

Abstract
Many cancers fail to respond to immunotherapy as a result of immune suppression by the tumor microenvironment. The exogenous expression of immune cytokines to reprogram the tumor microenvironment represents an approach to circumvent this suppression. The present studies describe the development of a novel dual nanoparticle (DNP) system for driving DNA expression vectors encoding inflammatory cytokines in tumor cells. The DNP system consists of a DNA expression vector-cationic nanocomplex (NC) surrounded by a diblock polymeric NP. Tumor necrosis factor alpha (TNF) was selected as the prototype cytokine for this system, based on its pleotropic inflammatory and anti-cancer activities. Our results demonstrate that the DNP system is highly effective in driving expression of TNF in tumor cells. We also demonstrate that the DNPs are effective in inducing apoptosis and anti-tumor activity. These findings support a novel immunotherapeutic approach for the intratumoral delivery of DNA vectors that express inflammatory cytokines.
Keywords: DNA expression vector, tumor necrosis factor, intratumoral delivery, nanocomplexes, nanoparticles
Graphical abstract
Nanocomplexes (NCs) were generated by incubation of the positively charged HCR peptide with the negatively charged pE425-TNF expression vector. To circumvent the instability of pTNF-NCs in plasma, a dual nanocomplex in a nanoparticle system was in turn generated by encapsulation of the pTNF-NCs in polymeric NPs. Intracellular delivery of the pTNF-NC in NPs results in sustained release of the pTNF-NC, import of pE425-TNF to the nucleus and expression of TNF. Our results further support the premise that this system is effective in the delivery of DNA expression vectors to tumors. Intratumoral expression of secreted cytokines, such as TNF, thus represents a novel approach for inducing anti-tumor activity and potentially reprogramming the tumor immune microenvironment.
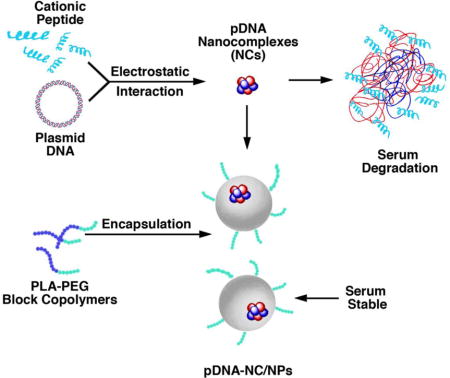
Background
A hallmark of diverse cancers is the capacity to circumvent immune destruction (1). Indeed, cancers are often infiltrated with immune cells that are ineffective in the recognition of tumor antigens and are in turn exploited to promote a protumorigenic microenvironment (2). Interestingly, however, the presence of immune cell infiltrates in what are referred to as “hot” tumors is associated with improved responsiveness to cytotoxic agents, anti-tumor vaccines and immune checkpoint inhibitors (3). These findings have emphasized the potential importance of reprogramming the microenvironment of “hot” and “cold” tumors with immune infiltrates that are effective in recognizing and destroying cancer cells.
Tumor necrosis factor alpha (TNF) was discovered as an endotoxin-inducible factor that induced necrosis of tumors (4). Since then, TNF has been one of the most extensively studied cytokines, based in part on the findings that it regulates proinflammatory responses, cell differentiation and cell death (5). We and others investigated the effectiveness of recombinant human TNF as a clinical anti-cancer agent; however, systemic side effects, such as fever, fatigue and hypotension, precluded its further development (6, 7). As a way of ameliorating the systemic toxicities, TNF has been approved in Europe for administration by isolated limb perfusion in the treatment of patients with locally advanced extremity soft tissue sarcoma (8). Another approach for the clinical development of TNF involved intratumoral delivery of an adenoviral vector encoding TNF under the control of a radio- and chemo-inducible promoter (TNFerade) (9). In early phase trials of TNFerade in combination with radiotherapy, significant clinical activity was observed in patients with metastatic melanoma, soft tissue sarcoma and locally advanced esophageal cancer (10). Anti-tumor activity was also observed in patients with pancreatic, rectal, and head and neck cancers (10). However, a phase III trial of TNFerade in patients with locally advanced pancreatic cancer failed to demonstrate a survival benefit (11), prompting the sponsor to discontinue the development of this agent.
The findings that TNF is an effective agent for the treatment of human cancers supported the notion that other approaches might be developed to appropriate the anti-tumor activity of TNF in a setting that limits systemic side effects. Therefore, the objective of the present study was to determine whether we could systemically deliver a TNF expression vector to tumors. In this context, systemic administration of vectors encoding cytokines, such as TNF, is a potential approach for reprogramming of the tumor immune microenvironment. However, this field has been limited by the lack of effective gene delivery systems (12–15).
To address this obstacle, we have developed a dual nanoparticle (DNP) system that includes a cationic peptide-DNA vector nanocomplex (NC). A histidine and cysteine modified arginine (HCR) peptide component of the NC has been designed for maintaining the balance of DNA condensation and intracellular release and nuclear localization (16). For delivery in vivo, the NC has been surrounded by a polymer diblock NP to circumvent the challenge of DNA-nanoparticle serum interactions that have limited the field (17). Our results demonstrate that this approach is highly effective in conferring TNF-induced anti-tumor activity in the absence of toxicity. Our findings further indicate that this DNP system maybe broadly applicable for expression of intratumoral cytokines that promote immune recognition and destruction.
Materials and Methods
Materials
Peptide CR5H7R4C (HCR; >95% purity) was custom synthesized by GL Biochem Ltd. (Shanghai, China). The peptide was dissolved in de-ionized water at a concentration of 5 mg/ml and stored in small aliquots at −80°C. An expression vector in which the human TNF cDNA is driven by a reactive oxygen species (ROS)-inducible promoter (pE425) derived from the EGR1 gene was constructed as described (10). The plasmids pEGFP-N3 (Clontech, Palo Alto, CA, USA) and pE425-TNF (10) were purified using the GenElute HP endotoxin free plasmid maxiprep kit (Sigma, St. Louis, MO, USA). PLA-PEG block co-polymer of 75 kDa PLA was designed and synthesized as reported (18).
Preparation of HCR-pDNA nanocomplexes (NCs)
HCR-pDNA nanocomplexes (NC) were prepared based on the electrostatic interaction of amino nitrogen (NH3+) of peptide per phosphate (PO4−) group of DNA at a charge ratio [Z(±)] of 10.0. The pDNA stock was diluted to a concentration of 20–40 ng/μl and added drop wise to an equal volume of HCR peptide dilutions while vortexing.
Preparation of pDNA-NC/NPs
NPs were prepared using a double emulsion evaporation method (19). Polymer solution and surfactant were prepared as: a) PEG-PLA block co-polymer dissolved in acetonitrile at a concentration of 6 mg/ml and b) Ploxomer F-127 dissolved at 3 mg/ml in de-ionized water with continuous stirring. pDNA-NCs were gently mixed with the PLA-PEG solution and then added slowly into the aqueous surfactant solution. This solution was continuously stirred for approximately 12 h to form a stable NP solution. Polymer only empty NPs and pDNA-NC/NPs and were stored at −80°C.
Characterization of pDNA-NCs and pDNA-NC/NPs
Size and zeta potential of pDNA-NCs and pDNA-NC/NPs were measured by Zetasizer Nano ZS (Malvern Instruments Ltd., Malvern, UK) at a fixed angle of 173° at 25°C. The hydrodynamic diameter and polydispersity index (PDI) values were analyzed in deionized water. The pDNA-NCs were monodisperse with a size range of 80±4 nm (Supplemental Table S1). The pDNA-NC/NPs were larger with a size range of 250±7 nm (Supplemental Table S1). pDNA-NCs had a positive zeta potential of +25 mV. Zeta potential of NPs was −30 mV. Morphology of the pDNA-NCs and pDNA-NC/NPs as imaged by Transmission Emission Microscopy (TEM) showed that the NPs are spherical in shape (Supplemental Figure S1A). pDNA-NC/NPs were stable in size and without aggregation even in presence of serum (Supplemental Figure S1B).
The loading efficiency of pDNA in pDNA-NC/NPs was approximately 60%. The release pattern of pDNA from pDNA-NC/NPs was analyzed by incubating the pDNA-NC/NPs in physiological buffer (1X PBS). The solution was centrifuged, and the supernatant was collected and analyzed for the presence of pDNA. The pellet was then again suspended in PBS. The supernatant was treated with 10 μg/μl heparin for 10 min and then analyzed by ethidium bromide exclusion assay. Approximately 60% of pDNA was released within 3 h from pDNA-NCs, whereas pDNA was released from pDNA-NC/NPs more slowly with a cumulative pDNA release of 30%, 45% and 60% for 12, 24 and 48 h, respectively (Supplemental Figure S2).
Cell culture
MCF-7 and MDA-MB-231 cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM), supplemented with 10% (v/v) fetal bovine serum (Life Technologies, Carlsbad, CA, USA).
In-vitro transfections
Cells were seeded in a 24-well plate format and the experiments were performed after 24 h when the confluency was ~70%. pDNA-NCs were prepared at a charge ratio of 10.0 and incubated for 1 h. pDNA-NCs (100 μl; 2 μg of pDNA/well) were added to the cells with 300 μl of serum free medium (OptiMEM; GIBCO, Invitrogen, Carlsbad, CA, USA). NPs were added to the cells at an increasing concentration of pDNA per well as 2 μg, 4 μg and 8 μg. The pDNA-NCs and pDNA-NC/NPs (8 μg pDNA/well) were also added to the cells in the presence of 10% serum to assess the effect of serum on the transfection efficiency. After 12 h of incubation, the medium containing pDNA-NCs and pDNA-NC/NPs was aspirated and cells were rinsed with 1X PBS and then supplemented with complete growth medium.
Analysis of GFP expression
The pEGFP N3 plasmid (Clontech, Palo Alto, CA, USA) was complexed with the HCR peptide and used to generate GFP-NCs and GFP-NC/NPs. Transfected cells were analyzed for GFP expression by fluorescence microscopy. Single cell suspensions were also analyzed for GFP expression using an Aria-III Flow Cytometer at an excitation wavelength of 488 nm.
Assessment of cell viability
Cell viability was measured using the MTT assay as described (18, 20).
Immunoblot analysis
Cell lysates were prepared in the presence of RIPA buffer (20). Soluble proteins were analyzed by immunoblotting using antibodies against human TNF and caspase-3 (Abcam, Cambridge, MA, USA). Anti-GAPDH (Santa Cruz Biotechnology, Dallas, TX, USA) was used as a protein loading control. Signals were detected by enhanced chemiluminescence (ECL, Thermo Fisher Scientific, Waltham, MA, USA).
Analysis of apoptosis
Cells were resuspended in Annexin-V buffer and stained with FITC-labeled Annexin-V and PI for 15 min. The cells were then analyzed using an Aria-III Flow Cytometer.
Immunocytochemistry
Cells were washed three times with PBS, incubated with anti-TNF and anti-caspase-3-CF antibodies for 1 h and then with FITC- and Cy5 fluorophore-labeled secondary antibodies for 1 h. Nuclei were stained with DAPI and the cells were analyzed by Confocal Laser Scanning Microscopy (CLSM).
Maximum Tolerated Dose (MTD) studies
Single intraperitoneal (IP) dosing of pTNF-NC/NPs was investigated in healthy female Balb/c mice to define the MTD. Five groups of Balb/c mice (5/group) received single IP injections of pTNF-NC/NPs at concentrations of 0.6 mg/kg, 1.2 mg/kg, 1.8 mg/kg and 2.4 mg/kg of pTNF DNA. PBS was administered as a control. Survival and changes in body weight were observed daily for 15 d.
Assessment of anti-tumor activity
Female Balb/c mice, 10–12 weeks of age (22–25 gms) were injected subcutaneously (sc) in the thigh of the right hind leg with ~1.8 × 106 Ehrlich breast tumor cells/mouse. When tumors reached a volume ~50 mm3 (day 0), the mice were randomly divided into 5 groups (n=6/group). Tumors were measured with a caliper twice a week. Tumor volume was calculated by the formula (L × W2)/2, where L is the longest and W is the shortest diameter (mm).
Histological analysis
Mice were sacrificed for histopathologic evaluation of tumor, heart, liver, kidney, spleen and lung. Samples were embedded in paraffin and 5 μm sections were stained with hematoxylin-eosin (H&E) for microscopic analysis.
Analysis of TNF and caspase-3-CF in tumors
Homogenized tumor cell suspensions were centrifuged and the supernatants were incubated with FITC-labeled anti-TNF and Cy5-labeled anti-caspase-3-CF for 10 min. The samples were then incubated with sepharose beads for 30 min. Following centrifugation, the pellet was resuspended and analyzed by fluorimetry.
Results
Intracellular GFP expression with transfection of GFP cDNA-NC/NPs
To assess the intracellular expression of an exogenous gene, we first generated CR5H7R4C peptide (HCR) nanocomplexes with the pEGFP-N3 vector expressing green fluorescence protein (GFP). The HCR-pGFP/NCs were then encapsulated into the polymeric NPs (pGFP-NC/NP). Exposure of MCF-7 cells to the pGFP-NC/NPs demonstrated a progressive increase in GFP expression over 96 h (Figure 1A). Moreover and importantly, GFP expression was substantially higher in cells treated with pGFP-NC/NPs, as compared to that obtained with pGFP-NCs or pGFP/NPs (Figure 1B). Additionally, there was little if any effect of the pGFP-NC/NPs on cell viability, a finding in contrast to transfection with pGFP-NCs (Figure 1C).
Figure 1. Expression of GFP by transfection of MCF-7 cells with pGFP-NC/NPs.
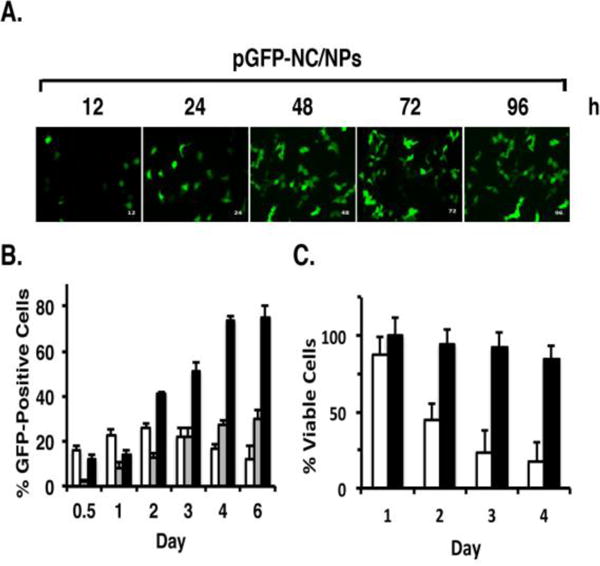
A. MCF-7 cells were treated with pGFP-NC/NPs for the indicated times. GFP expression was visualized by fluorescence microscopy. B. MCF-7 cells were treated with (i) pGFP-NCs (white bars), (ii) pGFP/NPs (grey bars), or (iii) pGFP-NC/NPs (black bars) for the indicated times. Single cell suspensions were analyzed by flow cytometry with the acquisition of 50,000 events. The results are expressed as the percentage GFP-positive cells (mean±SD) of three independent experiments. C. MCF-7 cells were treated with (i) pGFP-NCs (white bars), or (ii) pGFP-NC/NPs (black bars) for the indicated times and then analyzed by the MTT assay. The results are expressed as the percentage of viable cells (mean±SD of 3 determinations).
Generation of NC/NPs as a model system for in vitro induction of TNF expression and apoptosis
In these studies, we generated NCs containing the HCR peptide linked to the pE425-TNF (pTNF-NCs) (Figure 2A), which were then encapsulated in NPs (pTNF-NC/NPs). Significantly, treatment of MCF-7 cells with the pTNF-NC/NPs was associated with expression of the TNF transmembrane (21 kDa) and soluble (17 kDa) proteins (Figure 2B). In addition and in contrast to pGFP-NC/NPs, treatment with the pTNF-NC/NPs was effective in inducing cell death (Figure 2C).
Figure 2. Induction of TNF expression and cell death by pTNF-NC/NPs.
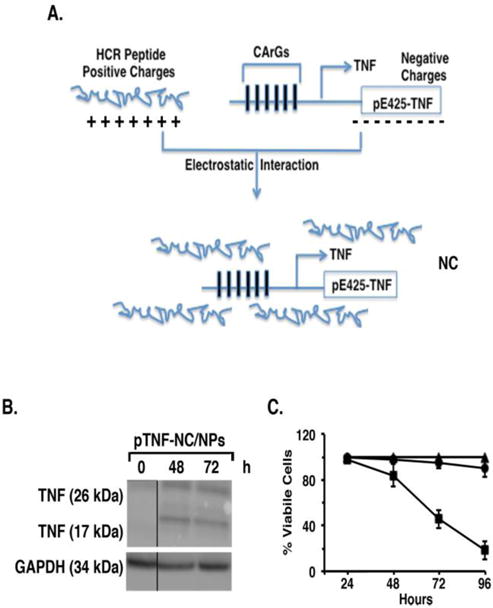
A. Schema of the interaction between the positively charged HCR peptide and the negatively charged pE425-TNF expression vector to form the nanocomplex. B. MCF-7 cells were treated with pTNF-NC/NPs for the indicated times. Total cell lysates were analyzed by immunoblotting with anti-TNF and anti-GAPDH antibodies. C. MCF-7 cells were either left untreated (triangles) and treated with pGFP-NC/NPs (circles) or pTNF-NC/NPs (squares). Cell viability was determined at the indicated times by MTT assays. Results are expressed as percentage of viable cells (mean±SD of 3 determinations).
MCF-7 cells responded to pTNF-NC/NPs with the induction of apoptosis as evidenced by Annexin V and PI staining (Figure 3A). Consistent with these results, we also found that treatment with the pTNF-NC/NPs is associated with activation of caspase-3 as determined by detection of the cleaved fragment (CF) by immunocytochemistry (Figure 3B) and immunoblot analysis (Figure 3C). These findings thus support a model in which pTNF-NC/NP induction of intracellular TNF expression is associated with apoptotic cell death.
Figure 3. Treatment with pTNF-NC/NPs induces apoptosis.
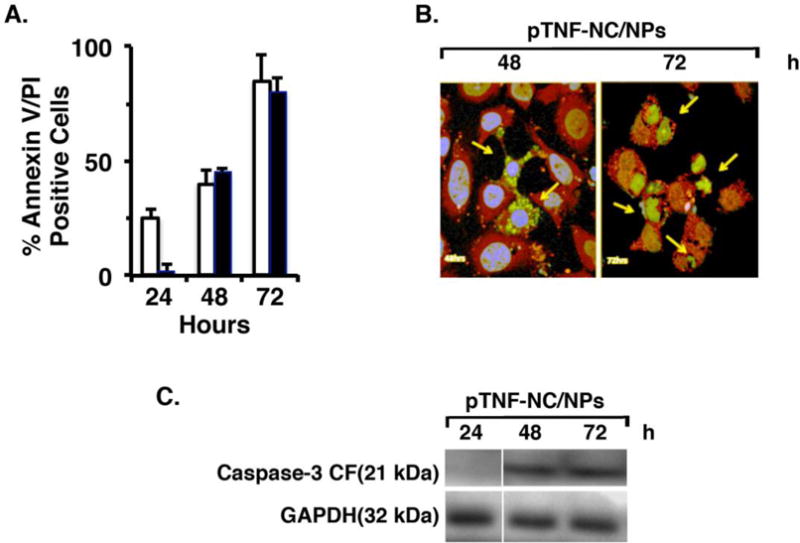
A. MCF-7 cells were treated with pTNF-NC/NPs for the indicated times and then analyzed for Annexin-V and PI staining by flow cytometry. The results are expressed as percentage (mean±SD of 3 determinations) of Annexin-V- (white bars) and PI- (black bars) positive cells. B. MCF-7 cells were treated with pTNF-NC/NPs for 48 (left) and 72 (right) h. The cells were then incubated with anti-TNF (green) and anti-caspase-3-cleaved fragment (CF; red) antibodies. Nuclei were stained with DAPI. Immunofluorescence images are shown with highlighting of TNF-expressing and apoptotic cells (arrows). C. MCF-7 cells were treated with pTNF-NC/NPs for the indicated times. Total cell lysates were analyzed by immunoblotting with anti-caspase-3-CF and anti-GAPDH antibodies.
Anti-tumor activity of pTNF-NC/NPs in vivo
Tolerability of pTNF-NC/NPs was first determined by intraperitoneal (IP) administration in Balb/c mice. Single pTNF-NC/NP doses of 0.6 and 1.2 mg/kg were well tolerated without significant weight loss or other overt signs of toxicity. Additionally, no signs of toxicity were observed in hematologic and blood chemistry profiles (Supplemental Tables S2 and S3) or by organ histopathology (data not shown).
Based on these results, we investigated the effects of pTNF-NC/NPs in mice bearing established syngeneic Ehrlich breast tumors. Notably, IP administration of pTNF-NC/NPs at a dose of 1.2 mg/kg × 6 was associated with substantial inhibition of tumor growth (Figure 4A); however, there was evidence of tumor regrowth after day 30. By contrast, growth inhibitory effects and prolonged tumor regressions were observed when the pTNF-NC/NPs were administered intratumorally (IT) at a dose of 1.2 mg/kg × 6 (Figure 4A). Moreover, the pTNF-NC/NPs were more effective that pTNF-NCs, supporting the importance of the DNP system (Figure 4B).
Figure 4. Anti-tumor activity of pTNF-NC/NPs.
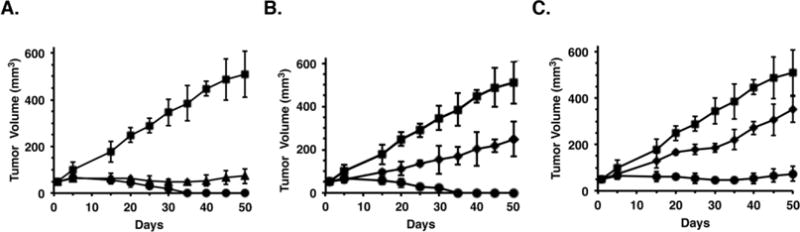
A. Balb/c mice (6 per group) with subcutaneous Ehrlich breast tumors (~50 mm3) were treated with 1.2 mg/kg pTNF IT (squares), 1.2 mg/kg pTNF-NC/NPs IP (triangles) or 1.2 mg/kg pTNF-NC/NPs IT (circles) on days 1, 5, 9, 13, 17 and 21. Tumor measurements were performed on the indicated days. The results are expressed as tumor volumes (mean±SD). B. Balb/c mice (6 per group) with subcutaneous Ehrlich tumors (~50 mm3) were treated IT with 1.2 mg/kg pTNF (squares), 1.2 mg/kg pTNF-NCs (diamonds) or 1.2 mg/kg pTNF-NC/NPs (circles) on days 1, 5, 9, 13, 17 and 21. Tumor measurements were performed on the indicated days. The results are expressed as tumor volumes (mean±SD). C. Balb/c mice (6 per group) with subcutaneous Ehrlich tumors (~50 mm3) were treated IP with 1.2 mg/kg pTNF (squares), 1.2 mg/kg pTNF-NCs (diamonds) or 1.2 mg/kg pTNF-NC/NPs (circles) on days 1, 5, 9, 13, 17 and 21. Tumor measurements were performed on the indicated days. The results are expressed as tumor volumes (mean±SD).
Analysis of tumor lysates demonstrated that IP and IT pTNF-NC/NP treatment is associated with an increase in TNF (Figure 5A) and activated caspase-3-CF (Figure 5B) expression as compared to that obtained with pTNF-NCs. In concert with these results, tumor sections from the pTNF-NC/NP-treated mice demonstrated an increase in apoptotic bodies and cell death as compared to that in tumors from mice treated with empty NPs or pTNF-NCs (Figure 5C). These findings supported the premise that pTNF-NC/NPs are effective against Ehrlich breast tumors when administered systemically or intratumorally.
Figure 5. Treatment with pTNF-NC/NPs results in intratumoral expression of TNF and activated caspase-3-CF.
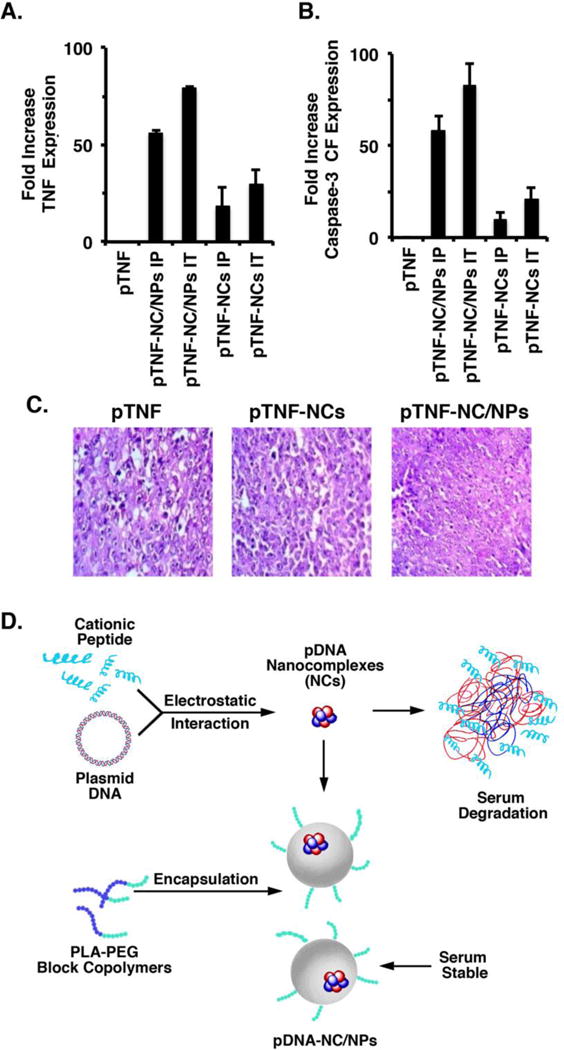
A–C. Balb/c mice with subcutaneous Ehrlich tumors (~50 mm3) were treated with 1.2 mg/kg pTNF IT, 1.2 mg/kg pTNF-NC/NPs (IT and IP) or 1.2 mg/kg pTNF-NCs (IT and IP) for 1, 5, 9, 13, 17 and 21 days. One mouse from each group was sacrificed on day 35 and the tumors were harvested. Lysates from the tumors were incubated with FITC-labeled anti-TNF and Cy5-labeled anti-caspase-3-CF, and analyzed by fluorimetry. The results are expressed as the relative fold-increase (mean±SD for 3 determinations) of TNF and caspase-3-CF as compared to that obtained for the empty NP-treated control tumor (assigned a value of 1). Tumor sections were also analyzed by H&E staining (C). In contrast to the H&E staining of empty NP- and pTNF-NC-treated tumors, the black circles highlight the presence of dead cells in the pTNF-NC/NP-treated tumors. D. Schematic representation of the pTNF-NCs and pTNF-NC/NPs. Nanocomplexes (NCs) were generated by incubation of the positively charged HCR peptide with the negatively charged pE425-TNF expression vector. To circumvent the instability of pTNF-NCs in plasma, a dual nanocomplex in a nanoparticle system was in turn generated by encapsulation of the pTNF-NCs in polymeric NPs. Intracellular delivery of the pTNF-NC in NPs results in sustained release of the pTNF-NC, import of pE425-TNF to the nucleus and expression of TNF. Our results further support the premise that this system is effective in the delivery of DNA expression vectors to tumors. Intratumoral expression of secreted cytokines, such as TNF, thus represents a novel approach for inducing anti-tumor activity and potentially reprogramming the tumor immune microenvironment.
Discussion
The therapeutic strategy of expressing exogenous genes in tumors growing in vivo has been substantially curtailed by the lack of effective delivery systems (12–15). One of the major challenges has been the systemic delivery of cationic peptide-DNA complexes to tumor cells at sufficient levels because of DNA degradation in serum (17, 21). To address this obstacle, we developed novel polymeric NPs with high molecular weight PLA for the systemic delivery of peptide cargoes to tumor cells (18, 20). Additionally, for effective DNA delivery, we designed a linear short HCR peptide that maintains DNA condensation, imparts endosomal escape properties and directs nuclear localization (16). The present work demonstrates that, using this novel HCR peptide, we could successfully generate cationic HCR-pDNA NCs (Figure 5D). Moreover, these ~80 nm pDNA-NCs were successfully encapsulated in our ~200 nm polymeric NPs to circumvent degradation of the NCs in plasma, which has been a significant hurdle in gene delivery systems (Figure 5D) (12–14). In this way, we have generated a “nanocomplex in a nanoparticle” or dual NP system for gene delivery (Figure 5D). In assessing the effectiveness of this system, we first prepared DNPs by incorporation of the GFP gene as a prototype model. Our results showed that treatment of MCF-7 cells with the pGFP-NC/NPs is associated with GFP expression, indicating that the HCR peptide-pGFP-NC functions in driving the exogenous GFP gene. Recent work has reported the encapsulation of polymer-doxorubicin NPs (~80 nm) into 2500 nm silica microparticles for the delivery of doxorubicin to tumors (22). That “nanoparticle in a nanoparticle” is thus substantially larger than the ~200 nm pDNA-NC/NPs described here and supports the potential of our DNPs for the delivery of DNA, as well as certain anti-cancer agents, into tumor cells.
Another obstacle for the delivery of DNA vectors is often the need to achieve high transfection or transduction efficiencies of the tumor cell population to have a significant therapeutic impact. This obstacle can be circumvented by the expression of a secreted protein, and is particularly applicable to reprogramming of the tumor microenvironment. In this context, TNF is a pleiotropic cytokine that promotes immune surveillance and directly kills cancer cells (5). Despite the promise of TNF as an anti-tumor agent, systemic TNF-induced toxicity precluded clinical development of this cytokine, other than in the setting of isolated limb perfusion for sarcomas (8). Noteworthy, however, are the findings that intratumoral administration of TNFerade has activity in the treatment of diverse human cancers (10), supporting the premise that a system for the delivery of TNF to tumors, while circumventing the systemic toxicity, could be an effective anti-cancer therapy. Accordingly, we constructed dual NC/NPs expressing the pE425-TNF vector. The pE425 promoter is activated by ROS in the response of tumor cells to radiation and chemotherapy (10). In addition, the pE425 promoter is selectively activated in cancer cells with increased ROS levels (23) and the production of TNF further drives ROS production in an autoinductive process (24). Thus, the pE425-TNF vector is particularly well suited for the induction of TNF in tumors. In this regard, we found that systemic pTNF-NC/NP treatment of Ehrlich breast tumors is associated with production of TNF and induction of apoptosis. Interestingly, similar effects with prolonged tumor regression were observed when the pTNF-NC/NPs were administered intratumorally, indicating that this dual NP system can be given systemically or directly into tumors. Moreover and importantly, the pTNF-NC/NPs induced little if any systemic toxicity.
The dual pDNA-NC/NP system reported here has certain potential advantages for cancer treatment. For instance, the pTNF-NC/NPs could be administered systemically or locally in combination with radiation and/or chemotherapy (10). In addition, treatment of tumors with pTNF-NC/NPs could be used to reprogram the immune microenvironment. In this way, pTNF-NC/NPs have the potential to convert “cold” to “hot” tumors and be combined with immune checkpoint inhibitors. This dual pDNA-NC/NP system could also be employed to reprogram immune surveillance in tumors by incorporating genes encoding cytokines, such as IL-12 and others, under control of the pE425 promoter.
Novelty
The systemic delivery of DNA expression vectors to tumors has been a major therapeutic challenge, in large part due to DNA-serum interactions. Indeed, to our knowledge, there is presently no effective way to systemically target tumors with the expression of exogenous genes.
The present studies have addressed this challenge by designing a peptide-DNA nanocomplex that is encapsulated in novel polymeric nanoparticles. As one model system, we investigated the systemic delivery of a DNA expression vector encoding TNF. The results show that our nanocomplex in a nanoparticle drives TNF expression in tumor cells growing in vitro and in syngeneic tumors in mice.
Our findings support the potential of this systemic DNA delivery system for expressing cytokines, such as TNF, or other proteins in tumors with the capacity to promote anti-tumor activity and immunity.
Supplementary Material
Acknowledgments
Financial Support: Research reported in this publication was supported by Grants from the National Cancer Institute of the National Institutes of Health under award numbers CA97098 and CA166480, and the Department of Science and Technology, Government of India.
Abbreviations
- NP
nanoparticle
- DNP
dual nanoparticle
- TNF
tumor necrosis factor alpha
- NC
nanocomplex
- HCR
histidine and cysteine modified arginine peptide
- GFP
green fluorescence protein
- caspase—cleaved fragment
caspase-3-CF
- IP
intraperitoneal
- IT
intratumoral
- PDI
polydispersity index
- TEM
transmission electron microscopy
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Potential Conflict of Interest: The authors declare competing financial interests, R.W., S.K., D.K. and H.S. hold equity in NanoProteagen. The other authors disclosed no potential conflicts of interest.
References
- 1.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011 Mar 4;144(5):646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 2.Balkwill F, Charles KA, Mantovani A. Smoldering and polarized inflammation in the initiation and promotion of malignant disease. Cancer Cell. 2005 Mar;7(3):211–217. doi: 10.1016/j.ccr.2005.02.013. [DOI] [PubMed] [Google Scholar]
- 3.Topalian SL, Drake CG, Pardoll DM. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell. 2015 Apr 13;27(4):450–461. doi: 10.1016/j.ccell.2015.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Carswell E, Old L, Kassel R, Green S, Fiore N, Williamson B. An endotoxin-induced serum factor that causes necrosis of tumors. Proc Natl Acad Sci USA. 1975;72:3666–3670. doi: 10.1073/pnas.72.9.3666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brenner D, Blaser H, Mak TW. Regulation of tumour necrosis factor signalling: live or let die. Nat Rev Immunol. 2015 Jun;15(6):362–374. doi: 10.1038/nri3834. [DOI] [PubMed] [Google Scholar]
- 6.Spriggs DR, Sherman ML, Michie H, Arthur KA, Imamura K, Wilmore D, et al. Recombinant human tumor necrosis factor administered as a 24-hour intravenous infusion. A phase I and pharmacologic study. J Natl Cancer Inst. 1988 Sep 7;80(13):1039–1044. doi: 10.1093/jnci/80.13.1039. [DOI] [PubMed] [Google Scholar]
- 7.Sherman ML, Spriggs DR, Arthur KA, Imamura K, Frei E, 3rd, Kufe DW. Recombinant human tumor necrosis factor administered as a five-day continuous infusion in cancer patients: phase I toxicity and effects on lipid metabolism. J Clin Oncol. 1988 Feb;6(2):344–350. doi: 10.1200/JCO.1988.6.2.344. [DOI] [PubMed] [Google Scholar]
- 8.Jakob J, Hohenberger P. Role of isolated limb perfusion with recombinant human tumor necrosis factor alpha and melphalan in locally advanced extremity soft tissue sarcoma. Cancer. 2016 Sep 1;122(17):2624–2632. doi: 10.1002/cncr.29991. [DOI] [PubMed] [Google Scholar]
- 9.Hallahan DE, Mauceri HJ, Seung LP, Dunphy EJ, Wayne JD, Hanna NN, et al. Spatial and temporal control of gene therapy using ionizing radiation. Nat Med. 1995 Aug;1(8):786–791. doi: 10.1038/nm0895-786. [DOI] [PubMed] [Google Scholar]
- 10.Weichselbaum RR, Kufe D. Translation of the radio- and chemo-inducible TNFerade vector to the treatment of human cancers. Cancer Gene Ther. 2009 Aug;16(8):609–619. doi: 10.1038/cgt.2009.37. [DOI] [PubMed] [Google Scholar]
- 11.Herman JM, Wild AT, Wang H, Tran PT, Chang KJ, Taylor GE, et al. Randomized phase III multi-institutional study of TNFerade biologic with fluorouracil and radiotherapy for locally advanced pancreatic cancer: final results. J Clin Oncol. 2013 Mar 1;31(7):886–894. doi: 10.1200/JCO.2012.44.7516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yin H, Kanasty RL, Eltoukhy AA, Vegas AJ, Dorkin JR, Anderson DG. Non-viral vectors for gene-based therapy. Nature reviews Genetics. 2014 Aug;15(8):541–555. doi: 10.1038/nrg3763. [DOI] [PubMed] [Google Scholar]
- 13.Amer MH. Gene therapy for cancer: present status and future perspective. Mol Cell Ther. 2014;2:27. doi: 10.1186/2052-8426-2-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Collins M, Thrasher A. Gene therapy: progress and predictions. Proc Biol Sci. 2015 Dec 22;282(1821):20143003. doi: 10.1098/rspb.2014.3003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim J, Wilson DR, Zamboni CG, Green JJ. Targeted polymeric nanoparticles for cancer gene therapy. J Drug Target. 2015;23(7–8):627–641. doi: 10.3109/1061186X.2015.1048519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mann A, Shukla V, Khanduri R, Dabral S, Singh H, Ganguli M. Linear short histidine and cysteine modified arginine peptides constitute a potential class of DNA delivery agents. Mol Pharm. 2014 Mar 3;11(3):683–696. doi: 10.1021/mp400353n. [DOI] [PubMed] [Google Scholar]
- 17.Zagorovsky K, Chou LY, Chan WC. Controlling DNA-nanoparticle serum interactions. Proc Natl Acad Sci U S A. 2016 Nov 16; doi: 10.1073/pnas.1610028113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hasegawa M, Sinha RK, Kumar M, Alam M, Yin L, Raina D, et al. Intracellular targeting of the oncogenic MUC1-C protein with a novel GO-203 nanoparticle formulation. Clin Cancer Res. 2015 May 15;21(10):2338–2347. doi: 10.1158/1078-0432.CCR-14-3000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shi J, Schellinger JG, Pun SH. Engineering biodegradable and multifunctional peptide-based polymers for gene delivery. J Biol Eng. 2013 Oct 24;7(1):25. doi: 10.1186/1754-1611-7-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kumar M, Gupta D, Singh G, Sharma S, Bhatt M, Prashant CK, et al. Novel polymeric nanoparticles for intracellular delivery of peptide cargos: antitumor efficacy of the BCL-2 conversion peptide NuBCP-9. Cancer Res. 2014 Apr 16;74(12):1–11. doi: 10.1158/0008-5472.CAN-13-2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kircheis R, Wightman L, Kursa M, Ostermann E, Wagner E. Tumor-targeted gene delivery: an attractive strategy to use highly active effector molecules in cancer treatment. Gene Ther. 2002 Jun;9(11):731–735. doi: 10.1038/sj.gt.3301748. [DOI] [PubMed] [Google Scholar]
- 22.Xu R, Zhang G, Mai J, Deng X, Segura-Ibarra V, Wu S, et al. An injectable nanoparticle generator enhances delivery of cancer therapeutics. Nat Biotechnol. 2016 Apr;34(4):414–418. doi: 10.1038/nbt.3506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gorrini C, Harris IS, Mak TW. Modulation of oxidative stress as an anticancer strategy. Nat Rev Drug Discov. 2013 Nov 29;12(12):931–947. doi: 10.1038/nrd4002. [DOI] [PubMed] [Google Scholar]
- 24.Blaser H, Dostert C, Mak TW, Brenner D. TNF and ROS crosstalk in inflammation. Trends Cell Biol. 2016 Apr;26(4):249–261. doi: 10.1016/j.tcb.2015.12.002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.