

Summary
Loss or inactivation of the histone H3K27 demethylase UTX occurs in several malignancies, including multiple myeloma (MM). Using an isogenic cell system, we found that loss of UTX leads to deactivation of gene expression ultimately promoting the proliferation, clonogenicity, adhesion and tumorigenicity of MM cells. Moreover, UTX-mutant cells showed increased in vitro and in vivo sensitivity to inhibition of EZH2, a histone methyltransferase that generates H3K27me3. Such sensitivity was related to a decrease in the levels of IRF4 and c-MYC, and an activation of repressors of IRF4 characteristic of germinal center B cells such as BCL6 and IRF1. Rebalance of H3K27me3 levels at specific genes through EZH2 inhibitors may be a therapeutic strategy in MM cases harboring UTX mutations.
Keywords: Multiple Myeloma, UTX, KDM6A, EZH2 inhibitors, IRF4, BCL6
Graphical abstract
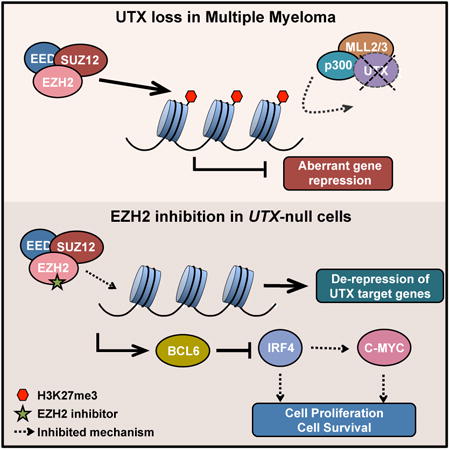
Ezponda et al. now demonstrate how the loss of UTX/KDM6A, a factor that regulates chromatin, contributes to multiple myeloma, conferring malignant properties to these cells. Moreover, they show how the use of EZH2 inhibitors, currently in clinical trials, specifically affect MM cells harboring UTX loss.
Introduction
Epigenetic regulation of gene expression encompasses a variety of mechanisms that control gene activity by modifying chromatin without perturbing DNA sequence. Advances in genome sequencing have revealed a wide range of mutations in chromatin regulators that lead to epigenetic deregulation in cancer. Alteration of methylation of lysine 27 on histone H3 (H3K27me) is a recurrent theme resulting from mutations in epigenetic regulators (Ezponda and Licht, 2014). Trimethylation of H3K27 (H3K27me3) is a repressive chromatin mark placed by the Polycomb Repressive Complex 2 (PRC2), catalyzed by it central component EZH2, and removed by the histone demethylases UTX/KDM6A and JMJD3/KDM6B. H3K27me has a critical role in spatial and temporal regulation of gene expression, controlling key cellular processes including animal body patterning, cell proliferation and stem cell differentiation. Alterations in cancer include both gains and losses of the levels of H3K27me, underscoring the importance of a tightly regulated balance for cell homeostasis. Alterations of H3K27me are due to mutations in chromatin writers (EZH2 an other PRC2 components mutations), erasers (UTX loss), alteration of other chromatin marks (MMSET overexpression), or mutation of the histone H3 itself (Ezponda and Licht, 2014).
The H3K27me2/3 demethylase UTX/KDM6A is encoded by the X-chromosome and escapes X-inactivation in females. This factor is part of a transcriptional activator complex including the MLL2/MLL3 H3K4 methyltransferases, and P300/CBP histone acetyl transferases (Shilatifard, 2012), suggesting a concerted mechanism by which repressive H3K27me3 is removed and replaced by the activation-associated H3K27 acetylation along with H3K4 methylation to activate transcription. Inactivating mutations/deletions encompassing the UTX locus occur in hematologic malignancies and solid tumors. UTX lesions tend to be homozygous in females and to be accompanied by the loss of its paralog UTY in males, suggesting a tumor suppressor role (van Haaften et al., 2009). Supporting this idea, loss of UTX promotes proliferation in many contexts, and accelerates NOTCH1-driven T-ALL onset in vivo (Ntziachristos et al., 2014, Van der Meulen et al., 2015, van Haaften et al., 2009, Wang et al., 2010). Nevertheless, the role of UTX in cancer seems to be tissue-specific as overexpression of UTX in breast cancer promotes proliferation and invasion (Kim et al., 2014). In agreement with this, UTX target genes seem to be very different among cell types, suggesting a cell-specific role (Kim et al., 2014, Ntziachristos et al., 2014, Wang et al., 2010).
UTX mutations/deletions are found in 3-4% of primary MM specimens (Pawlyn et al., 2016, van Haaften et al., 2009) and are common features of MM cell lines, with 30-40% of them presenting damaging lesions of this gene (www.cbioportal.org, www.keatslab.org). Most MM cell lines were established from extramedullary MM and plasma cell leukemia cases, suggesting that UTX loss may contribute to disease progression. Here, we characterized the effect of UTX loss in the biology and gene expression profile of MM. Moreover, we wished to determine whether such alterations could be targeted with the use of epigenetic drugs.
Results
Loss of UTX promotes the proliferation, clonogenicity and adhesion of MM cells
To model in vitro the loss of UTX in MM, we used a pair of cell lines derived from the same MM patient (Hardin et al., 1994, Ridley et al., 1993): ARP-1 is UTX wild-type, while ARD harbors an homozygous deletion encompassing the UTX locus, as determined by CGH and mRNA sequencing. UTX expression was validated by immunoblot (Fig. 1A). A detailed analysis of the cell lines, including spectral karyotyping and array-based CGH, detected some other differences between the cell lines, the most important being the step-wise rearrangement from Xp at the point of UTX loss in ARD (Xp/18q/1q) (Allen K, 2013). ARD cells were transduced with a lentiviral construct that enabled UTX re-expression in a doxycycline-inducible manner, and we selected the amount of doxycycline that generated UTX protein levels similar to those observed in ARP-1 cells (25 ng/ml, Fig. 1A).
Figure 1. Loss of UTX does not alter global levels of H3K27me3 but promotes the proliferation, clonogenicity, adhesion and tumorigenicity of MM cells.
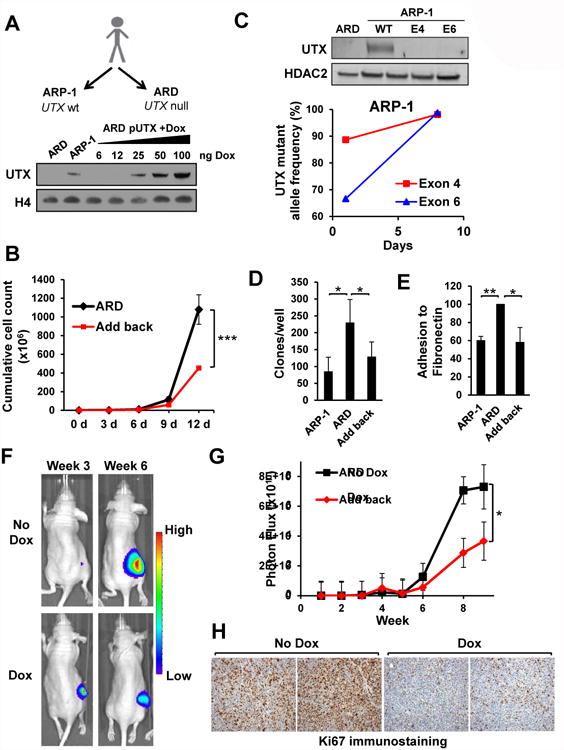
(A) Top: schematic of the isolation of ARP-1 and ARD cell lines from a MM patient and their UTX status. Bottom: ARD cells were stbaly transduced with a lentivirus harboring tetracycline-inducible UTX. Cells were treated with the indicated amounts of doxycycline (ng/ml) for 3 days and nuclear extracts were obtained and immunoblotted with the indicated antibodies. (B) The add-back system in ARD cells was grown in the absence (ARD) or presence (add-back) of doxycycline to re-express UTX. Cells were collected every three days, counted and the initial number of cells replated in fresh media with or without drug. The cumulative number of cells at each time point of three independent experiments +/- SD is represented. (C) CRISPR/Cas9-mediated gene editing was performed in ARP-1 cells targeting the UTX locus using gRNAs targeting exon 4 and exon 6. Top: nuclear extracts were obtained from ARD and ARP-1 cell lines as well as ARP-1 cells transduced with CRISPR/Cas9 systems targeting the UTX locus, and immunoblotted with the indicated antibodies. Bottom: mutant allele frequency as determined by next generation sequencing. (D) ARP-1 and ARD cells harboring the inducible system and with or without doxycyline were cultured in soft agar. The mean colony number per well of three biological triplicates +/- SD is presented. (E) Calcein-AM labeled ARP-1, ARD cells and the add-back system were cultured over fibronectin and adhesion determined by fluorescence intensity. Values are presented as percentage of those obtained for ARD cells. The average of three independent experiments +/- SD is presented. (F) ARD cells harboring the inducible UTX add-back system and stably expressing luciferase were subcutaneouly injected into NOD/SCID mice. Once tumors were formed the mice were randomized and exposed to normal water or water containing doxycycline to re-express UTX. Mice were monitored every week and tumor burden measured using luminescence. Representative images of tumors 3 and 6 weeks after injection are shown. (G) Total flux from mice described in A was quantified and presented over time (weeks) for each group, depicted as means +/-SE. (H) Representative micrographs of immunohistochemistry for Ki67 detection in tumors from control mice (No Dox) or mice treated with doxycycline (Dox). Mann-Whitney U test: * p<0.05; ** p<0.01. See also Figure S1 and S2.
In some cell types, loss of UTX was associated with altered levels of H3K27me3 (Herz et al., 2010, Ho et al., 2013). Mass spectrometry analysis of the inducible system and a panel of cell lines showed no difference in global H3K27me3 levels (Fig. S1A-C), suggesting that H3K27me3 changes due to UTX loss might be loci-specific. Re-expression of UTX resulted in a decrease of the proliferation of ARD cells (Fig. 1B) as determined by cumulative cell counts over a 12-day period. In accordance with this, we observed a modest 5% increase in the percentage of cells in G1 upon UTX repletion, and no increase in cell death (Fig. S1E-F). To further test the potential growth suppressor activity of UTX, we performed high-efficiency CRISPR-mediated knockout of the gene in the ARP-1 cell line (Fig. 1C) as well as in the UTX-wild-type cell lines AMO-1 and Karpas-620 (Fig. S1D) by electroporation of cells with guide RNAs complexed to CAS9 protein. The percentage of mutant alleles was quantified in the transduced cells by next generation sequencing and disruption of UTX protein expression was demonstrated by immunoblotting (Fig. 1C). In all three cell lines, the initial mutant allele fraction was about 50%, but after a week of growth in culture the mutant allele frequency rose to 90-100%, suggesting that UTX mutations confer a proliferative advantage to MM cells (Fig. 1C). While ARD cells were more clonogenic than ARP-1 cells, adding-back UTX partially reversed this phenotype (Fig. 1D, S1G). Moreover, ARD cells showed increased adhesion to fibronectin compared to ARP-1 cells, which was reversed when UTX expression was restored (Fig. 1E). None of these results were observed with the control plasmid (Fig. S1H-J).
We also analyzed another pair of isogenic MM cell lines: SCC1 (UTX wild-type) and SCC2 (UTX-mutant), that were subcloned from the KMS34 cell line. UTX status was analyzed by DNA, qPCR, and RNA-seq (data not shown). UTX add-back into SCC2 cells (Fig. S2A) did not alter global H3K27me3 levels or affect cell death, but decreased cell growth, adhesion to fibronectin and soft agar colony formation (Fig. S2B-F), confirming the effect of UTX loss on the biology of MM cells.
To determine the effect of UTX add-back in vivo, NOD-SCID mice were injected subcutaneously with luciferase-tagged ARD cells harboring the inducible UTX system. UTX re-expression led to a marked decrease in tumor growth (Fig. 1F-G), that was accompanied by a striking reduction of proliferation as measured by Ki67 staining (Fig. 1H), with no increase in apoptotic, cleaved caspase 3-positive cells. Collectively these data suggest that UTX loss may accelerate MM cell growth and alter clonogenic and adhesive properties that may accompany progression of disease.
Identification of the transcriptional program promoted by UTX loss in MM
To ascertain the subsets of genes altered following UTX loss in MM, we performed RNA-seq. While many genes (4,301) were differentially expressed between ARP-1 and ARD cells (FC>1.5, FDR<0.05), UTX add-back for 3 days altered the expression of 107 genes, with most of these (100 genes) being upregulated, consistent with the role of UTX in gene activation. At 6 days, 1,501 genes demonstrated altered expression, with only about half of them (749) being upregulated. A significant overlap (p <10-107, hypergeometric test) was observed between genes showing differential expression in ARD and ARP-1, and genes affected by re-expression of UTX for 3 and 6 days (Fig. 2A). This data is consistent with the idea that the loss of UTX leads to a failure in the activation of some genes, which can be rescued by re-expression of this factor, whereas other genes may gain expression likely through indirect mechanisms (Fig. 2B).
Figure 2. Identification of UTX target genes in MM.
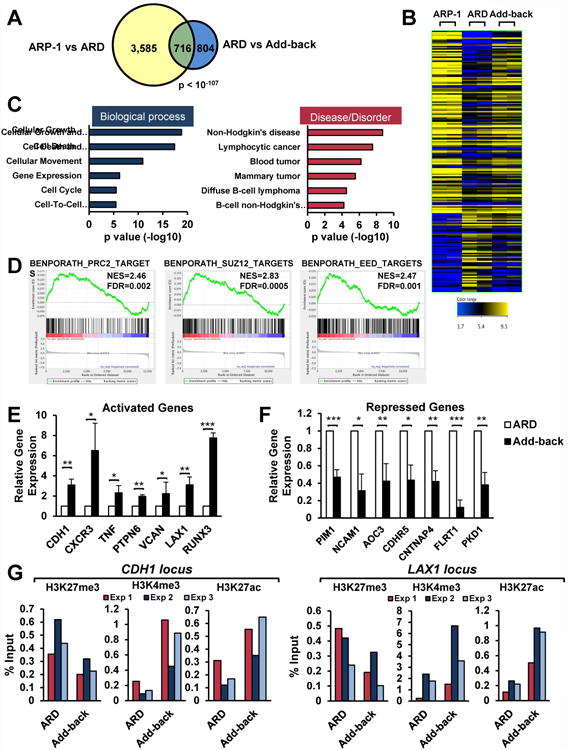
(A) Venn diagram showing a significant overlap of genes differentially expressed between ARP-1 and ARD and between ARD and UTX add-back. (B) Heatmap of RNA-seq analysis in ARP-1 (left), ARD (middle) and add-back (right) (n=2). The genes resulting from the overlap showed in A are depicted. (C) Ingenuity pathway analysis of the top six “Bio Functions” and “Diseases and Disorders” of the differentially expressed genes between ARD and add-back. (D) GSEA plots of datasets identified comparing ARD and add-back RNA-seq signatures. (E-F) Validation of genes responsive to UTX levels by real time PCR. Gene expression normalized to GAPDH, from three independent experiments (+/-SD), is presented relative to that observed in ARD cells. (G) ARD cells harboring the inducible system were treated with doxycyline for 6 days to add-back UTX. Chromatin from these cells was immunoprecipitated with anti-H3K27me3, anti-H3K4me3, or anti-H3K27ac antibodies. Immunoglobulin G was used as negative control. Eluted DNA was PCR amplified with primers designed at LAX1 and CDH1 loci. Three independent experiments are shown. Mann-Whitney U test: * p<0.05; ** p<0.01; *** p<0.001. See also Figure S3.
Consistent with the biological effects observed, genes regulated upon UTX add-back were enriched in the following six processes: cell growth and proliferation, cell death and survival, cell movement, cell cycle, gene expression and cell to cell signaling and interaction, this latter category including adhesion genes (Fig. 2C). Moreover, they were significantly enriched in several types of cancer, mainly hematological malignancies (Fig. 2C). Genes whose expression recovered upon UTX add-back included the epithelial marker E-cadherin (CDH1), whose loss has been linked to MM progression (Azab et al., 2012), the negative regulator of the JAK-STAT pathway PTPN6, and the cell cycle regulator RUNX3 (Fig. 2E). Among genes whose expression decreased upon UTX add-back were stimulators of cell growth such the oncogene PIM1, and a range of adhesion molecules (NCAM1, AOC3, FLRT1 or CDHR5) (Fig. 2F). The expression of many of these genes upon add-back was similar to that observed in UTX wild-type ARP-1 cells, highlighting the relevance of the loss of UTX to alter gene expression profiles and favor the malignant properties of ARD cells (Fig. S3A). Analysis of SCC1 and SCC2, confirmed that many of these genes represent UTX targets in MM cells (Fig. S3B).
Upregulated-genes upon UTX add-back were found by gene set enrichment analysis to correspond to sets of genes previously annotated to be targets of PRC2 and the PRC2 components EED and SUZ12 (Fig. 2D), suggesting that UTX loss leads to abnormal PRC2-mediated repression. Moreover, UTX-regulated genes were also corresponded to MLL2/KMT2D target genes (Fig. S3C), highlighting the known ability of these two proteins to cooperate to activate gene expression (Issaeva et al., 2007, Kim et al., 2014). Along these lines, chromatin immunoprecipitation (ChIP) analysis of some of these loci showed decreased presence of H3K27me3 and an enrichment of H3K4me3 and H3K27ac upon UTX add-back (Fig. 2F). All these findings suggest that the lack of UTX leads to gene repression through the action of PRC2. Restoration of UTX would then counteract PRC2 activity, and induce active chromatin marks by the complex harboring UTX, KMT2D and histone acetylases to ultimately reactivate gene expression.
Loss of UTX sensitizes MM cells to EZH2 inhibitors
We hypothesized that aberrant silencing promoted by UTX loss might be critical for the biology of UTX-mutant cells. Thus, those cells may be more dependent on the PRC2 activity to maintain such repression and, therefore, be more sensitive to EZH2 inhibitors (EZH2i). Therefore, we treated the ARD and ARP-1 cells with the EZH2i GSK343 (Verma et al., 2012), or the inactive compound GSK669. GSK343 treatment produced a global decrease in the levels of H3K27me3 in both cell lines (Fig. 3A-B), however, only UTX-null cells showed a marked decrease in viability (Fig. 3C). GSK343-treated ARD cells displayed a G0/G1 arrest, followed by an increase in apoptosis, whereas the agent did not affect ARP-1 cells (Fig. 3D-E, S4A). In a panel of MM cell lines treated with GSK343, all UTX-mutant cell lines showing decreased viability (Fig. 3F), cell cycle arrest and increased cell death (Fig. S4B-C). Intriguingly, when we tested our other isogenic cell system, both SCC1 and SCC2 were equally sensitive to EZH2i (Fig. S4D). The similar sensitivity may be explained by their genomic profiles, as discussed later.
Figure 3. Loss of UTX confers sensitivity to EZH2i.
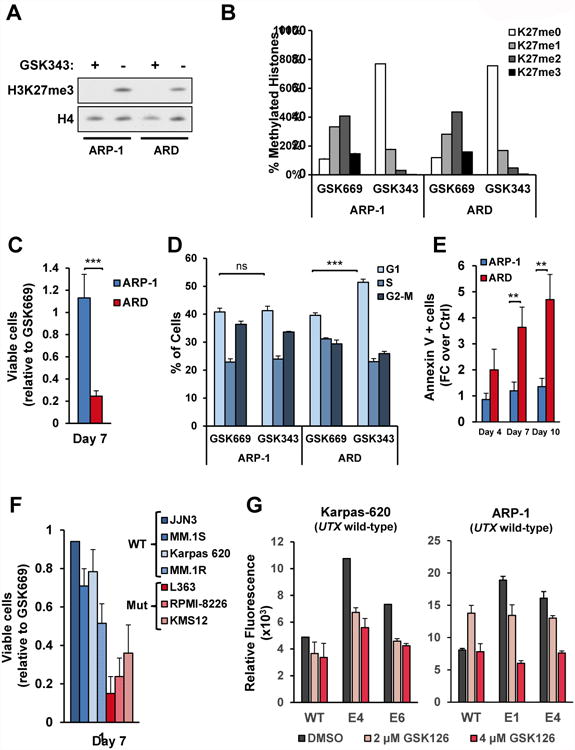
(A) Nuclear extracts from ARP-1 and ARD cells treated with GSK343 (+) or the control small molecule GSK669 (-) for 7 days were immunoblotted with the indicated antibodies. (B) Histones were extracted from ARP-1 and ARD cells treated with GSK669 or GSK343 for 7 days and analyzed by mass spectrometry. The percentage of each methylation state from histone H3K27 is represented in biological duplicates +/- SD. (C) ARP-1 and ARD cells were treated with GSK343 or GSK669 for 7 days. Viable cell counts of cells treated with GSK343 are presented relative to counts of cells treated with GSK669. The average from 4 biological independent experiments +/- SD is shown. (D) ARP-1 and ARD cells were treated with GSK343 or GSK669 for 4 days and the cell cycle profile was analyzed by flow cytometry using propidium iodide. The percentage of cells in each phase from 3 independent biological experiments +/- SD is presented. (E) ARP-1 and ARD cells were treated with GSK343 or GSK669 for 4, 7 or 10 days and Annexin V positive cells were detected by flow cytometry. Values from 3 biological replicates +/- SD are presented as relative to values obtained for GSK669. (F) A panel of UTX mutant and wild-type MM cell lines were treated as in B. Viable cell counts of cells treated with GSK343 are presented as relative to cell counts of cells treated with GSK669. The average from 3 biological independent experiments +/- SD is shown. (G) UTX-wild-type MM cell lines were CRISPR/Cas9-mediated gene edited in the UTX locus using gRNAs targeting two different exons. Cells were treated with GSK126 at the indicated concentrations for 6 days and the relative proliferation was measured using Alamar Blue. Bars represent tehcnical triplicates +/- SD. Biological replicates are found in Fig. S5E. Mann-Whitney U test: ** p<0.01; *** p<0.001. See also Figure S4, S5 and S6.
To validate the increased sensitivity of UTX-mutant cells to EZH2i, we used another EZH2i, GSK126 (McCabe et al., 2012b); and also observed decreased viability and increased cell death of most UTX-mutant cells (Fig. S5). Moreover, UTX-wild-type cells in which UTX gene was disrupted using CRISPR showed a dose-dependent increased sensitivity to GSK126 compared to control cells, further demonstrating the effect of UTX loss in promoting sensitivity to these compounds (Fig. 3G and S5E). As an additional way of inhibiting EZH2, we also transduced ARP-1 and ARD cells with a plasmid harboring wild-type histone H3.3, or H3.3 harboring a K27M mutation, which sequesters and inhibits EZH2 leading to decreased H3K27me3 levels (Lewis et al., 2013). Both ARD and ARP-1 cells showed a global reduction in H3K27me3 (Fig. S6A) upon transduction with the histone mutant, but only UTX-null cells showed decreased viability and cell death (Fig. S6B-C).
To determine whether EZH2i affects the differentiation state of MM cells, as it does in lymphoma (Knutson et al., 2012, Qi et al., 2012), we tested the surface expression of plasma cell (CD38 and CD138) and B-cell GC (CD20) markers. GSK343 treatment promoted a loss of CD38 and CD138 and a gain of CD20 in UTX-null cells, suggesting a de-differentiation process into a more primitive B-cell stage (Fig. S4E). Wild-type cells did not show these phenotypic changes or showed them to a much lesser extent (Fig. S4E). Analysis of additional UTX-mutant and wild-type MM cells confirmed the specificity of this effect for cells showing UTX loss (Fig. S4F).
Differential gene expression profile of UTX-mutant and wild-type cells in response to EZH2 inhibtion
To further study the effect of EZH2i on UTX-mutant MM cells, we performed RNA-seq on ARP-1 and ARD cells treated for 7 days with GSK343 or GSK669. As expected, most affected genes were upregulated upon inhibition of EZH2 (FC>1.5; p<0.05) (Fig. 4A, S7A). There was a highly significant overlap (p < 10-151, hypergeometric test) of genes differentially expressed upon EZH2i treatment of ARD or UTX add-back (Fig. 4B), suggesting that EZH2i can rescue many of the effects caused by UTX loss. Accordingly, the genes affected by EZH2i in UTX-null cells were enriched in very similar pathways to those regulated by UTX add-back (Fig. 4C), and were also involved in B-cell malignancies (Fig. S7B). Examples of genes altered upon both UTX add-back and EZH2 inhibition included E-cadherin, PTPN6, TNF, PIM1 and adhesion molecules (Fig. 4D).
Figure 4. EZH2i partially restore gene expression deregulation mediated by UTX loss and promote de-differentiation of UTX mutant cells.
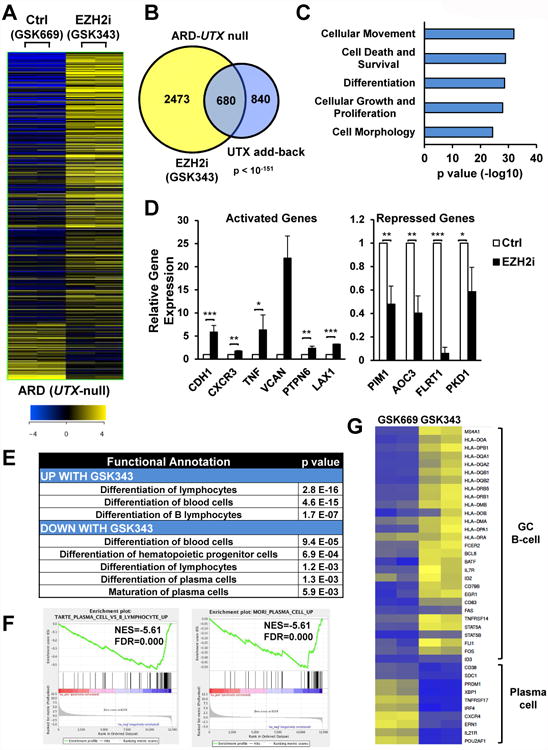
(A) Heatmap of RNA-seq analysis in ARD cells treated with the EZH2i GSK343 4 μM or an inactive control compound (GSK669) for 7 days (n=2) (FC>1.5; p<0.05). The significantly differentially expressed genes comparing both treatments are depicted. (B) Venn diagram showing a significant overlap of genes differentially expressed between ARD and UTX add-back and ARD treated with GSK343 and GSK669. (C) IPA of the top five “Bio Functions” of the differentially expressed genes between ARD treated with GK343 and control cells. (D) Validation of genes responsive to GSK343 by real time PCR. Gene expression normalized to GAPDH, from three independent experiments (+/-SD), is presented relative to that observed relative to that observed in GSK669-treated ARD cells. (E) IPA analysis of differentiation processes in ARD cells treated with GSK343. Function anotations and p-values are shown. (F) GSEA plots of datasets identified comparing ARD treated with GSK669 or GSK343. (G) Heatmap of GC B-cell and plasma cell markers in ARD cells treated with GSK669 or GSK343. Z-score is presented. Mann-Whitney U test: * p<0.05; ** p<0.01; *** p<0.001. See also Figure S7.
GSK343 treatment of ARD cells affected genes involved in differentiation of hematopoietic progenitor cells, lymphocytes and maturation of plasma cells (Fig. 4E). Accordingly, GSEA analysis showed that EZH2i treatment repressed genes that are up-regulated in plasma cells (Fig. 4F). For example, GSK343 promoted the loss of plasma cell markers such as PRDM1, CXCR4, CD38 or SDC1, and acquisition of germinal center (GC) B-cell markers, including HLA genes, MS4A1, STAT5 and BCL6 (Fig. 4G), suggesting that EZH2i evoke a de-differentiation process in MM cells, that is specific to UTX-mutant cells (Fig. S7C).
To ascertain the cause underlying the differential response of UTX-wild-type and mutant cells to EZH2i, we compared the gene expression profiles of ARP-1 and ARD cells after treatment with GSK343. About 1800 genes (FC>1.5; FDR<0.05) were exclusively deregulated in UTX-mutant cells, and were enriched in processes such as cell death and proliferation (Fig. 5A-B). Among these, we observed increased expression of cell cycle arrest and apoptotic genes such as RUNX3 and BIK (Fig. 5C-D). Intriguingly, there was a striking decrease in the levels of the transcription factor IRF4 and its downstream target c-MYC, an effect exclusive to UTX-null ARD cells (Fig. 5C-D, S7D). IRF4 expression is a hallmark of MM, and its knockdown is toxic to these cells (Shaffer et al., 2008). We confirmed that knockdown of IRF4 killed ARP-1 and ARD cells, suggesting that its decrease following EZH2i mediated the death of ARD cells (Fig. S7E-F). As EZH2 is a repressor, we hypothesized that EZH2i indirectly reduced IRF4 expression by induction of a negative regulator of this factor. Indeed, EZH2i treatment of ARD but not ARP-1 cells induced the expression of BCL6, IRF1 and BATF transcription factors (Fig. 5E, 6F-G), all reported to repress IRF4 expression (Ci et al., 2009, Piya et al., 2011, Kurachi et al., 2014). To ascertain the underlying cause of BCL6 exclusive activation in UTX-null cells upon treatment with EZH2i, we performed ChIP for H3K27me3 and H3K27ac in ARD and ARP-1 cells. The BCL6 locus showed a marked decrease in the levels of H3K27me3 in both cell lines upon treatment with EZH2i but, in agreement with the expression data, stronger levels of H3K27ac were observed in UTX-null ARD cells than in ARP-1 cells (Fig. 5F). At the IRF4 locus, a known previously characterized target of BCL6 repression, a decrease in H3K27ac was observed exclusively in ARD cells (Fig. 5F), correlating with the decreased expression observed at mRNA and protein levels. These data suggest that upon EZH2 inhibition, differential regulation of histone acetylation takes place in UTX-null and wild-type cells, leading to different gene expression regulation and ultimately different phenotypes. Finally, we performed drop out experiments to prove that the upregulation of IRF4 negative regulators was the cause of IRF4 knockdown and cell death in UTX-null cells.
Figure 5. EZH2i alter the expression of key MM survival genes exclusively in ARD cells.
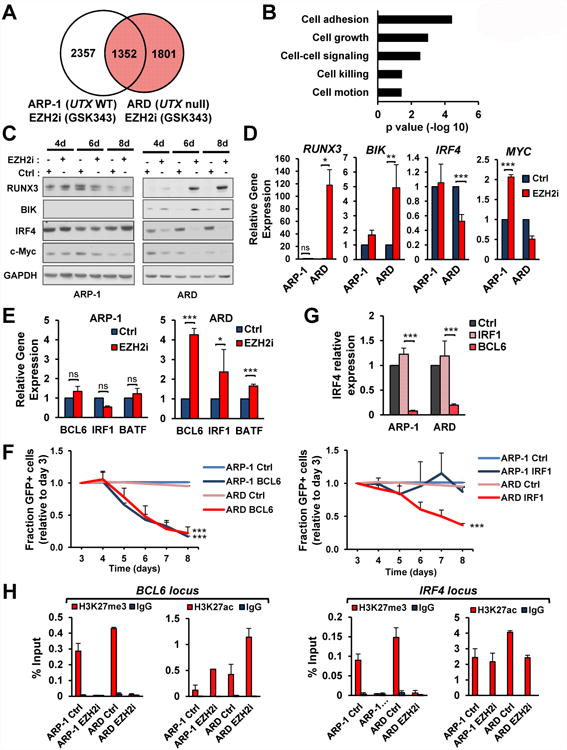
(A) Venn diagram showing the overlap of genes differentially expressed between ARP-1 and ARD treated with GSK343 or GSK669. Genes exclusively altered in ARD cells are shown in red. (B) Ingenuity pathway analysis of the top five “Bio Functions” of the genes altered in ARD cells and not in ARP-1 cells after treatment with GSK343 (shown in red in A). (C) ARD and ARP-1 cells were treated with GSK669 or GSK343 for the indicated times and total proteins were extracted and immunoblotted with the indicated antibodies. (D) RNA was extracted from cells treated as in C and qPCR was performed. Gene expression normalized to GAPDH, from three independent experiments (+/-SD), is presented relative to that observed in GSK669-treated cells. (E) Total RNA was obtained from cells treated as in D. Gene expression normalized to GAPDH, from three independent experiments (+/-SD), is presented relative to that observed in GSK669-treated cells. (F) ARD and ARP-1 cells were treated with GSK343 or with DMSO as control. Chromatin from these cells was immunoprecipitated with anti-H3K27me3 or anti-H3K27ac antibodies. Immunoglobulin G was used as negative control. Eluted DNA was PCR amplified with primers designed at BCL6 and IRF4 loci. Two independent experiments are shown. (G) ARP-1 and ARD cells were transduced with GFP-tagged plasmids harboring BCL6 or IRF1 coding sequence, or a control vector. The percentage of GFP+ cells was analyzed daily by flow cytometry. The average from three independent experiments (+SD), is presented relative to that observed at day 3 post-infection. (H) Cells transduced as in H were selected 3 days after infection by flow cytometry to isolate the GFP positive population. Total RNA was obtained and qPCR was performed. Gene expression normalized to GAPDH, from three independent experiments (+/-SD), is presented relative to that observed in cells transduced with empty vector (Ctrl). Mann-Whitney U test: * p<0.05; ** p<0.01; *** p<0.001. See also Figure S7.
Figure 6. GSK343-resistant ARD cells show low H3K27me3 levels but avoid critical gene expression changes promoted by EZH2i.
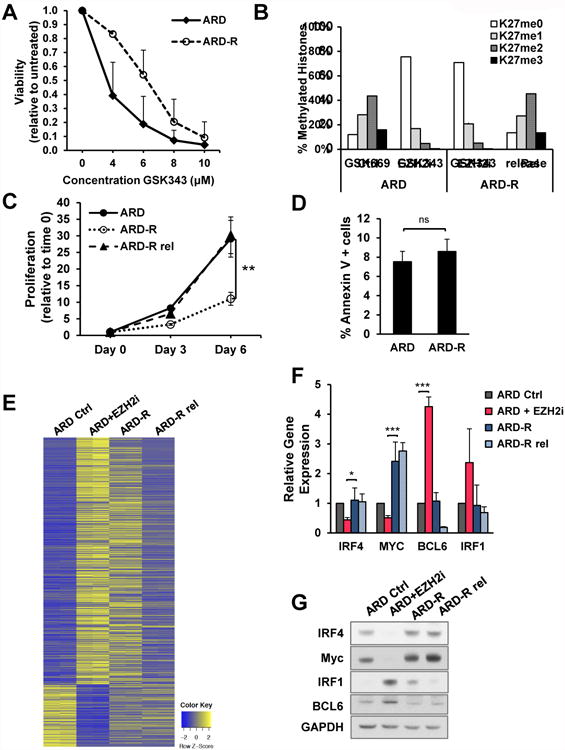
(A) Proliferation of parental and resistant cells treated with the indicated doses of GSK343 for 7 days (three biological replicates; +/-SD). (B) Histones were extracted from ARD cells grown in the presence of GSK669 or GSK343 for 7 days, GSK343-resistant cells growing contirnously in the presence of GSK343 4 μM, or resistant cells in released from the drug for 7 days. Histones were analyzed by mass spectrometry. The percentage of each methylation state from histone H3.1 and H3.2 variants is represented in technical triplicates +/- SD. (C) Proliferation of parental and resistant cells, and resistant cells upon release was measured at 3 and 6 days and values normalized to day 0 (three biological replicates; +/- SD). (D) The percentage of Annexin V positive cells was detected by flow cytometry. (three biological replicates; +/- SD). (E) Heatmap of RNA-seq analysis in ARD cells treated with the EZH2i GSK343 4 mM or a inactive control compound (GSK669) for 7 days, ARD-R cells and ARD-R after release from the drug for 7 days (n=2). The significantly differentially expressed genes comparing treatment with GSK669 and GSK343 are depicted. (F) The expression of the indicated genes was detected by real time PCR in cells described in (B). Gene expression normalized to GAPDH, from three independent experiments (+/-SD), is presented relative to that observed in control ARD cells. (G) Total protein extracts were isolated from cells described in (B) and immunoblotted with the indicated antibodies. Mann-Whitney U test: * p<0.05; ** p<0.01; *** p<0.001. See also Figure S7.
Overexpression of BCL6 or IRF1 from a vector co-expressing GFP in ARD cells resulted in a rapid loss of GFP+ cells from the population over time (Fig. 5G), whereas GFP+ cells persisted when co-expressed with BATF. Expression of BCL6 in ARD cells led to a decrease in IRF4 levels while IRF1 expression did not (Fig. 5H), suggesting that BCL6 induction in response to EZH2i was the underlying cause leading to repression of IRF4 and c-MYC, and loss of MM cell viability.
EZH2 inhibitor-resistant ARD cells show decreased H3K27me3 but bypass IRF4 inhibition
To further study the mechanism by which EZH2i affect MM cells, we generated a GSK343-resistant (ARD-R) cell line. The resistant cell line did not undergo cell death at 4 μM of GSK343, which promotes death of EZH2i-naive cells (Fig. 6A). Mass-spectrometry showed that ARD-R cells maintained low levels of H3K27me3, indicating that GSK343 was still effective in blocking EZH2 activity (Fig. 6B). ARD-R cells proliferate more slowly than parental cells, with growth recovering after removal of the drug from culture media (Fig. 6C-D).
RNA-seq analysis showed that ARD-R cells have an expression profile intermediate between GSK343 treated and untreated cells, and upon removal of drug the expression pattern reverts to that of untreated cells (Fig. 6E). Unlike acute treatment of ARD with EZH2i, ARD-R cells showed levels of IRF4, c-MYC, BCL6 and IRF1 similar to control cells (Fig. 6F-G), suggesting a failure in the EHZ2i-mediated gene activation that provokes MM cell death. RNA-seq also showed that while acute treatment of ARD cells with an EZH2i leads to a loss of plasma cell markers and gain of GC genes, the EZH2i-R cells regained plasma cell genes and showed a partial loss of expression of GC markers, suggesting a more widespread failure of EZH2i-mediated gene regulation. Upon removal of the drug, B-cell gene expression returned to levels similar to that of control cells (Fig. S7G).
EZH2 inhibition decreases growth of UTX-null MM cells in vivo
To test the effect of the EZH2i in vivo, luciferase-tagged ARP-1 and ARD cells were injected into the left and right flank, respectively, of immunosuppressed mice. Once tumors were established half of the mice were treated with GSK126. In control mice, ARP-1 and ARD tumors grew rapidly and animals were sacrificed at 4-5 weeks due to high tumor burden. No significant difference was detected in the size of ARP-1 and ARD tumors in untreated animals (Fig. 7A-C). In mice receiving GSK126, tumors formed by ARP-1 cells grew similarly to those of control mice, while ARD tumor volumes were dramatically reduced by bioluminescence signal and direct measurement (Fig. 7A-C). ARP-1 and ARD tumors from GSK126 treated mice displayed decreased H3K27me3 compared to levels in mice treated with vehicle (Fig. 7D). These results suggest that GSK126 effectively targeted EZH2 in UTX wild-type ARP-1 tumors, but in accordance with in vitro results, growth inhibition only occurred in UTX-deficient cells. Collectively, these data suggest that EZH2i may represent a rational therapy for the treatment of MM cases showing UTX inactivation.
Figure 7. EZH2i specifically decrease tumorigenicity of UTX-null cells in vivo.
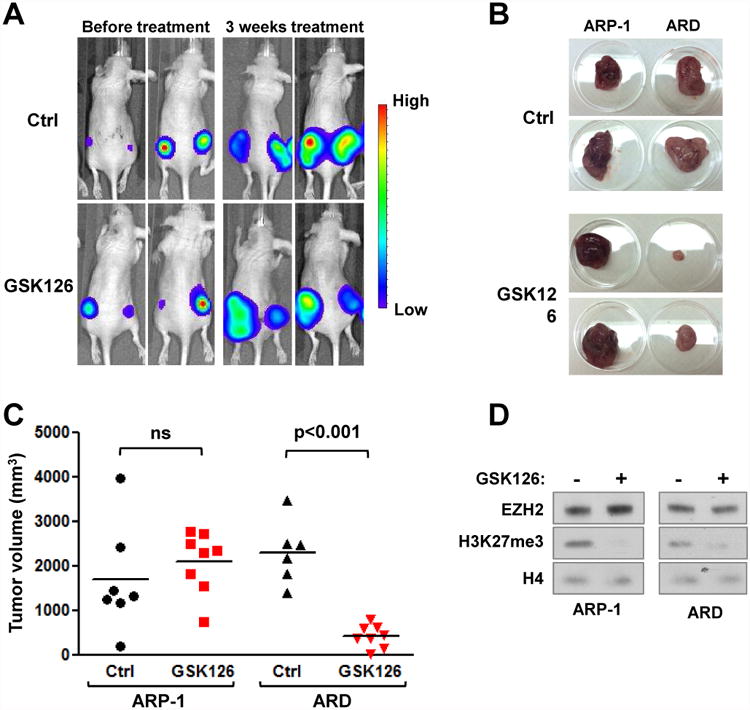
(A) ARP-1 and ARD cells stably expressing luciferase were subcutaneouly injected into NOD/SCID mice. Once tumors were formed were randomized treated with GSK126 50mg/kg or vehicle daily. Mice were monitored every week and tumor burden measured using luminescence. Representative images of mice before (week 0) and after 3 weeks of treatment are shown. (B-C) Mice were sacrifized and tumors were extracted and measured. Representative tumors are shown in B and volumes for all tumors are presented in C. (F) Nuclear proteins were extracted from mice treated with GSK126 or vehicle and blotted with the indicated antibodies.
Discussion
Loss of UTX occurs in a variety of cancers (Jankowska et al., 2011, Jones et al., 2012, van Haaften et al., 2009), being common in advanced urothelial carcinoma and high-risk acute lymphoid leukemia patients (Jankowska et al., 2011, Mar et al., 2012, Ross et al., 2014), suggesting it may be involved in the aggressive biological behavior of tumor progression. UTX disruption is present at low frequency in newly diagnosed patients but is common in MM cell lines (30-40%), many established from extramedullary MM and plasma cell leukemia. This background information and our data suggests that UTX loss may contribute to the progression and dissemination of MM. Loss of UTX leads to decreased levels of E-cadherin, a hallmark of epithelial to mesenchymal transition (EMT) in solid tumors, but a feature also linked to tumor progression in a mouse xenograft model of MM (Azab et al., 2012). UTX loss also promotes the expression of adhesion factors such as NCAM1, AOC3 or CDHR5, which may be involved in cell re-attachment upon dissemination. Moreover, UTX loss promotes the clonogenicity and presumably the self-renewal of MM cells, which is needed for the growth of individual cells once a new niche is reached. The effect of re-expression of UTX in MM cells was more profound in xenografted mice than in in vitro experiments, with a more marked decrease in proliferation noted. This is consistent with the ability of UTX to influence the expression of cell surface proteins that may be critical for tumor growth.
Intriguingly, not UTX loss, but UTX overexpression was associated with increased proliferation and invasiveness of breast cancer (Kim et al., 2014), suggesting that its role in tumorigenesis is context-specific. In accordance with this, UTX target genes also seem to be cell-type specific, as the genes regulated in response to UTX re-expression in UTX-null MM cells bore no similarity to those identified in human fibroblasts and breast cancer cells (Kim et al., 2014, Wang et al., 2010). UTX associates with tissue-specific transcription factors (TFs), directing the protein to specific genes (Miller et al., 2010, Shpargel et al., 2012). Hence, it is likely that the DNA binding transcription factors that interact with UTX in MM, yet to be identified, underlie tissue-specificity in the regulation of target genes in this disease.
The loss of UTX in MM cells leads to the loss of expression of a number of genes. This may be due to the failure of the activator complex of UTX, KMT2C/D and p300/CBP tp engage enhancer regions, opposing the action of the PRC2 complex. This idea is supported by our finding that nearly 45% of genes activated by UTX could also be activated by EZH2 inhibitors, which are in clinical trials and may soon enter clinical practice. EZH2 inhibition may have utility in rebalancing gene repression produced by gain of function mutations of EZH2 (Majer et al., 2012, McCabe et al., 2012a, Morin et al., 2010), loss of SWI/SNF chromatin remodelers (Wilson et al., 2010) and redistribution of EZH2 due to displacement by overexpression of MMSET/NSD2 in t(4:14) MM cells (Popovic et al., 2014). In our isogenic ARP-1/ARD cell system, we have observed a clear difference in EZH2i response, with UTX-null ARD cells being much more sensitive than UTX-wild-type ARP-1 cells. Moreover, we have shown the same results for a panel of MM cell lines with the use of different small molecules targeting EZH2. A notable exception was the isogenic pair KMS34-SCC1 and SCC2 cells, which are t(4:14)+, a genetic alteration that we previously showed also increased sensitivity to EZH2i (Popovic et al., 2014). Interestingly, in both t(4:14)+ and UTX-mutant cases, this enhanced sensitivity seems to be associated with the ability of EZH2i to decrease c-MYC levels, in t(4;14)+ cells due to de-repression of miR-126* which inhibits c-MYC expression (Min et al., 2013, Popovic et al., 2014), and in UTX-deficient cells by inhibition of IRF4 expression which normally stimulates c-MYC expression in MM cells. In a recent paper, the association between UTX status and response to EZH2i was not observed in MM cell lines (Hernando et al., 2016), suggesting that, similar to what we observed with the t(4:14) translocation, other genetic alterations may also affect sensitivity to these drugs. Importantly, a new set of cell lines we created in which UTX was disrupted by gene editing also showed increased sensitivity to EZH2i. Furthermore, the association between UTX loss and increased response to EZH2i was also observed in bladder cancer cells (Ler et al., 2017). This suggests several cancer types harboring loss of UTX may be susceptible of therapeutic intervention with these drugs.
The fact that EZH2i alter the differentiation state of MM cells is analogous to our previous finding that EZH2-dependent germinal center B (GCB) lymphoma cells are sensitive to EZH2i and differentiate towards plasma cells. By contrast, activated B-cell lymphoma cells are not sensitive to EZH2i, and display modest transcriptional changes upon treatment (McCabe et al., 2012b, Beguelin et al., 2013). EZH2i have been recently described to decrease the expression of oncogenes and promote the expression of miRNAs with tumor suppressor functions in MM cells (Alzrigat et al., 2017). In our study, EZH2i activated a large number of genes in both UTX-mutant and wild-type MM cells, but a substantial fraction of these genes were different, with enrichment of genes involved in cell death and regulation of proliferation found exclusively in UTX-mutant cells. Furthermore, EZH2i activated GC-specific genes only in UTX-deficient MM cells, which led to an apparent de-differentiation, induction of BCL6 and subsequent cell death. This implied that a subset of germinal center genes became rewired upon loss of UTX. Once a B-cell passes through the GC-stage, key TFs such as BCL6 decrease in expression and late B-cell factors such as PRDM1 and IRF4 are prominently expressed. PRDM1 represses the expression of BCL6, but other epigenetic regulatory processes are likely to contribute silencing these genes as B-cells mature. In UTX-deficient cells, we postulate that these more permanent silencing mechanisms are defective. In fact, our ChIP data shows how BCL6 seems to be primed for expression in UTX-null cells, showing higher H3K27ac levels at baseline and a more marked increase in this modification upon treatment with EZH2i than UTX-wild-type cells. The priming that BCL6 and some other GC genes may show in UTX-null cells may explain why they are able to de-differentiate upon EZH2i treatment. Further detailed studies of the nature of such chromatin changes at germinal center and later stages in B cell genes in normal differentiation and in UTX-deficient models will be required to further elucidate this process.
All together, our data demonstrates that loss of the histone demethylase UTX alters the transcriptional profile of MM cells contributing to a malignant phenotype. Importantly, the altered transcriptional poise of UTX-deficient cells offers a therapeutic opportunity in MM. Rebalancing UTX/PRC2 activity with the use of EZH2i reactivates tumor suppressor programs and alters differentiation states, ultimately leading to MM cell death. These data highly motivate testing these agents in MM and potentially other tumors displaying loss of UTX.
Experimental procedures
Cell Culture
The human MM cell lines (all gifts of M. Kuehl, National Cancer Institute) were grown in RPMI-1640 medium, supplemented with 10% fetal bovine serum (FBS) (XG-6, MM.1S, RPMI-8226, L363, KMS12, FR4, AMO-1 and MM.1R) or 20% FBS (Karpas-620); or IMDM medium containing 10% FBS (EJM). The isogenic MM cell line pairs ARP-1 and ARD and KMS34 SCC1 and SCC2 were cultured in advanced RPMI supplemented with 4% FBS and glutamax. The matching genotype of ARP-1 and ARD was confirmed by short tandem repeat (STR) profiling (Lab Corp Genetica). Additional details related to resistant cell line generation, gain and loss-of-function systems, protein and RNA isolation and detection, biological assays and ChIP studies can be found in the Supplemental Experimental Procedures.
RNA-Seq
For gene expression profiling studies, RNA was extracted from ARP-1 and ARD cells, the add-back system treated with doxycycline for 3, 6 and 9 days, and ARP-1 and ARD cells treated with 4 μM of GSK343 or GSK669 for 7 days. Total RNA was sequenced on HiSeq 2000. Sequence reads were aligned to human reference genome (hg19) with STAR (v 2.3.0e_r291) and read counts per gene were calculated using HTSeq (v0.6.0). The R (v3.1.1) Bioconductor package, edgeR (v3.8.5) was used to identify statistically significant differentially expressed genes. Before differential gene expression analysis, genes with less than one read per million of sequenced reads in three or more samples were filtered out. Candidate genes at significance level of Benjamini and Hochberg false discovery rate < 0.05 and fold change > 1.5 were entered into Ingenuity Pathway Analysis software to identify biological pathways involving these genes. The list of genes used in differential expression analysis was ranked based on log2 fold change and was imported to Gene Set Enrichment Analysis tool. Curated gene sets were obtained from MSigDB and were tested using GseaPreranked module for enrichment in the list of genes.
Statistical Analysis
Data obtained from proliferation, soft agar, adhesion, apoptosis, cell cycle, tumor growth in mice, fraction of GFP+ cells over time, real time PCR and ChIP assays were analyzed using the Student's T test or Mann-Whitney U test, according to a parametric o non-parametric distribution of data, respectively, Statistical analysis was done using the SPSS software package, version 15.0 (IBM).
Supplementary Material
Highlights.
UTX loss promotes MM proliferation, clonogenicity, adhesion andtumorigenicity.
UTX loss confers sensitivity to EZH2 inhibition, both in vitro and in vivo.
Enhanced EZH2i sensitivity is associated with a decrease in IRF4 and c-MYClevels.
EZH2i induces BCL6 expression leading to IRF4 and c-Myc repression inUTX-null cells.
Acknowledgments
This work was supported by awards by a Multiple Myeloma Research Foundation (MMRF) Fellow award (TE), the Epigenetics in Multiple Myeloma program grant (MMRF) (JDL), R01 CA180475 (JDL), a Leukemia and Lymphoma Society Specialized Center of Excellence grant (JDL), and P41 GM108569) (NLK).
Footnotes
Lead contact: Jonathan D. Licht, jdlicht@ufl.edu
Author contributions: T.E. D.D-R. and J.D.L designed the research and interpreted the data.
T.E. and D.D-R. performed the experiments with assistance from C.M.W., E.S., T.P., B.N., J.O., M. B., R.P., M. Y. S., X.H., and Y. Z.
T.E and N.V. performed the RNA-seq analyses.
T.E. and J.D.L. wrote the manuscript.
Accession Numbers: RNA-seq data reported in this paper has been deposited to the NCBI GEO and are available under accession number GEO: GSE103567.
Conflict of Interest: JDL and RP are co-inventors in USPO application 20170105997.
This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Allen K, Y V, Zhao Q, Edwards D, Chesi M, Bergsagel L, Carpten J, Keats J. Abstract 5209: Characterization of an isogenic model system for KDM6A/UTX loss in multiple myeloma. Cancer Res 2013 [Google Scholar]
- Alzrigat M, Parraga AA, Agarwal P, Zureigat H, Osterborg A, Nahi H, Ma A, Jin J, Nilsson K, Oberg F, et al. EZH2 inhibition in multiple myeloma downregulates myeloma associated oncogenes and upregulates microRNAs with potential tumor suppressor functions. Oncotarget. 2017;8:10213–10224. doi: 10.18632/oncotarget.14378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azab AK, Hu J, Quang P, Azab F, Pitsillides C, Awwad R, Thompson B, Maiso P, Sun JD, Hart CP, et al. Hypoxia promotes dissemination of multiple myeloma through acquisition of epithelial to mesenchymal transitionlike features. Blood. 2012;119:5782–94. doi: 10.1182/blood-2011-09-380410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beguelin W, Popovic R, Teater M, Jiang Y, Bunting KL, Rosen M, Shen H, Yang SN, Wang L, Ezponda T, et al. EZH2 is required for germinal center formation and somatic EZH2 mutations promote lymphoid transformation. Cancer Cell. 2013;23:677–92. doi: 10.1016/j.ccr.2013.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ci W, Polo JM, Cerchietti L, Shaknovich R, Wang L, Yang SN, Ye K, Farinha P, Horsman DE, Gascoyne RD, et al. The BCL6 transcriptional program features repression of multiple oncogenes in primary B cells and is deregulated in DLBCL. Blood. 2009;113:5536–48. doi: 10.1182/blood-2008-12-193037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ezponda T, Licht JD. Molecular pathways: deregulation of histone h3 lysine 27 methylation in cancer-different paths, same destination. Clin Cancer Res. 2014;20:5001–8. doi: 10.1158/1078-0432.CCR-13-2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardin J, Macleod S, Grigorieva I, Chang R, Barlogie B, Xiao H, Epstein J. Interleukin-6 prevents dexamethasone-induced myeloma cell death. Blood. 1994;84:3063–70. [PubMed] [Google Scholar]
- Hernando H, Gelato KA, Lesche R, Beckmann G, Koehr S, Otto S, Steigemann P, Stresemann C. EZH2 Inhibition Blocks Multiple Myeloma Cell Growth through Upregulation of Epithelial Tumor Suppressor Genes. Mol Cancer Ther. 2016;15:287–98. doi: 10.1158/1535-7163.MCT-15-0486. [DOI] [PubMed] [Google Scholar]
- Herz HM, Madden LD, Chen Z, Bolduc C, Buff E, Gupta R, Davuluri R, Shilatifard A, Hariharan IK, Bergmann A. The H3K27me3 demethylase dUTX is a suppressor of Notch- and Rb-dependent tumors in Drosophila. Mol Cell Biol. 2010;30:2485–97. doi: 10.1128/MCB.01633-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho AS, Kannan K, Roy DM, Morris LG, Ganly I, Katabi N, Ramaswami D, Walsh LA, Eng S, Huse JT, et al. The mutational landscape of adenoid cystic carcinoma. Nat Genet. 2013;45:791–8. doi: 10.1038/ng.2643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Issaeva I, Zonis Y, Rozovskaia T, Orlovsky K, Croce CM, Nakamura T, Mazo A, Eisenbach L, Canaani E. Knockdown of ALR (MLL2) reveals ALR target genes and leads to alterations in cell adhesion and growth. Mol Cell Biol. 2007;27:1889–903. doi: 10.1128/MCB.01506-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jankowska AM, Makishima H, Tiu RV, Szpurka H, Huang Y, Traina F, Visconte V, Sugimoto Y, Prince C, O'keefe C, et al. Mutational spectrum analysis of chronic myelomonocytic leukemia includes genes associated with epigenetic regulation: UTX, EZH2, and DNMT3A. Blood. 2011;118:3932–41. doi: 10.1182/blood-2010-10-311019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones DT, Jager N, Kool M, Zichner T, Hutter B, Sultan M, Cho YJ, Pugh TJ, Hovestadt V, Stutz AM, et al. Dissecting the genomic complexity underlying medulloblastoma. Nature. 2012;488:100–5. doi: 10.1038/nature11284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JH, Sharma A, Dhar SS, Lee SH, Gu B, Chan CH, Lin HK, Lee MG. UTX and MLL4 coordinately regulate transcriptional programs for cell proliferation and invasiveness in breast cancer cells. Cancer Res. 2014;74:1705–17. doi: 10.1158/0008-5472.CAN-13-1896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knutson SK, Wigle TJ, Warholic NM, Sneeringer CJ, Allain CJ, Klaus CR, Sacks JD, Raimondi A, Majer CR, Song J, et al. A selective inhibitor of EZH2 blocks H3K27 methylation and kills mutant lymphoma cells. Nat Chem Biol. 2012;8:890–6. doi: 10.1038/nchembio.1084. [DOI] [PubMed] [Google Scholar]
- Kurachi M, Barnitz RA, Yosef N, Odorizzi PM, Diiorio MA, Lemieux ME, Yates K, Godec J, Klatt MG, Regev A, et al. The transcription factor BATF operates as an essential differentiation checkpoint in early effector CD8+ T cells. Nat Immunol. 2014;15:373–83. doi: 10.1038/ni.2834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ler LD, Ghosh S, Chai X, Thike AA, Heng HL, Siew EY, Dey S, Koh LK, Lim JQ, Lim WK, et al. Loss of tumor suppressor KDM6A amplifies PRC2-regulated transcriptional repression in bladder cancer and can be targeted through inhibition of EZH2. Sci Transl Med. 2017;9 doi: 10.1126/scitranslmed.aai8312. [DOI] [PubMed] [Google Scholar]
- Lewis PW, Muller MM, Koletsky MS, Cordero F, Lin S, Banaszynski LA, Garcia BA, Muir TW, Becher OJ, Allis CD. Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science. 2013;340:857–61. doi: 10.1126/science.1232245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majer CR, Jin L, Scott MP, Knutson SK, Kuntz KW, Keilhack H, Smith JJ, Moyer MP, Richon VM, Copeland RA, et al. A687V EZH2 is a gain-of-function mutation found in lymphoma patients. FEBS Lett. 2012;586:3448–51. doi: 10.1016/j.febslet.2012.07.066. [DOI] [PubMed] [Google Scholar]
- Mar BG, Bullinger L, Basu E, Schlis K, Silverman LB, Dohner K, Armstrong SA. Sequencing histone-modifying enzymes identifies UTX mutations in acute lymphoblastic leukemia. Leukemia. 2012;26:1881–3. doi: 10.1038/leu.2012.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mccabe MT, Graves AP, Ganji G, Diaz E, Halsey WS, Jiang Y, Smitheman KN, Ott HM, Pappalardi MB, Allen KE, et al. Mutation of A677 in histone methyltransferase EZH2 in human B-cell lymphoma promotes hypertrimethylation of histone H3 on lysine 27 (H3K27) Proc Natl Acad Sci U S A. 2012a;109:2989–94. doi: 10.1073/pnas.1116418109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mccabe MT, Ott HM, Ganji G, Korenchuk S, Thompson C, Van Aller GS, Liu Y, Graves AP, Della Pietra A, 3rd, Diaz E, et al. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature. 2012b;492:108–12. doi: 10.1038/nature11606. [DOI] [PubMed] [Google Scholar]
- Miller SA, Mohn SE, Weinmann AS. Jmjd3 and UTX play a demethylase-independent role in chromatin remodeling to regulate T-box family member-dependent gene expression. Mol Cell. 2010;40:594–605. doi: 10.1016/j.molcel.2010.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min DJ, Ezponda T, Kim MK, Will CM, Martinez-Garcia E, Popovic R, Basrur V, Elenitoba-Johnson KS, Licht JD. MMSET stimulates myeloma cell growth through microRNA-mediated modulation of c-MYC. Leukemia. 2013;27:686–94. doi: 10.1038/leu.2012.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morin RD, Johnson NA, Severson TM, Mungall AJ, An J, Goya R, Paul JE, Boyle M, Woolcock BW, Kuchenbauer F, et al. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat Genet. 2010;42:181–5. doi: 10.1038/ng.518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ntziachristos P, Tsirigos A, Welstead GG, Trimarchi T, Bakogianni S, Xu L, Loizou E, Holmfeldt L, Strikoudis A, King B, et al. Contrasting roles of histone 3 lysine 27 demethylases in acute lymphoblastic leukaemia. Nature. 2014;514:513–7. doi: 10.1038/nature13605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawlyn C, Kaiser M, Heuck C, Melchor L, Wardell C, Murison A, Chavan S, Johnson DC, Begum DB, Dahir N, et al. The spectrum and clinical impact of epigenetic modifier mutations in myeloma. Clin Cancer Res. 2016 doi: 10.1158/1078-0432.CCR-15-1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piya S, Moon AR, Song PI, Hiscott J, Lin R, Seol DW, Kim TH. Suppression of IRF4 by IRF1, 3, and 7 in Noxa expression is a necessary event for IFN-gamma-mediated tumor elimination. Mol Cancer Res. 2011;9:1356–65. doi: 10.1158/1541-7786.MCR-11-0185. [DOI] [PubMed] [Google Scholar]
- Popovic R, Martinez-Garcia E, Giannopoulou EG, Zhang Q, Zhang Q, Ezponda T, Shah MY, Zheng Y, Will CM, Small EC, et al. Histone methyltransferase MMSET/NSD2 alters EZH2 binding and reprograms the myeloma epigenome through global and focal changes in H3K36 and H3K27 methylation. PLoS Genet. 2014;10:e1004566. doi: 10.1371/journal.pgen.1004566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi W, Chan H, Teng L, Li L, Chuai S, Zhang R, Zeng J, Li M, Fan H, Lin Y, et al. Selective inhibition of Ezh2 by a small molecule inhibitor blocks tumor cells proliferation. Proc Natl Acad Sci U S A. 2012;109:21360–5. doi: 10.1073/pnas.1210371110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridley RC, Xiao H, Hata H, Woodliff J, Epstein J, Sanderson RD. Expression of syndecan regulates human myeloma plasma cell adhesion to type I collagen. Blood. 1993;81:767–74. [PubMed] [Google Scholar]
- Ross JS, Wang K, Al-Rohil RN, Nazeer T, Sheehan CE, Otto GA, He J, Palmer G, Yelensky R, Lipson D, et al. Advanced urothelial carcinoma: next-generation sequencing reveals diverse genomic alterations and targets of therapy. Mod Pathol. 2014;27:271–80. doi: 10.1038/modpathol.2013.135. [DOI] [PubMed] [Google Scholar]
- Shaffer AL, Emre NC, Lamy L, Ngo VN, Wright G, Xiao W, Powell J, Dave S, Yu X, Zhao H, et al. IRF4 addiction in multiple myeloma. Nature. 2008;454:226–31. doi: 10.1038/nature07064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shilatifard A. The COMPASS family of histone H3K4 methylases: mechanisms of regulation in development and disease pathogenesis. Annu Rev Biochem. 2012;81:65–95. doi: 10.1146/annurev-biochem-051710-134100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shpargel KB, Sengoku T, Yokoyama S, Magnuson T. UTX and UTY demonstrate histone demethylase-independent function in mouse embryonic development. PLoS Genet. 2012;8:e1002964. doi: 10.1371/journal.pgen.1002964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Der Meulen J, Sanghvi V, Mavrakis K, Durinck K, Fang F, Matthijssens F, Rondou P, Rosen M, Pieters T, Vandenberghe P, et al. The H3K27me3 demethylase UTX is a gender-specific tumor suppressor in T-cell acute lymphoblastic leukemia. Blood. 2015;125:13–21. doi: 10.1182/blood-2014-05-577270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Haaften G, Dalgliesh GL, Davies H, Chen L, Bignell G, Greenman C, Edkins S, Hardy C, O'meara S, Teague J, et al. Somatic mutations of the histone H3K27 demethylase gene UTX in human cancer. Nat Genet. 2009;41:521–3. doi: 10.1038/ng.349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma SK, Tian X, Lafrance LV, Duquenne C, Suarez DP, Newlander KA, Romeril SP, Burgess JL, Grant SW, Brackley JA, et al. Identification of Potent, Selective, Cell-Active Inhibitors of the Histone Lysine Methyltransferase EZH2. ACS Med Chem Lett. 2012;3:1091–6. doi: 10.1021/ml3003346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JK, Tsai MC, Poulin G, Adler AS, Chen S, Liu H, Shi Y, Chang HY. The histone demethylase UTX enables RB-dependent cell fate control. Genes Dev. 2010;24:327–32. doi: 10.1101/gad.1882610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson BG, Wang X, Shen X, Mckenna ES, Lemieux ME, Cho YJ, Koellhoffer EC, Pomeroy SL, Orkin SH, Roberts CW. Epigenetic antagonism between polycomb and SWI/SNF complexes during oncogenic transformation. Cancer Cell. 2010;18:316–28. doi: 10.1016/j.ccr.2010.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.