

Abstract
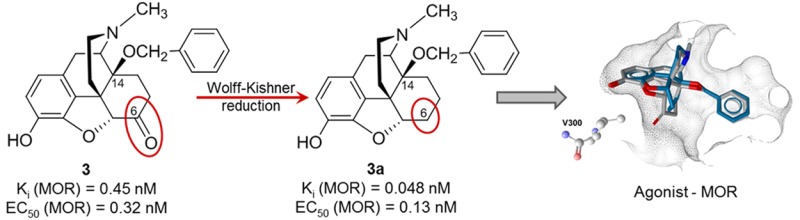
Position 6 of the morphinan skeleton plays a key role in the μ-opioid receptor (MOR) activity in vitro and in vivo. We describe the consequence of the 6-carbonyl group deletion in N-methylmorphinan-6-ones 1–4 on ligand–MOR interaction, signaling, and antinociception. While 6-desoxo compounds 1a, 2a, and 4a show similar profiles to their 6-keto counterparts, the 6-desoxo-14-benzyloxy substituted 3a displays significantly increased MOR binding and agonist potency and a distinct binding mode compared with its analogue 3.
It is more than 90 years ago since the structure of morphine, the naturally occurring alkaloid in the opium poppy Papaver somniferum, was clarified.1 Morphine has a long history of clinical use as an effective analgesic for the treatment of moderate to severe pain. It induces analgesia primarily via activation of the μ-opioid receptor (MOR).2 The morphine skeleton and its conversion to analogues have been continuously explored over the years. The driving force behind the synthetical efforts has been the search for an alternative to morphine that would produce powerful analgesia without its adverse effects (e.g., respiratory depression, sedation, constipation, analgesic tolerance, and addiction).2,3 Subsequently, the morphinan scaffold has been the basis of new drug developments, and ligands with distinct pharmacological properties are available for patient use or employed as research probes to explore opioid pharmacology in vitro and in vivo.2,4
Our laboratory has a long-standing focus in the field of opioid morphinan analgesics. The initial synthetical and pharmacological work led to the generation of N-methylmorphinan-6-ones with different substitution patterns at position 14.2b,4d,5 We established that the introduction of a 14-methoxy group into the clinically used analgesic oxymorphone (1),2a,2b (Figure 1) to yield 2 (14-O-methyloxymorphone)6a (Figure 1) not only increased MOR affinity by ∼9-fold but also produced a significant increase (40-fold) in the antinociceptive potency.6a,6b However, 2 induces the typical opioid-like side effects (respiratory depression, physical dependence, constipation, and motor dysfunction).6 Replacement of the 14-methoxy group in 2 with a benzyloxy substituent resulted in 3 (14-O-benzyloxymorphone)6b (Figure 1), which displayed a 5-fold increased antinociceptive potency when compared to 2.6b Moreover, this highly potent MOR agonist showed negligible inhibition of gastrointestinal motility, while it was much less constipating than morphine (2.5-fold) and 2 (7-fold).6b Chemical derivatization in the class of N-methylmorphinan-6-ones using 2 as the lead compound also targeted position 5, by introducing a 5-methyl group and giving rise to 4 (14-methoxymetopon)7 (Figure 1). Compound 4 is a highly efficacious analgesic in various pain models in animals.5,7 It was generally described to have an improved side effect profile by causing less respiratory depression, bradycardia, constipation, physical dependence, addiction potential, and development of analgesic tolerance in comparison to conventional MOR analgesics.5,7 Our recent molecular docking and molecular dynamics simulations study8 using the active structure of the MOR (PDB code 5C1M)9 in combination with structure–activity relationships (SAR) on N-methylmorphinan-6-ones revealed the subtle interplay between substituents at positions 5 and 14 in the morphinan scaffold in the ligand–MOR interaction by enabling identification of key structural elements that determine their distinct pharmacological profiles.8
Figure 1.

Structures of N-methylmorphinan-6-ones 1–4 and their 6-desoxo counterparts 1a–4a. Ph, phenyl.
Synthetic approaches have uncovered that functionalizing position 6 in N-methylmorphinan-6-ones results in a large diversity of activities.10−14 The 6-carbonyl group can be easily chemically converted into various functionalities, leading to hydrazones, oximes, carbazones, and semicarbazones,11 which display antinociceptive efficacies and a favorable side effect profile regarding respiratory depression and gastrointestinal motility.11 We and others have reported on the incorporation of amino acid residues at position 6 of N-methylmorphinan-6-ones.12 The 6-amino acid zwitterionic conjugates of 2 were established as MOR agonists inducing potent and long-lasting peripherally mediated antinociceptive effects after systemic administration.4,5a,13 The 6β-glycine substituted derivative of 2 was equipotent to fentanyl in the tail-flick assay in rats, acting via activation of peripherally located opioid receptors.13b We have also described derivatives of 2 with 6-amino and 6-guanidino substitution that showed high MOR affinity, selectivity, and efficacy and were highly active as antinociceptive agents.10 By targeting the chemically highly versatile 6-keto function of N-methylmorphinan-6-ones, we have reported on a chemically innovative modification giving rise to a novel class of morphinans with acrylonitrile incorporated substructures.14 The 6-cyano-N-methylmorphinans exhibit high affinity and selectivity for the MOR and potent in vitro and in vivo agonism.14
The present study was undertaken to evaluate the consequence of the deletion of the 6-carbonyl group of 14-hydroxy and 14-alkoxy substituted N-methylmorphinan-6-ones (1–4, Figure 1) resulting in the respective 6-desoxo-N-methylmorphinans 1a–4a (Figure 1) on binding and activation of the MOR and antinociceptive properties. Toward this aim, 1a–4a were prepared using Wolff–Kishner conditions similar to the earlier reported synthesis of 6-desoxonaltrexone15a and 6-desoxocyprodime15b from their corresponding 6-keto analogues naltrexone and cyprodime, respectively. Additionally, molecular modeling and SAR explorations aided by docking of investigated compounds to the active conformation of the MOR were performed to gain insights into their binding mode and interaction mechanisms.
Results and Discussion
Chemistry
The 6-desoxomorphinans 1a–4a reported herein were prepared from 1, 2,6a3,6b and 4,7 respectively, by Wolff–Kishner reduction as depicted in Scheme 1.
Scheme 1. Synthesis of Compounds 1a–4a.

Reagents and conditions: (a) hydrazine hydrate, triethylene glycol, 180 °C, 1.5 h; then KOH pellets, 180 °C, 2 h.
Pharmacology
Binding affinities at the human MOR, δ-opioid (DOR) and κ-opioid (KOR) receptors of N-methylmorphinan-6-ones 1–4 and their 6-desoxo counterparts 1a–4a (Figure 1) were first determined in competition binding assays using membranes from Chinese hamster ovary (CHO) cells stably transfected with one of the recombinant human opioid receptors (CHO-hMOR, CHO-hDOR, and CHO-hKOR cells) according to the described procedures.16 Binding affinities expressed as inhibition constant (Ki) values are shown in Table 1. All compounds displayed high potency to inhibit [3H][d-Ala2,Me-Phe4,Gly-ol5]enkephalin ([3H]DAMGO) binding to the human MOR in a concentration-dependent manner (Figure S1) with Ki values in the subnanomolar range (Table 1). The high binding affinities to the human MOR expressed in CHO cells showed by 1–4 corroborates our earlier findings at the rat MOR in brain tissue.6b Evaluation of N-methylmorphinan-6-ones and their 6-desoxo analogues revealed that the removal of the 6-keto function does not significantly affect binding affinity at the MOR when comparing 1 vs 1a, 2 vs 2a, and 4 vs 4a. In the case of 3 vs 3a only, a significant increase up to 9-fold in the MOR affinity was noticed. Generally, the 6-desoxo derivatives showed a similar binding affinity to the DOR as their 6-ketomorphinan counterparts, the exception being the pair 1a and 1, where a ∼3-fold decrease in affinity to the DOR was observed for 1a (Table 1). We also found that KOR affinities of 1a–4a were in the range of their parent compounds 1–4. The removal of the 6-keto group in 1 increased selectivity for MOR vs DOR of 1a, but it did not affect selectivity vs KOR. In the case of the 14-methoxy substituted 2 and 2a, a slight decrease in MOR vs KOR selectivity ratios was found, while selectivity for MOR vs DOR was not changed. The same observation on somewhat reduced MOR selectivity vs KOR was made when comparing 5-methyl substituted 4 and its 6-deoxo analogue 4a, while a decrease was also noticed vs DOR (Table 1). Interesting was the observation of the increase in MOR selectivity upon deletion of the 6-keto function in the 14-benzyloxy substituted 3 leading to 3a. We found that the nonselective 3 was converted into a MOR selective compound, 3a, which displayed about 9- and 6-fold higher MOR/DOR and MOR/KOR selectivity ratios, respectively, than 3 (Table 1).
Table 1. Opioid Receptor Binding, Functional Activities, Antinociceptive Potencies, and Physicochemical Properties of N-Methylmorphinan-6-ones 1–4 and Their 6-Desoxo Counterparts 1a–4a.
| opioid receptor binding, Ki (nM)a,c |
|
||||||||
|---|---|---|---|---|---|---|---|---|---|
| affinity |
selectivity |
functional activityb,c [35S]GTPγS MOR |
antinociceptive activityd hot-plate assay | ||||||
| compd | MOR | DOR | KOR | DOR/MOR | KOR/MOR | EC50 (nM) | % stim | ED50 (μg/kg, sc) (95% CI) | clogP e |
| 1 | 1.79 ± 0.34 | 70.0 ± 4.1 | 25.3 ± 8.4 | 39 | 14 | 7.80 ± 1.61 | 91.5 ± 5.0 | 382 (186–785) | 0.88 |
| 1a | 1.65 ± 0.5 | 201 ± 80 | 21.6 ± 3.9 | 122 | 13 | 8.99 ± 2.19 | 104 ± 7 | 383 (209–802) | 1.24 |
| 2 | 0.32 ± 0.08 | 8.80 ± 0.35 | 10.1 ± 3.1 | 28 | 32 | 1.45 ± 0.32 | 96.4 ± 5.0 | 14.2 (7.75–26.1) | 1.45 |
| 2a | 0.24 ± 0.08 | 8.64 ± 2.25 | 3.99 ± 0.63 | 36 | 17 | 1.37 ± 0.26 | 105 ± 6 | 17.3 (8.68–34.4) | 1.82 |
| 3 | 0.45 ± 0.10 | 1.05 ± 0.42 | 1.27 ± 0.24 | 2.3 | 2.8 | 0.32 ± 0.16 | 101 ± 5 | 2.38 (1.29–4.38) | 3.23 |
| 3a | 0.048 ± 0.011 | 0.89 ± 0.29 | 0.85 ± 0.05 | 21 | 18 | 0.13 ± 0.03 | 103 ± 8 | 2.41 (1.39–4.17) | 3.60 |
| 4 | 0.25 ± 0.04 | 19.8 ± 1.3 | 15.1 ± 0.9 | 79 | 60 | 3.28 ± 0.81 | 99.2 ± 2.8 | 27.1 (12.7–58.1) | 1.66 |
| 4a | 0.34 ± 0.08 | 12.9 ± 1.6 | 7.92 ± 4.47 | 38 | 23 | 2.95 ± 0.54 | 104 ± 3 | 25.1 (13.0–48.5) | 1.90 |
Determined in competition binding assays using membranes from CHO cells expressing human opioid receptors.
Determined in the [35S]GTPγS binding assay using CHO-hMOR cell membranes. Percentage stimulation (% stim) relative to the MOR full agonist DAMGO.
Values are the mean ± SEM of at least three independent experiments.
Determined in the hot-plate assay in mice at peak effect (30 min) after sc drug administration (n = 5–6 mice per group).
Calculated logP (clogP) using MarvinSketch 17.10 (ChemAxon, www.chemaxon.com).
Functional opioid activity of N-methylmorphinan-6-ones 1–4 and their 6-desoxo counterparts 1a–4a at the human MOR receptor was next evaluated, where ligand-induced stimulation of guanosine 5′-O-(3-[35S]thio)triphosphate ([35S]GTPγS) binding to membranes from CHO cells expressing the human MOR receptor was measured (Table 1) as described earlier.14c Efficacies are presented as percentage stimulation (% stim) relative to the prototypical MOR full agonist DAMGO (EC50 = 27.8 ± 3.7 nM and % stim = 221 ± 13%). While we have reported previously on the potent MOR agonism of 2–4 in bioassays using isolated organs (mouse vas deferens, guinea pig ileum)6b,7 and ligand-stimulated [35S]GTPγS binding in rat brain membranes,6c herein the first data on G protein activation upon ligand binding to the human MOR are presented. On the basis of in vitro functional activities, all test compounds produced a concentration-dependent increase in [35S]GTPγS binding (Figure S2) and showed high efficacy at the human MOR acting as full agonists (≥85% of the response to DAMGO), with the most potent agonists being 3 and 3a (Table 1). Our present data indicate that the removal of the 6-keto function did not largely influence the MOR agonism of 1 vs 1a, 2 vs 2a, and 4 vs 4a. However, an increase of ∼2.5-fold in the EC50 of 3a compared to 3 was observed (Table 1).
We have earlier reported on the replacement of the 6-keto group in N-methylmorphinans-6-ones with other functionalities (i.e., amino acids, cyano, amino, and guanidino) at C-6, which produced subtle alterations in the in vitro biological activities, i.e., MOR affinity and agonist potency. On the basis of previous and present findings, a 6-keto group is not a requirement for high affinity to the MOR of N-methylmorphinans displaying agonist activity. Earlier observations in the class of morphinans with opioid antagonism, including naltrexone and N-cycloproplymethyl-4,14-dimethoxymorphinan (cyprodime), revealed that deletion of the 6-keto group maintains the MOR antagonism while MOR selectivity is unchanged or decreased.15 The 6-desoxo-N-methylmorphinans 1a–4a were further assessed for antinociceptive properties after sc adminstration in a mouse model of acute thermal nociception, the hot-plate assay.6b,6c Antinociceptive potencies expressed as ED50 values (and 95% confidence intervals) were calculated at the peak of action and compared to those of the 6-ketomorphinans 1–4 (Table 1). All test compounds significantly increased latencies to heat stimulation in a time- and dose-dependent manner (Figures S3 and S4), with the peak of antinociception generally occurring at 30 min after sc administration to mice. Compared to morphine (ED50 = 2428 μg/kg, 95% CI of 1382–4268 in the hot-plate assay),14c all compounds were considerably more efficacious in inducing an antinociceptive response in mice, being up to 1020-fold more potent than morphine. Within the series, the 14-benzyloxy substituted 3 and 3a were the most active as antinociceptive agent, a finding that correlates well with their in vitro opioid activity profile (Table 1). Overall, as shown in Table 1 and Figure S5, the 6-desoxo compounds 1a–4a exhibit very similar antinociceptive potencies to their analogue 1–4 indicating that the absence of a 6-carbonyl group does not significantly affect the in vivo agonism (two-way ANOVA, P > 0.05 when comparing 1–4 to 1a–4a counterparts). In both series of N-methylmorphinan-6-ones 1–4 and 6-desoxo-N-methylmorphinans 1a–4a, it was apparent that compounds carrying a 14-methoxy (2, 4, 2a, and 4a) and a 14-benzyloxy (3 and 3a) group have enhanced affinity and in vitro and in vivo agonist potency to the MOR than the 14-hydroxy substituted 1 and 1a (Table 1).
Assessment of physicochemical properties has gained considerable importance over the past years, emerging as a significant aspect in drug development, especially for understanding of the behavior of bioactive molecules and correlation with pharmacological activities.17 The calculated log P (clogP) values of N-methylmorphinans 1–4 and 1a–4a are indicative of good capability to enter the CNS (Table 1).
Molecular Modeling
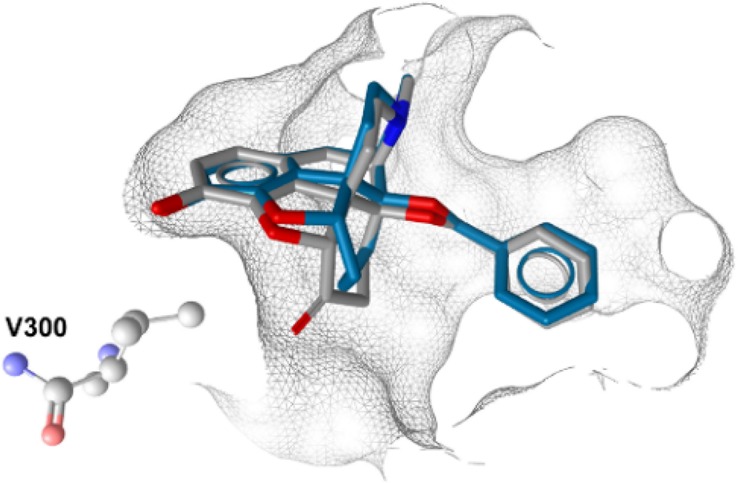
The X-ray crystal structure of the active MOR conformation (PDB code 5C1M)9 provides nowadays novel prospects in the drug discovery process.18 In order to further understand the effect of a 6-carbonyl group deletion in N-methylmorphinan-6-ones, we performed molecular docking using the active conformation of the MOR. Docking studies were carried out to unravel potential differences in receptor interactions of 1–4 and their 6-desoxo counterparts 1a–4a using GOLD19 as outlined in the Supporting Information. Differences and similarities in the experimentally determined binding affinities to the MOR (Table 1) are likely to emerge from the type and number of receptor contacts formed by these morphinans in those particularly similar docked orientations. Accounting for pharmacophore models inferred within LigandScout,20 receptor side chains in putative contact with the bound ligands are depicted in Figure 2 and listed in Table 2. In line with our recent findings from molecular docking,8 the aromatic ring of targeted morphinans was deduced to be embedded by hydrophobic residues (e.g., M151, V236, I296, and V300), which are responsible for hydrophobic interactions (Table 2 and Figures 2). The participation of the phenolic hydroxyl group at position 3 in a water-mediated hydrogen-bonding network with H297 was shown by all ligands. This is recognized as a conserved interaction between morphinan ligands and the binding pocket.8,9 In addition, the basic nitrogen forms a charge-enhanced hydrogen bond to the key residue D147. This observation is supported by earlier experimental data derived from site-directed mutagenesis studies demonstrating the crucial role of D147 for binding of small molecule agonists to the MOR.21 When comparing the retrieved docking poses of the 6-desoxo compounds 1a–4a to their corresponding 6-keto analogues 1–4, the observed differences were more or less subtle regarding critical noncovalent interactions, which are required for the recognition of these potent agonists by the MOR (Figure 2). We observed that 1 and 2 and their 6-desoxo analogues 1a and 2a, respectively, exhibit a similar binding mode regarding the orientation of these agonists relative to the receptor (Figure 2A and Figure 2B). Differences were noted when evaluating the binding mode of 3 along with its 6-desoxo counterpart 3a, as well as to the other morphinans (Figure 2C). Docking analysis showed that 3 and 3a adopt an orientation relative to the receptor distinct from the other analogues, represented by an additional hydrophobic interaction between I144 and the 14-O-benzyl group of 3 and 3a (Figure 2C and Table 2). As a consequence, this modified relative orientation in the binding pocket may be responsible for a steric clash, surmised between the 6-keto group of 3 and residue V300, which is essentially orientated in close proximity to the phenol of targeted morphinans (vide supra). However, in the case of 3 the side chain of V300 is getting rather close to the 6-keto substituted cyclohexane ring. This interaction was not observed for the 6-desoxo-14-benzyloxy substituted 3a, as the docking pose indicated that the corresponding methylene bridge at position 6 is nicely accommodated in the binding pocket (Figure 3).
Figure 2.
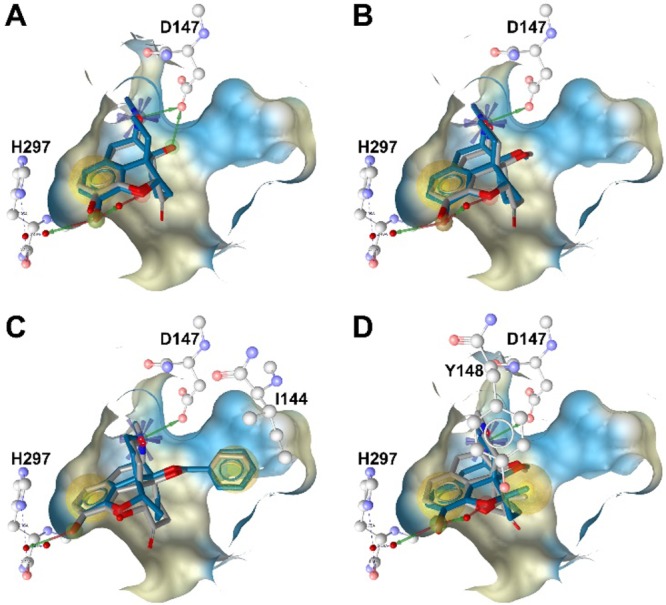
Docking of N-methylmorphinans-6-ones 1-4 (gray) and corresponding 6-desoxo counterparts 1a–4a (blue) to the active structure of the MOR: (A) overlay of 1 and 1a; (B) overlay of 2 and 2a; (C) overlay of 3 and 3a; (D) overlay of 4 and 4a. The key residues D147 and H297, involved in a hydrogen-bonding network, and residues I144 and Y148, involved in hydrophobic interactions, are depicted. Chemical features are color-coded: red/green arrow, hydrogen bond acceptor/donor; yellow sphere, hydrophobic interaction; blue asterisk, positively ionizable. Binding pocket surface is shown, colored according to aggregated hydrophilicity/hydrophobicity (blue/ochre).
Table 2. Ligand–MOR Interaction Pharmacophores Inferred from Molecular Docking Solutions of N-Methylmorphinan-6-ones 1–4 and Their 6-Desoxo Counterparts 1a–4a.
| hydrophobic
interactions |
hydrogen bonds |
|||
|---|---|---|---|---|
| compd | inferred from phenol | inferred from introduced group | charge-enhanced hydrogen bond | interactions mediated by water molecules |
| 1 | M151, V236, I296, V300 | NDa | D147 | H297 |
| 1a | M151, V236, I296, V300 | NDa | D147 | H297 |
| 2 | M151, V236, I296, V300 | NDa | D147 | H297 |
| 2a | M151, V236, I296, V300 | NDa | D147 | H297 |
| 3 | M151, V236, I296, V300 | I144b | D147 | H297 |
| 3a | M151, V236, I296, V300 | I144b | D147 | H297 |
| 4 | M151, V236, I296, V300 | NDa | D147 | H297 |
| 4a | M151, V236, I296, V300 | Y148c | D147 | H297 |
ND, not deduced.
Formed by the 14-benzyloxy group.
Formed by the 5-methyl group.
Figure 3.
Overlay of molecular docking solutions for 3 (gray) and 3a (blue) at the active structure of the MOR, illustrating the steric clash inferred to represent the interaction of the 6-keto group of 3 with V300, orientated in close proximity, and the absence of this interaction for 3a. Residue V300 is presented, along with the binding pocket surface shown in wireframe representation.
These features may contribute to the improved binding affinities to the MOR of 3a compared to 3 (Ki = 0.048 nM for 3a vs 0.45 nM for 3) experimentally determined in radioligand binding assays. The projected orientation in the binding pocket was only slightly different when comparing 5-methyl-14-methoxy-N-methylmorphinans 4 and 4a. Interestingly, the 6-desoxo analogue 4a was predicted to form an additional hydrophobic interaction by the 5-methyl group targeting the nearby Y148 as counterpart (Figure 2D and Table 2). Overall, the docking analysis pointed toward a subtle interplay of the substituents introduced to this series in positions 14 and 5, and in silico results are in accordance with pharmacological data.
Conclusions
The present study, combining synthetical, pharmacological, and molecular modeling approaches, established that the deletion of the 6-carbonyl group in targeted N- methylmorphinan-6-ones 1, 2, and 4 did not fundamentally affect binding, post-receptor-signaling, and antinociceptive activities, with the resulting 6-desoxo analogues evolving as potent MOR agonists. Notable was the observation that the 6-desoxo-14-benzyloxy 3a displays significantly increased MOR binding and agonist potency than its 6-keto counterpart 3. At the in silico level, the absence of the 6-carbonyl function in 3a depleted the steric clash shown by the 6-keto group of 3 with residue V300, thus explaining the improved binding affinities to the MOR of 3a compared to 3. These results expand the understanding on the impact of the 6-carbonyl to 6-CH2 conversion in N-methylmorphinans on ligand–MOR interaction and molecular mode of action and may aid in identifying new leads for development of opioid analgesics devoid of adverse effects for treatment of pain.
Experimental Section
Chemistry. General Methods
All chemicals were of reagent grade and obtained from standard commercial sources. Melting points were determined on a Kofler melting point microscope and are uncorrected. 1H NMR (200 MHz) spectra were recorded on a Varian Gemini 200 spectrometer using tetramethylsilane (TMS) as internal standard for CDCl3. IR spectra were taken on a Bruker Alpha FT-IR spectrometer (for detection, an ATR sensor was used). Mass spectra were recorded on a Varian MAT 44 S apparatus. Elemental analyses were performed at the Microanalytic Laboratory of the University of Vienna, Austria. For column chromatography (MPLC), silica gel 60 (0.040–0.063 mm, Fluka, Switzerland) was used. Compounds 1a–4a were used as bases for testing. The elemental analysis values were found to be within ±0.4% of the calculated values, indicating a purity of the tested compounds of >95%.
General Procedure for the Synthesis of 6-Desoxomorphinans 1a–4a
A mixture of 1, 2, 3, or 4 (0.60 mmol), hydrazine hydrate (27 mmol), and triethylene glycol (3 mL) was stirred at 130 °C for 1.5 h. After cooling, KOH pellets (6 mmol) were added and the mixture was stirred at 180 °C for 2 h. After cooling, the mixture was acidified with 2 N HCl, washed with Et2O (2 × 10 mL), rendered alkaline with NH4OHconc, and extracted with CH2Cl2 (3 × 10 mL). The organic layer was washed with H2O (3 × 10 mL), dried over Na2SO4, and evaporated to provide a solid which was purified by column chromatography (silica gel, CH2Cl2/MeOH/NH4OH, 98:1:1) to yield the final compounds.
3,14-Dihydroxy-4,5α-epoxy-17-methylmorphinan (1a)
Yield 11% as beige solid. Mp: 90–92 °C. IR (ATR) 3119 cm–1 (OH). 1H NMR (CDCl3): δ 6.71 (d, J = 8.0 Hz, H-C(1)), 6.58 (d, J = 8.0 Hz, H-C(2)), 4.72 (t, J = 8 Hz, H-C(5)), 2.37 (s, CH3N). MS (ESI) m/z 288.1 [M + 1]+. Anal. (C17H21NO3·0.2MeOH) C, H, N.
4,5α-Epoxy-3-hydroxy-14β-methoxy-17-methylmorphinan (2a)
Yield 22% as beige solid. Mp 82–86 °C. IR (ATR) 2935 cm–1 (OH). 1H NMR (CDCl3): δ 6.70 (d, J = 8.0 Hz, H-C(1)), 6.57 (d, J = 8.0 Hz, H-C(2)), 4.73 (t, J = 7.8 Hz, H-C(5)), 3.22 (s, CH3O), 2.37 (s, CH3N). MS (ESI) m/z 302.01 [M + 1]+. Anal. (C18H23NO3·0.6CH2Cl2) C, H, N.
14β-Benzyloxy-4,5α-epoxy-3-hydroxy-17-methylmorphinan (3a)
Yield 10% as white solid. Mp 79–83 °C. IR (ATR) 2923 cm–1 (OH). 1H NMR (CDCl3): δ 7.44–7.25 (m, 5 arom H), 6.70 (d, J = 7.4 Hz, H-C(1)), 6.58 (d, J = 8.0 Hz, H-C(2)), 4.76 (t, J = 7.6 Hz, H-C(5)), 4.63 (d, J = 11 Hz, CH2-phenyl), 4.30 (d, J = 11 Hz, CH2-phenyl), 2.36 (s, CH3N). MS (ESI) m/z 378.2 [M + 1]+. Anal. (C24H27NO3·0.2CH2Cl2) C, H, N.
5β,17-Dimethyl-4,5α-epoxy-3-hydroxy-14β-methoxymorphinan (4a)
Yield 16% as white solid. Mp 90–95 °C. IR (ATR) 2933 cm–1 (OH). 1H NMR (CDCl3): δ 6.68 (d, J = 8.0 Hz, H-C(1)), 6.55 (d, J = 8.0 Hz, H-C(2)), 3.19 (s, CH3O), 2.37 (s, CH3N), 1.58 (s, CH3-C(5)). MS (ESI) m/z 316.2 [M+1]+. Anal. (C19H25NO3·0.1CH2Cl2) C, H, N.
Acknowledgments
We thank Dr. Sonja Sturm for recording the mass spectra. This research was supported by grants from the Austrian Science Fund (FWF: TRP19-B18), the Tyrolean Research Fund (TWF: UNI-0404/1596), the Förderungsbeiträge Aktion D. Swarovski KG 2014, and the University of Innsbruck.
Glossary
Abbreviations Used
- CHO
Chinese hamster ovary
- DOR
δ-opioid receptor
- KOR
κ-opioid receptor
- MOR
μ-opioid receptor
- PDB
Protein Data Bank
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmedchem.7b01363.
Author Contributions
† M.D. and T.B.H. contributed equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- Benyhe S. Morphine: New Aspects in the Study of an Ancient Compound. Life Sci. 1994, 55, 969–979. 10.1016/0024-3205(94)00631-8. [DOI] [PubMed] [Google Scholar]
- a Pasternak G. W.; Pan Y. X. Mu Opioids and Their Receptors: Evolution of a Concept. Pharmacol. Rev. 2013, 65, 1257–1317. 10.1124/pr.112.007138. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Spetea M.; Asim M. F.; Wolber G.; Schmidhammer H. The μ Opioid Receptor and Ligands Acting at the μ Opioid Receptor, as Therapeutics and Potential Therapeutics. Curr. Pharm. Des. 2013, 19, 7415–7434. 10.2174/13816128113199990362. [DOI] [PubMed] [Google Scholar]
- a Skolnick P.; Volkow N. D. Re-energizing the Development of Pain Therapeutics in Light of the Opioid Epidemic. Neuron 2016, 92, 294–297. 10.1016/j.neuron.2016.09.051. [DOI] [PubMed] [Google Scholar]; b Grosser T.; Woolf C. J.; FitzGerald G. A. Time for Nonaddictive Relief of Pain. Science 2017, 355, 1026–1027. 10.1126/science.aan0088. [DOI] [PubMed] [Google Scholar]
- a Portoghese P. S. Stereochemical Factors and Receptor Interactions Associated With Narcotic Analgesics. J. Pharm. Sci. 1966, 55, 865–887. 10.1002/jps.2600550902. [DOI] [PubMed] [Google Scholar]; b Casy A. F.; Parfitt R. T.. Opioid Analgesics, Chemistry and Receptors; Plenum: New York, 1987. [Google Scholar]; c Fürst S.; Hosztafi S. The Chemical and Pharmacological Importance of Morphine Analogues. Acta Physiol. Hung. 2008, 95, 3–44. 10.1556/APhysiol.95.2008.1.1. [DOI] [PubMed] [Google Scholar]; d Schmidhammer H.; Spetea M.. Synthesis of 14-Alkoxymorphinans and Their Pharmacological Activities. In Chemistry of Opioids. Nagase H., Ed.; Topics in Current Chemistry, Vol. 299; Springer: Berlin Heidelberg, 2011; pp 63–91, DOI: 10.1007/128_2010_77. [DOI] [PubMed] [Google Scholar]; e Lewis J. W; Husbands S. M.. 14-Amino-4,5-epoxymorphinan Derivatives and Their Pharmacological Actions. In Chemistry of Opioids. Nagase H., Ed.; . Topics in Current Chemistry, Vol. 299; Springer: Berlin Heidelberg, 2011; pp 93–119. DOI: 10.1007/128_2010_89. [DOI] [PubMed] [Google Scholar]
- a Spetea M.; Schmidhammer H. Recent Advances in the Development of 14-Alkoxy Substituted Morphinans as Potent and Safer Opioid Analgesics. Curr. Med. Chem. 2012, 19, 2442–2457. 10.2174/092986712800269308. [DOI] [PubMed] [Google Scholar]; b Schmidhammer H.; Spetea M. Development of 5-Substituted N-Methylmorphinan-6-ones as Potent Opioid Analgesics with Improved Side-Effect Profile. Int. J. Med. Chem. 2012, 1–12. 10.1155/2012/208039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Schmidhammer H.; Aeppli L.; Atwell L.; Fritsch F.; Jacobson A. E.; Nebuchla M.; Sperk G. Synthesis and Biological Evaluation of 14-Alkoxymorphinans. 1. Highly Potent Opioid Agonists in the Series of (−)-14-Methoxy-N-methylmorphinan-6-ones. J. Med. Chem. 1984, 27, 1575–1579. 10.1021/jm00378a009. [DOI] [PubMed] [Google Scholar]; b Lattanzi R.; Spetea M.; Schüllner F.; Rief S. B.; Krassnig R.; Negri L.; Schmidhammer H. Synthesis and Biological Evaluation of 14-Alkoxymorphinans. 22. Influence of the 14-Alkoxy Group and the Substitution in Position 5 in 14-Alkoxymorphinan-6-ones on in Vitro and in Vivo Activities. J. Med. Chem. 2005, 48, 3372–3378. 10.1021/jm040894o. [DOI] [PubMed] [Google Scholar]; c Spetea M.; Bohotin C. R.; Asim M. F.; Stübegger K.; Schmidhammer H. In Vitro and in Vivo Pharmacological Profile of the 5-Benzyl Analogue of 14-Methoxymetopon, a Novel μ Opioid Analgesic with Reduced Propensity to Alter Motor Function. Eur. J. Pharm. Sci. 2010, 41, 125–135. 10.1016/j.ejps.2010.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fürst S.; Búzás B.; Friedmann T.; Schmidhammer H.; Borsodi A. Highly Potent Novel Opioid Receptor Agonist in the 14-Alkoxymetopon Series. Eur. J. Pharmacol. 1993, 236, 209–215. 10.1016/0014-2999(93)90591-5. [DOI] [PubMed] [Google Scholar]
- Noha S. M.; Schmidhammer H.; Spetea M. Molecular Docking, Molecular Dynamics and Structure-Activity Relationship Explorations of 14-Oxygenated N-Methylmorphinan-6-ones as Potent μ-Opioid Receptor Agonists. ACS Chem. Neurosci. 2017, 8, 1327–1337. 10.1021/acschemneuro.6b00460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W.; Manglik A.; Venkatakrishnan A. J.; Laeremans T.; Feinberg E. N.; Sanborn A. L.; Kato H. E.; Livingston K. E.; Thorsen T. S.; Kling R. C.; Granier S.; Gmeiner P.; Husbands S. M.; Traynor J. R.; Weis W. I.; Steyaert J.; Dror R. O.; Kobilka B. K. Structural Insights into μ-Opioid Receptor Activation. Nature 2015, 524, 315–321. 10.1038/nature14886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidhammer H.; Spetea M.; Windisch P.; Schütz J.; Riba P.; Al-Khrasani M.; Fürst S. Functionalization of the Carbonyl Group in Position 6 of Morphinan-6-ones. Development of Novel 6-Amino and 6-Guanidino Substituted 14-Alkoxymorphinans. Curr. Pharm. Des. 2013, 19, 7391–7399. 10.2174/138161281942140105164804. [DOI] [PubMed] [Google Scholar]
- a Pasternak G. W.; Hahn E. F. Long-acting Opiate Agonists and Antagonists: 14-Hydroxydihydromorphinone Hydrazones. J. Med. Chem. 1980, 23, 674–676. 10.1021/jm00180a019. [DOI] [PubMed] [Google Scholar]; b Krizsan D.; Varga E.; Hosztafi S.; Benyhe S.; Szücs M.; Borsodi A. Irreversible Blockade of the High and Low Affinity [3H]-Naloxone Binding Sites by C-6 Derivatives of Morphinane-6-ones. Life Sci. 1991, 48, 439–451. 10.1016/0024-3205(91)90500-B. [DOI] [PubMed] [Google Scholar]; c Monory K.; Greiner E.; Sartania N.; Sallai L.; Pouille Y.; Schmidhammer H.; Hanoune J.; Borsodi A. Opioid Binding Profiles of New Hydrazone, Oxime, Carbazone And Semicarbazone Derivatives Of 14-Alkoxymorphinans. Life Sci. 1999, 64 (22), 2011–2020. 10.1016/S0024-3205(99)00148-4. [DOI] [PubMed] [Google Scholar]; d Fürst Z.; Borsodi A.; Friedmann T.; Hosztafi S. 6-Substituted Oxycodone Derivatives have Strong Antinociceptive Effects and Block Irreversibly the Low Affinity [3H]-Naloxone Binding Sites in Rat Brain. Pharmacol. Res. 1992, 25, 31–32. 10.1016/1043-6618(92)90271-C. [DOI] [Google Scholar]; e Fürst S.; Hosztafi S.; Friedmann T. Structure-Activity Relationships of Synthetic and Semisynthetic Opioid Agonists and Antagonists. Curr. Med. Chem. 1995, 1, 423–440. [PubMed] [Google Scholar]
- a Botros S.; Lipkowski A. W.; Larson D. L.; Stark A. P.; Takemori A. E.; Portoghese P. S. Opioid Agonist and Antagonist Activities of Peripherally Selective Derivatives of Naltrexamine and Oxymorphamine. J. Med. Chem. 1989, 32, 2068–2071. 10.1021/jm00129a009. [DOI] [PubMed] [Google Scholar]; b Schütz J.; Brandt W.; Spetea M.; Wurst K.; Wunder G.; Schmidhammer H. Synthesis of 6-Amino Acid Substituted Derivatives of the Highly Potent Analgesic 14-O-Methyloxymorphone. Helv. Chim. Acta 2003, 86, 2142–2148. 10.1002/hlca.200390171. [DOI] [Google Scholar]
- a Spetea M.; Friedmann T.; Riba P.; Schütz J.; Wunder G.; Langer T.; Schmidhammer H.; Fürst S. In Vitro Opioid Activity Profiles of 6-Amino Acid Substituted Derivatives of 14-O-Methyloxymorphone. Eur. J. Pharmacol. 2004, 483, 301–308. 10.1016/j.ejphar.2003.10.049. [DOI] [PubMed] [Google Scholar]; b Fürst S.; Riba P.; Friedmann T.; Timar J.; Al-Khrasani M.; Obara I.; Makuch W.; Spetea M.; Schütz J.; Przewlocki R.; Przewlocka B.; Schmidhammer H. Peripheral Versus Central Antinociceptive Actions of 6-Amino Acid-Substituted Derivatives of 14-O-Methyloxymorphone in Acute and Inflammatory Pain in the Rat. J. Pharmacol. Exp. Ther. 2005, 312, 609–618. 10.1124/jpet.104.075176. [DOI] [PubMed] [Google Scholar]; c Spetea M.; Windisch P.; Guo Y.; Bileviciute-Ljungar I.; Schütz J.; Asim M. F.; Berzetei-Gurske I. P.; Riba P.; Király K.; Fürst S.; Al-Khrasani M.; Schmidhammer H. Synthesis and Pharmacological Activities of 6-Glycine Substituted 14-Phenylpropoxymorphinans, a Novel Class of Opioids with High Opioid Receptor Affinities and Antinociceptive Potencies. J. Med. Chem. 2011, 54, 980–988. 10.1021/jm101211p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Greiner E.; Schottenberger H.; Wurst K.; Schmidhammer H. Novel Class of Morphinans with Acrylonitrile Incorporated Substructures as Key Intermediates for Non-Oxygen-Bridged Opioid Ligands. J. Am. Chem. Soc. 2001, 123, 3840–3841. 10.1021/ja015550r. [DOI] [PubMed] [Google Scholar]; b Spetea M.; Greiner E.; Aceto M. D.; Harris L. S.; Coop A.; Schmidhammer H. Effect of a 6-Cyano Substituent in 14-Oxygenated N-Methylmorphinans on Opioid Receptor Binding And Antinociceptive Potency. J. Med. Chem. 2005, 48, 5052–5055. 10.1021/jm0580205. [DOI] [PubMed] [Google Scholar]; c Ben Haddou T.; Malfacini D.; Calo G.; Aceto M. D.; Harris L. S.; Traynor J. R.; Coop A.; Schmidhammer H.; Spetea M. Exploring Pharmacological Activities and Signaling of Morphinans Substituted in Position 6 as Potent Agonists Interacting with the μ Opioid Receptor. Mol. Pain 2014, 10, 48. 10.1186/1744-8069-10-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Wentland M. P.; Lou R.; Lu Q.; Bu Y.; Denhardt C.; Jin J.; Ganorkar R.; VanAlstine M. A.; Guo C.; Cohen D. J.; Bidlack J. M. Syntheses of Novel High Affinity Ligands for Opioid Receptors. Bioorg. Med. Chem. Lett. 2009, 19, 2289–2294. 10.1016/j.bmcl.2009.02.078. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Schmidhammer H.; Jennewein H. K.; Smith C. F. Synthesis and Biological Evaluation of 14-Alkoxymorphinans, V: 6-Deoxyocyprodime, an Opioid Antagonist with Decreased μ Receptor Selectivity in Comparison to Cyprodime. Arch. Pharm. (Weinheim, Ger.) 1991, 324, 209–211. 10.1002/ardp.19913240404. [DOI] [PubMed] [Google Scholar]
- Spetea M.; Berzetei-Gurske I. P.; Guerrieri E.; Schmidhammer H. Discovery and Pharmacological Evaluation of a Diphenethylamine Derivative (HS665), a Highly Potent and Selective κ Opioid Receptor Agonist. J. Med. Chem. 2012, 55, 10302–10306. 10.1021/jm301258w. [DOI] [PubMed] [Google Scholar]
- a Avdeef A.; Testa B. Physicochemical Profiling in Drug Research: A Brief Survey of the State-of-the-Art of Experimental Techniques. Cell. Mol. Life Sci. 2002, 59, 1681–1689. 10.1007/PL00012496. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Faller B.Physicochemical Profiling in Early Drug Discovery: New Challenges at the Age of High-Throughput Screen and Combinatorial Chemistry. In Chemistry and Molecular Aspects of Drug Design and Action; Rekka E. A., Kourounakis P. N., Eds.; CRC: Boca Raton, FL, U.S., 2008; pp 303–312, DOI: 10.1201/9781420008272.ch22. [DOI] [Google Scholar]
- a Manglik A.; Lin H.; Aryal D. K.; McCorvy J. D.; Dengler D.; Corder G.; Levit A.; Kling R. C.; Bernat V.; Hübner H.; Huang X.-P.; Sassano M. F.; Giguère P. M.; Löber S.; Duan D.; Scherrer G.; Kobilka B. K.; Gmeiner P.; Roth B. L.; Shoichet B. K. Structure-Based Discovery of Opioid Analgesics with Reduced Side Effects. Nature 2016, 537, 185–190. 10.1038/nature19112. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Kruegel A. C.; Gassaway M. M.; Kapoor A.; Váradi A.; Majumdar S.; Filizola M.; Javitch J. A.; Sames D. Synthetic and Receptor Signaling Explorations of the Mitragyna Alkaloids: Mitragynine as an Atypical Molecular Framework for Opioid Receptor Modulators. J. Am. Chem. Soc. 2016, 138, 6754–6764. 10.1021/jacs.6b00360. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Crowley R. S.; Riley A. P.; Sherwood A. M.; Groer C. E.; Shivaperumal N.; Biscaia M.; Paton K.; Schneider S.; Provasi D.; Kivell B. M.; Filizola M.; Prisinzano T. E. Synthetic Studies of Neoclerodane Diterpenes from Salvia divinorum: Identification of a Potent and Centrally Acting μ Opioid Analgesic with Reduced Abuse Liability. J. Med. Chem. 2016, 59, 11027–11038. 10.1021/acs.jmedchem.6b01235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdonk M. L.; Chessari G.; Cole J. C.; Hartshorn M. J.; Murray C. W.; Nissink J. W.; Taylor R. D.; Taylor R. Modeling Water Molecules in Protein-Ligand Docking using GOLD. J. Med. Chem. 2005, 48, 6504–6515. 10.1021/jm050543p. [DOI] [PubMed] [Google Scholar]
- Wolber G.; Langer T. LigandScout: 3-D Pharmacophores Derived from Protein-Bound Ligands and Their Use as Virtual Screening Filters. J. Chem. Inf. Model. 2005, 45, 160–169. 10.1021/ci049885e. [DOI] [PubMed] [Google Scholar]
- a Surratt C. K.; Johnson P. S.; Moriwaki A.; Seidleck B. K.; Blaschak C. J.; Wang J. B.; Uhl G. R. Mu Opiate Receptor. Charged Transmembrane Domain Amino Acids are Critical for Agonist Recognition and Intrinsic Activity. J. Biol. Chem. 1994, 269, 20548–20553. [PubMed] [Google Scholar]; b Mansour A.; Taylor J. L.; Fine J. L.; Thompson R. C.; Hoversten M. T.; Mosberg H. I.; Watson S. J.; Akil H. Key Residues Defining the μ-Opioid Receptor Binding Pocket: A Site-Directed Mutagenesis Study. J. Neurochem. 1997, 68, 344–353. 10.1046/j.1471-4159.1997.68010344.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.