

Abstract
Protein misfolding results in the accumulation of aggregated β-sheet-rich structures in Parkinson’s disease (PD) and Alzheimer’s disease (AD). The toxic oligomer hypothesis stipulates that prefibrillar assemblies, such as soluble oligomers or protofibrils, are responsible for the poor prognosis of these diseases. Previous studies demonstrated that a small molecule related to the natural compound orcein, O4, directly binds to amyloid-β fibrils and stabilizes them accelerating the formation of end-stage mature fibrils. Here we demonstrate a similar phenomenon during O4 treatment of α-synuclein (αsyn) aggregates, the protein responsible for PD symptoms. While the drug did not change the kinetics of aggregate formation as measured by the amyloidophilic dye Thioflavin T, O4 depleted αsyn oligomers and promoted the formation of SDS and proteinase K resistant aggregates consisting of large fibril clusters. These fibril clusters exhibited reduced toxicity to human neuronal model cells and reduced seeding activity in vitro. The effectiveness of O4 decreased when it was added to later points in the αsyn aggregation pathway, which suggests that the incorporation of O4 into fibril assemblies stabilizes them against chemical, enzymatic, and mechanic degradation. These findings suggest that small molecules, which stabilize amyloid fibrils, prevent fibril fragmentation and seeding and consequently prevent prion-like replication of misfolded αsyn. Inhibiting prion replication by fibril stabilization could thus be a therapeutic strategy for PD.
Keywords: synuclein, prion, O4, orcein, fibril, cluster
Graphical abstract
INRODUCTION
Parkinson’s disease (PD) pathology is characterized by the accumulation of Lewy bodies or fibrillar cellular inclusions in the axons of dopaminergic neurons. (1, 2) α-Synuclein (αsyn) is the most abundant protein in these inclusions; its misfolding and deposition in dopaminergic neurons is closely associated with PD pathology. (2) In its native state, αsyn is a 140 residue unfolded protein that can adopt an α-helical structure when bound to membranes. (3, 4) It has been discussed that the protein can form helix-bundles as part of the SNARE complex or, possibly, through self-association. (5, 6) Deposits of αsyn can propagate into tissue grafts in PD patients, and endogenous αsyn is recruited into fibrillar aggregates though a seeding mechanism in transgenic mice. (7, 8) Both findings strongly suggest that αsyn pathology can propagate by a prion mechanism. (9)
In vitro, αsyn produces fibrillar structures via a nucleation-dependent aggregation pathway (10, 11). Simple oligomers progress to become soluble protofibrils which further mature to become insoluble fibrils. (1, 12) In this process, post-nucleation species, such as β-sheet αsyn oligomers, protofibrils, and fibril fragments act as seeds for monomers, accelerating fibril formation. (13) Any process that generates seeds will cause αsyn to autocatalytically replicate via secondary nucleation. (14) This mechanism is a general feature of amyloidogenesis via nucleated polymerization and can be observed in other misfolded protein diseases such as Alzheimer’s disease (AD). (15)
Numerous studies have suggested that the aggregation intermediates in amyloidogenesis, i.e. oligomers and soluble protofibrils, are largely responsible for the poor prognosis of misfolded protein diseases such as AD and PD. (16–19) These findings suggest that depleting oligomeric intermediates should yield effective treatments for these diseases. An extensive number of small molecules have been identified to inhibit amyloidogenesis in AD and PD (reviewed in: (20)). Recently, studies on Lacmoid, Orcein and its derivatives, Congo Red, and ECGC have explored alternative therapeutic strategies, such as derailing amyloid formation and stabilizing mature fibrils and to deplete oligomeric aggregation intermediates. (1, 21–25)
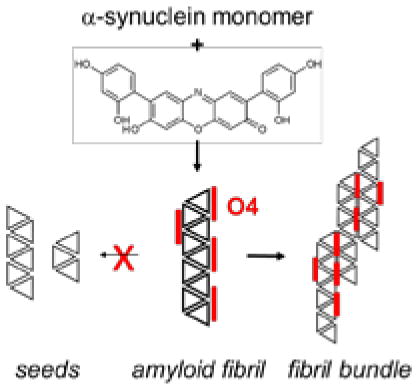
This study tests whether stabilization of mature fibrils could also be a therapeutic strategy to prevent prion replication through secondary nucleation mechanisms. We used the orcein-related small molecule, O4, which stabilizes mature amyloid-beta (Aβ), and tested whether the molecule stabilized αsyn fibrils in a similar manner. We also tested whether stabilization of αsyn fibrils could prevent their seeding capability, limiting αsyn toxicity. (20, 26) Our data demonstrate that O4 accelerated the αsyn fibrillation by stabilizing αsyn protofibrils, which consequently reduced the accumulation of αsynoligomers, decreased seeding capacity, and cellular toxicity in vitro.
EXPERIMENTAL PROCEDURES
α-Synuclein Expression
α-Synuclein was expressed in E. coli as described previously.(21, 27) Bacteria were grown at 37° C in LB and ampicillin (100μg/mL). αsyn expression was induced with IPTG once the OD 600 reached 0.5. The bacteria was then grown overnight at 30° C. The bacterial suspension was spun for 30 minutes at 3166 × g at 4°C. The resulting pellet was frozen at -80°C for an hour and then resuspended in lysis buffer (10mM Tris HCl, 1mM EDTA, pH 8) containing Roche complete protease inhibitor. The solution was then sonicated for 30 minutes, boiled for 20 minutes, and spun down at 4000 × g for 30 minutes at 4°C. The resulting αsyn pellet was collected and dissolved in 25 mM Tris, pH 7.7. The αsyn was purified using a FPLC Resource Q anion exchange column with a salt gradient from 0–1M NaCl. The protein was dialyzed overnight in ammonium bicarbonate buffer (10 mM, pH 7.5), frozen and then lyophilized for storage.
Thioflavin T (ThT) aggregation assays
αSyn (1mg/mL) was dissolved in 10 mM NaOH and sonicated (VWR® Symphony™ Ultrasonic Cleaner) for 20 minutes. The αsyn was then spun through a 50 kDa filter (Millipore) at 17,000 × g for 15 minutes. The concentration of monomerized αsyn was then determined by OD280 using an extinction coefficient of 5960/M/cm. The αsyn was then aggregated in a non-binding 96 well plate (Corning, #3651) at a concentration of 30μM with intermittent shaking in aggregation buffer (100mM NaP, pH 7.4, 10mM NaCl, 0.1% NaN3, and 20μM ThT). A 2 mm diameter glass bead was added to each well to accelerate the aggregation through stirring. The plate was kept at 37° C and agitated by orbital shaking once every minute for 15 seconds. The ThT fluorescence was recorded with an excitation wavelenth of 436 nm and an emission wavelenth of 485 nm in a fluorescence plate reader (Infinite F200, Tecan). O4 stock solutions (20 mM) were prepared in DMSO and diluted into the aggregation buffer at the final concentration. Equal amounts of DMSO were added to αsyn buffer control samples.
Light scattering aggregation assays
Monomeric αsyn (1mg/mL) was prepared as described above. The αsyn was then aggregated in a non-binding 96 well plate (Corning, #3651) at a concentration of 45μM with constant shaking in aggregation buffer (100mM NaP, pH 7.4, 10mM NaCl, 0.1% NaN3, and 20μM ThT). A 2 mm diameter glass bead was added to each well to accelerate the aggregation through stirring. At each time point, 10 μl sample was diluted 1:9 in water in a disposable cuvette for light scattering measurements in a Malvern Zetasizer Nano ZS. The count rate was recorded at a scattering angle of 173 degrees, attenuation 8. All solutions were cleared by microfiltration (0.2 μm) prior to use.
Circular dichroism spectroscopy
The αsyn samples (45μM) incubated under constant shaking at 37°C were diluted 1:1 in aggregation buffer (100mM NaP, pH 7.4, 10mM NaCl, 0.1% NaN3, and 20μM ThT) and sonicated for 10 minutes in a water bath sonicator (VWR® Symphony™ Ultrasonic Cleaner). A 1 mm path length cuvette was used for circular dichroism (CD) spectroscopy (Jasco Analytical Instruments).
Ultracentrifugation Assays
The αsyn samples (45μM) incubated under constant shaking at 37°C were centrifuged (TL-100, Beckman) at 70k RPM (205,000 × g) for 30 minutes. The supernatant was then analyzed by SDS-PAGE (Bio-Rad) at 160V for 30 minutes. The gel was then washed with water, fixed for 20 min (50% EtOH, 20% Acetic Acid), washed again with water and stained with EZBlue staining reagent (Sigma-Aldrich) overnight. Additional water washes were performed to de-stain the gel. The gel was imaged on a color scanner, and densitometric analysis was performed in ImageJ using the ImageJ measure function to measure the average pixel intensity of gel bands.
Quantification of SDS Resistant Aggregates
For filter retardation assays (FRA), 10μL of the 30 μM αsyn aggregated sample was diluted with 40μL of 2X NaP (200 mM Na-phosphate, pH 7.4, 20 mM NaCl) buffer and 50μL of denaturation buffer (4% SDS, 100mM DTT) and boiled for 5 minutes. A 0.2μm pore size cellulose acetate membrane was soaked in PBS + 0.1% SDS. A vacuum filter device was assembled and the membrane was wetted with PBS + 0.1% SDS. The αsyn samples (100μl) were added onto the cellulose acetate membrane, and PBS + 0.1% SDS was added in locations not containing the samples and a vacuum was applied. The membrane was then washed twice with PBS + 0.1% SDS. After disassembly, membranes were blocked with 5% milk powder in PBS. The SDS resistant αsyn aggregates on the membrane were stained with (1:500) anti-αsyn antibody 211 (Santa Cruz Biotech) in PBS + 5% milk and subsequently stained with goat-anti-mouse fluorescent antibody (Li-cor® IRDye 800CW; 1:15000) in PBS + 5% milk. The membrane was then washed with PBS + 0.05% Tween20 and imaged using a Li-cor® Odyssey system at 800 nm excitation.
PK Digestion and Western blotting
10 μL of 30 μM aggregated αsyn sample was incubated for 30 minutes at 37°C with varying concentrations of proteinase K (PK). After incubation, each sample was analyzed by SDS-PAGE and Western blotting on a nitrocellulose membrane (Biorad). The membrane was blocked with 5% milk powder in PBS and was then stained with 1:200 anti-αsyn antibody 211 in 5% milk and subsequently stained with goat-anti-mouse (Li-cor® IRDye 800CW; 1:15000) fluorescent antibody in 5% milk.
MTT Cell Toxicity Assays
SH-EP neuroblastoma cells (28) were plated on a 96 well plate with 20,000 cells per well and incubated overnight in DMEM media with 10% FBS. αSyn samples (originally 30 μM) were diluted in culture media to 2.5 – 0.3 μM monomer equivalent concentration. The diluted αsyn samples were then incubated with the SH-EP neuroblastoma cells for three days in 96 well plates with a volume of 100 μL. O4-treated and control samples contained the same amounts of DMSO (< 0.1%). After three days, 10 μL of 5mg/ml MTT (Thiazolyl Blue Tetrazolium Bromide) in PBS was added to all cells. 10μl Triton X-100 (Sigma) was added to designated “dead cells” as a negative control. The cells were then incubated for three hours at 37°C. 100μl denaturation buffer (10% Triton X-100, 0.1M HCl in isopropanol) was then added to the cells. The cells were incubated overnight and cell viability was calculated from OD570 measurements using the
Formation of α-Synuclein Seeds for Seeded Aggregation Assays
αSyn seeds were generated by sonication of αsyn aggregates at different aggregation time points for 10 minutes in a water bath sonicator (VWR® Symphony™ Ultrasonic Cleaner). 10% m/m αsyn seeds were then added to fresh αsyn monomers and aggregated with ThT for one week as described above.
TEM Imaging of α-synuclein
10μL of αsyn sample was placed on the copper mesh grid for 1 minute and then washed with water twice. 10μL of 4% uranyl acetate was then added to the copper mesh grid for 90 seconds and then removed. The copper mesh grid was then imaged using TEM (JEOL 2000FX).
RESULTS
O4 Induces SDS and Protease Resistant α-Synuclein Aggregates
To test how O4 affected αsyn amyloid formation, we used several assays to monitor different aspects of αsyn fibril formation. We first tested whether O4 affected the kinetics of the formation of β-sheet rich αsyn structures by monitoring the fluorescence of the amyloidophilic dye Thioflavin T (ThT). ThT increases in fluorescence upon binding to β-rich amyloid-like structures during the elongation phase.(29) When incubated in the presence of equimolar (1x) O4, endpoint ThT fluorescence was reduced (Fig. 1A). However, the presence of O4 did not influence the length of the lag phase of aggregation kinetics as can be easily seen when aggregation curves are normalized to the same amplitudes (Fig. 1B). These data suggest that O4 acted during the later stages of αsyn aggregation affecting protofibrils and fibrils formation rather than interfering with nucleation. It also suggest that O4 binds to the same binding site as ThT, as had been observed in the case of Aβ42, which would lead to competitive inhibition of ThT fluorescence. (26) Our interpretation also suggests that the αsyn aggregates retain their β-sheet structures in the presence of the drug. We measured the secondary structure of αsyn in the presence and absence of O4 by circular dichroism (CD; Fig. 1C, D). CD spectra of αsyn at t = 0 h are typical of an unstructured polypeptide, while spectra had minima at ~225 nm after incubation for 48 h. It should be noted that these minima are red-shifted from the minimum at 218 nm, which would be typical for β-sheet structures. Similar red-shifted minima have previously been observed in Aβ42 in the presence of O4(26) and in αsyn aggregates formed in the presence of other amyloid modulators, such as baicalein. (30, 31) The red shift may reflect an altered structure of αsyn in its aggregated state.
Figure 1.

O4 does not alter the formation of β-sheet aggregates. (A) ThT fluorescence of αsyn (30μM) aggregation in the presence and absence of 1:1 O4 to αsyn under intermittent shaking conditions. The results represented mean ThT fluorescence intensities (n=3). (B) Normalized ThT fluorescence signals of αsyn samples in (A). Time points for further analysis: 6 h, 20 h, 48 h, 80 h, and 168 h were denoted. (C, D) CD spectra of αsyn incubated with O4 for 0 h and 48 h, respectively.
Transmission electron microscopy (TEM) was used to visualize the structure of αsyn aggregates in the presence of O4. We sampled αsyn aggregates at time points corresponding to the different stages of aggregation: 6 h, 20 h, 48 h, 80 h, and 168 h (Fig. 2A). Time points were chosen corresponding to the different stages of αsyn aggregation. The 6 h time point was part of the lag phase. Time points of 20 hours and 48 hours were a part of the elongation phase. Time points after 48 hours were part of the end -stage fibrillar phase (Fig. 1B).
Figure 2.

O4 induces SDS and protease resistant αsyn aggregates. (A) Transmission electron microscopy (TEM) of αsyn time points denoted in Fig. 1 in the presence or absence of O4; scale bar 500 nm. (B and C) Effect of O4 on SDS resistant aggregate formation quantified by FRA. The fluorescent signal was generated by immunostaining using anti-αsyn antibodies on a cellulose acetate membrane. (D) Effect of O4 on αsyn resistance to proteinase K digestion on 1 week time point samples. An additional sample was formed by incubating the αsyn control sample with equimolar O4 for 30 minutes to observe short-term effects of O4 treatment. HMW= High molecular weight, T=trimer, D= dimer, and M=monomer.
TEM images confirmed the formation of αsyn fibrils that were about 200 nm in length and ~10 nm wide in the absence of O4 (Fig. 2A). Their formation coincided with the increase in ThT fluorescence (Fig. 1A, B) When comparing the later time points (48 h, 80 h and 168 h) of αsyn incubated with O4 to control conditions, we observed that O4 promotes the formation of fibril clusters characterized by a pronounced network of laterally linked αsyn fibrils (Fig. 2A). In contrast, these clusters formed later and were not as pronounced in αsyn samples aggregated in the absence of O4.
We then quantified the effect of O4 on the stability of αsyn aggregates to SDS denaturation in a membrane-filter retardation assay (FRA).(32) Aggregates that were resistant to SDS denaturation remained on the porous cellulose acetate membrane while denatured aggregates were washed through the membrane. SDS resistant aggregates were quantified by immunofluorescence staining using anti-αsyn antibody 211 (Fig. 2B). Compared to a DMSO vehicle control sample, O4 accelerated the formation of SDS resistant αsyn aggregates and significantly increased their amount at the endpoint of aggregation (Fig. 2B, C). These results demonstrate that O4 incubation with αsyn, promotes the formation of SDS resistant aggregates, as was the case for Aβ. The presence of fibril clusters in TEM coincided with SDS resistance, suggesting that these structures were responsible for the increased SDS resistance observed in the FRA.
Mature amyloid fibrils have increased resistance to protease digestion. (26, 33) We therefore assessed the effect of O4 on αsyn resistance to proteinase K (PK). All samples were aggregated for one week; equimolar O4 was added either at the beginning of incubation or at the end, 30 min before analysis (Fig. 2D). αSyn samples were then digested with PK at increasing concentrations (0, 0.2, 1 μg/mL). A Western blot was used to compare PK-resistance of samples incubated with O4 under the two conditions and αsyn control samples. Analysis of the Western blot suggested that O4 increases the resistance of αsyn aggregates to PK digestion. αSyn samples incubated with O4 for the full duration of aggregation showed pronounced high molecular weight bands (HMW) at 150 kDa, trimer bands (T) at 75 kDa, dimer bands (D) at 50 kDa, and monomer bands (M) at 25 kDa. The HMW αsyn resisted PK digestion at 1μg/mL in the O4 treated sample. Interestingly, αsyn fibrils that were incubated with O4 for 30 minutes at the end of the aggregation induced the formation of increased SDS-resistant dimers, trimers and HMW aggregates, but these aggregates had little resistance against PK digestion. This suggests that O4 stabilizes αsyn aggregates through a mechanism not related to fibril elongation, but likely by laterally linking αsyn fibrils and thus increasing stability to digestion or denaturation. The data also provide further support that O4 is responsible for the formation of the fibrillar clusters observed in the TEM (Fig. 2D) and that these fibril clusters correlate with the SDS resistant HMW αsyn band. It would be plausible that denser and thus more resistant fibril clusters form if O4 is present during aggregation as opposed to being added to preexisting fibrils.
Our results suggest that O4 may stabilize αsyn fibrils through the formation of fibril clusters. Consequently, O4 should deplete toxic oligomers and accelerate the formation of large aggregates. Therefore, we tested what effect O4 had on the presence of αsyn oligomers and large aggregates, respectively, and whether these species were correlated with cellular toxicity.
Induction of Large Fibril Clusters Rescues the Formation of Toxic α-Synuclein Oligomers
We probed the αsyn aggregation course for oligomer formation and toxicity. Oligomers were shown to rapidly form under constant agitation (34). To test whether O4 specifically depletes αsyn oligomers and increases the formation of large aggregates, we incubated αsyn under constant agitation, which accelerated aggregation compared to the previous experiment (Fig. 3A–D). We monitored the formation of β-sheet structures by ThT fluorescence (Fig. 3A, B) while quantifying the formation of large αsyn aggregates by light scattering (Fig. 3C) and by ultracentrifugation (Fig. 3D), respectively. As observed before, O4 had little influence on the kinetics of β-sheet formation as measured by ThT fluorescence. When monitoring aggregation by light scattering, O4 accelerated aggregation kinetics, especially when present at 5 x molar excess. Since light scattering is a function of particle size, these results suggest that large αsyn aggregates form faster in the presence of O4 than in its absence. To confirm this interpretation we separated aggregated αsyn from soluble protein by ultracentrifugation at 200,000 × g and quantified soluble αsyn by SDS-PAGE and Coomassie staining (Fig. 3D and Suppl. Fig. 1). The αsyn in the supernatant did not form SDS resistant aggregates (Suppl. Fig 1). This allowed us to directly quantify aggregation without any detection bias, which may result from aggregate size dependence of light scattering or of antibody affinity in FRA. Ultracentrifugation confirmed that O4 accelerated the formation of insoluble aggregates (Fig. 3D). However, the effect was only moderate, which is in accordance with our model of O4 binding to and stabilizing αsyn fibrils, which are insoluble even in the absence of the drug.
Figure 3.

O4 Incubation rescues αsyn toxicity in vitro. (A) Time course of ThT fluorescence of αsyn under constant shaking condition. (B) ThT fluorescence data from (A) normalized to end point fluorescence. –(C) Aggregation time course monitored by light scattering. (D) Fraction of soluble αsyn analyzed densitometrically from SDS-PAGE after centrifugation at 200,000 × g. (E and F) Effects of O4 on the formation of αsyn oligomers as assessed by anti-oligomer antibody, A11, dot blot analysis. (G) Assessment of αsyn cytotoxicity using the MTT metabolic assay. SH-EP cells were incubated with αsyn time point samples (2.5μM) used in the A11 dot blot analysis for three days. *P<0.05, **P<0.01, ***P<0.001 (Student’s T-Test, n = 5. (H) Normalized MTT and A11 fluorescent signal data to compare time course dynamics.
We then performed dot blot assays using the antibody A11 that specifically detects oligomeric amyloid intermediates (35) at different time points (Fig. 3E–H) to probe if fibril stabilization depletes αsyn oligomers. A11 binding was then juxtaposed with MTT reduction data in human neuroblastoma (SH-EP) cells generated form the same sample set (Fig. 3G and Fig. 3H). MTT reduction is a frequently used marker for mitochondrial activity (36). Toxic αsyn species inhibit mitochondrial activity and increase the formation of reactive oxygen species (36). Untreated samples showed a rise in oligomers between 2–6 h, which was suppressed by O4 (Fig. 2B). After 9 h incubation, the A11 signal in both O4 treated and untreated αsyn control samples was no longer detected, which suggested that the oligomers had matured into β-sheet rich fibrils (Fig. 3E, F). Correspondingly, maximal ThT fluorescence was observed at this time (Fig. 3A) and insoluble αsyn aggregates were formed (Fig. 3D). When the A11 data was juxtaposed to the MTT data of the same time points (Fig. 3G, H), the increase in αsyn oligomers corresponded to a decrease in cellular viability and vice versa. O4 suppressed oligomer formation and was able to significantly rescue cellular viability. This strongly suggests that O4 depleted toxic oligomers and other aggregation intermediates detected by the A11 antibody and promoted the formation of mature fibrils and fibril clusters.
O4 Reduces Seeding Activity
Recently, it was also observed that misfolded αsyn can replicate by an autocatalytic mechanism akin to prion replication (37). This autocatalytic replication is a crucial step both in the aggregation process in vitro (14) and in the spread of toxic αsyn species in cellular tissue (9). These observations link autocatalytic replication and toxicity. Secondary nucleation is the key process in prion replication. It can be achieved either by fibril fragmentation, which generates new seeds, or by secondary seeding on the surface of existing fibrils. (14) Since O4 promotes the formation of large fibril clusters, which would inhibit both forms of secondary nucleation, one would expect that it also inhibits seeding activity and the prion-like replication of αsyn fibrils.
To test this hypothesis, the seeding activities of O4 treated αsyn samples were investigated. Different αsyn seeds were created by taking αsyn samples at time points from 6 – 72 h during the aggregation, which was performed in the presence of O4 at molar ratios of 0x, 1x, or 5x (Fig. 4A). The samples were then sonicated for 10 minutes, added as seeds (10% m/m) to fresh monomerized αsyn, and incubated for three days (Fig. 4B–E). ThT fluorescence was used to map the aggregation kinetics.
Figure 4.

O4 Reduces seeding activity. (A) αsyn aggregation (30μM) monitored by Thioflavin T (ThT) fluorescence in the presence of 1:1 O4 to αsyn and 5:1 O4 to αsyn. Samples were taken at 6 h, 20 h, 48 h, and 72 h and sonicated to create seeds. (B - E); αsyn aggregation (30μM) in the presence of untreated seeds and O4 treated seeds created from the 6h, 20 h, 48 h, and 72 h samples, respectively (10% m/m). The results represented mean ThT fluorescence intensities (n=3).
Potent seeds annulled the lag phase of αsyn aggregation, while impotent seeds had a lesser effect on the lag phase. Seeds that were generated from the early plateau phase of aggregation (48 h) had the most seeding activity, while the seeds generated from the growth phase (20 h) and late plateau phase (72 h), respectively, were slightly less active (Fig. 4C, E) and αsyn from the lag phase (6 h) had no seeding activity (Fig. 4B). Addition of O4 during seed generation completely removed seeding activity of αsyn at all phases of the aggregation process at 5x molar excess and inhibited seeding at equimolar concentration (Fig. 4A–E). These data indicate that the stabilization of αsyn fibrils by O4can remove seeding activity in vitro.
α-Synuclein Seeding Activity Correlates to Toxicity
We then tested if the reduction in seeding activity by O4 correlated to reduced toxicity in cell culture. In a first experiment, we incubated the same time point samples used in the EM and FRA experiments (Fig. 2) with human neuroblastoma (SH-EP) cells for 72 h and measured cell metabolic rate by MTT reduction.
The results showed significant reduction in cell metabolic activities of αsyn control samples taken after 20 h, 48 h, and 80 h, (Fig. 5A). A minimum in cellular viability was observed at the 48 h time point, which corresponded to the αsyn species with highest seeding potency. According to the ThT fluorescence data (Fig. 1A, B) and the TEM data (Fig. 2A), these time points correspond to the formation of β-sheet rich aggregates and early fibrils. The incubation of αsyn with O4 significantly reduced the cellular toxicity compared to the αsyn control samples, and cellular viability was rescued during the most toxic stages of αsyn aggregation (between 20 h to 80 h).
Figure 5.

Time dependent effect of O4 on forming SDS resistant aggregates and cellular toxicity. (A) Assessment of time point αsyn cytotoxicity using the MTT metabolic assay. SH-EP cells were incubated with αsyn time point samples (1.25μM) in the presence or absence of O4 for three days. The time points selected correspond to the same time points in Figure 1. *P<0.05, **P<0.01, ***P<0.001 (Student’s T-Test, n = 5). (B, C, and D) αsyn aggregation (30μM) in the presence of untreated seeds and O4 treated seeds created from the 20 h, 48 h, and 72 h samples, respectively (10% m/m). (E) ThT fluorescence of αsyn (30μM) aggregation with O4 added at different time points. The aggregation period lasted for 1 week. The results represented mean ThT fluorescence (n=3). (F) Effect of adding O4 at different time points during the aggregation period on SDS resistant aggregates quantified by FRA. The * on the 168 h time point indicates O4 treatment for 30 minutes. (G) Assessment of αsyn cytotoxicity using the MTT metabolic assay using the same samples (0.3 μM) used in the FRA (F). *P<0.05, **P<0.01, ***P<0.001 (Student’s T-Test, n = 3).
It should be noted that, while toxic αsyn species were mostly lost after 18 h under constant agitation, the protein partially retained the capacity to inhibit metabolic activity for a full week under the slower aggregation regime using intermittent agitation (Fig. 1, Fig. 2). It should also be noted that metabolic inhibition in cells tracked the seeding competency of the αsyn. Thus, seeding competent αsyn species may play a large role in toxicity to the neuronal model cells.
The results also suggest that by destroying αsyn seeding activity, a compound might rescue toxicity. To test this hypothesis, we probed at which stage of aggregation O4 treatment was effective in rescuing metabolic inhibition by αsyn and whether treatment of preexisting αsyn aggregates prevented seeding and rescue d toxicity.
Time Dependency of O4 induced SDS Resistance, Seeding Potency, and Cellular Toxicity
We tested whether a brief treatment with O4 could inactivate existing seeds or whether the presence of O4 during the seeding process itself inhibited seeded aggregation. Seeds generated from αsyn samples at 6 h, 20 h, 48 h, and 72 h were treated with O4 at a 5x molar ratio (equivalent αsyn monomer concentration) for 30 minutes. These seeds were then diluted into fresh monomeric αsyn (10% m/m), so that the final O4 concentration was the same as in the reactions containing αsyn seed that had been generated in 5x O4 from the beginning (Fig. 4A and Fig. 5B–D). O4 was able to partially inhibit seeding activities under these conditions. Notably, its effect was greatest on the most active seeds (48 h) when compared to seeds generated later in the aggregation process (72 h) (Fig. 5B–D). These data support the model that O4 accelerates the formation of stable seeding-incompetent fibril clusters from oligomers and early fibrils and thereby inactivates early seeding competent species.
To test the effect of O4 treatment of preformed seeds on toxicity and fibril stability, αsyn was aggregated for one week under the intermittent agitation regime. O4 was added at different stages of aggregation (0 h, 6 h, 20 h, 48 h, 3 d, and 7d) (Fig. 5E). The time points corresponded to the αsyn species characterized in Figs. 1 and 2. They covered the lag phase, early and late growth phase, and early and late plateau phase of ThT aggregation kinetics. Addition of O4 caused an immediate drop in ThT fluorescence likely due to fluorescence quenching and/or competitive binding at the ThT binding site.(26) These samples were then further incubated until the end of one full week (Fig. 5E) and tested for the formation of SDS resistant aggregates as a proxy for fibril cluster formation (Fig. 5F). FRA data of these samples showed that O4 significantly increased the formation of SDS resistant aggregates when compared to the untreated αsyn. Notably, even treatment of the final 7 d aggregation sample with O4 for 30 min, induced partial resistance to SDS denaturation (168*; Fig. 5F) confirming the previous observation from SDS-PAGE (Fig. 2D).
To determine how this O4 activity translated to cellular toxicity, an MTT assay was performed on the same samples (Fig. 5G). O4 completely rescued metabolic inhibition when added during the first 20 h of the aggregation process and conferred partial rescue at 48 h and 3 d. However, no rescue in MTT reduction was achieved in the 7 d sample that was only treated for 30 minutes(168 h*; Fig. 5G).
These three data sets taken together demonstrate three points: 1) metabolic inhibition by αsyn aggregate species correlated with their seeding capacity in vitro; 2) Treatment with O4 prior to fibril formation or in the early stages of fibrillization, induced SDS stable fibril clusters, destroyed seeding activity, and rescued toxicity; 3) The effectiveness of O4 treatment decreased during fibril formation the later it was added. However, the formation of these clusters did not necessarily correlate to increased cellular viability and a partial transformation of fibrils into SDS-stable clusters was not sufficient to rescue toxicity.
DISCUSSION
Previous studies demonstrated that O4 stabilization of Aβ accelerated the formation of mature amyloid fibrils with increased resistance against SDS denaturation and PK digestion. This in turn decreased cellular toxicity by depleting Aβ oligomers. (26) We demonstrate a similar effect of O4 in αsyn fibril formation. The ability of O4 to increase αsyn stability against SDS denaturation and PK digestion suggests that O4 increases the stability of αsyn fibrils. Interestingly, large bundles and tangles of fibrils were observed in the presence of O4, which are the likely cause for increased stability. We observed a decrease in ThT fluorescence with time that was absent in when monitoring aggregation by light scattering or by protein solubility. This may reflect the formation of large fibril clusters that either bind ThT less efficiently or scatter the ThT fluorescence (Fig. 1B; Fig. 3C, D). These structures would not only be more stable against fragmentation but also provide fewer points of secondary nucleation by lateral association of monomers to the fibrils (14), which is reflected in their decreased seeding activity (Fig. 6). This observation corresponds to our observation that addition of O4 at the endpoint of aggregation while conferring some resistance to SDS denaturation (Fig. 2D, Fig. 5F), does not produce αsyn that is resistant to PK digestion (Fig. 2D) and does little to rescue metabolic toxicity (Fig. 5G), while presence of O4 during the aggregation process induces formation does both. One may speculate that both properties emerge as a result of the formation of large, tightly packed fibril bundles. Preformed αsyn fibrils may be sterically hindered from forming these bundles after the addition of O4.
Figure 6.

Model of autocatalytic amyloid formation through fibril fragmentation and secondary nucleation. O4 promotes the formation of fibril bundles and inhibits fragmentation and seeding.
In this study, we test an alternative therapeutic strategy using O4 which disrupts αsyn amyloidogenesis by promoting the formation of mature fibrillar aggregates. Previous small molecule studies that target αsyn amyloidogenesis include the use of Lacmoid, PcTS-Cu2+, Chlorazole black E, Congo Red, EGCG and Rosmarinic Acid. These molecules have been found to prevent the formation of amyloid fibrils by derailing amyloidogenesis or by disaggregation of aggregation intermediates. Studies on Lacmoid suggested that it disrupted the formation of fibrillar aggregates by stabilizing random coil formation through its binding to exposed hydrophobic surfaces in αsyn and Aβ. (23, 25) This would decrease the attractive forces between aggregates disrupt aggregation. Similarly, ECGC was suggested to remodel fibrillar oligomers to stunt monomer addition. Congo Red has also been suggested to redirect amyloidogenesis to produce amorphous aggregates. (1, 21–23).
Similar to the small molecules listed above, O4 preferentially binds to amyloid aggregates. However, our data, as well as previous findings on Aβ, suggest that O4 does not directly disrupt amyloidogenesis. As such, it doesn’t interfere with the formation of ThT binding aggregates as was observed in the case of Lacmoid, Congo Red, and ECGC. In analogy to Aβ, we hypothesize that the increase in fibril stability reduces the accumulation of toxic oligomers (26), which was confirmed by the decrease in structures detected by the anti-oligomer antibody A11. The increase in SDS-resistant fibrils and depletion of oligomers corresponded to decrease in αsyn’s ability to inhibit mitochondrial activity, supporting the amyloid oligomer hypothesis (35). However, it should be noted that toxic αsyn species persist longer than the signal from the A11 antibody (Fig. 3H) and that O4can still rescue toxicity at these later time points. This observation raises several possible interpretations: 1) toxic oligomers persist at low levels even in the presence of fibrils in the absence of O4 but not in its presence; 2) Other aS species, such as small fibrils or protofibrils are also toxic to the cell and O4 sequesters these species into large fibrils; or 3) O4 interacts with the cells to protect them from toxic species. A more detailed analysis of the interaction of amyloid aggregation intermediates with neuronal cells will be needed to address this question.
Our data strongly support the hypothesis that O4 inhibits αsyn toxicity by a similar mechanism to the one we previously found in Aβ, where O4 decreased oligomers and increased mature fibrillar aggregates, which corresponded to decreased cellular toxicity. (26). However, despite the similarities between the effects of O4 on αsyn and Aβ, studies using O4 treatment on the islet amyloid polypeptide (IAPP) found in type 2 diabetes mellitus-related amyloid have yielded slightly different results. Here, similar to the effect of EGCG, O4 was shown to redirect the IAPP aggregation pathway towards off-pathway globular aggregates that were shown to be less cytotoxic, instead of promoting the formation of mature fibrillar aggregates and fibril bundles. (24)
Prion-like propagation of misfolded αsyn through seeding is central to the spread of PD pathology.(9) We therefore tested if O4, in addition to depleting oligomers, could inhibit propagation of misfolded αsyn through seeding (Fig. 4). When comparing seeding ability of αsyn seeds generated in the presence or absence of O4, seeds generated by sonication from O4 treated fibrils had lost a large degree of their seeding activity compared to untreated αsyn fibrils. This suggests that O4 reduces αsyn’s susceptibility to fragmentation, which corresponded to the increased resistance to SDS denaturation and proteinase K digestion. Interestingly, short-term treatment of αsyn fibrils with O4 also resulted in increased fibril SDS resistance, decreased seeding potency, and decreased cellular toxicity. However, O4’s effect on these parameters in the short-term was not as pronounced compared to O4’s effect if the compound was present during fibril formation (Fig. 5B–C).
A possible explanation may be that a heterogeneous population of αsyn fibrils exists in solution, only some of which are seeding competent. Binding of O4 to fibrils or fibril bundles that already have limited seeding activity would then reduce the compound’s effective concentration towards the seeding competent species. We observed that the effect of short O4 (30 minute) treatment is less effective for seeds generated from the later (72 h) plateau phase of aggregation than for seeds from the growth phase (20 h) or early plateau phase (48 h) of aggregation, which would support this hypothesis.
Experimental evidence indicates that accelerated Aβ fibrillization reduces oligomer levels and functional deficits in mouse models of Alzheimer’s disease. (38) O4 is chemically and functionally similar to Orcein and Lacmoid, which historically have been used as a histological dyes, and food coloring (20, 26). Its pharmacological properties remain to be explored; its molecular weight and propensity to form colloid may be less than desirable for drug development. (23) However, our in vitro data suggest that stabilization of amyloid fibrils by small molecules may be a viable strategy to prevent seeding and prion replication. Future studies involving O4’s effect on other amyloid plaques will elucidate the therapeutic potential of preventing prion replication of misfolded protein pathology in PD and other amyloid diseases. O4 may serve as a model compound to further test this therapeutic strategy in vivo.
Supplementary Material
Acknowledgments
Funding Sources
This research was in part financially supported by the DRC at Washington University (NIH Grant No. 5 P30 DK020579), the German Science Foundation (DFG, BI 1409/1-1) and by the German Ministry for Science and Education (BMBF, NGFN-Plus 01GS08132, GERAMY 01GM1107C).
The SH-EP cell line was a kind gift from R. König, F Westermann and M. Schwab (DKFZ, Heidelberg, Germany). We gratefully acknowledge the use of the Nano Research Facility Washington University in St. Louis. This research was in part financially supported by the DRC at Washington University (NIH Grant No. 5 P30 DK020579), the German Science Foundation (DFG, BI 1409/1-1) and by the German Ministry for Science and Education (BMBF, NGFN-Plus 01GS08132, GERAMY 01GM1107C).
ABBREVIATIONS
- PD
Parkinson’s disease
- AD
Alzheimer’s disease
- Aβ
amyloid-β
- αsyn
α-synuclein
- SDS
Sodium Dodecyl Sulphate
- EGCG
Epigallocatechin gallate
- ThT
Thioflavin T
- FRA
Membrane-filter retardation assay
- DMSO
Dimethyl sulfoxide
- TEM
Transmission electron microscopy
- PK
Proteinase K
- T
Trimer band
- D
Dimer band
- M
Monomer band
- HMW
High molecular weight band
- MTT
Thiazolyl Blue Tetrazolium Bromide
- OD
Optical Density
- IAPP
islet amyloid polypeptide
- IPTG
Isopropyl β-D-1-thiogalactopyranoside
- FPLC
Fast protein liquid chromatography
- PBS
Phosphate-buffered saline
- DMEM
Dulbecco’s Modified Eagle’s medium
- FBS
Fetal bovine serum
- A11
Anti-oligomer antibody
Footnotes
Author Contributions
The manuscript was written through contributions of all authors. HL, KG and MG performed experiments. JB conceived experiments; HL and JB co-wrote the manuscript. All authors have given approval to the final version of the manuscript.
References
- 1.Rao JN, Dua V, Ulmer TS. Characterization of alpha-synuclein interactions with selected aggregation-inhibiting small molecules. Biochemistry. 2008;47:4651–4656. doi: 10.1021/bi8002378. [DOI] [PubMed] [Google Scholar]
- 2.Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. Alpha-synuclein in Lewy bodies. Nature. 1997;388:839–840. doi: 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- 3.Jao CC, Hegde BG, Chen J, Haworth IS, Langen R. Structure of membrane-bound alpha-synuclein from site-directed spin labeling and computational refinement. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:19666–19671. doi: 10.1073/pnas.0807826105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lashuel HA, Overk CR, Oueslati A, Masliah E. The many faces of alpha-synuclein: from structure and toxicity to therapeutic target. Nature reviews Neuroscience. 2013;14:38–48. doi: 10.1038/nrn3406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Burre J, Sharma M, Tsetsenis T, Buchman V, Etherton MR, Sudhof TC. Alpha-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science. 2010;329:1663–1667. doi: 10.1126/science.1195227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang W, Perovic I, Chittuluru J, Kaganovich A, Nguyen LT, Liao J, Auclair JR, Johnson D, Landeru A, Simorellis AK, Ju S, Cookson MR, Asturias FJ, Agar JN, Webb BN, Kang C, Ringe D, Petsko GA, Pochapsky TC, Hoang QQ. A soluble alpha-synuclein construct forms a dynamic tetramer. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:17797–17802. doi: 10.1073/pnas.1113260108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hansen C, Angot E, Bergstrom AL, Steiner JA, Pieri L, Paul G, Outeiro TF, Melki R, Kallunki P, Fog K, Li JY, Brundin P. alpha-Synuclein propagates from mouse brain to grafted dopaminergic neurons and seeds aggregation in cultured human cells. The Journal of clinical investigation. 2011;121:715–725. doi: 10.1172/JCI43366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Luk KC, Kehm VM, Zhang B, O’Brien P, Trojanowski JQ, Lee VM. Intracerebral inoculation of pathological alpha-synuclein initiates a rapidly progressive neurodegenerative alpha-synucleinopathy in mice. The Journal of experimental medicine. 2012;209:975–986. doi: 10.1084/jem.20112457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Olanow CW, Brundin P. Parkinson’s disease and alpha synuclein: is Parkinson’s disease a prion-like disorder? Mov Disord. 2013;28:31–40. doi: 10.1002/mds.25373. [DOI] [PubMed] [Google Scholar]
- 10.Conway KA, Harper JD, Lansbury PT., Jr Fibrils formed in vitro from alpha-synuclein and two mutant forms linked to Parkinson’s disease are typical amyloid. Biochemistry. 2000;39:2552–2563. doi: 10.1021/bi991447r. [DOI] [PubMed] [Google Scholar]
- 11.Conway KA, Harper JD, Lansbury PT. Accelerated in vitro fibril formation by a mutant alpha-synuclein linked to early-onset Parkinson disease. Nature medicine. 1998;4:1318–1320. doi: 10.1038/3311. [DOI] [PubMed] [Google Scholar]
- 12.Uversky VN, Lee HJ, Li J, Fink AL, Lee SJ. Stabilization of partially folded conformation during alpha-synuclein oligomerization in both purified and cytosolic preparations. The Journal of biological chemistry. 2001;276:43495–43498. doi: 10.1074/jbc.C100551200. [DOI] [PubMed] [Google Scholar]
- 13.Harper JD, Lansbury PT., Jr Models of amyloid seeding in Alzheimer’s disease and scrapie: mechanistic truths and physiological consequences of the time-dependent solubility of amyloid proteins. Annual review of biochemistry. 1997;66:385–407. doi: 10.1146/annurev.biochem.66.1.385. [DOI] [PubMed] [Google Scholar]
- 14.Cohen SI, Linse S, Luheshi LM, Hellstrand E, White DA, Rajah L, Otzen DE, Vendruscolo M, Dobson CM, Knowles TP. Proliferation of amyloid-beta42 aggregates occurs through a secondary nucleation mechanism. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:9758–9763. doi: 10.1073/pnas.1218402110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Goedert M. NEURODEGENERATION. Alzheimer’s and Parkinson’s diseases: The prion concept in relation to assembled Abeta, tau, and alpha-synuclein. Science. 2015;349:1255555. doi: 10.1126/science.1255555. [DOI] [PubMed] [Google Scholar]
- 16.Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416:535–539. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- 17.Lashuel HA, Hartley D, Petre BM, Walz T, Lansbury PT., Jr Neurodegenerative disease: amyloid pores from pathogenic mutations. Nature. 2002;418:291. doi: 10.1038/418291a. [DOI] [PubMed] [Google Scholar]
- 18.Haass C, Selkoe DJ. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid beta-peptide. Nature reviews Molecular cell biology. 2007;8:101–112. doi: 10.1038/nrm2101. [DOI] [PubMed] [Google Scholar]
- 19.Hong DP, Han S, Fink AL, Uversky VN. Characterization of the non-fibrillar alpha-synuclein oligomers. Protein and peptide letters. 2011;18:230–240. doi: 10.2174/092986611794578332. [DOI] [PubMed] [Google Scholar]
- 20.Bieschke J. Natural compounds may open new routes to treatment of amyloid diseases. Neurotherapeutics. 2013;10:429–439. doi: 10.1007/s13311-013-0192-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bieschke J, Russ J, Friedrich RP, Ehrnhoefer DE, Wobst H, Neugebauer K, Wanker EE. EGCG remodels mature alpha-synuclein and amyloid-beta fibrils and reduces cellular toxicity. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:7710–7715. doi: 10.1073/pnas.0910723107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wobst HJ, Sharma A, Diamond MI, Wanker EE, Bieschke J. The green tea polyphenol (−)-epigallocatechin gallate prevents the aggregation of tau protein into toxic oligomers at substoichiometric ratios. FEBS letters. 2015;589:77–83. doi: 10.1016/j.febslet.2014.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lendel C, Bertoncini CW, Cremades N, Waudby CA, Vendruscolo M, Dobson CM, Schenk D, Christodoulou J, Toth G. On the mechanism of nonspecific inhibitors of protein aggregation: dissecting the interactions of alpha-synuclein with Congo red and lacmoid. Biochemistry. 2009;48:8322–8334. doi: 10.1021/bi901285x. [DOI] [PubMed] [Google Scholar]
- 24.Gao M, Estel K, Seeliger J, Friedrich RP, Dogan S, Wanker EE, Winter R, Ebbinghaus S. Modulation of human IAPP fibrillation: cosolutes, crowders and chaperones. Phys Chem Chem Phys. 2015;17:8338–8348. doi: 10.1039/c4cp04682j. [DOI] [PubMed] [Google Scholar]
- 25.Abelein A, Bolognesi B, Dobson CM, Graslund A, Lendel C. Hydrophobicity and conformational change as mechanistic determinants for nonspecific modulators of amyloid beta self-assembly. Biochemistry. 2012;51:126–137. doi: 10.1021/bi201745g. [DOI] [PubMed] [Google Scholar]
- 26.Bieschke J, Herbst M, Wiglenda T, Friedrich RP, Boeddrich A, Schiele F, Kleckers D, Lopez del Amo JM, Gruning BA, Wang Q, Schmidt MR, Lurz R, Anwyl R, Schnoegl S, Fandrich M, Frank RF, Reif B, Gunther S, Walsh DM, Wanker EE. Small-molecule conversion of toxic oligomers to nontoxic beta-sheet-rich amyloid fibrils. Nat Chem Biol. 2012;8:93–101. doi: 10.1038/nchembio.719. [DOI] [PubMed] [Google Scholar]
- 27.Hoyer W, Antony T, Cherny D, Heim G, Jovin TM, Subramaniam V. Dependence of alpha-synuclein aggregate morphology on solution conditions. Journal of molecular biology. 2002;322:383–393. doi: 10.1016/s0022-2836(02)00775-1. [DOI] [PubMed] [Google Scholar]
- 28.Muth D, Ghazaryan S, Eckerle I, Beckett E, Pohler C, Batzler J, Beisel C, Gogolin S, Fischer M, Henrich KO, Ehemann V, Gillespie P, Schwab M, Westermann F. Transcriptional repression of SKP2 is impaired in MYCN-amplified neuroblastoma. Cancer research. 2010;70:3791–3802. doi: 10.1158/0008-5472.CAN-09-1245. [DOI] [PubMed] [Google Scholar]
- 29.Sulatskaya AI, Maskevich AA, Kuznetsova IM, Uversky VN, Turoverov KK. Fluorescence quantum yield of thioflavin T in rigid isotropic solution and incorporated into the amyloid fibrils. PloS one. 2010;5:e15385. doi: 10.1371/journal.pone.0015385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sivanesam K, Byrne A, Bisaglia M, Bubacco L, Andersen N. Binding Interactions of Agents That Alter alpha-Synuclein Aggregation. RSC advances. 2015;5:11577–11590. doi: 10.1039/C5RA00325C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jiang M, Porat-Shliom Y, Pei Z, Cheng Y, Xiang L, Sommers K, Li Q, Gillardon F, Hengerer B, Berlinicke C, Smith WW, Zack DJ, Poirier MA, Ross CA, Duan W. Baicalein reduces E46K alpha-synuclein aggregation in vitro and protects cells against E46K alpha-synuclein toxicity in cell models of familiar Parkinsonism. Journal of neurochemistry. 2010;114:419–429. doi: 10.1111/j.1471-4159.2010.06752.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wanker EE, Scherzinger E, Heiser V, Sittler A, Eickhoff H, Lehrach H. Membrane filter assay for detection of amyloid-like polyglutamine-containing protein aggregates. Methods in enzymology. 1999;309:375–386. doi: 10.1016/s0076-6879(99)09026-6. [DOI] [PubMed] [Google Scholar]
- 33.Neumann M, Kahle PJ, Giasson BI, Ozmen L, Borroni E, Spooren W, Muller V, Odoy S, Fujiwara H, Hasegawa M, Iwatsubo T, Trojanowski JQ, Kretzschmar HA, Haass C. Misfolded proteinase K-resistant hyperphosphorylated alpha-synuclein in aged transgenic mice with locomotor deterioration and in human alpha-synucleinopathies. The Journal of clinical investigation. 2002;110:1429–1439. doi: 10.1172/JCI15777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kostka M, Hogen T, Danzer KM, Levin J, Habeck M, Wirth A, Wagner R, Glabe CG, Finger S, Heinzelmann U, Garidel P, Duan W, Ross CA, Kretzschmar H, Giese A. Single particle characterization of iron-induced pore-forming alpha-synuclein oligomers. The Journal of biological chemistry. 2008;283:10992–11003. doi: 10.1074/jbc.M709634200. [DOI] [PubMed] [Google Scholar]
- 35.Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW, Glabe CG. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science. 2003;300:486–489. doi: 10.1126/science.1079469. [DOI] [PubMed] [Google Scholar]
- 36.Hsu LJ, Sagara Y, Arroyo A, Rockenstein E, Sisk A, Mallory M, Wong J, Takenouchi T, Hashimoto M, Masliah E. alpha-synuclein promotes mitochondrial deficit and oxidative stress. Am J Pathol. 2000;157:401–410. doi: 10.1016/s0002-9440(10)64553-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Saborio GP, Permanne B, Soto C. Sensitive detection of pathological prion protein by cyclic amplification of protein misfolding. Nature. 2001;4111:810–813. doi: 10.1038/35081095. [DOI] [PubMed] [Google Scholar]
- 38.Cheng IH, Scearce-Levie K, Legleiter J, Palop JJ, Gerstein H, Bien-Ly N, Puolivali J, Lesne S, Ashe KH, Muchowski PJ, Mucke L. Accelerating amyloid-beta fibrillization reduces oligomer levels and functional deficits in Alzheimer disease mouse models. The Journal of biological chemistry. 2007;282:23818–23828. doi: 10.1074/jbc.M701078200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.