

Abstract
Cas9 is an RNA-guided DNA cleavage enzyme being actively developed for genome editing and gene regulation. To be cleaved by Cas9, a double stranded DNA, or the protospacer, must be complementary to the guide region, typically 20-nucleotides in length, of the Cas9-bound guide RNA, and adjacent to a short Cas9-specific element called Protospacer Adjacent Motif (PAM). Understanding the correct juxtaposition of the protospacer- and PAM-interaction with Cas9 will enable development of versatile and safe Cas9-based technology. We report identification and biochemical characterization of Cas9 from Acidothermus cellulolyticus (AceCas9). AceCas9 depends on a 5′-NNNCC-3′ PAM and is more efficient in cleaving negative supercoils than relaxed DNA. Kinetic as well as in vivo activity assays reveal that AceCas9 achieves optimal activity when combined with a guide RNA containing a 24-nucleotide complementarity region. The cytosine-specific, DNA topology-sensitive, and extended guide-dependent properties of AceCas9 may be explored for specific genome editing applications.
Keywords: Cas9, PAM, RNA-guided DNA cleavage, CRISPR
Graphical Abstract
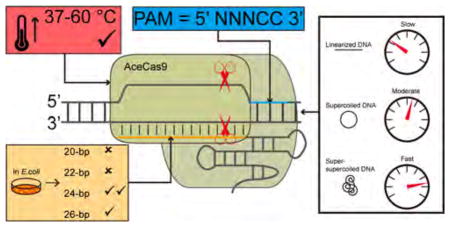
Cas9 is an RNA-guided DNA cleavage enzyme utilized by some bacteria to defend against invading species, such as bacteriophages, through the Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)-mediated pathway. A functional Cas9 is comprised of a polypeptide and two short RNAs, a CRISPR RNA (crRNA) and a trans-activating crRNA (tracrRNA).1 In previously characterized Cas9s, the crRNA and tracrRNA could be covalently linked into a chimeric RNA called single guide RNA (sgRNA) with no loss of function.2–5 The ability of Cas9 to target any double stranded DNA (dsDNA) for cleavage using customizable guides has enabled development of multiple Cas9-based biotechnologies.6–10 Introducing cells with Cas9 and a sgRNA results in site-specific double-stranded breaks (DSB) that trigger either nonhomologous end joining (NHEJ) or homology directed repair (HDR).11–13 These DNA repair processes can be utilized to either disrupt protein-encoding genes (gene knockout and gene knockdown) or to substitute a gene of choice using a desired DNA template through homologous recombination (gene editing).13–15 In addition, catalytically inactive Cas9 (dCas9) has been applied in transcriptional regulation and other applications through its programmable binding to specific DNA elements.16–21 The power of the CRISPR-Cas9 effector nuclease as a genome-editing tool has been demonstrated in multiple organisms and cell types.12–14,22–25 However, currently available Cas9s, both natural and engineered, still lack diversity in substrate selection. Furthermore, these Cas9s continue to show off-target cleavage activity.23,26–28 Broadening the choice of Cas9s while improving Cas9 specificity is a major goal in the field of Cas9-based biotechnology.
Cas9 requires two elements within the DNA target for cleavage: (1) an 18–24 base pairs (protospacer) complementary to the guide region of the sgRNA and (2) a Cas9-specific 3–8 nucleotides (nts) adjacent to the protospacer known as PAM (Protospacer Adjacent Motif).5,29–34 Structural and fluorescence studies suggest that specific recognition of the short PAM nucleotides by Cas9 leads to unwinding of the protospacer and formation of a structural motif called the R-loop in which the sgRNA base pairs with the targeting strand of the protospacer. Subsequently, the HNH- and RuvC-like nuclease domains of Cas9 reposition to the targeting and the nontargeting strand of the dsDNA, respectively, to generate a DSB.33,35,36 In most CRISPR-Cas9 systems, the absence of a PAM consensus from self DNA, such as the CRISPR repeats, prevents self-destruction. Notably, though both the complementarity to sgRNA and the PAM of a DNA substrate are required for its cleavage by Cas9, low-level activities have been observed for DNA containing deviations from these elements (the protospacer, in particular), leading to cleavage of unintended targets.26,27,37 The processes of Cas9 assembly, dsDNA unwinding, and interaction with protospacers and PAM all contribute to the control of substrate cleavage38,39 that, if understood, may be harnessed for improvement of Cas9-based biotechnology.
Most of the well characterized Cas9s require PAM sequences comprised of guanine nucleotide(s) located 2–3 bases from the 3′ end of the nontargeting strand of the proto-spacer2,4,5,30,34,40,41 (Table S1). Despite low sequence homology of the PAM interacting domain (PID) among the known Cas9s, crystal structures of Streptococcus pyogenes (Spy) Cas9 (type II-A, 5′-NGG-3′),42 Staphylococcus aureus (Sa) Cas9 (type II-A, 5′-NNGRRT-3′)43 and Francisella novicida (Fn) Cas9 (type II-B, 5′-NGG-3′)44 bound to their DNA substrates revealed a similar mechanism of PAM recognition that involves guanine base-specific contacts primarily by arginine residues.42–44 Mutation of these arginine residues reduced Cas9 activity or altered the PAM sequence recognized by specific Cas9 enzymes.42–45 Given the wide sequence variation in PID, it is possible that some Cas9 have evolved to either position arginine differently or rely on other polar residues in accommodating different PAM than currently known. Identification and characterization of additional PAM-Cas9 interactions will contribute to the knowledge of Cas9 activity control.
Previous studies show that recognition of the protospacer by Cas9 plays an important role in substrate cleavage. The guide region of the sgRNA (spacer), typically 20-nt in length, serves as the key component in substrate recognition of the Cas9 enzyme by base pairing with the targeting strand of the protospacer.33,46–48 Both the length and sequence of the spacer impact the enzyme efficiency as well as specificity. Cas9 uses its large nucleic acid recognition (REC) domain to nearly enwrap the DNA-spacer heteroduplex. However, the REC-mediated heteroduplex recognition has moderate fidelity, as disruption of base pairing between the spacer of sgRNA and the targeting strand at both the PAM-distal and PAM-adjacent ends is tolerated by Cas9 both in vitro and in vivo.2,26–28 Tolerance of mismatched DNA-spacer by Cas9 is a source of unwanted off-target cleavage in cells. Efforts to increase specificity of the widely used SpyCas9 were made by engineering regions in both sgRNA and Cas9, by reducing its expression level, or by employing double sgRNA with Cas9 nickase (reviewed in Nuñez et al., 2016).49 Furthermore, reducing the heteroduplex base pairs from 20 to 17–18-nts was found to enhance SpyCas9’s ability to discriminate mismatches.37 Recently, mutations of positively charged residues within the hetero-duplex binding REC50 and nuclease domains51 were shown to also increase the specificity of SpyCas9. Note that these engineering strategies led to reduced Cas9 nonspecific binding affinity for the substrate, raising the possibility that reduction of enzyme activity could in general benefit specific cleavage by Cas9.
We sought to identify naturally occurring Cas9s that recognize different PAM sequences than are currently known and that possess desired biochemical properties for high specificity. We characterized Acidothermus cellulolyticus (Ace-Cas9) (type II-C) that, unlike previously characterized Cas9s, recognizes a 5′-NNNCC-3′ PAM and has optimal activity with a sgRNA of 24-nt spacer both in vitro and in vivo. Kinetic studies indicate that AceCas9 has a preference for negatively supercoiled DNA is significantly more specific for relaxed than for supercoiled protospacers.
RESULTS
Purification and Characterization of AceCas9 Ribonu-cleoprotein Particle
Following early bioinformatics methods in identifying Cas9 orthologs,52 we identified Cas9 from a thermophile, A. cellulolyticus 11B (optimal growth temperature 55 °C).53 We were able to express and purify the recombinant AceCas9 to homogeneity by using three chromatography steps (see Materials and Methods) (Figure 1). To identify its associated tracrRNA, we used the available A. cellulolyticus consensus direct repeat sequence on CRISPRdb54 to identify a match to 16 bases (with one mismatch) in the noncoding region downstream of the cas9 gene following putative transcription start sites (Figure 1A) The resulting tracrRNA is 83-nt in length and predicted by mfold55 to contain two stem loops (a 6-bp stem loop I and a 10-bp stem loop II) (Figure 1A). On the basis of previous Cas9 studies that sgRNA is fully functional,2,12,13 we generated a sgRNA by linking the tracrRNA and crRNA through a 5′-GAAA-3′ tetraloop (Figure 1A). The AceCas9:sgRNA ribonucleoprotein particle (RNP) was able to shift a Hexachloro-fluorescein phosphoramidite (HEX)- or 6-carboxyfluorescein (6-FAM)-labeled DNA oligo complementary to the 20-nt spacer (Figure 1B).
Figure 1.

Identification and purification of the Type II-C Cas9 from A. cellulolyticus (AceCas9). (A) (Top) CRISPR locus in A. cellulolyticus 11B and the sequences of CRISPR RNA (crRNA) and trans-activating crRNA (tracrRNA). Black rectangles (R), indicate consensus direct repeat sequence and colored diamonds (S) indicate spacer sequences. The red arrow indicates the putative transcription direction for tracrRNA. (Bottom) Schematic representation of R-loop formation between the dsDNA target and the constructed single guide RNA (sgRNA). The guide region of sgRNA (or spacer) is shown in red and base pairs with the targeting DNA strand of the protospacer DNA. Four guide sequences ranging from 20-nts to 26-nts used in this study are listed. (B) (Top) Domain organization for AceCas9. Conserved catalytic residues are indicated in red. RuvC refers to the RuvC nuclease domain (dark green), ABH refers to the Arginine-rich Bridge Helix (light green), REC refers to the heteroduplex Recognition lobe (gray), HNH refers to the HNH nuclease domain (dark blue), PID refers to the PAM-interacting domains and is composed of 3 segments: β-hairpin (β-H, orange), Topo-homology domain (TOPO, blue), and the C-terminal domain (CTD, light blue). (Bottom left) SDS-PAGE analysis of the wild-type (WT) and H591A mutant AceCas9 following Nickel affinity chromatography (Ni-NTA), ion-exchange (IEC) and size-exclusion chromatography (S200). (Bottom right) Binding of AceCas9:sgRNA or H591A:sgRNA complex to single stranded targeting DNA labeled by HEX. Black line indicates the merged lanes from two different gels.
AceCas9 Has a Double Cytosine PAM
Sequence search did not yield matches in various databases using Nucleotide BLAST56 and CRISPRTarget57 to any of the 23 spacer sequences in A. cellulolyticus CRISPR locus. Therefore, the PAM sequences for AceCas9 could not be identified by comparing the flanking regions of the potential protospacers. We designed and constructed a DNA library bearing random sequences in the PAM region as the substrates for AceCas9 cleavage (Figure S1). The PAM region contains seven randomized base pairs (5′-NNNNNNN-3′ with a 25% probability for each base pair at any given position) and is located downstream of a 20-nt protospacer inserted into the pUC19 plasmid (Figure S1A). The potential AceCas9 cleavage site is located ~500 base pairs from that of BamHI, which allowed us to generate DNA fragments from AceCas9:sgRNA/ BamHI cleavage (Figure S1B) for cloning and Sanger sequencing (see Materials and Methods). We successfully cloned and sequenced 34 ~500-bp fragments, among which 18 contained the PAM-protospacer insertion (Figure S1C). Strikingly, the 18 clones that contained the insertion all had cytosine at positions 4 and 5 downstream of the protospacer on the nontargeting DNA strand, suggesting that 5′-NNNCC-3′ is a functional PAM for AceCas9 (Figure S1C).
To observe the PAM-activated DNA cleavage activity of AceCas9, we synthesized a series of dsDNA oligos containing the 5′-NNNCC-3′ PAM and its variants. The dsDNA was either labeled with HEX on the targeting strand or with 6-FAM on the nontargeting strand (Figure 2 and Figure S2). In a typical cleavage experiment, we used 1.2–20 molar excess of nonlabeled DNA to ensure formation of the dsDNA substrate for AceCas9 (Figure 2A). Wild-type AceCas9 cleaved dsDNA oligos efficiently only when the correct PAM was present (Figure 2B, Figure S2A and S2B), while HNH-inactivated AceCas9 (H591A AceCas9) can only cleaved the nontargeting DNA strand of the same oligo (Figure S2A and S2B). Compensatory mutations of nucleotide sequences at PAM positions 4 or 5 greatly reduced cleavage activity by AceCas9 (Figure 2B and Figure S2A). Intriguingly, the cleavage defect on these PAM mutants could be rescued if the protospacer contained a bulge at position −1 (Figure 2B) or when the two guanine nucleotides base paired with dC4 and dC5 were unchanged (Figure 2B and Figure S2A). These results suggest a critical dependence of AceCas9 on the C-G base pair at positions 4 and 5 but, more importantly, the guanine bases paired with dC4 and dC5 in DNA cleavage.
Figure 2.

Experimental confirmation of the 5′-NNNCC-3′ PAM for AceCas9 on oligo DNA substrates. (A) Cleavage results of double stranded DNA oligos (dsDNA) containing the 5′-NNNCC-3′ PAM. (Left) examination of HEX-labeled targeting DNA strand and its annealed dsDNA product in a nondenaturing gel. (Right) cleavage of dsDNA labeled with fluorophore 6-FAM on its nontargeting strand (red) and cleavage of dsDNA labeled with fluorophore HEX on its targeting strand (green). (B) Cleavage results of dsDNA oligos containing variations in PAM. (Top) Sequences and names of the wild-type and mutant oligo DNA. Mutations are indicated in red. (Bottom) Results of oligo DNA cleavage visualized on a 15% denaturing polyacrylamide gel. The two wild-type (WT) dsDNA are distinguished by the position of the fluorescence labels. The dsDNA with nontargeting strand labeled by 6-FAM is denoted as WT*, whereas that with targeting strand labeled by HEX is denoted as WT. Other dsDNA variants are indicated by nucleotides in red.
We subsequently tested AceCas9 activity on plasmid substrates. Consistently, AceCas9 also cleaved the plasmid substrate containing the 5′-NNNCC-3′ PAM, and not those with dC4/5 to G or dC4/5 to T mutations (Figure 3A). The cleavage sites on the plasmid were mapped by DNA sequencing to the phosphodiester bond between the third and the fourth nucleotides upstream of the PAM (Figure 3B). The cleavage specifically required magnesium, but tolerated manganese and generated a nick with copper (Figure S3A). Furthermore, due to its thermostability, AceCas9 is active at temperatures 25–60 °C (Figure S3B).
Figure 3.

Experimental confirmation of the 5′-NNNCC-3′ PAM for AceCas9 on plasmid DNA substrates. (A) (Top left) Schematic representation of the pUC19 plasmid substrate for AceCas9 with protospacer and the PAM regions colored orange and blue, respectively. Names of the plasmids corresponding to the sequence variations (red) in the PAM region are shown to the right. (Top right) Sequences and names of a series of PAM mutations. Sequence alterations are colored red. (Bottom) Cleavage results by Ace-Cas9:sgRNA at 37 and 50 °C for the wild-type and various mutant plasmids visualized by 0.5% agarose gel. (B) The predicted site of cleavage (red arrows) by AceCas9 by sequencing the cleavage product on both strands of the wild-type plasmid. The sequences for the protospacer (yellow) and for the PAM (blue) are typed below the sequence traces. Asterisk denotes extra adenosine resulted from DNA sequencing. Related to Figure S1.
DNA Unwinding Limits AceCas9 Cleavage Efficiency
To learn the cleavage efficiency of AceCas9, we subjected both oligo and plasmid substrates to single-turnover kinetics analysis (Figure 4 and Table 1). Under this condition, the measured cleavage rate depends on steps following AceCas9 binding to the substrates that may include unwinding the DNA, conformational change in the catalytic domains and the cleavage step itself.58 Although AceCas9 cleaved ssDNA the fastest (kcleavage = 0.65 ± 0.10 min−1), the fraction of cleavage plateaued at ~50% (Figure 4A), suggesting the possible presence of nonproductive substrates. Consistently, dsDNA oligo (kcleavage = 0.104 ± 0.005 min−1) was cleaved to ~70% (Figure 4A) and the circular plasmid substrate (kcleavage = 0.26 ± 0.02 min−1) was cleaved to more than 90% (Figure 4C). To understand which functional step in AceCas9 cleavage limits its cleavage efficiency, we measured rate constant for the dsDNA containing a bulge at −1 position (Figure 4B) and found that its cleavage was nearly 1.7-fold (kcleavage = 0.179 ± 0.007 min−1) of the fully base-paired dsDNA, suggesting that DNA unwinding is likely the rate-limiting step (Figure 4A).
Figure 4.

Kinetic analysis of DNA cleavage by AceCas9. Single turnover cleavage assays with AceCas9:sgRNA were performed at 50 °C in triplicate. The fraction of cleaved were quantified and fitted to a single exponential function to extract pseudo-first-order rate constant, kcleave, for each reaction. Related to Figure S4. (A) Comparison of kinetic rate constants between a ssDNA substrate (kcleave ± SD = 0.65 ± 0.10 min−1) and a dsDNA substrate (kcleave ± SD = 0.104 ± 0.003 min−1). (B) Comparison of kinetic rate constants for the dsDNA containing one nucleotide bulge with PAM (kcleave ± SD = 0.179 ± 0.007 min−1) and without the PAM (kcleave ± SD = 0.098 ± 0.009 min−1). (C) Comparison of rate constants for BamHI-linearized plasmid DNA (kcleave ± SD = 0.10 ± 0.01 min−1), supercoiled plasmid DNA (kcleave ± SD = 0.26 ± 0.02 min−1), and gyrase-treated supercoiled DNA (kcleave ± SD = 0.30 ± 0.02 min−1). (D) Comparison of rate constants for supercoiled plasmid DNA when using sgRNA with 20-nt spacer (kcleave ± SD = 0.26 ± 0.02 min−1) and that with 24-nt spacer (kcleave ± SD = 1.04 ± 0.03 min−1).
Table 1.
Rate Constant of AceCas9 Cleaving Various Substrates
| 20-nt spacer sgRNA
| |||
|---|---|---|---|
| substrate | rate constant (min−1) | S.D.a (min−1) | goodness of fit (R square) |
| dsDNA + PAM | 0.104 | 0.005 | 0.9852 |
| T(−1)C bulge dsDNA + PAM | 0.179 | 0.007 | 0.9904 |
| T(−1)C bulge dsDNA − PAM | 0.098 | 0.009 | 0.9349 |
| ssDNA | 0.65 | 0.10 | 0.8348 |
| Supercoiled DNA plasmid (37 °C) | 0.093 | 0.003 | 0.9950 |
| Supercoiled DNA plasmid (50 °C) | 0.26 | 0.02 | 0.9761 |
| Gyrase-treated supercoiled DNA plasmid (50 °C) | 0.30 | 0.02 | 0.9649 |
| BamHI-treated linearized DNA plasmid (50 °C) | 0.10 | 0.01 | 0.9484 |
| 24-nt spacer sgRNA
| |||
|---|---|---|---|
| substrate | rate constant (min−1) | S.D.a (min−1) | goodness of fit (R square) |
| Supercoiled DNA plasmid (37 °C) | 0.28 | 0.01 | 0.9538 |
| Supercoiled DNA plasmid (50 °C) | 1.04 | 0.03 | 0.9684 |
| BamHI-treated linearized DNA plasmid (50 °C) | 0.53 | 0.05 | 0.9798 |
SD denotes standard deviation of experiments in triplicate.
We then tested if enhanced complementarity between the spacer and protospacer can elevate the rate of DNA unwinding, thus that of AceCas9 cleavage by use of a sgRNA containing a 24-nt spacer (Figure 1A). The single-turnover rate of cleavage indeed increased by four times (kcleavage = 1.04 ± 0.03 min−1) of that with the sgRNA containing the 20-nt spacer (kcleavage = 0.26 ± 0.02 min−1) (Figure 4D). This result shows that increased spacer-protospacer pairing, from 20-nt to 24-nt, improves the overall DNA cleavage efficiency. Note that the pseudo-first-order rate constant of AceCas9 with the 24-nt spacer sgRNA is comparable to that of SpyCas9 on plasmid DNA substrates measured at 37 °C (kcleavage = 0.89 ± 0.12 min−1).2
AceCas9 Cleaves Negatively Supercoiled DNA Efficiently
Comparison of the cleavage rate for dsDNA oligo to that for the plasmid substrate showed that AceCas9 prefers plasmid DNA. To examine if it is simply the length of DNA substrate, which could facilitate one-dimensional diffusion, that accelerated the rate of cleavage for the plasmid DNA, we carried out cleavage kinetics on the same plasmid pretreated with BamHI by use of the 20-nt spacer sgRNA (Figure 4C). Surprisingly, the rate constant of linearized 3-kb DNA (kcleavage = 0.10 ± 0.01 min−1) was very similar to that of dsDNA oligo (kcleavage = 0.104 ± 0.005 min−1), thereby ruling out one-dimensional diffusion as the possible cause for the observed difference between oligo and plasmid DNA. Instead, the negative supercoil in the circular DNA must act as a favorable factor for AceCas9 function. To further explore the dependence of AceCas9 on DNA topology, we increased the number of topoisomers of the pUC19 substrate by treating it with E. coli gyrase before performing single turnover kinetics with AceCas9. The gyrase-treated plasmid (Figure 4C) indeed resulted in faster rate of cleavage (kcleavage = 0.30 ± 0.02 min−1) than the untreated plasmid (Figure 4C and Table 1). A similar rate difference when cleaving the supercoiled and linearized DNA plasmid by AceCas9 with the 24-nt guide sgRNA was observed, suggesting DNA topology plays the same role in substrate binding when the guide region is extended (Table 1).
AceCas9 Has Low Off-Target Cleavage Activity for Relaxed DNA
To investigate the specificity of AceCas9 for the protospacer, we generated single or double mutations at positions −1, −4, −8, −19, and −20 of the protospacer and subjected these plasmids to AceCas9 cleavage with the 20-nt spacer sgRNA (Figure 5A). We quantified the fraction of cleaved at the end of 1 h incubation of AceCas9:sgRNA with each plasmid (Figure 5B) and compared this value to that of the wild-type plasmid (Figure 5C). At 50 °C where AceCas9 activity is optimal, all plasmids mutants but position −4, pPSG(−4)A, were cleaved nearly to completion (Figures 5B and 5C). The pPSG(−4)A was cleaved poorly, reflecting a critical role of position −4 and possibly its surrounding in substrate recognition by AceCas9. The similar pattern of cleavage activity was observed when the reaction temperature was lowered to 37 °C (Figure 5C), despite an overall reduction in AceCas9 activity (Figure 5B). Significantly, when these mutant plasmids were prelinearized with BamHI, in which case, the wild-type plasmid had reduced cleavage (Figure 5B), all mutations, except that at −20, resulted in detectable cleavage after reacting with molar excessive AceCas9 at both 50 and 37 °C (Figure 5B). The shift in cleavage specificity is the most pronounced for position −1 where DNA unwinding occurs (Figures 5B and 5C), suggesting that the energy stored in supercoiling helps to drive the initial steps of R-loop formation.
Figure 5.

Plasmid protospacer specificity of AceCas9. 3 nM of plasmid substrates were incubated with 500 nM of AceCas9:sgRNA for 1 h and the cleavage products were separated and visualized on a 1.0% agarose gel. Fraction of cleavage was calculated based on integrated band intensities. (A) Sequences and names of a series of protospacer mutants in the pUC19 substrate for AceCas9:sgRNA. Mutated base pairs are shown in bold letters. (B) Comparison of DNA cleavage by AceCas9:sgRNA between the wild-type and mutants for the BamHI-prelinearized and supercoiled plasmids and for reaction temperatures of 50 and 37 °C. (C) Quantified cleavage activities from reactions shown in (B). For quantification, the intensity of the 3kb linearized DNA plasmid and that of the 2.5 kb large cleavage product bands were obtained by integration and the fraction of cleavage was calculated by taking the ratio of the two. Similarly, the intensity of the supercoiled DNA plasmid and that of the linearized cleavage product were used to determine fraction of cleavage for the supercoiled substrates. The fraction of cleavage for the wild-type plasmid was normalized to 100%.
AceCas9 Cleaves DNA Efficiently in Escherichia coli
To confirm the DNA cleavage activity by AceCas9 under in vivo condition, we performed a bacteria-based cell survival assay (Figure 6).45,59 We transformed the plasmid coexpressing AceCas9 (or SpyCas9) and its sgRNA into E. coli competent cells harboring a target (selection) plasmid that expresses the toxic ccdB gene under the control of BAD promoter (inducible with arabinose). Successful cleavage of the ccdB plasmid by Cas9 would render the growth of E. coli cells when induced by arabinose (Figure 6A and 6B). The efficiency of DNA cleavage in E. coli can thus be measured as the percentage of colonies with arabinose versus those without arabinose. As expected, the cells transformed with SpyCas9 and its sgRNA targeting ccdB have about 96% survival (Figure 6C). The cells transformed with AceCas9 and its 24-nt spacer sgRNA also reached nearly 75% survived. Strikingly, cells transformed with AceCas9 and the 20-nt or a 22-nt spacer sgRNA had less than 0.01% survival (Figure 6C), reflecting the lower rate of cleavage for the 20-nt spacer sgRNA in vitro. Further extension of the spacer length to 26-nt resulted in about 14% cell survival but did not improve cleavage efficiency (Figure 6C).
Figure 6.

In vivo activity of AceCas9. (A) A bacterial-based positive-selection assay was used to determine the activity of AceCas9 with sgRNA that contains either the 20-nt (top tow) or the 24-nt (bottom row) spacer with 1:1000 cell dilutions. The AceCas9:sgRNA plasmids were transformed into competent cells with the ccdB plasmid that has a unique protospacer sequence and either a functional PAM (PAMWT, 5′-NNNCC-3′) or a double mutant of PAM (PAMC(4,5)T, 5′-NNNTT-3′). Cells were selected in the presence of antibiotics chloramphenicol (C+) for AceCas9:sgRNA coexpression plasmid, or with both chloramphenicol and arabinose (C+, Arabinose) to induce the expression of toxic ccdB gene. The right most plate illustrates low survival cells without dilution for the 20-nt spacer targeting wild-type plasmid, 24-nt spacer targeting PAMC(4,5)T, or SpyCas9 targeting a noncognate protospacer (ΔPS). (B) The cell survival assay of AceCas9 with 24-nt spacer sgRNA in comparison with SpyCas9 targeting the same plasmid. (C) Percent of Escherichia coli survival in the presence of arabinose were determined for SpyCas9 with a 20-nt spacer (96% ± 25%), AceCas9 with a 20-nt spacer (<0.01%), AceCas9 with a 22-nt spacer (<0.01%), AceCas9 with a 24-nt spacer (75% ± 6%), and AceCas9 with 26-nt spacer (13.6% ± 0.4%) sgRNA that target the ccdB plasmid with PAMWT. Percent survival were also determined for AceCas9 with 24-nt spacer (<0.001%) and 26-nt spacer (<0.001%) sgRNA target the ccdB plasmid with PAMC(4,5)T. Each experiment was performed in triplicate.
We tested in E. coli if the 5′-NNNCC-3′ PAM is specifically recognized by AceCas9 by carrying out the survival assay with the ccdB plasmid containing a 5′-NNNTT-3′ PAM in place of a 5′-NNNCC-3′ PAM. We observed nearly no survival colonies (<0.001% survival) under the conditions we used for the wild-type target (Figure 6A and 6C), confirming that 5′-NNNCC-3′ is a functional PAM for AceCas9 in E. coli cells.
DISCUSSION
Currently known Cas9 substrates require a guanine-containing adjacent motif to be cleaved. Those for AceCas9, in contrast, are associated with a cytosine adjacent motif. Mutational studies indicated that AceCas9 is highly specific for cytosine at position 4 or 5 under a variety of conditions. This specificity for PAM is unprecedented, as other Cas9s tolerated mutation to similar nucleotides (i.e., G to A for SpyCas9), and may thus be explored for applications requiring targeting cytosine adjacent DNA. AceCas9 differs from another type II-C Cas9, NmCas9,60 and resembles type II-A SpyCas92 in that it does not require a fully base paired PAM region, suggesting that the interaction between the PAM and the PID of Cas9 are, in general, diverse and potentially species-rather than subtype-specific.
Our kinetic analysis revealed a unique dependence of AceCas9 on negatively supercoiled DNA. The fact that dsDNA was cleaved more slowly than ssDNA suggests that DNA unwinding is the rate-limiting step for AceCas9. Consistently, the rate of cleavage increased when the first protospacer base pair was disrupted. Ma et al. also reported similar observations on another type II-C Cas9, CdiCas9.61 AceCas9 is able to lower the energetic barrier of DNA unwinding by harnessing the energy stored in DNA topology, as its rate of cleavage for a supercoiled plasmid DNA is about 2.6-fold that for a linearized plasmid DNA or a dsDNA oligo. Unwinding circular DNA introduces positive supercoiling, and thus strains in the DNA double helix structure. Negative supercoiling allows faster duplex rotation resulting from the superhelical torque, thereby alleviating the structural strain during unwinding and providing a favorable energy for AceCas9 to reach its final conformation for catalysis.
The bacteria-based positive-selection assay results further support our hypothesis that elongated spacer improves the catalytic activity of AceCas9 due to its role in unwinding dsDNA. The measured DNA cleavage efficiency in E. coli agrees with the rate of cleavage obtained in vitro. The 4-fold rate increase of the 24-nt spacer from that of the 20-spacer guided AceCas9 is translated into a more than 12 500-fold increase in DNA cleavage efficiency in cells. The narrow dependence of AceCas9 on the 24-nt spacer sgRNA provides an opportunity to improve the off-target activities of Cas9-derived tools.
The dependence of AceCas9 activity on DNA topology has not been detected for other characterized Cas9s. However, this effect was observed for Cascade, a type I-E CRISPR-Cas system62 and for SpyCas9 in cleaving an artificial plasmid containing consecutive targets.63 For a typical single target plasmid, cleavage of plasmids by SpyCas9 has a compatible if not slightly lower rate constant2,48 than that of oligo dsDNA.61,64 This suggests a mechanistic distinction between the more sophisticated type II-A Cas9 and the simpler type II-C Cas9. The type II-A Cas9 may be able to keep the effect of DNA unwinding localized, while the type II-C allows it to spread along the DNA molecule in order to take advantage of energy stored in DNA topology.
Significantly, our biochemical data showed that AceCas9 exhibits high specificity for the protospacer on relaxed DNA. Whereas several mutations of the protospacer within a supercoiled plasmid were largely tolerated by AceCas9, those within its prelinearized counterpart were not. The disparate activity on different DNA topologies is especially striking for the first base pair of the protospacer, where DNA unwinding and target-sgRNA pairing are initiated. This suggests that negative supercoiling in the plasmid specifically enhances these steps. The absence of favorable superhelicity in prelinearized DNA renders AceCas9 unable to overcome mismatches between the DNA and the sgRNA in the PAM proximal region. This property of AceCas9 makes it applicable to chromatin domain specific editing.
AceCas9 is functional in vitro at 25 °C through 60 °C with activity being optimal at elevated temperatures. A low temperature environment may also be explored for increased specificity of AceCas9. Our in vitro cleavage experiments indeed provided some evidence that AceCas9 is less tolerant to mutations within the protospacer at 37 °C than at 50 °C. It remains to be tested if temperature alone or in combination with DNA topology may be used to control AceCas9 specificity in vivo.
EXPERIMENTAL PROCEDURES
Cloning of AceCas9 and AceCas9 Mutant
A. cellulolyticus cas9 gene fused at the 3′ end with the sequence encoding a hexahistidine tag was PCR-amplified from the genomic DNA of Acidothermus cellulolyticus strain 11B (ATCC 43068D-5) and cloned into pET28b (Novagen) to yield pETAceCas9. The H591A mutant of A. cellulolyticus cas9 gene was generated by site-directed mutagenesis kit Q5 (New England Biolabs) using appropriate primers purchased from IDT DNA and Eurofins Genomics. The clones encoding Cas9 and its H591A mutant were verified by Sanger sequencing (Eurofins Genomics).
Protein Expression and Purification
E. coli Rosetta (DE3) cells transformed with pETAceCas9 were grown in LB medium with appropriate antibiotics until OD600 reached 0.4–0.6 before induction by 0.3 mM Isopropyl β-D-1-thiogalactopyranoside (IPTG) followed by continued incubation at 16.5 °C for 18 h. Cells were harvested by centrifugation at 4250 rpm for 30 min, resuspended in a lysis buffer (50 mM Tris pH 8.0, 1 M NaCl, 1 mM imidazole pH 8.0, 10% glycerol, 14.3 mM β-mercaptoethanol [βME] and 0.3 mM phenylmethylsulfonyl fluoride [PMSF]), and lysed by sonication. The lysate was clarified by centrifugation at 22 000 rpm at 4 °C for 30 min and the cleared supernatant was passed through a Ni-NTA agarose column equilibrated with the lysis buffer. The column was washed by 15–20 column volumes of the lysis buffer, followed by another 15–20 column volumes of a wash buffer (30 mM Tris pH 8.0, 250 mM NaCl, 5 mM imidazole pH 8.0, 5% glycerol). AceCas9 was eluted by an elution buffer that contained 30 mM Tris pH 8.0, 150 mM NaCl, 350 mM imidazole pH 8.0, 5% glycerol. To remove nucleic acids that associated with AceCas9, the Ni-NTA elutant was loaded into an HiTrap Heparin HP (GE Healthcare) column equilibrated with an ion exchange buffer (30 mM Tris pH 7.5, 100 mM KCl, 5% glycerol, 14.3 mM βME), and eluted with a linear salt gradient. Heparin column elutant was concentrated and then loaded on to a size-exclusion column HiLoad 26/60 Superdex 200 (GE Healthcare) equilibrated with a gel filtration buffer (30 mM HEPES pH 7.5, 200 mM KCl, 14.3 mM βME). The peak fractions were concentrated and buffer exchanged to a storage buffer (30 mM HEPES pH 7.5, 200 mM KCl, 10 mM (Tris(2-carboxyethyl)phosphine) [TCEP]) using Amicon Ultra-15 Centrifugal Filter Units (Millipore), prior to being flash frozen in liquid nitrogen and stored at −80 °C. Expression and purification of AceCas9 mutant followed the same procedure.
In Vitro Transcription and Purification of sgRNA
400 μM of an oligo with T7 promoter sequence was annealed in equimolar with a DNA oligo that encodes the reverse complement sequence of sgRNA fused with that of T7 promoter (Table S2) in a 10× PCR buffer at 94 °C for 4 min followed by slow cooling. The sgRNA was synthesized by in vitro transcription by adding 1 μM of annealed dsDNA template into 50 mM Tris-HCl pH 8.0, 40 mM MgCl2, 10 mM dithiothreitol (DTT), 2 mM spermidine, 5 mM ATP pH 8, 5 mM UTP pH 8, 6.2 mM CTP pH 8, 6.2 mM GTP pH 8, 0.1% Triton X-100, and 0.15 mg/mL T7 RNA polymerase. The transcription reaction was incubated at 37 °C water bath overnight, and quenched by addition of EDTA to a final concentration at 65 mM. The sgRNA was extracted by phenol:chloroform extraction, and purified by either ethanol precipitation or size exclusion chromatography (HiLoad Superdex 75 16/60, GE Healthcare) at 4 °C in 20 mM Tris pH 7.4, 150 mM KCl. The concentration was determined by 260 nm absorption using an ND-1000 spectrophotometer (NanoDrop). When necessary, sgRNA was concentrated by Vivaspin 20 (Sartorius) before being stored at −80 °C.
PAM Library Construction
A predetermined protospacer sequence followed by 7-bp degenerate sequences (Table S2) was cloned into the high copy number plasmid pUC19 vector ~500-bp upstream of the BamHI cleavage site. The plasmid library was transformed into chemical competent E. coli DH5α cells by standard heat shock transformation procedure and incubated in LB agar plates containing 75 μg/mL ampicillin. More than 500 single colonies were picked and pooled for growth in fresh LB medium in the presence of ampicillin prior to DNA extraction. Sequences of the extracted DNA library plasmids were verified by Sanger sequencing (Eurofins Genomics) using the standard M13 forward and M13 reverse primers to ensure the presence of the 7-bp degenerate region. To cleave the DNA library, 300 ng of plasmid DNA library was incubated with preannealed 1 μM AceCas9 and 5 μM sgRNA in a 20 μL volume containing a cleavage buffer (20 mM Tris pH 7.5, 150 mM KCl, 2 mM DTT, 10 mM MgCl2, 5% Glycerol) at 37 °C for 1.5 h. 44 units (2.2 μL of 20000 U/mL) of BamHI (or SbfI) was then added to the reaction at 37 °C and incubated for an additional 1.5 h. The reaction was stopped by the addition of EDTA to a final concentration of 50 mM and 6X NEB Purple Loading dye before being resolved on a 1% 0.8× TAE agarose gel. The cleavage product that contained the PAM sequence was gel extracted by E.Z.N.A. gel extraction kit (Omega Biotek), end repaired and ligated with index primers using NEB Ultra II Library Prep Kit and NEBNext Multiplex Oligos for Illumina (New England Biolabs). The treated fragments were amplified by PCR and subsequently cloned into pCR4Blunt-TOPO vector (Thermo Fisher Scientific). 34 independent clones were subjected to Sanger sequencing using the standard T3 and T7 primers. Sequences with the inserted PCR product, which contains parts of the protospacer sequence and the 7-bp degenerate region, were identified based on pUC19 backbone sequence downstream of library sequence. The region composed of 7-bp of the PAM sequence and 15-bp neighboring sequences in both directions was extracted and aligned using WebLogo.65
Oligonucleotide DNA Cleavage Assay
Single stranded oligonucleotide DNAs or those containing HEX- or 6-FAM-labels were purchased from Eurofins Genomics or IDT (Table S2). The labeled DNA strand was annealed with a nonlabel strand in a 1:20 (HEX) or 1:1.2 (6-FAM) molar ratio at 75 °C for 5 min, followed by slow cooling to room temperature to generate dsDNA. For cleavage, 500 nM AceCas9 was preincubated with equimolar of sgRNA in a cleavage buffer for 15 min at 37 °C and cooled to room temperature. The ssDNA or dsDNA substrates were then added to the tube to a final concentration 3 nM to initiate the reaction. The reaction tube was incubated for 1 h at 50 °C, before being stopped by the addition of 2× formamide gel loading buffer (90% formamide, 50 mM EDTA pH 8.0). The samples were heated at ~90 °C for 2 min prior being resolved on a 15% urea denaturing polyacrylamide (19:1) TBE gel. The gel was scanned and visualized by Typhoon Trio (GE Healthcare) using appropriate filters/excitation wavelengths.
Plasmid DNA Cleavage Assay
The same protospacer as that in the dsDNA and PAM were introduced into pUC19 to construct the plasmid substrate for AceCas9 (Table S2). For cleavage reaction, 100 ng (~6 nM) of plasmid DNA was incubated with preannealed 500 nM AceCas9:sgRNA in a cleavage buffer at 50 °C for 1 h. The reaction was quenched by the addition of 5× gel loading buffer (25 mM Tris pH 7.5, 250 mM EDTA pH 8, 1% SDS, 0.05% w/v bromophenol blue, 30% glycerol) and resolved on a 0.5% 1× TBE agarose gel with EZ-Vision In-Gel solution (Amresco). To test metal requirement, 10 mM MgCl2 in regular cleavage buffer was replaced by the following divalent salts: MnCl2, CuCl2, NiCl2, CdCl2, CaCl2, CoCl2, ZnCl2, BaCl2. The gels were imaged by ChemiDoc XRS System (Bio-Rad) and analyzed by ImageQuant (GE Health-care).
Generation of Negatively Supercoiled DNA by DNA Gyrase
To create a more negatively supercoiled DNA for single-turnover kinetic assay, DNA gyrase from E. coli (NEB) were used. Briefiy, 1 μL gyrase was added to 1 μg of plasmid DNA in 30 μL 1× manufacture supplied buffer (NEB) and the mixture was incubation in a 37 °C water bath for 30 min. The gyrase-treated DNA was then purified by ethanol precipitation and resuspeneded in ddH2O. To confirmed its superhelicity, the plasmid is resolved in a 0.5% 1× TBE agarose gel (with final concentration of 10 μg/mL chloroquine from Sigma-Aldrich) and stained with SYBR Gold (Thermo Fisher Scientific) for 30 min before visualized by ChemiDoc XRS System (Bio-Rad).
Single-Turnover Kinetic Assay
For kinetic assays with oligonucleotides, 500 nM AceCas9 sgRNP was preannealed in the presence of a cleavage buffer in a master mix for triplicate reactions at 37 °C for 15 min, then cooled down on ice before adding ice cold, HEX-labeled dsDNA (final concentration at 3 nM). The reaction mixture was aliquoted into 33 prechilled Eppendorf tubes on ice and then quickly transferred to a 50 °C water bath to initiate reactions. Samples were removed in triplicate and placed on ice at each time point. To ensure reactions were quenched thoroughly, prechilled 2× formamide gel loading buffer was added to each tube immediately afterward. At the end of the reaction, samples were mixed and heated at ~90 °C for 2 min before being loaded to a 15% urea denaturing polyacrylamide gel. The gel was scanned by Typhoon Trio (GE Healthcare) using green laser at 532 nm with 520 nm band-pass filter. Intensities of the bands corresponding to uncleaved and cleaved DNA were integrated using the ImageQuant TL software (GE Healthcare) and were fitted to a simple exponential progress curve using Prism 6 (GraphPad Software) to extract kcleavage.
Kinetic assays with DNA plasmids followed the same procedure with the following modifications. ~3.6 μg (~6 nM) of plasmid DNA was added to the preannealed AceCas9:sgRNA, mixed, and aliquoted into prechilled Eppendorf tubes on ice before initiating reactions. Reactions were quenched by the addition of 5× gel loading buffer immediately after placing tubes on ice. Samples were loaded directly into a 0.5% 1× TBE agarose gel with EZ-Vision In-Gel solution (Amresco) and resolved at 6.5 mA/cm for ~1 h. The gel was scanned by ChemiDoc XRS System (Bio-Rad), analyzed by ImageQuant (GE Healthcare) and were fitted to a simple exponential progress curve using Prism 6 (GraphPad Software) to extract kcleavage.
Bacteria-Based Positive Selection Assay
The bacteria-based positive selection assay was performed as previously described45 with minor modifications. Briefly, a positive selection plasmid encoding ccdB toxin (gift from Dr. David Edgell) was isolated and mutagenized to contain a target site with PAM and variants by Q5 SDM kit (NEB). Each selection plasmid variant was transformed into E. coli BW25141(λDE3) cells (gift from D. Edgell) and made competent for electroporation. A coexpression plasmid for AceCas9 and its sgRNA was constructed by inserting AceCas9 and sgRNA encoding genes into the multiple cloning sites of the pCACYduet vector. ~100 ng of AceCas9:sgRNA coexpression plasmid (or its variants) were transformed into the ccdB-harboring cells by electroporation. Cells were recovered in 500 μL of SOB media and shake at ~250 rpm for 5 min at 37 °C, before being diluted 5-fold with SOB media and further incubated for 60 min. Harvested culture were further diluted ~1000-fold (or no dilution for controls) and plated on various agar plates. Experiment with SpyCas9 was performed as above by use of a modified BPK764 plasmid (Addgene plasmid #65767 deposited by Keith Joung).45
Supplementary Material
Acknowledgments
We thank A. Cocozaki, J. Pennington, G. Yu for discussion; B.P. Kleinstiver and B. Miller for advice on in vivo studies; D. Edgell for providing cells and plasmid for in vivo studies; J. Terrell for assistance in preparing DNA plasmids; B. Washburn and C. Pye of the Molecular Cloning Facility at FSU for the cloning experiments. This work was supported by American Heart Association predoctoral fellowship 15PRE25330004 to T.K.M.T., and by NIH grant R01 GM099604 to H.L.
Footnotes
Author Contributions
T.K.M.T., T.H.H. and H.L. designed the experiments. T.K.M.T. performed the identification and preparation of Cas9, sgRNA, and plasmid substrates, identification of PAM, DNA oligo experiments and in vivo bacterial assay. T.H.H. and E.C.D. performed plasmid cleavage assays. T.K.M.T., T.H.H., and H.L. analyzed the data. T.K.M.T. and H.L. wrote the manuscript, and all authors edited the manuscript.
Notes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acssynbio.7b00050.
Data for PAM determination, gels for oligo cleavage assays, plasmid cleavage assays, single-turnover kinetic assays, reported PAM sequence, and oligos DNA that were used (PDF)
References
- 1.Deltcheva E, Chylinski K, Sharma CM, Gonzales K, Chao Y, Pirzada ZA, Eckert MR, Vogel J, Charpentier E. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature. 2011;471:602–7. doi: 10.1038/nature09886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–21. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hou Z, Zhang Y, Propson NE, Howden SE, Chu L, Sontheimer EJ, Thomson JA. Efficient genome engineering in human pluripotent stem cells using Cas9 from Neisseria meningitidis. Proc Natl Acad Sci U S A. 2013;110:15644–9. doi: 10.1073/pnas.1313587110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Esvelt KM, Mali P, Braff JL, Moosburner M, Yaung SJ, Church GM. Orthogonal Cas9 proteins for RNA-guided gene regulation and editing. Nat Methods. 2013;10:1116–21. doi: 10.1038/nmeth.2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ran FA, Cong L, Yan WX, Scott DA, Gootenberg JS, Kriz AJ, Zetsche B, Shalem O, Wu X, Makarova KS, Koonin EV, Sharp PA, Zhang F. In vivo genome editing using Staphylococcus aureus Cas9. Nature. 2015;520:186–91. doi: 10.1038/nature14299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Beisel CL, Gomaa AA, Barrangou R. A CRISPR design for next-generation antimicrobials. Genome Biol. 2014;15:516. doi: 10.1186/s13059-014-0516-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Choi PS, Meyerson M. Targeted genomic rearrangements using CRISPR/Cas technology. Nat Commun. 2014;5:3728. doi: 10.1038/ncomms4728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shalem O, Sanjana NE, Zhang F. High-throughput functional genomics using CRISPR-Cas9. Nat Rev Genet. 2015;16:299–311. doi: 10.1038/nrg3899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Torres R, Martin MC, Garcia A, Cigudosa JC, Ramirez JC, Rodriguez-Perales S. Engineering human tumour-associated chromosomal translocations with the RNA-guided CRISPR-Cas9 system. Nat Commun. 2014;5:3964. doi: 10.1038/ncomms4964. [DOI] [PubMed] [Google Scholar]
- 10.Wang Z, Pan Q, Gendron P, Zhu W, Guo F, Cen S, Wainberg MA, Liang C. CRISPR/Cas9-Derived Mutations Both Inhibit HIV-1 Replication and Accelerate Viral Escape. Cell Rep. 2016;15:481–489. doi: 10.1016/j.celrep.2016.03.042. [DOI] [PubMed] [Google Scholar]
- 11.Cho SW, Kim S, Kim Y, Kweon J, Kim HS, Bae S, Kim JS. Analysis of off-target effects of CRISPR/Cas-derived RNA-guided endonucleases and nickases. Genome Res. 2014;24:132–141. doi: 10.1101/gr.162339.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini La, Zhang F. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–23. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM. RNA-guided human genome engineering via Cas9. Science. 2013;339:823–6. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Charpentier E, Marraffini LA. Harnessing CRISPR-Cas9 immunity for genetic engineering. Curr Opin Microbiol. 2014;19:114–9. doi: 10.1016/j.mib.2014.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hsu PD, Lander ES, Zhang F. Development and Applications of CRISPR-Cas9 for Genome Engineering. Cell. 2014;157:1262–1278. doi: 10.1016/j.cell.2014.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zalatan JG, Lee ME, Almeida R, Gilbert LA, Whitehead EH, LaRussa M, Tsai JC, Weissman JS, Dueber JE, Qi LS, Lim WA. Engineering complex synthetic transcriptional programs with CRISPR RNA scaffolds. Cell. 2015;160:339–350. doi: 10.1016/j.cell.2014.11.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Komor AC, Kim YB, Packer MS, Zuris JA, Liu DR. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature. 2016;533:420–424. doi: 10.1038/nature17946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gilbert LA, Larson MH, Morsut L, Liu Z, Brar GA, Torres SE, Stern-Ginossar N, Brandman O, Whitehead EH, Doudna JA, Lim WA, Weissman JS, Qi LS. CRISPR-Mediated Modular RNA-Guided Regulation of Transcription in Eukaryotes. Cell. 2013;154:442–451. doi: 10.1016/j.cell.2013.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Qi LS, Larson MH, Gilbert LA, Doudna JA, Weissman JS, Arkin AP, Lim WA. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell. 2013;152:1173–83. doi: 10.1016/j.cell.2013.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guilinger JP, Thompson DB, Liu DR. Fusion of catalytically inactive Cas9 to FokI nuclease improves the specificity of genome modification. Nat Biotechnol. 2014;32:577–582. doi: 10.1038/nbt.2909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tsai SQ, Wyvekens N, Khayter C, Foden JA, Thapar V, Reyon D, Goodwin MJ, Aryee MJ, Joung JK. Dimeric CRISPR RNA-guided FokI nucleases for highly specific genome editing. Nat Biotechnol. 2014;32:569–576. doi: 10.1038/nbt.2908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Terns RM, Terns MP. CRISPR-based technologies: prokaryotic defense weapons repurposed. Trends Genet. 2014;30:111–8. doi: 10.1016/j.tig.2014.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Malina A, Mills JR, Cencic R, Yan Y, Fraser J, Schippers LM, Paquet M, Dostie J, Pelletier J. Repurposing CRISPR/Cas9 for in situ functional assays. Genes Dev. 2013;27:2602–2614. doi: 10.1101/gad.227132.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jiang W, Bikard D, Cox D, Zhang F, Marraffini LA. RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nat Biotechnol. 2013;31:233–9. doi: 10.1038/nbt.2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bikard D, Euler CW, Jiang W, Nussenzweig PM, Goldberg GW, Duportet X, Fischetti Va, Marraffini La. Exploiting CRISPR-Cas nucleases to produce sequence-specific antimicrobials. Nat Biotechnol. 2014;32:1146–50. doi: 10.1038/nbt.3043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hsu PD, Scott DA, Weinstein JA, Ran FA, Konermann S, Agarwala V, Li Y, Fine EJ, Wu X, Shalem O, Cradick TJ, Marraffini LA, Bao G, Zhang F. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol. 2013;31:827–32. doi: 10.1038/nbt.2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pattanayak V, Lin S, Guilinger JP, Ma E, Doudna JA, Liu DR. High-throughput profiling of off-target DNA cleavage reveals RNA-programmed Cas9 nuclease specificity. Nat Biotechnol. 2013;31:839–43. doi: 10.1038/nbt.2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fu Y, Foden JA, Khayter C, Maeder ML, Reyon D, Joung JK, Sander JD. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat Biotechnol. 2013;31:822–6. doi: 10.1038/nbt.2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Deveau H, Barrangou R, Garneau JE, Labonté J, Fremaux C, Boyaval P, Romero Da, Horvath P, Moineau S. Phage response to CRISPR-encoded resistance in Streptococcus thermophilus. J Bacteriol. 2008;190:1390–400. doi: 10.1128/JB.01412-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fonfara I, LeRhun A, Chylinski K, Makarova KS, Lécrivain AL, Bzdrenga J, Koonin EV, Charpentier E. Phylogeny of Cas9 determines functional exchangeability of dual-RNA and Cas9 among orthologous type II CRISPR-Cas systems. Nucleic Acids Res. 2014;42:2577–90. doi: 10.1093/nar/gkt1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Horvath P, Romero DA, Coûté-Monvoisin AC, Richards M, Deveau H, Moineau S, Boyaval P, Fremaux C, Barrangou R. Diversity, activity, and evolution of CRISPR loci in Streptococcus thermophilus. J Bacteriol. 2008;190:1401–1412. doi: 10.1128/JB.01415-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tsui TK, Li H. Structure Principles of CRISPR-Cas Surveillance and Effector Complexes. Annu Rev Biophys. 2015;44:229–255. doi: 10.1146/annurev-biophys-060414-033939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang Y, Heidrich N, Ampattu BJ, Gunderson CW, Seifert HS, Schoen C, Vogel J, Sontheimer EJ, Fonfara I, LeRhun A, Chylinski K, Makarova KS, Lécrivain AL, Bzdrenga J, Koonin EV, Charpentier E. Processing-independent CRISPR RNAs limit natural transformation in Neisseria meningitidis. Mol Cell. 2013;50:488–503. doi: 10.1016/j.molcel.2013.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jiang F, Zhou K, Ma L, Gressel S, Doudna JA. A Cas9-guide RNA complex preorganized for target DNA recognition. Science (Washington, DC, U S) 2015;348:1477–1481. doi: 10.1126/science.aab1452. [DOI] [PubMed] [Google Scholar]
- 36.Jackson RN, Wiedenheft B. A Conserved Structural Chassis for Mounting Versatile CRISPR RNA-Guided Immune Responses. Mol Cell. 2015;58:722. doi: 10.1016/j.molcel.2015.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fu Y, Sander JD, Reyon D, Cascio VM, Joung JK. Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. Nat Biotechnol. 2014;32:279–84. doi: 10.1038/nbt.2808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Makarova KS, Haft DH, Barrangou R, Brouns SJJ, Charpentier E, Horvath P, Moineau S, Mojica FJM, Wolf YI, Yakunin AF, van der Oost J, Koonin EV. Evolution and classification of the CRISPR-Cas systems. Nat Rev Microbiol. 2011;9:467–77. doi: 10.1038/nrmicro2577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Makarova KS, Wolf YI, Alkhnbashi OS, Costa F, Shah SA, Saunders SJ, Barrangou R, Brouns SJJ, Charpentier E, Haft DH, Horvath P, Moineau S, Mojica FJM, Terns RM, Terns MP, White MF, Yakunin AF, Garrett RA, van derOost J, Backofen R, Koonin EV. An updated evolutionary classification of CRISPR-Cas systems. Nat Rev Microbiol. 2015;13:722–736. doi: 10.1038/nrmicro3569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gasiunas G, Barrangou R, Horvath P, Siksnys V. Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc Natl Acad Sci U S A. 2012;109:E2579–86. doi: 10.1073/pnas.1208507109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lopez-Sanchez MJ, Sauvage E, Da Cunha V, Clermont D, Ratsima Hariniaina E, Gonzalez-Zorn B, Poyart C, Rosinski-Chupin I, Glaser P. The highly dynamic CRISPR1 system of Streptococcus agalactiae controls the diversity of its mobilome. Mol Microbiol. 2012;85:1057–71. doi: 10.1111/j.1365-2958.2012.08172.x. [DOI] [PubMed] [Google Scholar]
- 42.Anders C, Niewoehner O, Duerst A, Jinek M. Structural basis of PAM-dependent target DNA recognition by the Cas9 endonuclease. Nature. 2014;513:569–73. doi: 10.1038/nature13579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nishimasu H, Cong L, Yan WX, Ran FA, Zetsche B, Li Y, Kurabayashi A, Ishitani R, Zhang F, Nureki O. Crystal Structure of Staphylococcus aureus Cas9. Cell. 2015;162:1113–1126. doi: 10.1016/j.cell.2015.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hirano H, Gootenberg JS, Horii T, Abudayyeh OO, Kimura M, Hsu PD, Nakane T, Ishitani R, Hatada I, Zhang F, Nishimasu H, Nureki O. Structure and Engineering of Francisella novicida Cas9. Cell. 2016;164:950–61. doi: 10.1016/j.cell.2016.01.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kleinstiver BP, Prew MS, Tsai SQ, Topkar VV, Nguyen NT, Zheng Z, Gonzales AP, Li Z, Peterson RT, Yeh JR, Aryee MJ, Joung JK. Engineered CRISPR-Cas9 nucleases with altered PAM specificities. Nature. 2015;523:481–485. doi: 10.1038/nature14592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jiang F, Doudna JA. The structural biology of CRISPR-Cas systems. Curr Opin Struct Biol. 2015;30:100–11. doi: 10.1016/j.sbi.2015.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nishimasu H, Ran FA, Hsu PD, Konermann S, Shehata SI, Dohmae N, Ishitani R, Zhang F, Nureki O. Crystal structure of Cas9 in complex with guide RNA and target DNA. Cell. 2014;156:935–49. doi: 10.1016/j.cell.2014.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sternberg SH, Redding S, Jinek M, Greene EC, Doudna Ja. DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature. 2014;507:62–7. doi: 10.1038/nature13011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nuñez JK, Harrington LB, Doudna JA. Chemical and Biophysical Modulation of Cas9 for Tunable Genome Engineering. ACS Chem Biol. 2016;11:681–8. doi: 10.1021/acschembio.5b01019. [DOI] [PubMed] [Google Scholar]
- 50.Kleinstiver BP, Pattanayak V, Prew MS, Tsai SQ, Nguyen NT, Zheng Z, Joung JK. High-fidelity CRISPR-Cas9 nucleases with no detectable genome-wide off-target effects. Nature. 2016;529:490–5. doi: 10.1038/nature16526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Slaymaker IM, Gao L, Zetsche B, Scott DA, Yan WX, Zhang F. Rationally engineered Cas9 nucleases with improved specificity. Science (Washington, DC, U S) 2016;351:84–88. doi: 10.1126/science.aad5227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chylinski K, LeRhun A, Charpentier E. The tracrRNA and Cas9 families of type II CRISPR-Cas immunity systems. RNA Biol. 2013;10:726–37. doi: 10.4161/rna.24321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mohagheghi A, Grohmann K, Himmel M, Leighton L, Updegraff DM. Isolation and Characterization of Acid-othermus cellulolyticus gen. nov., sp nov., a New Genus of Thermophilic, Acidophilic, Cellulolytic Bacteria. Int J Syst Bacteriol. 1986;36:435–443. [Google Scholar]
- 54.Grissa I, Vergnaud G, Pourcel C. The CRISPRdb database and tools to display CRISPRs and to generate dictionaries of spacers and repeats. BMC Bioinf. 2007;8:172. doi: 10.1186/1471-2105-8-172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–10. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 57.Biswas A, Gagnon JN, Brouns SJJ, Fineran PC, Brown CM. CRISPRTarget: bioinformatic prediction and analysis of crRNA targets. RNA Biol. 2013;10:817–27. doi: 10.4161/rna.24046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Campbell FE, Cassano AG, Anderson VE, Harris ME. Pre-steady-state and stopped-flow fluorescence analysis of Escherichia coli ribonuclease III: insights into mechanism and conformational changes associated with binding and catalysis. J Mol Biol. 2002;317:21–40. doi: 10.1006/jmbi.2002.5413. [DOI] [PubMed] [Google Scholar]
- 59.Chen Z, Zhao H. A highly sensitive selection method for directed evolution of homing endonucleases. Nucleic Acids Res. 2005;33:e154–e154. doi: 10.1093/nar/gni148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhang Y, Rajan R, Seifert HS, Mondragón A, Sontheimer EJ. DNase H Activity of Neisseria meningitidis Cas9. Mol Cell. 2015;60:242–55. doi: 10.1016/j.molcel.2015.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ma E, Harrington LB, O’Connell MR, Zhou K, Doudna JA. Single-Stranded DNA Cleavage by Divergent CRISPR-Cas9 Enzymes. Mol Cell. 2015;60:398–407. doi: 10.1016/j.molcel.2015.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Westra ER, Nilges B, vanErp PBG, van derOost J, Dame RT, Brouns SJJ. Cascade-mediated binding and bending of negatively supercoiled DNA. RNA Biol. 2012;9:1134–8. doi: 10.4161/rna.21410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Farasat I, Salis HM. A Biophysical Model of CRISPR/Cas9 Activity for Rational Design of Genome Editing and Gene Regulation. PLoS Comput Biol. 2016;12:e1004724. doi: 10.1371/journal.pcbi.1004724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wright AV, Sternberg SH, Taylor DW, Staahl BT, Bardales JA, Kornfeld JE, Doudna JA. Rational design of a split-Cas9 enzyme complex. Proc Natl Acad Sci U S A. 2015;112:2984–9. doi: 10.1073/pnas.1501698112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Crooks GE, Hon G, Chandonia JM, Brenner SE. WebLogo: A sequence logo generator. Genome Res. 2004;14:1188–1190. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.