination at a fixed concentration ratio of chaetocin:MLN8237 at 1:3. Thus, the combination is more effective than individual treatments (Figure 3B; CI values of 0.307, 0.139, 0.0632, and 0.0369 for Fa 50, 75, 90 and 95, respectively) for 3D cultures, which are increasingly favored for drug testing [17].
Figure 3. Combination treatment of 3D spheroids, organoids and orthotopic xenografts increases efficacy of PDAC growth reduction.
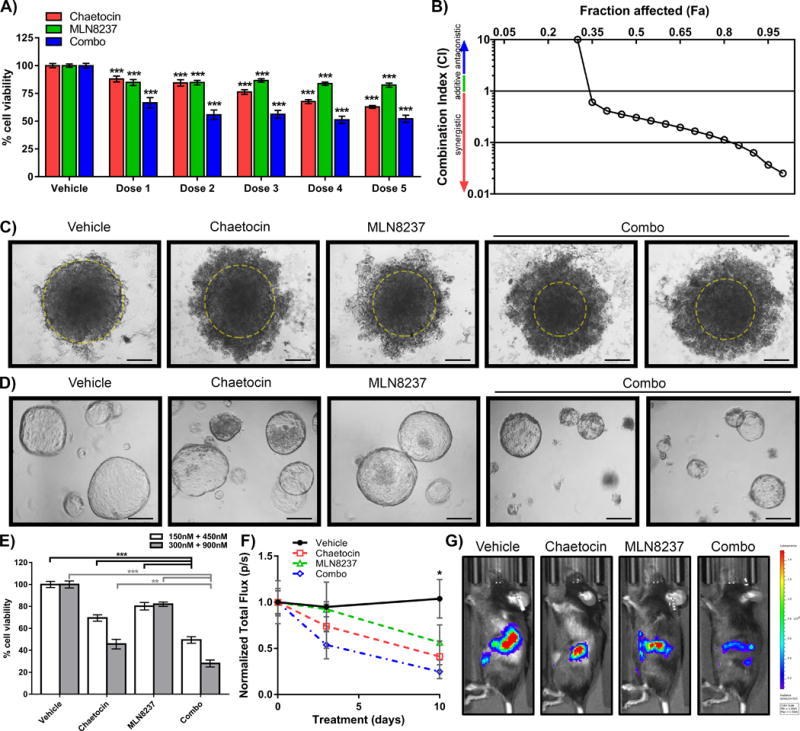
A) PANC-1 cells were grown in methylcellulose as spheroids to mimic a 3D tumor model and treated with various concentrations of vehicle, chaetocin or MLN8237 alone and in combination (Dose 1: 50nM chaetocin, 150nM MLN8237; Dose 2: 150nM chaetocin, 450nM MLN8237; Dose 3: 300nM chaetocin, 900nM MLN8237; Dose 4: 450nM chaetocin, 1350nM MLN8237; Dose 5: 600nM chaetocin, 1800nM MLN8237). APH activity was measured by colorimetric readings and mean cell viability (mean±SEM, n=3) calculated as a percentage of vehicle-treated control. Each experiment was performed in triplicate and statistical significance compared to control was calculated by 1 way ANOVA with multiple comparisons, *** indicates p-value ≤0.0005. B) The CI for the spheroid model indicated that the drugs act synergistically. C) Representative images after 72hrs of treatment are presented. Yellow circle delineates edge of relatively dense core of the spheroid. Scale=25μm. D) Pancreatic duct cells were isolated from the pancreas of Ela-Kras mice and grown in matrigel domes to form 3D organoids. Representative images after 72hrs of treatment (300nM CH, 900nM MLN, and 300nM CH +900nM MLN8237) are presented. Scale=25μm. E) After 72hrs of treatment with vehicle, chaetocin (150nM, 300nM), MLN8237 (450nM, 900nM) and combination (150nM+450nM or 300nM+900nM, CH+MLN8237 respectively), Ela-Kras organoid viability was assessed by luminescent readings normalized to the vehicle-treated organoids. Each experiment was performed in triplicate, results expressed as mean ± SEM, and statistical analyses were performed using 1-way ANOVA and multiple comparisons. ** indicates p-value ≤0.005, *** indicates p-value ≤0.0005. F) C57Bl/6 mice were orthotopically injected with syngeneic Pan02 cells constitutively expressing luciferase and allowed to recover and grow tumors for 1 week before initiating vehicle, chaetocin or MLN9237 alone and combination treatments. To monitor in vivo tumor growth, mice were injected with D-luciferin at 0, 3, and 10 days of treatment and total flux (photons/second, p/s) measured for each mouse in the Xenogen IVIS-200 imaging system. Total flux was normalized to day 0 and averaged (mean±SEM) across mice per condition and statistical significance was calculated by multiple t-tests at each day using the Holm-Sidak correction for multiple comparisons. * indicates p- value ≤0.05 for comparison of combination treatment to vehicle control. Treatments with individual drugs did not produce statistically significant effects. G) Representative images of mice injected with D-luciferin at day 10.
We also evaluated these drugs, in a more relevant 3D culture model [18], namely organoids. Pancreatic ducts were isolated from Ela-Kras mice [19] and propagated in matrigel to form organoid structures. Organoids derived from these murine models of PDAC can generate lesions that resemble pancreatic intraepithelial neoplasias (PanINs) and progress to invasive PDAC, thereby offering a useful tool for evaluating therapeutic approaches [18]. Organoids were treated with 150nM or 300nM chaetocin, 450nM or 900nM MLN8237 or their combination (150nM + 450nM; 300nM + 900nM) for 72 hours, and viability of cells was evaluated by CellTiter-Glo assay (Figure 3D–E). With 150nM chaetocin and 450nM MLN8237, the combination showed a significant reduction in viability (50.5±3.0% compared to vehicle) in relationship to MLN8237 (19.8±3.4% reduction compared to vehicle) and chaetocin alone (30.6±2.9% reduction compared to vehicle; Figure 3E). Similarly, 300nM chaetocin and 900nM MLN8237 significantly reduced viability (69.2±3.5% compared to vehicle) in relationship to MLN8237 (16.3±3.3% reduction compared to vehicle) and chaetocin alone (54.3±4.4% reduction compared to vehicle; Figure 3E). Representative images of the organoids demonstrate appreciable reduction in size and disruption of the 3D structure in the combination treatment (Figure 3D).
Lastly, we evaluated the effect of the AurkA and pan-H3K9me HMT inhibitors using orthotopic xenografts. Pan02 cells stably expressing luciferase were injected into the pancreas of C57BL/6 mice. These cells allow accurate monitoring of time-dependent in vivo growth of xenografts. Xenografts were treated with vehicle control, chaetocin alone, MLN8237 alone or their combination (N=5/group) and monitored by bioluminescence. Mean bioluminescence values are shown at 0, 3, and 10 days of treatment (Figure 3F). While individual treatments did not result in a statistically significant reduction in tumor growth (normalized total flux compared to start of treatment 1.04±0.21 p/s for control animals versus 0.41±0.17 p/s for chaetocin alone, p>0.05; or 0.57±0.19 p/s for MLN8237 alone, p>0.05), the group receiving the combination displayed significant reduction of tumor growth (0.25±0.08 p/s, p<0.05) after 10 days of treatment (representative images, Figure 3G). Therefore, the MLN8237-chaetocin combination is more effective than each of the drugs alone for inhibiting cell growth both, in vitro and in vivo.
The MLN8237-chaetocin combination inhibits PDAC growth via a cellular mechanism involving mitotic catastrophe
Since the AurkA and HP1-HMT pathways are both involved in the regulation of cell division, we examined the mechanisms by which antagonizing these pathways leads to PDAC cell growth inhibition. Using fluorescence-based measurement of live-cell protease activity and luminogenic-based substrate to detect caspase-3/7 activation, we confirmed reduction of cell viability along with a corresponding increase of apoptotic indicators in PANC-1 cells treated with vehicle, 30nM chaetocin, 90nM MLN8237 or their combination. After 72 hours, relative cell viability (Figure 4A) of combination-treated cells (1.53±0.20) was significantly reduced compared to vehicle (2.65±0.06, p≤0.0005), chaetocin alone (2.03±0.08, p≤0.05), or MLN8237 alone (2.41±0.07, p≤0.0005), while relative caspase 3/7 activation (Figure 4B) was significantly increased in combination-treated cells (7.74±0.34) compared to vehicle (3.17±0.21, p≤0.0005), chaetocin alone (6.08±0.25, p≤0.0005), or MLN8237 alone (4.53±0.43, p≤0.0005). Similar observations were made with L3.6 cells (Supplementary Figure 1D–E). Subsequently, we measured how these inhibitors impact the cell cycle using fluorescent activated cell sorting (FACS). We found that while chaetocin and MLN8237 alone caused modest accumulation of cells with G2/M DNA content (35.6±0.04% and 22.0±3.3%, respectively versus 17.6±0.03% in vehicle control treated cells), their combination induced a substantial shift of the cells to G2/M (67.2±1.4%) with an increase in subG1 population (Figure 4C–D). Interestingly, confocal and electron microscopy demonstrated that the effect of the combination was due to chromosomal instability, mitotic aberrations, and ultimately induction of mitotic cell death (Figure 4E–G). For instance, immunofluorescence with anti-α-tubulin for spindle poles and anti-γ-tubulin for centrosomes showed that cells treated with either drug exhibited mitotic aberrations, including multipolar spindles, centrosome disruption or lagging, unorganized chromosomes (Figure 4E–F). While cells with vehicle treatment displayed mitotic abnormalities in only 15.9% of the mitotic population (n=208), 31.6% of chaetocin-treated (n=171), and 42.6% of MLN8237-treated (n=169) cells in mitosis had these types of aberrations. Cells with complete disruption of the mitotic spindle and chromosome alignment were categorized as undergoing mitotic catastrophe. None of the vehicle-treated cells exhibited this level of mitotic disturbance. However, 9.4% of chaetocin-treated cells and 33.1% of MLN8237-treated cells showed mitotic catastrophe. Interestingly, the combination of chaetocin and MLN8237 demonstrated a significant portion of cells in mitotic catastrophe rather than mitotic aberrations (81.8% mitotic catastrophe versus 11.8% mitotic aberrations; n=187; Figure 4E). Similarly, PANC-1 cells treated by the combination show increased mitotic catastrophe (29.9% mitotic catastrophe, n=147, Supplementary Figure 1F). Scanning electron microscopy confirmed that while chaetocin and MLN8237 individually caused multinucleated cells (Figure 4G, black arrows), their combination resulted in multiple nuclei per cell and a significant proportion of cells that had died in mitosis, as evidenced by a highly disorganized cytoplasm, altered vesicular organelles, and presence of electron dense condensed chromosomes (Figure 4G, red arrow). Biochemically, the combined treatment with MLN8237 and chaetocin resulted in increased levels of P-S10-H3 and P-S139-H2A.X, as measured by western blot (Figure 4H). The coinciding increase of both histone marks is characteristic of mitotic catastrophe [31], further confirming this phenomenon. Notably, we also checked tissue from our treatment of orthotopic xenografts for P-S139-H2A.X positive cells by IHC to confirm this observation in vivo. Congruently, we found that orthotopic xenografts from mice receiving the combination displayed a higher number of P-S139-H2A.X positive cells (Figure 4I–J; 8.4±0.51 positive cells/field) compared to control (1.8±0.37 positive cells/field; p=0.0001), chaetocin alone (3.4±0.6 positive cells/field; p=0.0002), or MLN8237 alone (1.4±0.24 positive cells/field; p<0.0001), confirming that the combination had the expected effect in vivo. Collectively, our data demonstrate that the cellular mechanism underlying PDAC growth inhibition observed with combined MLN8237 and chaetocin treatment is through induction of mitotic catastrophe. Thus, these results prompted the search for molecular mechanisms to explain this effect by focusing on how these drugs modify the transcriptional response by RNA-sequencing (RNA-seq), as well as cell cycle checkpoint pathways that are activated by chromosomal instability.
Figure 4. Chaetocin–MLN8237 combination synergizes to trigger a G2/M shift and significant mitotic catastrophe.
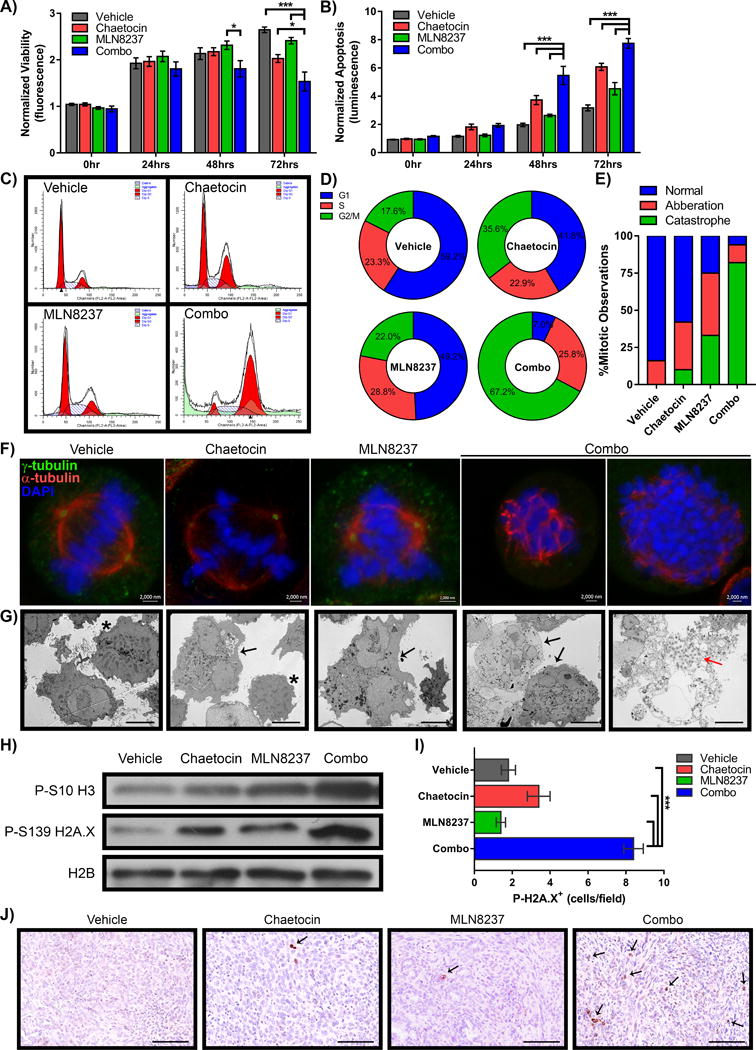
PANC-1 cells were plated and treated for 0, 24, 48 and 72hrs with vehicle, chaetocin (30nM), MLN8237 (90nM) or combination (30 + 90nM, CH + MLN8237 respectively). Levels of cell A) viability and B) apoptosis were observed by protease and caspase 3/7 cleavage of fluorescent and luminescent substrates respectively. Values were normalized to 0hr vehicle for each treatment and statistical significance calculated by 2-way ANOVA with Tukey’s multiple comparisons test. * indicates p- value ≤0.05, *** indicates p-value ≤0.0005. C) FACS analysis of PANC-1 cells treated for 48hrs with chaetocin, MLN8237 and combination treatments resulted in slightly increased G2/M arrest for single treatments and nearly complete G2/M arrest and cell death (sub G1 events shown in light green) for the combination treatment. Representative FACS cell cycle graphs illustrate the significant shift and D) average percentage (n=2) of events in each phase of cell cycle, G1, S, or G2/M are graphed with relative percentages indicated in each segment. E) L3.6 cells were treated with vehicle, chaetocin, MLN8237 or combination for 48hrs, fixed and stained antibodies to γ-tubulin (green), α-tubulin (red), and DNA stained with DAPI (blue) to consider mitotic progression. Over 150 mitotic cells were observed and quantified as normal, aberration, or catastrophe and resultant graph indicates the percentage of the total number of cells counted. F) Representative images of cells counted as normal mitosis (vehicle), multiple spindle poles (chaetocin and MLN8237 alone), and total disruption of the spindle apparatus in mitotic catastrophe during combination treatment. Scale=2000nm. G) EM images of PANC-1 cells after 72hrs of treatment demonstrated multinucleated cells (black arrows). Normal mitoses are indicated by (*). Evidence of mitotic cell death/mitotic catastrophe is shown in the combination treatment (red arrow). Scale=10μM. H) PANC-1 lysates after 48hrs of treatment indicate that the relative levels of P-S10 H3 and P-S139 H2A.X (indicators of mitotic catastrophe) are increased in the combination treatment by Western blot, with H2B as loading control. Pancreatic tissue from orthotopic xenografts was stained by IHC for P-S139 H2A.X as a measure of mitotic catastrophe in vivo. I) Quantification of average positive staining cells per field are graphed ±SEM with 5 field views per sample. Statistical significance was performed using a Student’s t-test to compare combination treatment to all other conditions, *** indicates p-value ≤0.0005. J) Representative fields illustrate the increased number of positively stained cells (black arrows) in the combination treated animals as compared to vehicle, chaetocin or MLN8237 alone. Scale=0.1mm.
RNA-seq defines molecular markers of the MLN8237-chaetocin response
We utilized RNA-Seq to define molecular markers for the effect of the MLN8237-chaetocin combination. PDAC cells were treated with individual drugs or their combination and compared to vehicle. Treatment was limited to 24 hours to extract RNA before significant mitotic catastrophe occurred to capture the transcriptional effect. Figure 5A shows the heat map from triplicate experiments. Supplementary Table 3 lists the specific values for expression of differentially expressed genes (DEG) found in response to treatment. A Venn diagram (Figure 5B) compared the DEG in chaetocin alone and the combination treatment. Here, 548 genes are indicated as changed specifically as the result of the combination and not found to be changing in the chaetocin alone treatment. Furthermore, MLN8237 alone did not result in any significant DEG at the dose used for all experiments, with the exception of two pseudogenes. Notably, the majority (83%) of the 548 DEG in response to the combination were up-regulated, consistent with inhibition of the repressive H3K9me pathway, which represents one arm of the treatment and is expected to de-repress gene expression. A bar graph with genes grouped by ontological function and number of DEG activated or repressed by combination treatment, colored red and green, respectively, is shown (Figure 5C). We also found that the treatment regulates genes, which promoters contain xenobiotic response elements involved in drug metabolism, including CYP1A1, CYP1B1, CYP2B7P, CYP7B1, CYP4F12, CYP2E1, and CYP2C8, serving as an internal positive control for drug exposure. Interestingly, 36% (199/548) of DEG are non-coding RNAs, constituting an overrepresentation beyond the expected number. Among these transcripts, 77% of them belong to the group of the recently reported non-coding regulatory RNAs, known as mitotic chromosome-associated RNAs (mCARs), which appear to be critical structural components of chromosomes [32]. In the similar functional category, the entire histone 1 cluster (HIST1H4I, HIST1H4H, HIST1H4E, HIST1H3J, HIST1H3D, HIST1H3C, HIST1H2BO, HIST1H2BN, HIST1H2BJ, HIST1H2BG, HIST1H2AM, HIST1H2AG and HIST1H2AD) was also represented. The percentage of DEG associated to chromosome structure in response to the drug combination was higher than expected for a non-specific effect and may likely reflect a cellular response to alterations in these organelles, which are observed during mitotic catastrophe. In addition to chromosomal markers, the remaining DEG participate in chromatin organization, transcription, and epigenetics, cell adhesion and extracellular matrix, cell signaling, cytoskeleton, metabolism, mitochondria homeostasis, membrane proteins, cytoskeleton, channels and transporters, as well as cell cycle, senescence, and apoptosis (Figure 5C). Whether the changes in gene expression result from effects of abnormal gene regulation in response to the drugs or reflect a role in mitotic catastrophe is less clear. Nevertheless, these experiments provide a number of potential markers, which can serve to monitor the effectiveness of this combination.
Figure 5. RNA-seq defines molecular markers of the MLN8237-chaetocin response.
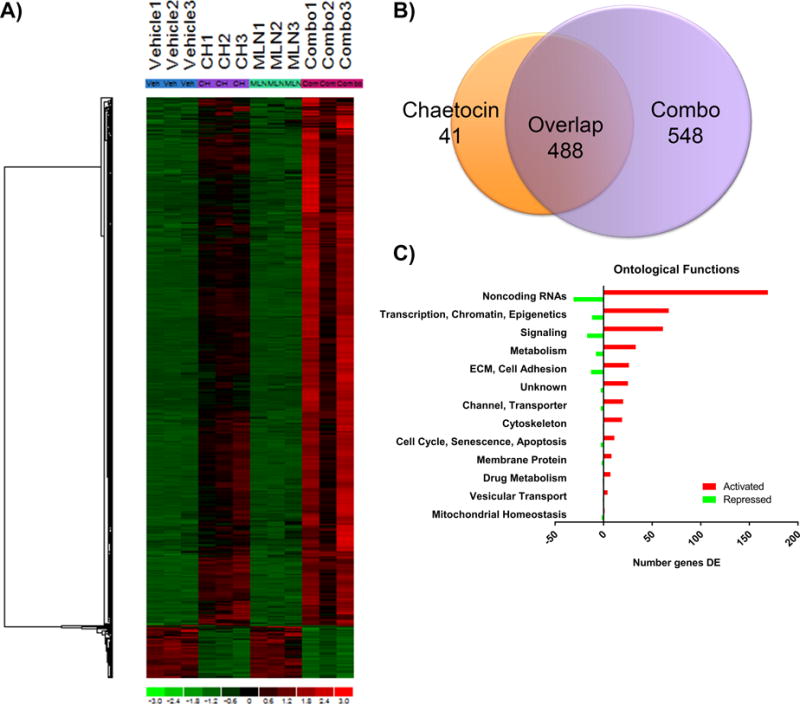
RNA-seq was performed on PANC-1 cells that were treated for 24hrs with vehicle, chaetocin (30nM), MLN8237 (90nM) or combination (30 + 90nM, CH + MLN8237 respectively). A) Hierarchical clustering of all genes identified across the four conditions indicates large clusters of genes increased after cells were treated with the combination of drugs. B) A Venn diagram of the two conditions that induced differential gene expression, chaetocin alone and combination, illustrates 548 unique genes that changed expression only in the combination therapy. C) Separation of the 548 DEG by ontological function demonstrates that genes include a large number of non-coding RNAs and transcriptional, chromatin, epigenetic elements. Red and green bars depict up-regulated and down-regulated genes in each category, respectively.
Mitotic catastrophe induced by combined MLN8237-chaetocin treatment is characterized by aberrant mitotic checkpoint signaling and decreased centromeric H3K9 methylation
To unravel molecular mechanisms that may explain how synergism of the MLN8237-chaetocin combination triggers mitotic death, we investigated the integrity of G2/M checkpoint pathways. ATM (Ataxia Telangiectasia Mutated) and ATR (ATM and Rad3-related) are part of a unique family of serine-threonine kinases functioning as transducers of DNA signals [33]. We hypothesized that, in the combination treatment, these pathways may be affected due to the observed centromere changes, mitotic instability, prolonged cell cycle arrest, and ultimately, mitotic catastrophe. Testing this hypothesis followed the rationale that these kinases orchestrate a network of cellular processes that maintains genomic integrity by signaling to downstream kinases, such as Chk1. Chk1 functions in survival and cell cycle checkpoints, in particular cell cycle arrest before mitosis in response to DNA damage [34]. Activated Chk1 phosphorylates the dual specificity phosphatase Cdc25C on Ser216, which sequesters this phosphatase in a complex within the cytoplasm, thereby preventing Cdc25C from activating Cdk1 through removal of the inhibitory phosphorylation. As a result, the Cdk1/Cdc2-cyclin B complex is maintained in an inactive state to block entry into mitosis. In MLN8237-treated cells, ATM and ATR along with P-S345 Chk1 were present, presumably leading to high levels of P-S216 Cdc25c, its inactive form (Figure 6A). Cdk1 was also inactive, as reflected by high levels of P-Y15 Cdk1/cdc2. High levels of Wee1, which phosphorylates Y15-Cdk1/cdc2 leading to its inactivation, likely, reinforced this effect (Figure 6A). Therefore, treatment with MLN8237 leads to signals that are known to render the Cdk1-Cyclin B complex unable to mediate cell division, accompanied by G2/M arrest (Figure 6B) [31]. Chaetocin treatment alone had ATM and ATR levels similar to control and MLN8237-treated cells (Figure 6A). There were slight decreases in the amounts of P-S345 Chk1 and P-S216 Cdc25c in comparison to control and MLN8237, although not to the extent observed in the combination. However, the slight decrease in inactive P-S216 Cdc25c did not result in the presence of active P-T48 Cdc25c (Figure 6A). The presence of inactive P-Y15 Cdk1/cdc2 remained, while a low level of active P-T161 Cdk1/cdc2 was also detected. Levels of cyclin B stayed constant. Overall, the molecular response to chaetocin alone was not markedly distinct from control-treated cells in key mitotic checkpoints. However, chaetocin with MLN8237 triggered an aborted response to bypass this G2/M arrest, which ultimately resulted in mitotic catastrophe and death. At the molecular level, lysates from the MLN8237-chaetocin combination demonstrated Chk1 inactivation by decreased levels of both, total and phosphorylated form of this protein (Figure 6A). Chk1 inactivation was also evidenced by a decrease in P-S216 Cdc25c (inactive) with a corresponding increase in P-T48 Cdc25c (active). This Cdc25c activation led to Cdk1/cdc2 dephosphorylation (Y15), allowing activation of the Cdk1/cdc2-Cyclin B complex, as observed by increased P-T161 Cdk1/cdc2 (Figure 6A). Concordantly, low levels of Wee1 likely strengthened this effect along with increased Cyclin B (Figure 6A). Central to these G2/M arrest pathways is p53 [35], which levels were significantly ablated by the combination (Figure 6A), a phenomenon that has been implicated in mitotic catastrophe [36, 37]. Notably, p53 downregulation occurs at the protein level without changes at the transcript level (Supplementary Table 4) in real time PCR, which yielded a fold change for p53: chaetocin, 1.130; MLN8237, 1.039; combination, −1.219 each compared to vehicle, or in RNA-seq (log fold change: chaetocin, −0.169; MLN8237, −0.156; combination, −0.415 each compared to vehicle). Similarly, none of the other mitotic checkpoint proteins were altered at the transcript level (Supplementary Table 4), indicating that these changes are post-transcriptional events. As a consequence, combination treatment results in a signal for inducing mitotic progression (Figure 6B), but since both the AurkA and H3K9me pathways are inhibited, this response appears to abort with cells ultimately entering mitotic catastrophe and cell death due to the structural disorder and chromosomal instability (Figure 4C–G). As chaetocin is an inhibitor of all known H3K9me HMTs, we checked the levels of the dimethyl (H3K9me2) and trimethyl (H3K9me3) post-translational marks of K9H3. As expected, levels of both, H3K9me2 and H3K9me3 decreased with chaetocin alone, as observed by western blot (Figure 6C). Interestingly, levels of H3K9me2 were also reduced with MLN8237, and both of these post-translational modifications were reduced even more in the combination compared to either of the individual drugs (Figure 6C). In mitotic cells, the H3K9me3 mark co-localizes with pericentromeric heterochromatin [29]. Using immunofluorescence, we observed a decrease in pericentromeric H3K9me3 in mitotic cells treated with chaetocin or MLN8237 alone compared to control, which was almost completely absent in cells treated with their combination (Figure 6D, Supplementary Figure 1G), congruent with our western blot findings. In summary, the molecular signature in the combination treatment reflects one of cell cycle progression combined with signals indicative of mitotic catastrophe, namely the concurring increase of both P-S10-H3 and P-S139-H2A.X histone marks (Figure 4H), and decreased centromeric H3K9 methylation (Figure 6D), suggesting that mitotic cell death results from aberrant checkpoint signaling along with severe chromatin-DNA damage.
Figure 6. Combined AurkA and pan-H3K9-HMT inhibition triggers uncoordinated checkpoint responses and reduces the centromeric H3K9me mark.
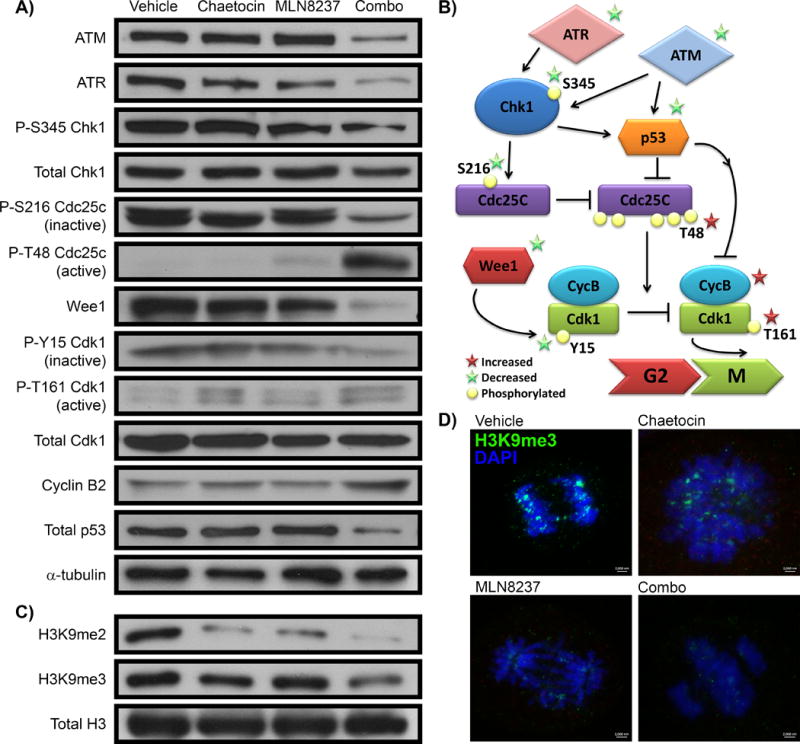
A) Combination of chaetocin and MLN8237 treatments triggered dysregulation of a variety of proteins involved in cellular checkpoint response. Protein levels were observed by western blot with α-tubulin illustrating equal loading of samples across all treatments. B) Pathway diagram illustrates the ATM/ATR-Chk1-p53 pathway for G2M cell arrest. Increases and decreases in nodes affected by the combination treatment are marked with red and green stars, respectively. C) Treatments synergize to reduce the H3K9me mark in cells. Total levels of H3K9me2 and H3K9me3 are indicated by western blot of the vehicle, chaetocin, MLN8237, and combination treatments of PDAC cells. D) Representative images of H3K9me3 immunofluorescence staining in L3.6 cells demonstrated a global decrease of H3K9me3 upon treatment with the combination of drugs. Scale=2000nm.
Discussion
Epigenomic-based pharmacology has the potential to serve as a robust tool to improve the future treatment of PDAC. The results of the current study significantly contribute to the field of experimental therapeutics by demonstrating how to inhibit PDAC growth through combined targeting of an oncogene and epigenetic regulator (genetic-to-epigenetic crosstalk). We tested the efficiency of this approach by jointly targeting AurkA and one of its epigenetic targets, the H3K9me pathway. We also dissected cellular and molecular mechanisms by which this combination leads to a cytotoxic effect. Thus, it is important to consider how our evidence supports a model by which these types of therapies may work and how similar approaches can be translated to other genetic-to-epigenetic pathways controlling cancer growth.
Antitumor agents have the possible cellular outcomes of cytotoxicity by cell death or cytostasis through cell cycle arrest and/or senescence [38]. While senescent cells have the benefit of suppressing tumorigenesis, unfortunately, they leave cells metabolically active that can potentially contribute to progression and therapy resistance. MLN8237 (alisertib), a MLN8054 derivative, works as highly specific ATP-competitive drug that binds AurkA and inhibits T288 phosphorylation [39]. As a result of AurkA inhibition, MLN8237 blocks G2/M cell-cycle progression, induces polyploidy, and results in senescence [40]. Since AurkA is overexpressed in many cancers, including PDAC, it continues to be a feasible target for therapy. While several AurkA inhibitors have been developed, MLN8237 remains the most promising due to minor to moderate side effects. There are numerous ongoing Phase I and II trials with MLN8237 as a single agent or in combination with other drugs in advanced solid and hematologic malignancies [41]. Combination of MLN8237 with cytotoxic drugs, including taxanes, cisplatin, vincristine and gemcitabine among others, potentiates anticancer activity. However, even though Phase I and II studies have offered encouraging results, the first Phase III study with MLN8237 was discontinued due to unsatisfactory efficacy, leaving some yet unanswered questions for MLN8237 use in clinical settings. Therefore, although MLN8237 holds promise as a therapeutic agent, there remains much speculation that this drug will ultimately better serve in combination with other agents if it is to gain clinical approval. In particular, the optimal endpoint of such therapy, rather than senescence, would be to effectively eliminate malignant cells through cell death.
Our data demonstrate that the combined inhibition of AurkA, and the H3K9me pathway is synergistic (Figures 2–3). An enhanced effect of the combination to inhibit PDAC growth is also observed with 3D spheroid and organoid cultures, as well as xenografts in vivo (Figure 3). This is consistent with the fact that these targets are in the same biological pathway that aids to the proper completion of mitosis [12]. Upon cell cycle arrest caused by AurkA inhibition, the mitotic machinery is exposed longer to targeting by the H3K9me pathway, which during mitosis regulates centromere structure. Histones associated with centromeric heterochromatin are typically hypoacetylated [42] and methylated on H3K9 [29]. H3K9me levels rapidly increase upon mitotic entry, reaching maximum in metaphase [13]. Congruent with previous reports [13], disruption of this H3K9me-mediated centromeric pathway in mitosis (Figure 6C–D, Supplementary Figure 1G), which in the current study is achieved through the combined use of a pan-H3K9me HMT and an AurkA inhibitor, leads to mitotic aberrations (Figure 4E–F, Supplementary Figure 1F), abnormal mitotic checkpoint responses (Figure 6A–B), and cell death by mitotic catastrophe. Furthermore, the cellular response to the drug combination is marked by expression changes of genes involved in drug metabolism, mitotic chromosome-associated non-coding regulatory RNAs (mCARs), as well as genes associated to chromosome structure, as observed by RNA-seq (Figure 5). Thus, inhibition of the H3K9me epigenetic pathway, specifically in mitosis, offers a unique therapeutic approach not characteristically considered when utilizing epigenetic agents, which have more commonly been tested for modulating gene expression during interphase.
Mitotic catastrophe is an oncosuppressive mechanism defined as a mode of cell death resulting from aberrant mitosis [43]. Several drugs, including spindle assembly inhibitors, DNA damaging agents, and radiation, induce mitotic catastrophe. Microtubule-hyperpolymerizing agents, such as paclitaxel and docetaxel, and depolymerizing agents of the Vinca-alkaloid type (vinblastine, vincristine and vindesine) are mitotic inhibitors that influence microtubule dynamics by binding to tubulin dimers, leading to spindle disturbances [44]. There are other inducers of mitotic catastrophe that are undergoing evaluation in preclinical and clinical settings, including inhibitors of checkpoint kinases (CHK), polo-like kinases (PLK), poly(ADP-ribose) polymerase (PARP) and survivin. Mitotic catastrophe ensues from a combination of dysfunctional cell-cycle checkpoints, particularly those related to DNA structure and spindle assembly, along with cellular damage [45]. Here, we find that the combination of AurkA and H3K9me-HMT inhibition elicits an aberrant response of checkpoint proteins, bypassing the Chk1-Cdc25c-Cdk1-dependent G2/M arrest observed with MLN8237 alone (Figure 6A–B), in the presence of DNA damage marked by high P-S139-H2A.X levels (Figure 4H–J) to culminate in mitotic catastrophe. The phenomenon of mitotic catastrophe is a desirable outcome in cancer therapy, and thus, the current study, which achieves this effect by targeting a genetic-to-epigenetic pathway, provides a model for effective combinatorial therapeutic approaches for PDAC, and likely other cancers.
In lieu of these findings, it is tempting to speculate how inhibition of other genetic-to-epigenetic pathways may exert similar effects. For instance, another well known repressive methyl-histone mark, H3K27Me, mediated by the Polycomb complex, is also retained through mitosis and required for proper cell cycle progression [46, 47]. Similarly, the H4K20me1 demethylase, PHF8, influences G2/M transition and its loss leads to highly defective mitosis [48]. Brd4, a member of the BET bromodomain family that has elicited significant attention as cancer targets, localizes to chromosomes during mitosis and plays an essential role in mitotic spindle assembly [49]. Interestingly, a recent study found that the combination of the BET bromodomain inhibitor JQ1 and vincristine, an anti-microtubule drug that arrests cells at mitosis, synergistically induces the formation of aberrant mitotic spindles, cell cycle arrest at G2/M and death in neuroblastoma cells [50]. Thus, inhibition of various epigenetic pathways has the possibility to be exploited for the efficient development of therapeutic modalities in the future.
In summary, the current study advances our understanding of how epigenetic inhibitors can be utilized in novel therapeutic approaches. Our results outline a model for combined targeting of a genetic-to-epigenetic pathway to inhibit PDAC cell growth via a cytotoxic mechanism that involves perturbation of normal mitotic progression to end in mitotic catastrophe, an attractive outcome in oncology. Furthermore, it suggests an important and provocative consideration for harnessing the capacity of cell cycle inhibitors in efforts to enhance the future use of epigenetic inhibitors.
Supplementary Material
Implications.
These results outline a model for the combined inhibition of a genetic-to-epigenetic pathway to inhibit cell growth and suggest an important and provocative consideration for harnessing the capacity of cell cycle inhibitors to enhance the future use of epigenetic inhibitors.
Acknowledgments
We want to express our sincere gratitude to Dr. Raul Urrutia for critical insight and valuable scientific discussions and Kristin Mantz from the Mayo Clinic Microscopy and Cell Analysis Core facility for her assistance with the Mikroscan Slide Scanner.
Financial Support: This work was supported by funding from the National Institutes of Health (grant R01 CA178627 to GL), the Mayo Clinic Division of Gastroenterology and Hepatology, Department of Medicine, as well as the Mayo Clinic Center for Cell Signaling in Gastroenterology (P30DK084567) and the Mayo Clinic SPORE in Pancreatic Cancer (P50 CA102701).
Abbreviations
- PDAC
pancreatic adenocarcinoma
- AurkA
Aurora kinase A
- HMTs
histone methyltransferases
- mCARs
mitotic chromosome-associated non-coding regulatory RNAs
- CH
Chaetocin
- CI
combination index
- Fa
Fraction Affected
- APH
acidic phosphatase
- PanINs
pancreatic intraepithelial neoplasias
- photons/second, p/s
total flux
- IACUC
Instiutional Animal Care and Use Committe
- FACS
fluorescent activated cell sorting
- RNA-seq
RNA-sequencing
- ECL
enhanced chemiluminescence
- DEG
differentially expressed genes
- HRP
horse radish peroxidase
- Ataxia Telangiectasia Mutated
ATM
- ATM and Rad3-related
ATR
- TIS
therapy-induced senescence
- CHK
checkpoint kinases
- PLK
polo-like kinases
- PARP
poly(ADP-ribose) polymerase
- IVIS-200
in vivo imaging system
- ATCC
American Type Culture Collection
- DTP
Developmental Therapeutics Program
- DCTD
Division of Cancer Treatment and Diagnosis
- SEM
Tumor Repository, Standard Error of the Mean
- PNPP
p-nitrophenyl phosphate
- PVDF
polyvinylidene difluoride
- i.p.
intraperitoneal
Footnotes
Conflict of Interest: The authors declare that they have no competing interests.
References
- 1.Iovanna J, Mallmann MC, Goncalves A, Turrini O, Dagorn JC. Current knowledge on pancreatic cancer. Front Oncol. 2012;2:6. doi: 10.3389/fonc.2012.00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sharma C, Eltawil KM, Renfrew PD, Walsh MJ, Molinari M. Advances in diagnosis, treatment and palliation of pancreatic carcinoma: 1990–2010. World J Gastroenterol. 2011;17(7):867–897. doi: 10.3748/wjg.v17.i7.867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yu X, Zhang Y, Chen C, Yao Q, Li M. Targeted drug delivery in pancreatic cancer. Biochim Biophys Acta. 2010;1805(1):97–104. doi: 10.1016/j.bbcan.2009.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang F, Li H, Yan XG, Zhou ZW, Yi ZG, He ZX, Pan ST, Yang YX, Wang ZZ, Zhang X, Yang T, Qiu JX, Zhou SF. Alisertib induces cell cycle arrest and autophagy and suppresses epithelial-to-mesenchymal transition involving PI3K/Akt/mTOR and sirtuin 1-mediated signaling pathways in human pancreatic cancer cells. Drug Des Devel Ther. 2015;9:575–601. doi: 10.2147/DDDT.S75221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Neel NF, Stratford JK, Shinde V, Ecsedy JA, Martin TD, Der CJ, Yeh JJ. Response to MLN8237 in pancreatic cancer is not dependent on RalA phosphorylation. Mol Cancer Ther. 2014;13(1):122–133. doi: 10.1158/1535-7163.MCT-12-1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Marumoto T, Zhang D, Saya H. Aurora-A - a guardian of poles. Nat Rev Cancer. 2005;5(1):42–50. doi: 10.1038/nrc1526. [DOI] [PubMed] [Google Scholar]
- 7.Li D, Zhu J, Firozi PF, Abbruzzese JL, Evans DB, Cleary K, Friess H, Sen S. Overexpression of oncogenic STK15/BTAK/Aurora A kinase in human pancreatic cancer. Clin Cancer Res. 2003;9(3):991–997. [PubMed] [Google Scholar]
- 8.Zhu J, Abbruzzese JL, Izzo J, Hittelman WN, Li D. AURKA amplification, chromosome instability, and centrosome abnormality in human pancreatic carcinoma cells. Cancer Genet Cytogenet. 2005;159(1):10–17. doi: 10.1016/j.cancergencyto.2004.09.008. [DOI] [PubMed] [Google Scholar]
- 9.Hata T, Furukawa T, Sunamura M, Egawa S, Motoi F, Ohmura N, Marumoto T, Saya H, Horii A. RNA interference targeting aurora kinase a suppresses tumor growth and enhances the taxane chemosensitivity in human pancreatic cancer cells. Cancer Res. 2005;65(7):2899–2905. doi: 10.1158/0008-5472.CAN-04-3981. [DOI] [PubMed] [Google Scholar]
- 10.Gorgun G, Calabrese E, Hideshima T, Ecsedy J, Perrone G, Mani M, Ikeda H, Bianchi G, Hu Y, Cirstea D, Santo L, Tai YT, Nahar S, Zheng M, Bandi M, Carrasco RD, et al. A novel Aurora-A kinase inhibitor MLN8237 induces cytotoxicity and cell-cycle arrest in multiple myeloma. Blood. 2010;115(25):5202–5213. doi: 10.1182/blood-2009-12-259523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Manfredi MG, Ecsedy JA, Chakravarty A, Silverman L, Zhang M, Hoar KM, Stroud SG, Chen W, Shinde V, Huck JJ, Wysong DR, Janowick DA, Hyer ML, Leroy PJ, Gershman RE, Silva MD, et al. Characterization of Alisertib (MLN8237), an investigational small-molecule inhibitor of aurora A kinase using novel in vivo pharmacodynamic assays. Clin Cancer Res. 2011;17(24):7614–7624. doi: 10.1158/1078-0432.CCR-11-1536. [DOI] [PubMed] [Google Scholar]
- 12.Grzenda A, Leonard P, Seo S, Mathison AJ, Urrutia G, Calvo E, Iovanna J, Urrutia R, Lomberk G. Functional impact of Aurora A-mediated phosphorylation of HP1gamma at serine 83 during cell cycle progression. Epigenetics Chromatin. 2013;6(1):21. doi: 10.1186/1756-8935-6-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McManus KJ, Biron VL, Heit R, Underhill DA, Hendzel MJ. Dynamic changes in histone H3 lysine 9 methylations: identification of a mitosis-specific function for dynamic methylation in chromosome congression and segregation. J Biol Chem. 2006;281(13):8888–8897. doi: 10.1074/jbc.M505323200. [DOI] [PubMed] [Google Scholar]
- 14.Bruns CJ, Harbison MT, Kuniyasu H, Eue I, Fidler IJ. In vivo selection and characterization of metastatic variants from human pancreatic adenocarcinoma by using orthotopic implantation in nude mice. Neoplasia. 1999;1(1):50–62. doi: 10.1038/sj.neo.7900005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 1984;22:27–55. doi: 10.1016/0065-2571(84)90007-4. [DOI] [PubMed] [Google Scholar]
- 16.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9(7):671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Longati P, Jia X, Eimer J, Wagman A, Witt MR, Rehnmark S, Verbeke C, Toftgard R, Lohr M, Heuchel RL. 3D pancreatic carcinoma spheroids induce a matrix-rich, chemoresistant phenotype offering a better model for drug testing. BMC Cancer. 2013;13:95. doi: 10.1186/1471-2407-13-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Boj SF, Hwang CI, Baker LA, Chio II, Engle DD, Corbo V, Jager M, Ponz-Sarvise M, Tiriac H, Spector MS, Gracanin A, Oni T, Yu KH, van Boxtel R, Huch M, Rivera KD, et al. Organoid models of human and mouse ductal pancreatic cancer. Cell. 2015;160(1–2):324–338. doi: 10.1016/j.cell.2014.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Grippo PJ, Nowlin PS, Demeure MJ, Longnecker DS, Sandgren EP. Preinvasive pancreatic neoplasia of ductal phenotype induced by acinar cell targeting of mutant Kras in transgenic mice. Cancer Res. 2003;63(9):2016–2019. [PubMed] [Google Scholar]
- 20.Fernandez-Zapico ME, Lomberk GA, Tsuji S, DeMars CJ, Bardsley MR, Lin YH, Almada LL, Han JJ, Mukhopadhyay D, Ordog T, Buttar NS, Urrutia R. A functional family-wide screening of SP/KLF proteins identifies a subset of suppressors of KRAS-mediated cell growth. Biochem J. 2011;435(2):529–537. doi: 10.1042/BJ20100773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gebelein B, Urrutia R. Sequence-Specific Transcriptional Repression by KS1, a Multiple-Zinc-Finger-Kruppel-Associated Box Protein. Molecular and Cellular Biology. 2001;21(3):928–939. doi: 10.1128/MCB.21.3.928-939.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mathison A, Grzenda A, Lomberk G, Velez G, Buttar N, Tietz P, Hendrickson H, Liebl A, Xiong YY, Gores G, Fernandez-Zapico M, Larusso NF, Faubion W, Shah VH, Urrutia R. Role for Kruppel-like transcription factor 11 in mesenchymal cell function and fibrosis. PLoS One. 2013;8(9):e75311. doi: 10.1371/journal.pone.0075311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dowsett M, Nielsen TO, A’Hern R, Bartlett J, Coombes RC, Cuzick J, Ellis M, Henry NL, Hugh JC, Lively T, McShane L, Paik S, Penault-Llorca F, Prudkin L, Regan M, Salter J, et al. Assessment of Ki67 in breast cancer: recommendations from the International Ki67 in Breast Cancer working group. J Natl Cancer Inst. 2011;103(22):1656–1664. doi: 10.1093/jnci/djr393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kalari KR, Nair AA, Bhavsar JD, O’Brien DR, Davila JI, Bockol MA, Nie J, Tang X, Baheti S, Doughty JB, Middha S, Sicotte H, Thompson AE, Asmann YW, Kocher JP. MAP-RSeq: Mayo Analysis Pipeline for RNA sequencing. BMC Bioinformatics. 2014;15:224. doi: 10.1186/1471-2105-15-224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14(4):R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26(1):139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Harrington E, Bebbington D, Moore J, Rasmussen R, Ajose-Adeogun A, Nakayama T, Graham J, Demur C, Hercend T, Diu-Hercend A, Su M, Golec J, Miller K. VX-680, a potent and selective small-molecule inhibitor of the Aurora kinases, suppresses tumor growth in vivo. Nat Med. 2004;10(3):262–267. doi: 10.1038/nm1003. [DOI] [PubMed] [Google Scholar]
- 28.Xie L, Kassner M, Munoz R, Que Q, Kiefer J, Zhao Y, Mousses S, Yin H, Von Hoff D, Han H. Kinome-wide siRNA screening identifies molecular targets mediating the sensitivity of pancreatic cancer cells to Aurora kinase inhibitors. Biochemical Pharmacology. 2012;83(4):452–461. doi: 10.1016/j.bcp.2011.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Saksouk N, Simboeck E, Dejardin J. Constitutive heterochromatin formation and transcription in mammals. Epigenetics Chromatin. 2015;8:3. doi: 10.1186/1756-8935-8-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Iwasa E, Hamashima Y, Fujishiro S, Higuchi E, Ito A, Yoshida M, Sodeoka M. Total synthesis of (+)-chaetocin and its analogues: their histone methyltransferase G9a inhibitory activity. J Am Chem Soc. 2010;132(12):4078–4079. doi: 10.1021/ja101280p. [DOI] [PubMed] [Google Scholar]
- 31.On KF, Chen Y, Tang Ma H, Chow JPH, Poon RYC. Determinants of Mitotic Catastrophe on Abrogation of the G2 DNA Damage Checkpoint by UCN-01. Mol Cancer Ther. 2011;10(5):784–794. doi: 10.1158/1535-7163.MCT-10-0809. [DOI] [PubMed] [Google Scholar]
- 32.Meng Y, Yi X, Li X, Hu C, Wang J, Bai L, Czajkowsky DM, Shao Z. The non-coding RNA composition of the mitotic chromosome by 5′-tag sequencing. Nucleic Acids Res. 2016;44(10):4934–4946. doi: 10.1093/nar/gkw195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Marechal A, Zou L. DNA damage sensing by the ATM and ATR kinases. Cold Spring Harb Perspect Biol. 2013;5(9) doi: 10.1101/cshperspect.a012716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sanchez Y, Wong C, Thoma RS, Richman R, Wu Z, Piwnica-Worms H, Elledge SJ. Conservation of the Chk1 checkpoint pathway in mammals: linkage of DNA damage to Cdk regulation through Cdc25. Science. 1997;277(5331):1497–1501. doi: 10.1126/science.277.5331.1497. [DOI] [PubMed] [Google Scholar]
- 35.Taylor WR, Stark GR. Regulation of the G2/M transition by p53. Oncogene. 2001;20(15):1803–1815. doi: 10.1038/sj.onc.1204252. [DOI] [PubMed] [Google Scholar]
- 36.Huang X, Tran T, Zhang L, Hatcher R, Zhang P. DNA damage-induced mitotic catastrophe is mediated by the Chk1-dependent mitotic exit DNA damage checkpoint. Proc Natl Acad Sci USA. 2005;102(4):1065–1070. doi: 10.1073/pnas.0409130102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jurvansuu J, Fragkos M, Ingemarsdotter C, Beard P. Chk1 Instability Is Coupled to Mitotic Cell Death of p53-deficient Cells in Response to Virus-induced DNA Damage Signaling. J Mol Biol. 2007;372(2):397–406. doi: 10.1016/j.jmb.2007.06.077. [DOI] [PubMed] [Google Scholar]
- 38.Lee M, Lee JS. Exploiting tumor cell senescence in anticancer therapy. BMB Rep. 2014;47(2):51–59. doi: 10.5483/BMBRep.2014.47.2.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Katsha A, Belkhiri A, Goff L, El-Rifai W. Aurora kinase A in gastrointestinal cancers: time to target. Mol Cancer. 2015;14:106. doi: 10.1186/s12943-015-0375-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu Y, Hawkins OE, Su Y, Vilgelm AE, Sobolik T, Thu Y-M, Kantrow S, Splittgerber RC, Short S, Amiri KI, Ecsedy JA, Sosman JA, Kelley MC, Richmond A. Targeting aurora kinases limits tumour growth through DNA damage-mediated senescence and blockade of NF-κB impairs this drug-induced senescence. EMBO Mol Med. 2013;5(1):149–166. doi: 10.1002/emmm.201201378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Durlacher CT, Li ZL, Chen XW, He ZX, Zhou SF. An update on the pharmacokinetics and pharmacodynamics of alisertib, a selective Aurora kinase A inhibitor. Clin Exp Pharmacol Physiol. 2016;43(6):585–601. doi: 10.1111/1440-1681.12571. [DOI] [PubMed] [Google Scholar]
- 42.Jeppesen P, Mitchell A, Turner B, Perry P. Antibodies to defined histone epitopes reveal variations in chromatin conformation and underacetylation of centric heterochromatin in human metaphase chromosomes. Chromosoma. 1992;101(5–6):322–332. doi: 10.1007/BF00346011. [DOI] [PubMed] [Google Scholar]
- 43.Galluzzi L, Vitale I, Abrams JM, Alnemri ES, Baehrecke EH, Blagosklonny MV, Dawson TM, Dawson VL, El-Deiry WS, Fulda S, Gottlieb E, Green DR, Hengartner MO, Kepp O, Knight RA, Kumar S, et al. Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ. 2012;19(1):107–120. doi: 10.1038/cdd.2011.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Denisenko TV, Sorokina IV, Gogvadze V, Zhivotovsky B. Mitotic catastrophe and cancer drug resistance: A link that must to be broken. Drug Resist Updat. 2016;24:1–12. doi: 10.1016/j.drup.2015.11.002. [DOI] [PubMed] [Google Scholar]
- 45.Castedo M, Perfettini JL, Roumier T, Andreau K, Medema R, Kroemer G. Cell death by mitotic catastrophe: a molecular definition. Oncogene. 2004;23(16):2825–2837. doi: 10.1038/sj.onc.1207528. [DOI] [PubMed] [Google Scholar]
- 46.Zaret KS. Genome reactivation after the silence in mitosis: recapitulating mechanisms of development? Dev Cell. 2014;29(2):132–134. doi: 10.1016/j.devcel.2014.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Aoto T, Saitoh N, Sakamoto Y, Watanabe S, Nakao M. Polycomb group protein-associated chromatin is reproduced in post-mitotic G1 phase and is required for S phase progression. J Biol Chem. 2008;283(27):18905–18915. doi: 10.1074/jbc.M709322200. [DOI] [PubMed] [Google Scholar]
- 48.Lim HJ, Dimova NV, Tan MK, Sigoillot FD, King RW, Shi Y. The G2/M regulator histone demethylase PHF8 is targeted for degradation by the anaphase-promoting complex containing CDC20. Mol Cell Biol. 2013;33(21):4166–4180. doi: 10.1128/MCB.00689-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dey A, Nishiyama A, Karpova T, McNally J, Ozato K. Brd4 marks select genes on mitotic chromatin and directs postmitotic transcription. Mol Biol Cell. 2009;20(23):4899–4909. doi: 10.1091/mbc.E09-05-0380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu PY, Sokolowski N, Guo ST, Siddiqi F, Atmadibrata B, Telfer TJ, Sun Y, Zhang L, Yu D, McCarroll J, Liu B, Yang RH, Guo XY, Tee AE, Itoh K, Wang J, et al. The BET bromodomain inhibitor exerts the most potent synergistic anticancer effects with quinone-containing compounds and anti-microtubule drugs. Oncotarget. 2016;7(48):79217–79232. doi: 10.18632/oncotarget.12640. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.