

ABSTRACT
Mesothelioma is an incurable cancer for which effective therapies are required. Aberrant MET expression is prevalent in mesothelioma, although targeting using small molecule-based therapeutics has proven disappointing. Chimeric antigen receptors (CARs) couple the HLA-independent binding of a cell surface target to the delivery of a tailored T-cell activating signal. Here, we evaluated the anti-tumor activity of MET re-targeted CAR T-cells against mesothelioma. Using immunohistochemistry, MET was detected in 67% of malignant pleural mesotheliomas, most frequently of epithelioid or biphasic subtype. The presence of MET did not influence patient survival. Candidate MET-specific CARs were engineered in which a CD28+CD3ζ endodomain was fused to one of 3 peptides derived from the N and K1 domains of hepatocyte growth factor (HGF), which represents the minimum MET binding element present in this growth factor. Using an NIH3T3-based artificial antigen-presenting cell system, we found that all 3 candidate CARs demonstrated high specificity for MET. By contrast, these CARs did not mediate T-cell activation upon engagement of other HGF binding partners, namely CD44v6 or heparan sulfate proteoglycans, including Syndecan-1. NK1-targeted CARs demonstrated broadly similar in vitro potency, indicated by destruction of MET-expressing mesothelioma cell lines, accompanied by cytokine release. In vivo anti-tumor activity was demonstrated following intraperitoneal delivery to mice with an established mesothelioma xenograft. Progressive tumor regression occurred without weight loss or other clinical indicators of toxicity. These data confirm the frequent expression of MET in malignant pleural mesothelioma and demonstrate that this can be targeted effectively and safely using a CAR T-cell immunotherapeutic strategy.
KEYWORDS: chimeric antigen receptor, cancer, Immunotherapy, mesothelioma, MET, NK1
Abbreviations
- AAPC
artificial antigen-presenting cell
- BLI
bioluminescence imaging
- CAR
chimeric antigen receptor
- ffLuc
firefly luciferase
- GAG
glycosaminoglycans
- HGF
hepatocyte growth factor
- HSPG
heparan sulfate proteoglycans
- IL
interleukin
- i.p.
intraperitoneal
- K
Kringle
- MPM
malignant pleural mesothelioma
- NK1
N-terminal and Kringle 1 domains
- RFP
red fluorescence protein
- UT
untransduced
Introduction
Mesothelioma derives from malignant transformation of mesothelial cells that line body cavities. Tumors most commonly originate from the pleural space and are primarily attributed to inhalation of asbestos fibers, followed by a prolonged latency period. Despite advances in surgical and radiation-based approaches and the advent of pemetrexed-based chemotherapy regimens, mesothelioma remains incurable. Furthermore, incidence continues to increase worldwide,1 highlighting the need for improved treatments of this intractable cancer.
Conceptually, immunotherapy represents an attractive therapeutic approach for mesothelioma, given the close inter-relationship between this cancer and the immune system.2 Immune checkpoint blockade has elicited promising, albeit somewhat inconsistent results in early phase clinical trials.3,4 An alternative strategy that achieves striking potency against some hematologic malignancies entails adoptive immunotherapy using chimeric antigen receptor (CAR)-engineered T-cells. Chimeric antigen receptors are fusion molecules that couple the binding of a native cell surface target to the delivery of a bespoke T-cell activating signal.5 Pre-clinical studies using several mesothelioma models have demonstrated efficacy of CAR T-cells directed against mesothelin,6, 7 and fibroblast-activation protein.8,9 Clinical trials of intrapleural10,11 or intravenously administered CAR T-cell immunotherapy12 are currently ongoing in patients with this cancer.
A number of pivotal receptor tyrosine kinases are commonly dysregulated in mesothelioma.13 These also represent attractive targets for CAR-based immunotherapy, given their contribution to disease pathogenesis and the potential for regional delivery of engineered T-cells as a device to maximize therapeutic index.14 In keeping with this, we have recently demonstrated that ErbB re-targeted CAR T-cells achieve significant anti-tumor activity combined with excellent safety in pre-clinical models of malignant pleural mesothelioma (MPM).15 The MET receptor tyrosine kinase is also aberrantly expressed in a large proportion of mesotheliomas16-21 and has been proposed as an attractive therapeutic target in this disease.18,22,23 Disappointingly however, a phase II trial of the selective MET inhibitor tivantinib has recently been terminated (https://clinicaltrials.gov/ct2/show/NCT01861301; accessed March 25th, 2017). Consequently, more potent MET-targeted approaches such as CAR T-cell immunotherapy warrant investigation in this disease.
Hepatocyte growth factor (HGF; also known as scatter factor) is the only known MET ligand. It has a plasminogen-like structure that comprises an N-terminal and 4 Kringle (K) domains, followed by a catalytically inactive serine proteinase module. The shortest naturally occurring splice variant of HGF, named NK1, comprises the N and K1 domains alone.24 Initially described as a MET antagonist,25 conditional agonistic activity has more recently been attributed to NK1.26 This requires binding of a glycosaminoglycan (GAG) such as heparan sulfate27 (or soluble heparin), which promotes NK1 dimerization and also facilitates the formation of a ternary complex with NK1 and MET.28
Given these binding properties, we have evaluated NK1 as a targeting moiety to generate a MET-specific CAR. Three variants of NK1 have been compared in which functional activity or stability have been enhanced by protein engineering. We report here that all 3 CAR candidates mediate MET-dependent T-cell activation and destruction of mesothelioma cells. Moreover, adoptive immunotherapy using MET re-targeted CAR T-cells promotes tumor regression in a mesothelioma xenograft model, without toxicity.
Results
Expression of MET by malignant pleural mesotheliomas
To characterize MET receptor expression, tissue microarrays were prepared from 114 MPMs of epithelioid (59.8%), sarcomatoid (22.0%) or biphasic (18.3%) morphology (histologic subtype available for 82 tumors). There was a small but insignificant trend for improved survival in patients with epithelioid subtype tumors (Fig. 1A). Analysis of a larger series of MPM that contained the subset presented here confirmed, as expected, that epithelioid tumors were associated with improved patient survival.15
Figure 1.
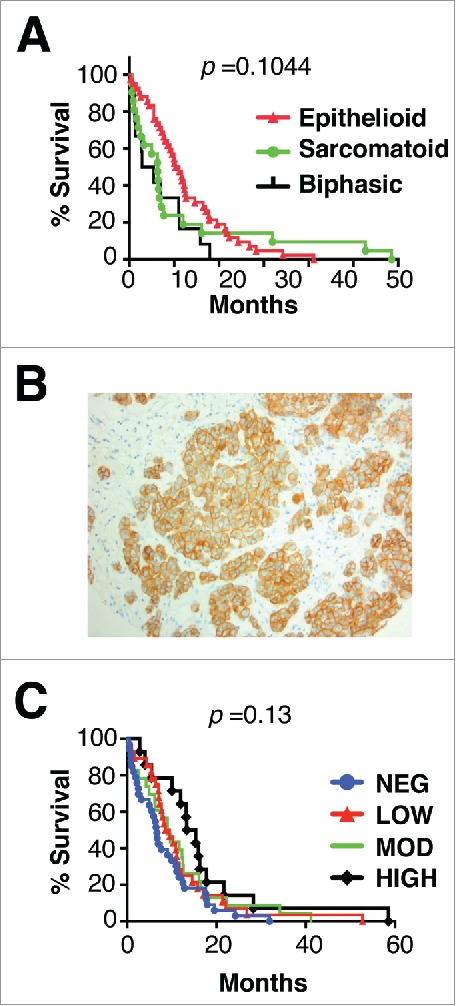
Histology, MET receptor expression and survival analysis of patients with malignant pleural mesothelioma. (A) Kaplan Meier plot for survival of patients with mesothelioma of the specified histologic subtypes. (B) Representative example of MET staining pattern seen in mesothelioma tissue microarray sections. Magnification × 200. (C) Kaplan Meier plots for survival of patients with mesothelioma according to level of expression of MET. p value was generated by Log-rank (Mantel Cox) test.
Expression of MET was detected by immunohistochemistry in 67.6% of MPM, either at low (28.1%), intermediate (24.6%) or high intensity (14.9%). Receptor distribution was cytoplasmic with membranous accentuation (Fig. 1B). MET expression was more frequent in epithelioid (71.4%) and biphasic tumors (66.7%), although it was also commonly found in sarcomatoid tumors (44.4%). All tumors in which MET expression was of high intensity were of epithelioid or biphasic subtypes. Neither the presence, nor the intensity of MET expression was associated with alteration in patient survival (Fig. 1C).
Engineering of candidate MET-specific chimeric antigen receptors
To exploit the frequent expression of MET in MPM, we set out to engineer a CAR that targets this receptor. Three derivatives of the NK1 splice variant of HGF were evaluated for their ability to re-target a second generation (CD28+CD3ζ)29 CAR specifically against MET. In 1K1,28 2 basic amino acids within the low affinity heparan-binding region of the K1 domain have been replaced with acidic residues, yielding a variant with enhanced MET agonistic properties (Fig. 2A). The M2.2 variant was isolated using a mutagenesis approach and contains 8 alterations distributed across the N and K1 domains, one of which has been reverted (D127N) to restore MET agonistic activity (M2.2rev).30 M2.2rev exhibits enhanced stability as does the cysteine-containing M2.2 D127N variant (cM2.2rev; Fig. 2A).30 All 3 were placed downstream of the HGF signal peptide and were coupled to a described previously CD28+CD3ζ second generation CAR framework29 in which a Myc epitope tag had been substituted for the MYPPPY motif within the CD28 ectodomain (Fig. 2B).31 The resultant CARs, named 1–28z, M-28z and cM-28z respectively, were expressed in human T-cells by retroviral-mediated gene transfer. Comparison was made with a pan-ErbB targeted CAR (T-28z, targeted with a promiscuous ErbB ligand named T1E)32 and a negative control in which the targeting moiety consisted of a scrambled 20mer peptide sequence (C-28z).31 Since all CARs contain a Myc epitope tag, cell surface expression was compared by flow cytometry after incubation of transduced cells with the 9e10 antibody (Fig. 2C). Stable expression of candidate MET-specific CARs was also confirmed in human T-cells by western blotting (Fig. 2D). In some experiments, CARs were co-expressed with a chimeric cytokine receptor named 4αβ in which IL-4 receptor α ectodomain has been fused to the transmembrane and endodomain of the IL-2/15 receptor β chain (Fig. S1). Culture of 4αβ-expressing CAR T-cells in IL-4 leads to selective enrichment of transduced cells, with retention of type 1 polarity,31-33 providing a convenient device to enrich for transduced T-cells during in vitro expansion.
Figure 2.
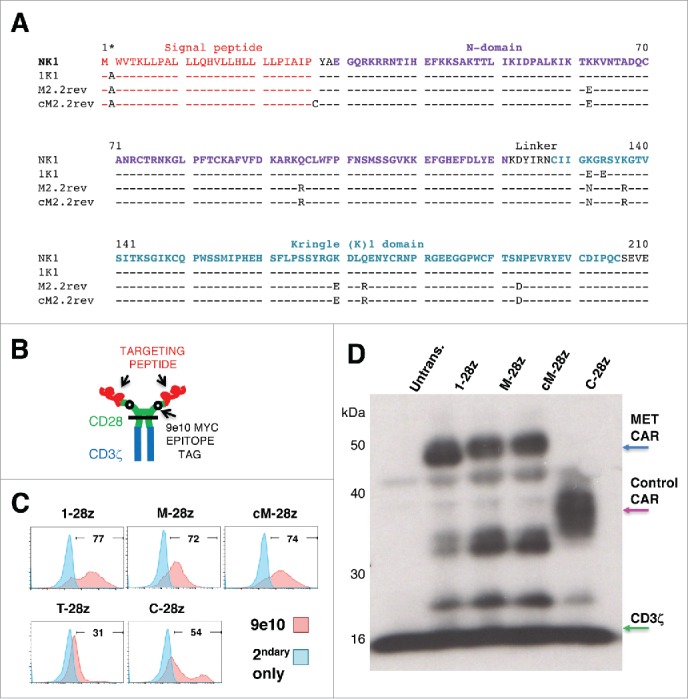
Sequence and expression of candidate MET-specific CARs in human T-cells. (A) Targeting moieties were derived from the NK1 splice variant of HGF and contained the indicated mutations. Both M2.2 and cM2.2 contain a D127N revertant mutation which restores the naturally occurring sequence found in human NK1. (B) Cartoon structure of CARs. The horizontal line indicates the transmembrane domain. (C) Representative examples of cell surface expression of the indicated CARs. Detection was performed by flow cytometry after incubation with the anti-Myc 9e10 antibody. Percentage positivity has been calculated with respect to staining by secondary antibody alone. Data are representative of 10 independent replicates. (D) Expression of CARs in human T-cells was also detected by western blotting, performed under reducing conditions and probed with an anti-CD3ζ antibody. Arrowed CAR bands are of the predicted molecular mass while the endogenous T-cell receptor-associated CD3ζ band (predicted molecular mass 18 kDa) serves as a loading control.
Characterization of specificity of candidate CARs using NIH3T3 artificial antigen presenting cells
In many settings, HGF-dependent activation of MET is dependent on the co-expression of CD44v6, which are non-heparin/heparan sulfated isoforms of CD44 that bind HGF with micromolar affinity.34 Hepatocyte growth factor has also been reported to bind to GAGs and proteoglycans. Notable examples include heparan sulfate proteoglycans (HSPG),35 such as Syndecan-1.36
To investigate the target specificity of the candidate MET CARs described above, artificial antigen-presenting cells (AAPC) were engineered whereby NIH3T3 mouse fibroblasts were modified to express human MET, CD44v6 or both of these molecules (Fig. 3A).37 T-cells were engineered to express candidate MET-specific (1–28z, M-28z, cM-28z), the C-28z control CAR or T-28z, which targets ErbB dimers (absent in NIH3T3 cells) (Fig. 3B). We observed that 1–28z+, M-28z+ and cM-28z+ human T-cells elicited comparable killing of MET-expressing NIH3T3 cells, in a dose- and time-dependent manner (Fig. 3C). Activation of MET re-targeted CAR T-cells was accompanied by secretion of low levels of interferon (IFN)-γ (Fig. 3D). As expected, ErbB-specific (T-28z) and control (C-28z) CAR T-cells were inactive in these assays. Co-expression of CD44v6 was not required for cytotoxicity or cytokine release, although there was a significant increase in IFN-γ release by MET re-targeted CAR T-cells when CD44v6 and MET were co-expressed (Fig. 3D).
Figure 3.
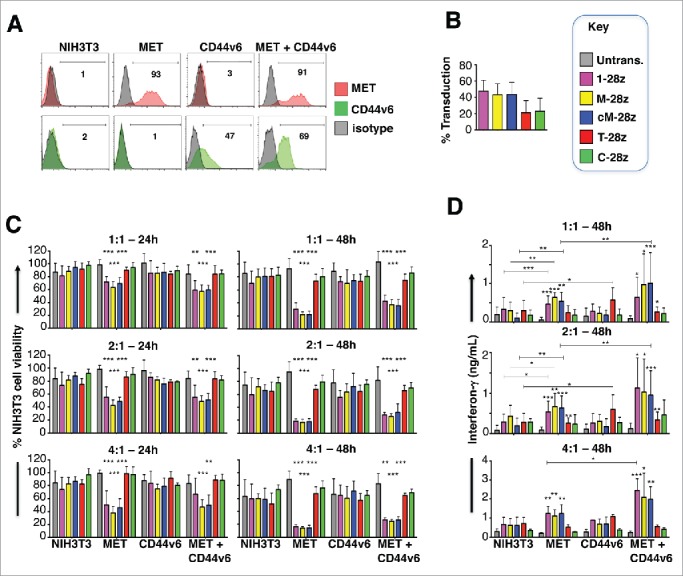
Assessment of specificity of candidate MET targeted CARs using NIH3T3-based artificial antigen presenting cells (AAPC) – I. (A) NIH3T3 fibroblasts were engineered to express human MET, CD44v6 or both molecules. Expression was detected by flow cytometry. Percentage positivity has been calculated with respect to staining by an isotype control antibody. Data are representative of 3 independent experiments. (B) Transduction efficiency of human T-cells engineered to express the indicated candidate MET-specific CARs (1–28z, M-28z, cM-28z), control CARs targeted against ErbB dimers (T-28z) or control CAR targeted with a scrambled 20mer peptide (C-28z). (C) CAR-engineered T-cells were co-cultivated with the indicated NIH3T3-based AAPC for 24 or 48 hours at the specified effector:target ratios. After removal of T-cells by careful washing with PBS, residual viability of NIH3T3 cells was determined by MTT assay, making comparison with a parallel culture of the corresponding NIH3T3 cells alone. (D) Interferon-γ content of supernatants harvested from these co-cultures was quantified by ELISA. Data in (B)-(D) show mean ± SD of 5 independent replicate experiments. *p < 0.05; ** p < 0.01; ***p < 0.001, making comparison with untrans(duced) T-cells or, where indicated, between CAR T-cells under the appropriate horizontal line.
Although unmodified NIH3T3 cells express HSPG (Fig. S2A),38,39 they were not killed (Fig. 3C) nor did they promote significant IFN-γ release (Fig. 3D) by any of the CAR T-cell populations under study. To test potential HSPG reactivity more stringently, we engineered NIH3T3 cells to over-express the human HSPG, Syndecan-1 (Fig. 4A), which is known to bind HGF and is commonly expressed in MPM and derived cell lines (Fig. 5B).40 When co-cultivated with MET re-targeted CAR T-cells (Fig. 4B), destruction of MET-expressing, but not Syndecan-1 expressing, NIH3T3 cells was observed (Fig. 4C). Moreover, release of IFN-γ was only detected when these CAR T-cells were co-cultivated with MET-expressing but not Syndecan-1 expressing NIH3T3 cells (data not shown). As expected, control CAR T-cells were inactive in these assays.
Figure 4.
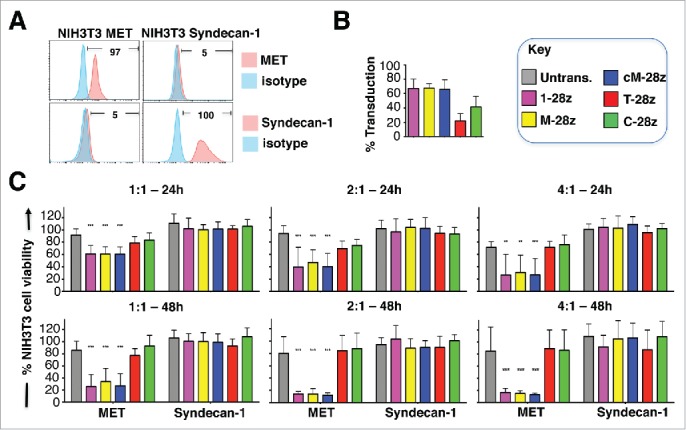
Assessment of specificity of candidate MET targeted CARs using NIH3T3-based artificial antigen presenting cells (AAPC) – II. (A) NIH3T3 fibroblasts were engineered to express human MET or Syndecan-1. Expression was detected by flow cytometry. Percentage positivity has been calculated with respect to staining by an isotype control antibody. Data are representative of 3 independent experiments. (B) Transduction efficiency of human T-cells engineered to express the indicated candidate MET-specific CARs (1–28z, M-28z, cM-28z), control CARs targeted against ErbB dimers (T-28z) or control CAR targeted with a scrambled 20mer peptide (C-28z). (C) CAR-engineered T-cells were co-cultivated with the indicated NIH3T3-based AAPC for 24 or 48 hours at the specified effector:target ratios. After removal of T-cells by careful washing with PBS, residual viability of NIH3T3 cells was determined by MTT assay, making comparison with a parallel culture of the corresponding NIH3T3 cells alone. Data in (B)-(C) show mean ± SD of 3 independent replicate experiments. ***p < 0.001, making comparison with untrans(duced) T-cells.
Figure 5.
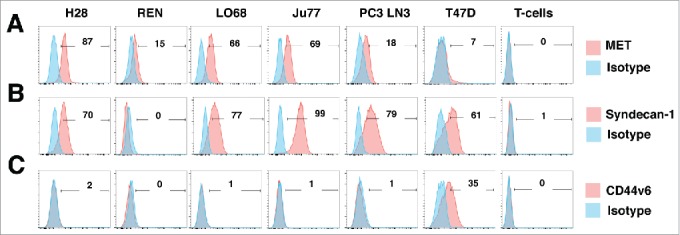
Characterization of human malignant pleural mesothelioma cell lines. The indicated human MPM cell lines were analyzed by flow cytometry for expression of MET (A), Syndecan-1 (B) or CD44v6 (C). PC3 LN3 cells were used as a positive control for MET and Syndecan-1 expression, while T47D cells were used as a positive control for CD44v6 expression. Untransduced T-cells were also analyzed for expression of these molecules. Data are representative of 3 independent experiments, all of which yielded similar findings.
MET re-targeted CAR T-cells exert in vitro anti-tumor activity against mesothelioma
Next, we evaluated anti-tumor activity of MET re-targeted CAR T-cells using a panel of human mesothelioma cell lines in which this receptor was naturally expressed, namely H28, REN, LO68 and Ju77 (Fig. 5A). All tumor cells co-expressed high levels of HSPGs (Fig. S3), generally including Syndecan-1 (Fig. 5B).
CAR T-cells engineered to express 1–28z, M-28z and cM-28z at comparable, albeit lower efficiency (Fig. 6A) mediated equipotent killing of MET-expressing H28 mesothelioma cells (Fig. 6B). While M-28z+ and cM-28z+ T-cells exerted greater cytotoxic activity against REN and LO68 tumor cells (Fig. 6B), this difference was not maintained when T-cells were transduced more efficiently (Fig. 7A–B). Tumor cell destruction was accompanied by release of IFN-γ (Fig. 6C), IL-2 (Fig. S4) and some other cytokines (e.g. IL-13, GM-CSF; Fig. S5) by MET re-targeted CAR T-cells. Although REN, LO68 and Ju77 tumor cells proved more resistant to cytotoxic destruction than H28 cells, all 3 were effectively destroyed by MET re-targeted CAR T-cells over a longer time course (Fig. S6).
Figure 6.
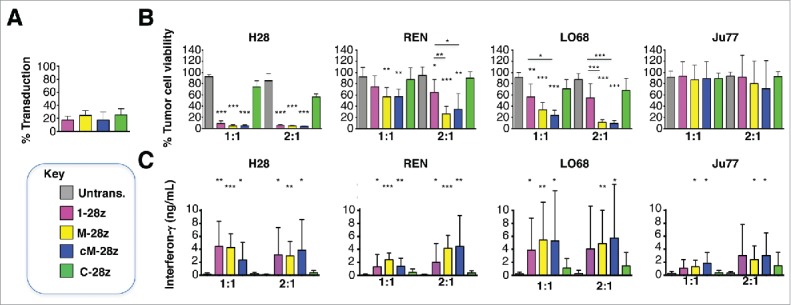
In vitro cytotoxic activity of MET re-targeted CAR T-cells against malignant pleural mesothelioma cell lines. CAR engineered T-cells (transduction efficiency shown in A) were co-cultivated with malignant pleural mesothelioma cells for 48 hours at the indicated effector:target ratios. All CAR T-cells co-expressed the 4αβ chimeric cytokine receptor. (B) After removal of T-cells, residual viable tumor was quantified by MTT assay, making comparison with a parallel culture of tumor cells alone (set to 100%). (C) Supernatants collected from these cultures were analyzed for interferon-γ content. Data show mean ± SD of 3 independent replicate experiments. *p < 0.05; ** p < 0.01; ***p < 0.001, making comparison with untrans(duced) T-cells or, where indicated, between CAR T-cells under the appropriate horizontal line.
Figure 7.
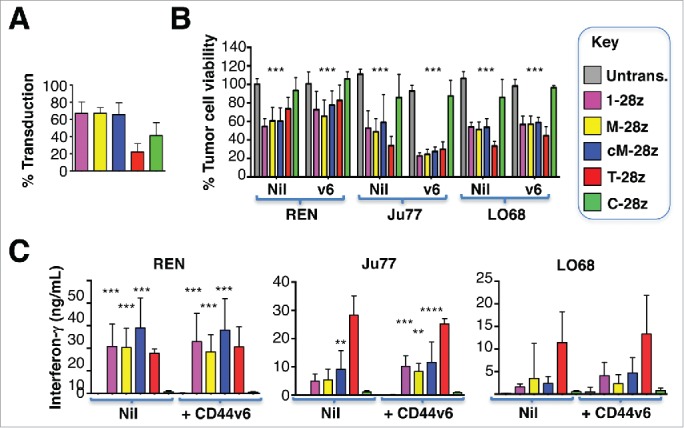
Ectopic overexpression of CD44v6 does not render mesothelioma tumor cells more amenable to destruction by MET re-targeted CAR T-cells. Mesothelioma tumor cell lines were engineered to over-express CD44v6 (v6). (A) Transduction efficiency of human T-cells, engineered to express the indicated CARs. (B) CAR-engineered T-cells were co-cultivated with unmodified or v6-engineered malignant pleural mesothelioma cells at a 1:1 effector:target ratio and for the indicated interval. An MTT assay was performed after 72 hours to quantify residual tumor cell viability. (C) Supernatants were harvested from these co-cultivations after 48 hours and were analyzed for interferon-γ content by ELISA. All data shown are mean ± SD of 3 independent replicate experiments. *p < 0.05; ** p < 0.01; ***p < 0.001 making comparison with untrans(duced) T-cells.
Given the variability in tumor cell susceptibility to MET re-targeted CAR T-cells, we explored if CD44v6 status might be of relevance in this regard. None of the mesothelioma cell lines under study naturally expressed CD44v6 (Fig. 5C). Ectopic overexpression of CD44v6 in these tumor cells did not reproducibly influence their susceptibility to cytotoxic destruction (Fig. 7B), nor did it lead to significant alteration in the release of IFN-γ by activated CAR T-cells (Fig. 7C). In these experiments, T-28z represents a positive control, given the established ability of ErbB re-targeted CAR T-cells to destroy MPM tumor cells.15 Moreover, addition of exogenous unfractionated heparin did not influence cytotoxic activity of MET re-targeted CAR T-cells against several tumor cell lines (data not shown). In summary, these data indicate that MET was both necessary and sufficient for target cell engagement and destruction by CAR T-cells containing an NK1-based targeting moiety.
Activated T-cells also expressed cell surface HSPGs (Fig. S2B), although they lacked expression of Syndecan-1 (Fig. 5B). Furthermore, although MET-expressing T-cell populations have been described,41 we did not detect MET expression on the cell surface of CAR-engineered T-cells (Fig. 5A), in agreement with previous reports.42
MET re-targeted CAR T-cells exert in vivo anti-tumor activity against an established mesothelioma xenograft
To test in vivo function, an intraperitoneal (i.p.) xenograft model was established in NOD SCID γc null mice. REN mesothelioma cells were engineered to co-express tandem dimer tomato red fluorescent protein (RFP) and firefly luciferase (ffLuc) and were flow sorted for RFP expression (Fig. S7A). Inoculation of 5 – 50 × 104 ffLuc/RFP+ REN cells led to rapid tumor engraftment and exponential growth within the peritoneal cavity (Fig. S7B), recapitulating the propensity of mesothelioma to undergo intra-cavitary dissemination.
Given the broadly similar in vitro anti-tumor activity and specificity of all 3 candidate CARs, one (cM-28z) was advanced for in vivo testing. Mice with an established ffLuc/RFP+ REN tumor burden were treated i.p. with cM-28z+ T-cells, making comparison with C-28z+ (Fig. 8A) or untransduced control T-cells (n = 5 each). T-cells were administered as a single low (L; 2.5 × 106 cells) or high dose (H; 10 × 106 cells – total T-cell number) and tumor status was monitored thereafter by serial bioluminescence imaging. Tumor progression in the absence of specific therapeutic intervention was monitored in mice that received PBS (n = 8). Intraperitoneal administration of a low dose of cM-28z+ T-cells caused a transient and minor retardation of tumor progression, followed by sustained disease advancement (Fig. 8B). By contrast, administration of the higher dose of CAR T-cells led to a progressive and significant reduction in tumor burden (Fig. 8C). Bioluminescence images of representative animals using the same scale throughout are shown in Fig. 8D. Treatment was extremely well tolerated without weight loss or other clinical indicators of toxicity (Fig. 8E). Furthermore, histologic analysis of liver, spleen and intestine harvested from treated animals revealed no evidence of pathology (data not shown). Survival analysis was not possible since mice treated with T-cells developed signs compatible with graft versus host disease between days 49–69 (loss of fur, reduced mobility, hunched posture and reduced weight and/or mobility).
Figure 8.
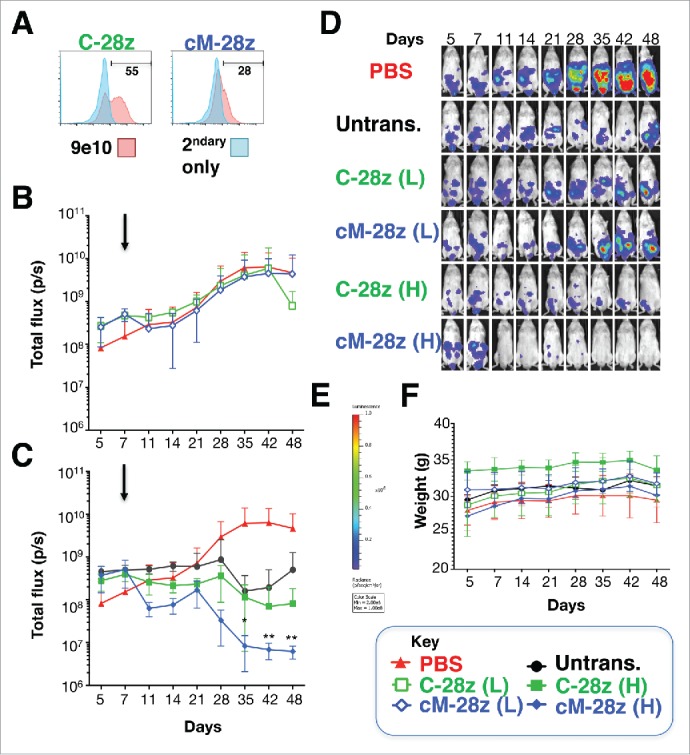
In vivo anti-tumor activity of MET re-targeted CAR T-cells against an established malignant pleural mesothelioma xenograft. (A) T-cells were engineered to express the indicated test (cM-28z) or control CAR (C-28z), co-expressed with 4αβ. NOD SCID γc null mice were inoculated i.p. with 5 × 104 ffLuc/ RFP+ REN tumor cells. Following assignment to groups with comparable mean established tumor burden, mice were treated in a blinded fashion 7 d after tumor implantation (arrowed) with cM-28z+ or C-28z+ CAR T-cells, administered to groups of 5 mice at (L)ow or (H)igh doses of 2.5 × 106 or 10 × 106 T-cells respectively. Comparison was made with untrans(duced) T-cells (10 × 106 cells; n = 5 mice) or PBS (n = 8 mice). Serial bioluminescence imaging of animals that received low dose (B) or high doses (C) of the indicated T-cell populations are shown. The PBS group is shown on each panel for reference. (D) Bioluminescence images of representative mice are shown on the same imaging scale (E) over the course of the experiment. (F) Serial weights of mice. All data are mean ± SD of n = 5–8 mice. *p < 0.05; ** p < 0.01 making comparison with PBS-treated mice.
Discussion
Expression of the MET receptor tyrosine kinase has been reported to occur commonly in mesothelioma.16-19 In keeping with this, we detected MET in two thirds of tumors diagnosed at our center, with a predilection for epithelioid mesotheliomas. To exploit aberrant MET expression in this disease, we set out to engineer a CAR that specifically engages this receptor. Targeting was achieved using 3 derivatives of the smallest natural ligand of the MET receptor, namely the NK1 splice variant of HGF. Using an NIH3T3-based AAPC system, we demonstrated that all candidate CARs could elicit MET-dependent T-cell activation and cytotoxic activity. Similar findings were obtained using a panel of MET-expressing mesothelioma cell lines. In each case, CAR T-cell effector function was dependent upon duration of tumor/ CAR T-cell co-culture.
In addition to MET, HGF also binds to 2 discrete co-factor groups, namely CD44v6 isoforms34 and a range of GAGs and derived proteoglycans.43 Consequently, it was imperative to explore whether NK1-based CARs could also engage these molecules, either to trigger or to modulate CAR T-cell activation. Using NIH3T3 cells engineered to constitutively express human CD44v6, we observed that NK1-based CARs did not mediate T-cell activation upon encounter with this potential target in the absence of MET. Moreover, T-cell activation was not enhanced in human mesothelioma cells in which CD44v6 was ectopically overexpressed. CD44v6 has been reported to bind HGF with micromolar affinity.34 However, binding has not been mapped to NK1 and thus insufficient or absent engagement of CD44v6 by the targeting moiety may account for these findings. Alternatively, the CD44v6 binding epitope on NK1 may be occluded in the CAR format, or the binding site may have been altered by one or more of the mutations contained within 1K1, M2.2rev or cM2.2rev.
The question of CAR T-cell binding to GAGs and proteoglycans also requires careful consideration. The best characterized binding partners of HGF are heparin, heparan sulfate and HSPGs, although weaker binding to other GAGs such as chondroitin sulfate and dermatan sulfate has also been reported.44 Interaction between HGF and these GAGs may assist in the formation of a ternary complex with MET. The primary binding site for all of these GAGs lies within the N domain of HGF,45 an element that is retained in the NK1-based CARs tested here. Nonetheless, although unmodified NIH3T3 fibroblasts naturally express large quantities of cell surface HSPG,38 these cells did not trigger the activation of any CAR under study. One concern is the possibility that mouse-derived NIH3T3 cells may not produce HSPG that are representative of those found in human cells. To address this, we expressed human Syndecan-1 in these cells, given the established ability of this HSPG to bind HGF.36 Once again, no evidence of CAR-mediated targeting of these cells was observed.
Several explanations can be envisioned for the inability of GAGs or proteoglycans to promote the MET-independent activation of NK1-targeted CARs. First, while solid phase assays attributed high (nanomolar) affinity to GAG/ NK1 interactions,46 more recent studies suggest that when this interaction is studied in solution, the true Kd lies within the micromolar range.44,45 The latter may be insufficient to promote the activation of CAR-engineered T-cells. An alternative possibility stems from the fact that T-cells produce a range of GAGs and proteoglycans,47 especially when activated.48,49 The predominant side chain is chondroitin sulfate, which has recently been shown to bind HGF.44 Consequently, the GAG binding site of the CAR may be saturated given the large local concentration of these potential binding partners, priming the receptor to engage in high affinity binding to MET alone. Lastly, it is possible that mutation of several lysine resides to glutamic acid in the engineered cM2.2rev variant reduce its binding affinity toward heparin.
Given the comparable in vitro anti-tumor activity of all 3 NK1-targeted candidates, one CAR was advanced to in vivo evaluation in tumor-bearing mice. These studies demonstrated that MET re-targeted CAR T-cells elicited the dose-dependent and progressive regression of an established mesothelioma tumor burden. A relatively high dose of 2.5 million CAR-engineered T-cells was required to control the REN xenograft, in keeping with its more modest expression of MET (Fig. 5A). While background alloreactivity was evident in these studies, significantly enhanced tumor control was observed with MET re-targeted CAR T-cells. Although human HGF and NK1 are fully active on the mouse MET receptor,50 treatment was well tolerated without any weight loss or histologic evidence of end-organ toxicity.
Our study is not the first to engineer a MET-specific CAR. Frigault et al. described a second generation candidate MET CAR in which targeting was achieved using an scFv isolated from the 5D5 hybridoma.42 This CAR exhibited constitutive activity leading to tonic signaling in T-cells, resulting in acquisition of an exhausted phenotype and compromised anti-tumor function. In this context, constitutive CAR activity has been ascribed to the propensity of some scFvs to undergo antigen-independent clustering,51 an attribute that would not occur using a ligand-based targeting moiety. Tonic signaling could be overcome by expression of the MET CAR at lower levels in the engineered T-cell population. However, anti-tumor activity of the resultant CAR T-cells was insignificantly improved, when compared with the alloreactive effect of CAR T-cells directed against an irrelevant antigen.42
An important consideration is the expression of MET in numerous organs (e.g., lung, kidney, and liver), raising concerns that ‘on target/off cancer’ toxicity could compromise this therapeutic strategy. Four points are of relevance in the evaluation and mitigation of this risk. First, intra-tumoral delivery of CAR T-cells mediates therapeutic benefit in the absence of significant toxicity.52 In an ongoing clinical trial, we have shown that doses of up to 300 million ErbB re-targeted CAR T-cells can be delivered using the intra-tumoral route in patients with head and neck cancer, without dose-limiting toxicity or detectable systemic absorption of the T-cells.53 Second, intra-cavitary delivery of CAR T-cells is likely to also prove safer than their systemic administration. This is supported by the normal histologic analysis of MET-expressing mouse organs analyzed in this study, despite targeting of CARs with a ligand that is known to bind to the murine MET ortholog. Third, we are not the first group to demonstrate the safety of MET targeting using a directly cytolytic approach. A MET-specific monoclonal antibody that elicits potent antibody-dependent cell mediated cytotoxicity achieved anti-tumor activity combined with excellent safety when administered to cynomolgus monkeys, in which both target binding and Fc-mediated interactions are fully retained.54 Finally, should MET targeting prove unduly toxic despite the above considerations, combinatorial strategies in which optimal CAR T-cell activation is contingent upon co-engagement of MET and a second target may provide a strategy that more effectively balances efficacy with safety.55,56
In summary, data presented in this study demonstrate that MET can be effectively targeted using ligand-directed CAR T-cells. Clinical evaluation of MET re-targeted CAR T-cells warrants consideration in this disease, using intra-cavitary delivery to maximize tumor delivery.14 Inclusion of a suicide gene (such as inducible caspase 9)57 would be recommended in such a protocol, as is currently under study with mesothelin-targeted CAR T-cells (https://clinicaltrials.gov/ct2/show/NCT02414269, accessed 07–15–2017).
Materials and methods
All mandatory laboratory health and safety procedures have been complied with over the course of this work.
DNA constructs
All recombinant DNA constructs were expressed using the SFG retroviral vector. Codon optimized cDNAs encoding CAR targeting moieties were synthesized by Genscript. All contained an alanine codon after the start codon to preserve the Nco1 restriction site. Fragments were substituted by molecular cloning for the smaller Nco1/Not1 fragment within SFG T-28z (previously referred to as Tm28z).31 Codon optimized cDNAs encoding human MET, CD44v6 and Syndecan-1 were synthesized by Genscript and were cloned as Nco1/ Xho1 fragments in the SFG retroviral vector. The ffLuc/ RFP retroviral vector, C-28z control CAR (referred to previously as C20–28z) and IL-4 receptor-α/ IL2/15 receptor β chimeric cytokine receptor (4αβ) have all been described previously.31,33
Cell culture and gene transfer
This study was approved by the South-East London Research Ethics Committee 1 (reference 09/H0804/92). Peripheral blood T-cells from healthy donors were activated using CD3+CD28-coated paramagnetic beads and subjected to RetroNectin-enhanced retroviral-mediated gene transfer and culture as described.31 Source and culture conditions for mesothelioma tumor cell lines have been described.15 T47D cells were obtained from the American Tissue Culture Collection while PC3 LN3 cells were provided by Prof Suzanne Eccles (Institute of Cancer Research, London, UK). Human tumor cell lines were subject to retroviral transduction using supernatant (0.44 µM filtered) collected from PG13 retroviral packaging cells while mouse target cells were transduced using supernatant collected from Phoenix Eco retroviral packaging cells (both obtained from the European Collection of Authenticated Cell Cultures).
Immunohistochemistry of tumors
Access to patient tissue samples and data are regulated under the Human Tissue Act and Guy's and St Thomas' Hospital Thoracic Cancer Biobank Access Committee (License number 12121) in accordance with NHS Research Ethics Committee conditions. Mesothelioma tissue microarrays (TMAs) were acquired from the Departmental Tissue Bank. Sections (3μm) were cut from formalin fixed paraffin-embedded TMA blocks of 114 mesotheliomas obtained at video-associated thoracoscopic surgery or open pleural biopsy and were prepared on appropriately coated slides. Dewaxing and blockade of endogenous peroxidase were performed as described previously.15 Microarrays were processed using a Ventana Benchmark Ultra using methods recommended by the manufacturers. In brief, antigen was retrieved using Cell Conditioning solution 1 (Ventana). Slides were stained with CONFIRM anti-total c-MET (SP44) rabbit monoclonal antibody C-MET for 16 minutes at 37°C. Antibody binding was visualized using the Benchmark ultraView Universal DAB detection kit. Slides were lightly counterstained with hematoxylin. Breast carcinoma sections were used as positive and negative controls. For all negative controls, normal horse serum was used instead of primary antibody. Following staining, TMA slides were scored for intensity of staining by an independent histopathologist, who reported results as negative (absence of staining), low intensity (+), moderate intensity (++) or high intensity (+++).
Flow cytometry analysis
Expression of CARs was detected using 9e10 hybridoma supernatant (generated in house) followed by PE-conjugated goat anti-mouse IgG (Dako), making comparison with cells incubated with secondary antibody alone. Expression of 4αβ was detected using PE-conjugated anti-CD124 (BD PharMingen). Human MET and CD44v6 were detected using FITC-conjugated rat and mouse IgG1 antibodies respectively (eBioscience). Human Syndecan-1 was detected using APC-conjugated rat IgG1 anti-human CD138 (BioLegend). Heparan sulfate was detected using FITC-conjugated 10e4 (Stratech Scientific Ltd.). Isotype controls from the same supplier were used where indicated. Flow cytometry was performed using an LSRFortessa cytometer and analyzed with FloJo software.
Western blotting
CAR-transduced or unmodified control T-cells (0.5 × 107 cells) were lysed in RIPA buffer (50 mM Tris, pH 7.5, 150 mM NaCl, 0.1% sodium dodecyl sulfate, 0.5% sodium deoxycholate, 1% Triton X100, 1 mM phenylmethylsulfonyl fluoride) containing cOmplete protease inhibitor cocktail tablets (Roche) as per manufacturer's instruction. Cell lysates were separated under denaturing conditions on a 4–20% gradient gel (Invitrogen), transferred to PVDF membrane (Amersham Biosciences) and probed with mouse anti-human CD3ζ antibody (BD Biosciences) followed by HRP-conjugated goat anti-mouse IgG (Dako) both diluted 1:1000 in TBS-Tween/5% skimmed milk. Bands were developed using an ECL Western Blotting Detection kit (Amersham Biosciences) according to manufacturer's instruction.
Cytotoxicity, cytokine release and luciferase assays
Target cells were plated overnight (either 2 × 104 cells in 96 well plates or 2 × 105 cells in 24 well plates). Test or control T-cells were added on the next day at the specified ratio. Anti-tumor activity was quantified by MTT assay, as described.31 Interferon (IFN)-γ and IL-2 was measured in supernatants collected after 48 hours using Ready Set Go ELISA kits (eBioscience), as per manufacturer's protocol. In vitro luciferase testing of tumor cells was performed as described.31 A human cytokine antibody array was performed according to the manufacturer's instructions (Abcam).
Animal studies
In vivo experimentation complied with UK Home Office guidelines, under project license 70/7794. NOD SCID γc null mice were inoculated by i.p. injection with ffLuc/RFP+ REN cells. Tumor engraftment was confirmed by bioluminescence imaging (BLI) using the IVIS Lumina Imaging platform and Living Image software (PerkinElmer). Mice were sorted into groups with similar mean total flux BLI measurements. Treatment groups were assigned numbers and all treatment and imaging was performed thereafter in a blinded fashion. Following i.p. delivery of CAR T-cells, tumor tracking was performed using BLI.58 Mice were culled when symptomatic due to disease progression, or when experimental endpoints had been met.
Statistical analysis
Analysis was performed using Excel for Mac 2011 (Berkshire, UK), GraphPad Prism 6.0, (GraphPad software) or IBM SPSS Statistics 22 for Mac. Comparison between groups was performed using one-way or 2-way ANOVA, followed by Tukey's multiple comparisons test. For statistical comparison of 2 groups, data sets were analyzed by Student's t test. Significance in Kaplan Meier curves was determined using the Log-rank (Mantel Cox) test.
Supplementary Material
Disclosure of potential conflicts of interest
JM is chief scientific officer of Leucid Bio, which is a spinout company focused on development of cellular therapeutic agents.
Acknowledgments
We thank other members of the CAR Mechanics group for useful discussions.
Funding
This research was supported by the British Lung Foundation (APHD12–11), the June Hancock Mesothelioma Research Fund (JHMRF 2014), the Experimental Cancer Medicine Center at King's College London, the King's Health Partners/ King's College London Cancer Research UK Cancer Centre and by the National Institute for Health Research (NIHR) Biomedical Research Centre based at Guy's and St Thomas' NHS Foundation Trust and King's College London (grant number IS-BRC-1215–20006). The views expressed are those of the authors and not necessarily those of the NHS, the NIHR or the Department of Health.
References
- 1.Bianchi C, Bianchi T. Global mesothelioma epidemic: Trend and features. Indian J Occup Environ Med. 2014;18:82-8. doi: 10.4103/0019-5278.146897. PMID:25568603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Izzi V, Masuelli L, Tresoldi I, Foti C, Modesti A, Bei R. Immunity and malignant mesothelioma: from mesothelial cell damage to tumor development and immune response-based therapies. Cancer Lett. 2012;322:18-34. doi: 10.1016/j.canlet.2012.02.034. PMID:22394996. [DOI] [PubMed] [Google Scholar]
- 3.Thapa B, Watkins DN, John T. Immunotherapy for malignant mesothelioma: reality check. Expert Rev Anticancer Ther. 2016;16:1167-1176. doi: 10.1080/14737140.2016.1241149. PMID:27669108. [DOI] [PubMed] [Google Scholar]
- 4.Lievense LA, Sterman DH, Cornelissen R, Aerts JG. Checkpoint Blockade in Lung Cancer and Mesothelioma. Am J Respir Crit Care Med. 2017;196:274-282. doi: 10.1164/rccm.201608-1755CI. PMID:28252315. [DOI] [PubMed] [Google Scholar]
- 5.Whilding LM, Maher J. CAR T-cell immunotherapy: The path from the by-road to the freeway? Mol Oncol. 2015;9:1994-2018. doi: 10.1016/j.molonc.2015.10.012. PMID:26563646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Adusumilli PS, Cherkassky L, Villena-Vargas J, Colovos C, Servais E, Plotkin J, Jones DR, Sadelain M. Regional delivery of mesothelin-targeted CAR T cell therapy generates potent and long-lasting CD4-dependent tumor immunity. Sci Transl Med. 2014;6:261ra151. doi: 10.1126/scitranslmed.3010162. PMID:25378643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cherkassky L, Morello A, Villena-Vargas J, Feng Y, Dimitrov DS, Jones DR, Sadelain M, Adusumilli PS. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J Clin Invest. 2016;126:3130-44. doi: 10.1172/JCI83092. PMID:27454297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schuberth PC, Hagedorn C, Jensen SM, Gulati P, van den Broek M, Mischo A, Soltermann A, Jungel A, Marroquin Belaunzaran O, Stahel R, et al.. Treatment of malignant pleural mesothelioma by fibroblast activation protein-specific re-directed T cells. J Transl Med. 2013;11:187. doi: 10.1186/1479-5876-11-187. PMID:23937772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moon EK, Wang LC, Dolfi DV, Wilson CB, Ranganathan R, Sun J, Kapoor V, Scholler J, Pure E, Milone MC, et al.. Multifactorial T-cell hypofunction that is reversible can limit the efficacy of chimeric antigen receptor-transduced human T cells in solid tumors. Clin Cancer Res. 2014;20:4262-73. doi: 10.1158/1078-0432.CCR-13-2627. PMID:24919573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mayor M, Zeltsman M, McGee E, Adusumilli PS. A regional approach for CAR T-cell therapy for mesothelioma: from mouse models to clinical trial. Immunotherapy. 2016;8:491-4. doi: 10.2217/imt-2016-0008. PMID:27140404. [DOI] [PubMed] [Google Scholar]
- 11.O'Hara M, Stashwick C, Haas AR, Tanyi JL. Mesothelin as a target for chimeric antigen receptor-modified T cells as anticancer therapy. Immunotherapy. 2016;8:449-60. doi: 10.2217/imt.16.4. PMID:26973126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Beatty GL, Haas AR, Maus MV, Torigian DA, Soulen MC, Plesa G, Chew A, Zhao Y, Levine BL, Albelda SM, et al.. Mesothelin-specific chimeric antigen receptor mRNA-engineered T cells induce anti-tumor activity in solid malignancies. Cancer Immunol Res. 2014;2:112-20. doi: 10.1158/2326-6066.CIR-13-0170. PMID:24579088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jeannon JP, Ofu E, Balfour A, Bowman J, Simo R. The natural history of untreated squamous cell carcinoma of the head and neck: how we do it. Clin Otolaryngol. 2011;36:384-8. doi: 10.1111/j.1749-4486.2011.02325.x. PMID:21848553. [DOI] [PubMed] [Google Scholar]
- 14.Papa S, van Schalkwyk M, Maher J. Clinical Evaluation of ErbB-Targeted CAR T-Cells, Following Intracavity Delivery in Patients with ErbB-Expressing Solid Tumors. Methods Mol Biol. 2015;1317:365-82. doi: 10.1007/978-1-4939-2727-2_21. PMID:26072418. [DOI] [PubMed] [Google Scholar]
- 15.Klampatsa A, Achkova DY, Davies DM, Parente-Pereira AC, Woodman N, Rosekilly J, Osborne G, Thayaparan T, Bille A, Sheaf M, et al.. Intracavitary ‘T4 immunotherapy’ of malignant mesothelioma using pan-ErbB re-targeted CAR T-cells. Cancer Lett. 2017;393:52-9. doi: 10.1016/j.canlet.2017.02.015. PMID:28223167. [DOI] [PubMed] [Google Scholar]
- 16.Tolnay E, Kuhnen C, Wiethege T, Konig JE, Voss B, Muller KM. Hepatocyte growth factor/scatter factor and its receptor c-Met are overexpressed and associated with an increased microvessel density in malignant pleural mesothelioma. Journal of cancer research and clinical oncology 1998;124:291-6. doi: 10.1007/s004320050171. PMID:9692834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kawaguchi K, Murakami H, Taniguchi T, Fujii M, Kawata S, Fukui T, Kondo Y, Osada H, Usami N, Yokoi K, et al.. Combined inhibition of MET and EGFR suppresses proliferation of malignant mesothelioma cells. Carcinogenesis. 2009;30:1097-105. doi: 10.1093/carcin/bgp097. PMID:19380521. [DOI] [PubMed] [Google Scholar]
- 18.Jagadeeswaran R, Ma PC, Seiwert TY, Jagadeeswaran S, Zumba O, Nallasura V, Ahmed S, Filiberti R, Paganuzzi M, Puntoni R, et al.. Functional analysis of c-Met/hepatocyte growth factor pathway in malignant pleural mesothelioma. Cancer Res. 2006;66:352-61. doi: 10.1158/0008-5472.CAN-04-4567. PMID:16397249. [DOI] [PubMed] [Google Scholar]
- 19.Levallet G, Vaisse-Lesteven M, Le Stang N, Ilg AG, Brochard P, Astoul P, Pairon JC, Bergot E, Zalcman G, Galateau-Salle F. Plasma cell membrane localization of c-MET predicts longer survival in patients with malignant mesothelioma: a series of 157 cases from the MESOPATH Group. Journal of thoracic oncology: official publication of the International Association for the Study of Lung Cancer. 2012;7:599-606. doi: 10.1097/JTO.0b013e3182417da5. PMID:22246193. [DOI] [PubMed] [Google Scholar]
- 20.Bronte G, Incorvaia L, Rizzo S, Passiglia F, Galvano A, Rizzo F, Rolfo C, Fanale D, Listi A, Natoli C, et al.. The resistance related to targeted therapy in malignant pleural mesothelioma: why has not the target been hit yet? Critical Reviews in Oncology/ Hematology. 2016;107:20-32. doi: 10.1016/j.critrevonc.2016.08.011. PMID:27823648. [DOI] [PubMed] [Google Scholar]
- 21.Thayaparan T, Spicer JF, Maher J. The role of the HGF/Met axis in mesothelioma. Biochemical Society transactions. 2016;44:363-70. doi: 10.1042/BST20150252. PMID:27068941. [DOI] [PubMed] [Google Scholar]
- 22.Kanteti R, Dhanasingh I, Kawada I, Lennon FE, Arif Q, Bueno R, Hasina R, Husain AN, Vigneswaran W, Seiwert T, et al.. MET and PI3K/mTOR as a potential combinatorial therapeutic target in malignant pleural mesothelioma. PloS one. 2014;9:e105919. doi: 10.1371/journal.pone.0105919. PMID:25221930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Leon LG, Gemelli M, Sciarrillo R, Avan A, Funel N, Giovannetti E. Synergistic activity of the c-Met and tubulin inhibitor tivantinib (ARQ197) with pemetrexed in mesothelioma cells. Curr Drug Targets. 2014;15:1331-40. doi: 10.2174/1389450116666141205160924. PMID:25483224. [DOI] [PubMed] [Google Scholar]
- 24.Cioce V, Csaky KG, Chan AM, Bottaro DP, Taylor WG, Jensen R, Aaronson SA, Rubin JS. Hepatocyte growth factor (HGF)/NK1 is a naturally occurring HGF/scatter factor variant with partial agonist/antagonist activity. J Biol Chem 1996;271:13110-5. doi: 10.1074/jbc.271.22.13110. PMID:8662798. [DOI] [PubMed] [Google Scholar]
- 25.Lokker NA, Godowski PJ. Generation and characterization of a competitive antagonist of human hepatocyte growth factor, HGF/NK1. J Biol Chem 1993;268:17145-50. PMID:8349603. [PubMed] [Google Scholar]
- 26.Jakubczak JL, LaRochelle WJ, Merlino G. NK1, a natural splice variant of hepatocyte growth factor/scatter factor, is a partial agonist in vivo. Molecular and cellular biology 1998;18:1275-83. doi: 10.1128/MCB.18.3.1275. PMID:9488442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Simon Davis DA, Parish CR. Heparan sulfate: a ubiquitous glycosaminoglycan with multiple roles in immunity. Front Immunol. 2013;4:470. doi: 10.3389/fimmu.2013.00470. PMID:24391644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lietha D, Chirgadze DY, Mulloy B, Blundell TL, Gherardi E. Crystal structures of NK1-heparin complexes reveal the basis for NK1 activity and enable engineering of potent agonists of the MET receptor. EMBO J. 2001;20:5543-55. doi: 10.1093/emboj/20.20.5543. PMID:11597998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Maher J, Brentjens RJ, Gunset G, Riviere I, Sadelain M. Human T-lymphocyte cytotoxicity and proliferation directed by a single chimeric TCRzeta /CD28 receptor. Nat Biotechnol. 2002;20:70-5. doi: 10.1038/nbt0102-70. PMID:11753365. [DOI] [PubMed] [Google Scholar]
- 30.Jones DS 2nd, Tsai PC, Cochran JR. Engineering hepatocyte growth factor fragments with high stability and activity as Met receptor agonists and antagonists. Proc Natl Acad Sci U S A. 2011;108:13035-40. doi: 10.1073/pnas.1102561108. PMID:21788476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Whilding LM, Parente-Pereira AC, Zabinski T, Davies DM, Petrovic RM, Kao YV, Saxena SA, Romain A, Costa-Guerra JA, Violette S, et al.. Targeting of Aberrant alphavbeta6 Integrin Expression in Solid Tumors Using Chimeric Antigen Receptor-Engineered T Cells. Mol Ther. 2017;25:259-73. doi: 10.1016/j.ymthe.2016.10.012. PMID:28129120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Davies DM, Foster J, Van Der Stegen SJ, Parente-Pereira AC, Chiapero-Stanke L, Delinassios GJ, Burbridge SE, Kao V, Liu Z, Bosshard-Carter L, et al.. Flexible targeting of ErbB dimers that drive tumorigenesis by using genetically engineered T cells. Mol Med. 2012;18:565-76. doi: 10.2119/molmed.2011.00493. PMID:22354215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wilkie S, Burbridge SE, Chiapero-Stanke L, Pereira AC, Cleary S, van der Stegen SJ, Spicer JF, Davies DM, Maher J. Selective expansion of chimeric antigen receptor-targeted T-cells with potent effector function using interleukin-4. J Biol Chem. 2010;285:25538-44. doi: 10.1074/jbc.M110.127951. PMID:20562098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Volz Y, Koschut D, Matzke-Ogi A, Dietz MS, Karathanasis C, Richert L, Wagner MG, Mely Y, Heilemann M, Niemann HH, et al.. Direct binding of hepatocyte growth factor and vascular endothelial growth factor to CD44v6. Biosci Rep. 2015;35. doi: 10.1042/BSR20150093. PMID:26181364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hartmann G, Prospero T, Brinkmann V, Ozcelik C, Winter G, Hepple J, Batley S, Bladt F, Sachs M, Birchmeier C, et al.. Engineered mutants of HGF/SF with reduced binding to heparan sulphate proteoglycans, decreased clearance and enhanced activity in vivo. Current biology: CB 1998;8:125-34. doi: 10.1016/S0960-9822(98)70059-4. PMID:9443912. [DOI] [PubMed] [Google Scholar]
- 36.Derksen PW, Keehnen RM, Evers LM, van Oers MH, Spaargaren M, Pals ST. Cell surface proteoglycan syndecan-1 mediates hepatocyte growth factor binding and promotes Met signaling in multiple myeloma. Blood. 2002;99:1405-10. doi: 10.1182/blood.V99.4.1405. PMID:11830493. [DOI] [PubMed] [Google Scholar]
- 37.Latouche JB, Sadelain M. Induction of human cytotoxic T lymphocytes by artificial antigen-presenting cells. Nat Biotechnol. 2000;18:405-9. doi: 10.1038/74455. PMID:10748520. [DOI] [PubMed] [Google Scholar]
- 38.Bellosta P, Iwahori A, Plotnikov AN, Eliseenkova AV, Basilico C, Mohammadi M. Identification of receptor and heparin binding sites in fibroblast growth factor 4 by structure-based mutagenesis. Molecular and cellular biology. 2001;21:5946-57. doi: 10.1128/MCB.21.17.5946-5957.2001. PMID:11486033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nasimuzzaman M, Persons DA. Cell Membrane-associated heparan sulfate is a receptor for prototype foamy virus in human, monkey, and rodent cells. Mol Ther. 2012;20:1158-66. doi: 10.1038/mt.2012.41. PMID:22434139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Heidari-Hamedani G, Vives RR, Seffouh A, Afratis NA, Oosterhof A, van Kuppevelt TH, Karamanos NK, Metintas M, Hjerpe A, Dobra K, et al.. Syndecan-1 alters heparan sulfate composition and signaling pathways in malignant mesothelioma. Cell Signal. 2015;27:2054-67. doi: 10.1016/j.cellsig.2015.07.017. PMID:26210886. [DOI] [PubMed] [Google Scholar]
- 41.Komarowska I, Coe D, Wang G, Haas R, Mauro C, Kishore M, Cooper D, Nadkarni S, Fu H, Steinbruchel DA, et al.. Hepatocyte Growth Factor Receptor c-Met Instructs T Cell Cardiotropism and Promotes T Cell Migration to the Heart via Autocrine Chemokine Release. Immunity. 2015;42:1087-99. doi: 10.1016/j.immuni.2015.05.014. PMID:26070483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Frigault MJ, Lee J, Basil MC, Carpenito C, Motohashi S, Scholler J, Kawalekar OU, Guedan S, McGettigan SE, Posey AD Jr., et al.. Identification of chimeric antigen receptors that mediate constitutive or inducible proliferation of T cells. Cancer Immunol Res. 2015;3:356-67. doi: 10.1158/2326-6066.CIR-14-0186. PMID:25600436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Catlow KR, Deakin JA, Wei Z, Delehedde M, Fernig DG, Gherardi E, Gallagher JT, Pavao MS, Lyon M. Interactions of hepatocyte growth factor/scatter factor with various glycosaminoglycans reveal an important interplay between the presence of iduronate and sulfate density. J Biol Chem. 2008;283:5235-48. doi: 10.1074/jbc.M706589200. PMID:18156180. [DOI] [PubMed] [Google Scholar]
- 44.Johansson CM. Biophysical characterisation of the hepatocyte growth factor – glycosaminglycan interaction. PhD Thesis submission to the University of Edinburgh 2011. https://www.era.lib.ed.ac.uk/handle/1842/9904.
- 45.Zhou H, Casas-Finet JR, Heath Coats R, Kaufman JD, Stahl SJ, Wingfield PT, Rubin JS, Bottaro DP, Byrd RA. Identification and dynamics of a heparin-binding site in hepatocyte growth factor. Biochemistry 1999;38:14793-802. doi: 10.1021/bi9908641. PMID:10555961. [DOI] [PubMed] [Google Scholar]
- 46.Merkulova-Rainon T, England P, Ding S, Demerens C, Tobelem G. The N-terminal domain of hepatocyte growth factor inhibits the angiogenic behavior of endothelial cells independently from binding to the c-met receptor. J Biol Chem. 2003;278:37400-8. doi: 10.1074/jbc.M212768200. PMID:12847110. [DOI] [PubMed] [Google Scholar]
- 47.Kaur S, Kuznetsova SA, Pendrak ML, Sipes JM, Romeo MJ, Li Z, Zhang L, Roberts DD. Heparan sulfate modification of the transmembrane receptor CD47 is necessary for inhibition of T cell receptor signaling by thrombospondin-1. J Bio Chem. 2011;286:14991-5002. doi: 10.1074/jbc.M110.179663. PMID:21343308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jones KS, Petrow-Sadowski C, Bertolette DC, Huang Y, Ruscetti FW. Heparan sulfate proteoglycans mediate attachment and entry of human T-cell leukemia virus type 1 virions into CD4+ T cells. J Virol. 2005;79:12692-702. doi: 10.1128/JVI.79.20.12692-12702.2005. PMID:16188972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fadnes B, Husebekk A, Svineng G, Rekdal O, Yanagishita M, Kolset SO, Uhlin-Hansen L. The proteoglycan repertoire of lymphoid cells. Glycoconj J. 2012;29:513-23. doi: 10.1007/s10719-012-9427-9. PMID:22777011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ross J, Gherardi E, Mallorqui-Fernandez N, Bocci M, Sobkowicz A, Rees M, Rowe A, Ellmerich S, Massie I, Soeda J, et al.. Protein engineered variants of hepatocyte growth factor/scatter factor promote proliferation of primary human hepatocytes and in rodent liver. Gastroenterology. 2012;142:897-906. doi: 10.1053/j.gastro.2011.12.006. PMID:22178588. [DOI] [PubMed] [Google Scholar]
- 51.Long AH, Haso WM, Shern JF, Wanhainen KM, Murgai M, Ingaramo M, Smith JP, Walker AJ, Kohler ME, Venkateshwara VR, et al.. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat Med. 2015;21:581-90.doi: 10.1038/nm.3838. PMID:25939063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.van der Stegen SJ, Davies DM, Wilkie S, Foster J, Sosabowski JK, Burnet J, Whilding LM, Petrovic RM, Ghaem-Maghami S, Mather S, et al.. Preclinical in vivo modeling of cytokine release syndrome induced by ErbB-retargeted human T cells: identifying a window of therapeutic opportunity? J Immunol. 2013;191:4589-98. doi: 10.4049/jimmunol.1301523. PMID:24062490. [DOI] [PubMed] [Google Scholar]
- 53.Papa S, Adami AA, Metoudi M, Achkova DY, van Schalkwyk M, Parente-Pereira AC, Bosshard-Carter L, Whilding LM, van der Stegen S, Davies D.M., et al.. Online Proceedings of the Annual Meeting of the American Association for Cancer Research. http://wwwabstractsonlinecom/pp8/ – !/4292/presentation/12333. 2017. [Google Scholar]
- 54.Hultberg A, Morello V, Huyghe L, De Jonge N, Blanchetot C, Hanssens V, De Boeck G, Silence K, Festjens E, Heukers R, et al.. Depleting MET-Expressing Tumor Cells by ADCC Provides a Therapeutic Advantage over Inhibiting HGF/MET Signaling. Cancer Res. 2015;75:3373-83. doi: 10.1158/0008-5472.CAN-15-0356. PMID:26141862. [DOI] [PubMed] [Google Scholar]
- 55.Wilkie S, van Schalkwyk MC, Hobbs S, Davies DM, van der Stegen SJ, Pereira AC, Burbridge SE, Box C, Eccles SA, Maher J. Dual Targeting of ErbB2 and MUC1 in Breast Cancer Using Chimeric Antigen Receptors Engineered to Provide Complementary Signaling. J Clin Immunol. 2012;32:1059-70. doi: 10.1007/s10875-012-9689-9. PMID:22526592. [DOI] [PubMed] [Google Scholar]
- 56.Kloss CC, Condomines M, Cartellieri M, Bachmann M, Sadelain M. Combinatorial antigen recognition with balanced signaling promotes selective tumor eradication by engineered T cells. Nat Biotechnol. 2013;31:71-5. doi: 10.1038/nbt.2459. PMID:23242161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Straathof KC, Pule MA, Yotnda P, Dotti G, Vanin EF, Brenner MK, Heslop HE, Spencer DM, Rooney CM. An inducible caspase 9 safety switch for T-cell therapy. Blood. 2005;105:4247-54. doi: 10.1182/blood-2004-11-4564. PMID:15728125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wilkie S, Picco G, Foster J, Davies DM, Julien S, Cooper L, Arif S, Mather SJ, Taylor-Papadimitriou J, Burchell JM, et al.. Retargeting of human T cells to tumor-associated MUC1: the evolution of a chimeric antigen receptor. J Immunol. 2008;180:4901-9. doi: 10.4049/jimmunol.180.7.4901. PMID:18354214. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.