

Abstract
Platelet‐derived growth factor (PDGF) can promote vascular smooth muscle cells (VSMCs) to switch from the quiescent contractile phenotype to synthetic phenotype, which contributes to atherosclerosis. We aimed to investigate the role of microRNA let‐7g in phenotypic switching. Bioinformatics prediction was used to find let‐7g target genes in the PDGF/mitogen‐activated protein kinase kinase kinase 1 (MEKK1)/extracellular signal‐regulated kinase (ERK)/Krüppel‐like factor‐4 (KLF4) signalling pathway that affects VSMC phenotypic switching. The luciferase reporter assay and let‐7g transfection were used to confirm let‐7g target genes. Two contractile proteins alpha‐smooth muscle actin (α‐SMA) and calponin were VSMC‐specific genes and were measured as the indicators for VSMC phenotype. Lentivirus carrying the let‐7g gene was injected to apolipoprotein E knockout (apoE−/−) mice to confirm let‐7g's effect on preventing atherosclerosis. Through the PDGF/MEKK1/ERK/KLF4 signalling pathway, PDGF‐BB can inhibit α‐SMA and calponin. The PDGFB and MEKK1 genes were predicted to harbour let‐7g binding sites, which were confirmed by our reporter assays. Transfection of let‐7g to VSMC also reduced PDGFB and MEKK1 levels. Moreover, we showed that let‐7g decreased phosphorylated‐ERK1/2 while had no effect on total ERK1/2. KLF4 can reduce VSMC‐specific gene expression by preventing myocardin–serum response factor (SRF) complex from associating with these gene promoters. The immunoprecipitation assay showed that let‐7g decreased the interaction between KLF4 and SRF. Further experiments demonstrated that let‐7g can increase α‐SMA and calponin levels to maintain VSMC in the contractile status. Injection of lentivirus carrying let‐7g gene increased let‐7g's levels in aorta and significantly decreased atherosclerotic plaques in the apoE−/− mice. We demonstrated that let‐7g reduces the PDGF/MEKK1/ERK/KLF4 signalling to maintain VSMC in the contractile status, which further reduce VSMC atherosclerotic change.
Keywords: microRNA let‐7g, PDGF, vascular smooth muscle cell, α‐SMA, calponin atherosclerosis
Introduction
Proliferation and phenotypic switching of vascular smooth muscle cells (VSMCs) is an important pathogenesis for the development of atherosclerotic plaque 1. Oxidized low‐density lipoprotein (oxLDL) can induce platelet‐derived growth factor (PDGF) secretion 2 and increase PDGF receptor β (PDGFRβ) activation in VSMCs 3. Platelet‐derived growth factor exists as PDGF‐AA or PDGF‐BB homodimer, or PDGF‐AB heterodimer. It has also been found that PDGF‐BB and PDGFRβ are expressed in VSMCs within atherosclerotic lesions, and that inhibition of PDGF‐BB and PDGFRβ reduces atherosclerotic lesion size 4, 5, 6, 7, 8, 9. Platelet‐derived growth factor can promote VSMCs to switch from the quiescent contractile phenotype (also known as differentiated state) to the ‘synthetic’ phenotype (also known as dedifferentiated state) by inhibiting expression of VSMC‐specific marker genes such as alpha‐smooth muscle actin (α‐SMA) and calponin 10, 11. Vascular smooth muscle cells of synthetic phenotype is a key element in atherosclerotic development because this type of VSMCs can migrate, proliferate and generate extracellular matrix proteins 12.
The switching of VSMC phenotypes between the contractile type and synthetic type is regulated by Krüppel‐like factor‐4 (KLF4), myocardin and serum response factor (SRF). Myocardin is a coactivator of SRF and KLF4 can affect both myocardin and SRF. Krüppel‐like factor‐4 can suppress myocardin expression 13. In addition, KLF4 can also inhibit myocardin/SRF complex from binding to the CArG box 13 that is found in the promoters of most VSMC‐specific marker genes. For example, the expression of α‐SMA and calponin that are VSMC‐specific contractile proteins is regulated by myocardin and SRF 14. Platelet‐derived growth factor‐BB up‐regulates KLF4 through the mitogen‐activated protein kinase kinase kinase 1 (MEKK1)/extracellular signal‐regulated kinase (ERK)/SP‐1 pathway 15, 16. Accordingly, PDGF‐BB mediates VSMC phenotypic switching through down‐regulation of myocardin 15.
MicroRNAs (miRNAs) are a family of small (~22 nucleotides) noncoding RNA that negatively regulate the expression of target genes 17. Previous studies from our group have shown that microRNA let‐7g can prevent the uptake of oxLDL into VSMC by inhibiting LOX‐1 expression 18. Our group also demonstrated that let‐7g can improve endothelial functions by targeting the transforming growth factor β (TGF‐β) pathway and by increasing sirtuin‐1 (SIRT‐1) expression 19. This study aimed to examine let‐7g's roles in the PDGF/MEKK1/ERK/KLF4 pathway and in regulating VSMC‐specific marker genes during VSMC phenotypic switching.
Materials and methods
Materials
Primary human aortic smooth muscle cells (HASMC), medium 231 and smooth muscle cell growth supplement (SMGS) were purchased from Cascade Biologics (Portland, OR, USA). Other cell culture‐related reagents were purchased from GIBCO‐BRL (Grand Island, NY, USA). Trizol® reagent, Lipofectamine 2000, secondary antibodies were purchased from Invitrogen (Carlsbad, CA, USA). SYBR® Green PCR Master Mix, MultiScribe(tm) Reverse Transcriptase Kit, TaqMan® let‐7g and U44 Assays and let‐7g mimic were purchased from Applied Biosystems (Foster City, CA, USA). Primer sets were synthesized from Mission Biotech. Protein G plus/protein A‐agarose and PCR Master Mix were purchased from Thermo Fisher Scientific Inc (Waltham, MA, USA). Unless otherwise specified, all other reagents were of analytical grade. Recombinant human PDGF‐BB was purchased from R&D Systems (Minneapolis, MN, USA). Antibodies against α‐tubulin, α‐SMA and calponin were purchased from Abcam (Cambridge, UK). Antibodies recognizing phospho‐ERK1/2 (Thr202/Tyr204) and ERK1/2 were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Antibodies against KLF4 and SRF were obtained from Proteintech (Chicago, IL, USA).
Let‐7g expressing lentivirus
A lentivirus vector named pCDH‐CMV‐MCS‐EF1 containing a CMV‐driven EGFP reporter was purchased from LabLife Co., Ltd. (Cambridge, MA, USA). The full‐length sequence of let‐7g was amplified from human genomic DNA by PCR. Primer pairs for constructing lentivirus vectors were let‐7g forward: TATGAATTCAGCTTTGCTGCCAAGCCTCTGCTGTG and let‐7g reverse: TATGGATCCCCTAAGAAGAAAAAGACTTCCTCCCC. The PCR products were annealed and inserted between the EcoRI and BamHI sites of the plasmid. The positive clones were identified as lentiviral vectors and pCDH‐let‐7g. Correct insertions of pCDH‐let‐7g plasmids were confirmed by restriction mapping and DNA sequencing to ensure accuracy.
Lentivirus production
Recombinant lentiviruses were produced by cotransfecting 293T cells with the lentivirus expression plasmid and packaging plasmids using Lipofectamine 2000. Infectious lentiviruses were harvested at 72 hrs post‐transfection, centrifuged to eliminate cell debris and then filtered through 0.22‐μm cellulose acetate filters (Millipore, Billerica, MA, USA). Infectious titre was determined by counting GFP positive‐293T cells using the fluorescence microscope. Virus titres were at the range of 108 transducing units/ml medium. Determination of the lentiviral titre allowed us to estimate the multiplicity of infection (MOI) and thus to deduce the infectious activity of the viral stocks. The pCDH mock vector was also packaged and used as a negative control, which has no significant homology to mouse gene sequences.
Lentiviral vector transduction in cultured HASMCs
On the day of transduction, cells were replated at 5 × 103 cells/well in 96‐well plates along with recombinant lentivirus encoding for let‐7g gene at different MOIs in serum‐free growth medium containing 5 mg/ml polybrene at 37°C and 5% CO2. After 4 hrs, serum containing growth medium was added to the cells, and there was complete replacement of growth medium after 48 hrs. Then, after days post‐transfection, reporter gene expression was examined using fluorescent microscopy and quantitative real‐time PCR (qPCR).
Apolipoprotein E knockout mice study
The apolipoprotein E knockout (apoE−/−) mice were originally purchased from Jackson Laboratory (Bar Harbor, MI, USA). ApoE−/− mice aged 8 weeks were fed with the Western diet containing 0.25% cholesterol and 15% cacao butter (Testdiet® 5TJN), and were allocated to receive either the pCDH‐let‐7g (n = 6) treatment or placebo PBS/control lentivirus (n = 6) by tail vein hydrodynamic injection (1 × 107 TU diluted in 1% w/v body weight PBS, typically 0.2–0.25 ml). Mice were injected once per week for 12 weeks. Weight gain was monitored every week, and food intake was monitored twice daily during the period of each study. Mice were maintained in a temperature‐controlled (25°C) facility with a strict 12‐hr light: dark cycle. All animals were provided free access to food and water throughout the experiment.
After the 12‐week treatment, mice were killed using an overdose of isoflurane anaesthesia. Mice were perfused with PBS, followed by 10% sucrose in PBS. The blood and aorta were collected for further studies. The blood biochemical tests were conducted by Kaohsiung Medical University Hospital. To detect atherosclerotic plaques, aortas were stained by oil red, and the lesion areas were quantified by densitometer. For measuring the let‐7g levels, total RNA was extracted from aorta tissues. Let‐7g and U6B levels were, respectively, determined by quantitative real‐time PCR. The U6B levels were used as internal control.
Cell culture and transfection
Human aortic smooth muscle cells were grown in medium 231 supplemented with SMGS at 37°C in a humidified atmosphere of 95% air/5% CO2. Cells between passages 6 and 10 were used in all experiments. For transfection experiment, cells were seeded onto six‐well plate at a density of 2 × 105 cells/well. Cells achieving 95% confluence were transfected with let‐7g mimic or let‐7g inhibitor using the HiPerFect Transfection Reagent (Qiagen, Hilden, Germany).
RNA isolation and qPCR
Total RNA extraction was carried out using Trizol® according to the manufacturer instructions. RNA quality was confirmed using A260/A280 readings. The cDNA was synthesized from 1 μg total RNA using a random primer and the MultiScribe(tm) Reverse Transcriptase Kit. For quantitative real‐time PCR, specific primers for human or mouse MEKK1, PDGFB, α‐SMA, calponin and GAPDH are listed in Table S1. For let‐7g and U44 detection, cDNA was synthesized from TaqMan® MicroRNA Assays. The cDNA was diluted 1:30 with PCR grade water and stored at −20°C. Relative quantification of gene expression was performed with pre‐optimized conditions using the ABI 7500 real‐time PCR machine (Applied Biosystems). The expression ratios were calculated as the normalized CT difference between the control and pCDH‐let‐7g with the adjustment for the amplification efficiency relative to the expression level of the U44.
Luciferase reporter assay
To conduct the luciferase reporter assay, we amplified the PDGFB and MEKK1 3′‐untranslated region (3′‐UTR) regions by PCR using specific primers of which the forward primer was SpeI‐site‐linked and the reverse primer MluI‐site‐linked. Human aortic smooth muscle cell genomic DNA was used as the template. PCR products were digested with SpeI and MluI and cloned downstream of the luciferase gene in the pMIR‐REPORT luciferase vector (Ambion, Austin, TX, USA). These vectors were sequenced and named pMIR‐PDGFB‐3UTR and pMIR‐MEKK1‐3UTR. To further make sure that let‐7g directly targets to the PDGFB and MEKK1 3′‐UTR, we conducted the site‐directed mutagenesis to mutate the let‐7g target site in the PDGFB and MEKK1 3′‐UTR. The site‐directed mutagenesis was carried out using the QuikChangeH Site‐Directed Mutagenesis Kit (Stratagene, La Jolla, CA, USA). The pMIR‐PDGFB‐3UTR and pMIR‐MEKK1‐3UTR were used as a template. For reporter assays, the cells were transiently transfected with wild‐type or mutant reporter plasmid and let‐7g expressing lentivirus using Lipofectamine 2000. pEGFP plasmids were cotransfected and acted as the internal control. Reporter assay was performed at 24 hrs post‐transfection using the Luciferase Assay System (Promega, Madison, WI, USA).
Western blot analysis
For the Western blot analysis, HASMCs were lysed with buffer containing 20 mM Tris–HCl (pH 7.4), 150 mM NaCl, 1% Tween 20, 0.1% SDS, 1 mM ethylenediaminetetraacetic acid and complete protease inhibitor cocktail (Roche Applied Science, Mannheim, Germany). Twenty micrograms of proteins was subject to SDS‐PAGE, and then, electrophoresed proteins were transferred to polyvinylidene difluoridemembrane (Millipore). The immunoblots were incubated with primary antibodies overnight at 4°C. After washing, the immunoblots were incubated with horseradish peroxidase‐conjugated secondary antibodies for 1 hr at 4°C. The peroxidase‐conjugated secondary antibody was visualized using enhanced chemiluminescence (ECL) chemiluminescent detection system (PerkinElmer, Shelton, CT, USA) according to the manufacturer's instructions.
Immunofluorescence microscopy
Human aortic smooth muscle cells were treated with PDGF‐BB (5 ng/ml) for 48 hrs before let‐7g transfection (5 nM) for 24 hrs on poly (l‐lysine)‐coated glass coverslips in 6‐well plates. Then, cells were fixed in 4% formaldehyde for 15 min. and permeabilized with 0.2% Triton X‐100 for 5 min. The coverslips were washed with 0.05% TBST three times and blocked with 5% bovine serum albumin for 1 hr at 37°C. Immunofluorescence staining was performed using anti‐calponin (1:200) and anti‐α‐SMA (1:200) antibodies. The cells were counterstained with 4′,6‐diamidino‐2‐phenylindole (DAPI) to identify the nuclei.
Co‐immunoprecipitation assay
Human aortic smooth muscle cell extract was precipitated with 1 μg of KLF4 or 1 μg of SRF antibodies for 4 hrs at 4°C, and antibody–protein complex was incubated with protein G plus/protein A‐agarose (Santa Cruz Biotechnology) for overnight at 4°C. The immune complexes were centrifuged and washed four times with buffer containing 20 mM Tris‐HCl, 150 mM NaCl, 0.1% Triton X‐100 and 10% glycerol. Agarose bead was then subject to the Western blotting analyses.
Statistical analysis
Student's t‐test was used to compare all experimental results. A P‐value less than 0.05 was considered significant.
Result
Let‐7g down‐regulates PDGF‐BB‐activated MEKK1/ERK/KLF4 pathway
Through the PDGF/MEKK1/ERK/KLF4 signalling pathway, PDGF‐BB can inhibit myocardin‐SRF complex from binding bind to the CArG box in the promoters of VSMC‐specific genes 20, 21. Based on bioinformatics prediction, potential let‐7g binding sites may exist in the 3′‐UTRs of the PDGFB and MEKK1 genes (Fig. 1A). Using the reporter assays, we showed that let‐7g could directly target wild‐type sequences of PDGFB and MEKK1 genes and suppress the luciferase activity, while let‐7g had no effects on the mutant sequences (Fig. 1B and C).
Figure 1.
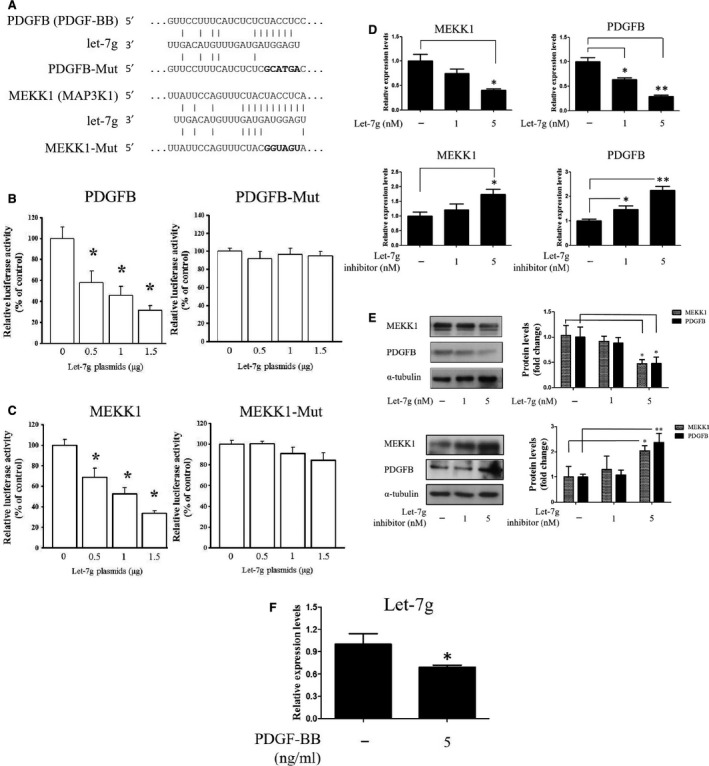
Identification of PDGFB and MEKK1 as let‐7g direct target genes. (A) Schematic diagram shows the let‐7g‐binding site in PDGFB and MEKK1 3′‐UTR regions. (B and C) The effects of let‐7g on luciferase activity. Cells were transfected with pMIR‐REPORT luciferase vector that carries wild‐type or mutant type PDGFB and MEKK1 3′‐UTR, and let‐7g‐expressing vector. The luciferase activity was measured at 24 hrs in triplicate. (D and E) Cells were transfected with let‐7g or its inhibitor, and gene expression was measured at 24 hrs. The mRNA and protein levels of MEKK1 and PDGFB were reduced by let‐7g and increased by let‐7g inhibitor. (F) The let‐7g level was determined in the PDGF‐BB‐treated cells. Data are means ± S.E.M. from three experiments; *P < 0.05; **P < 0.01.
To further confirm that let‐7g's knockdown effect on these 2 genes, transient transfection of let‐7g mimic into HASMCs caused a reduction of their protein and mRNA levels at 24 hrs, whereas transfection of let‐7g inhibitor caused the opposite effect (Fig. 1D and E). These results indicated that MEKK1 and PDGFB are direct targets of let‐7g in human cells. It has been shown that let‐7g expression was altered in PDGF‐treated glioblastoma cells 22. In our model, let‐7g expression was suppressed in PDGF‐BB‐treated HASMCs (Fig. 1F).
Because MEKK1 can regulate ERK1 and ERK2 (ERK1/2) 23, we then investigated the effect of let‐7g on the PBDGF‐BB‐induced phosphorylation of ERK1/2. As shown in Figure 2A, let‐7g reduced phosphorylated‐ERK1/2 while had no effect on total ERK1/2. Previous studies have reported that ERK1/2 are the upstream kinases of KLF4 21 and KLF4 can down‐regulate myocardin and prevent myocardin‐SRF complex from associating with VSMC‐specific gene promoters 16. We therefore conducted the co‐immunoprecipitation assay to test whether let‐7g inhibits the interaction between SRF and KLF4. The result showed that let‐7g decreased the interaction between KLF4 and SRF in the presence of PDGF‐BB (Fig. 2B and C).
Figure 2.
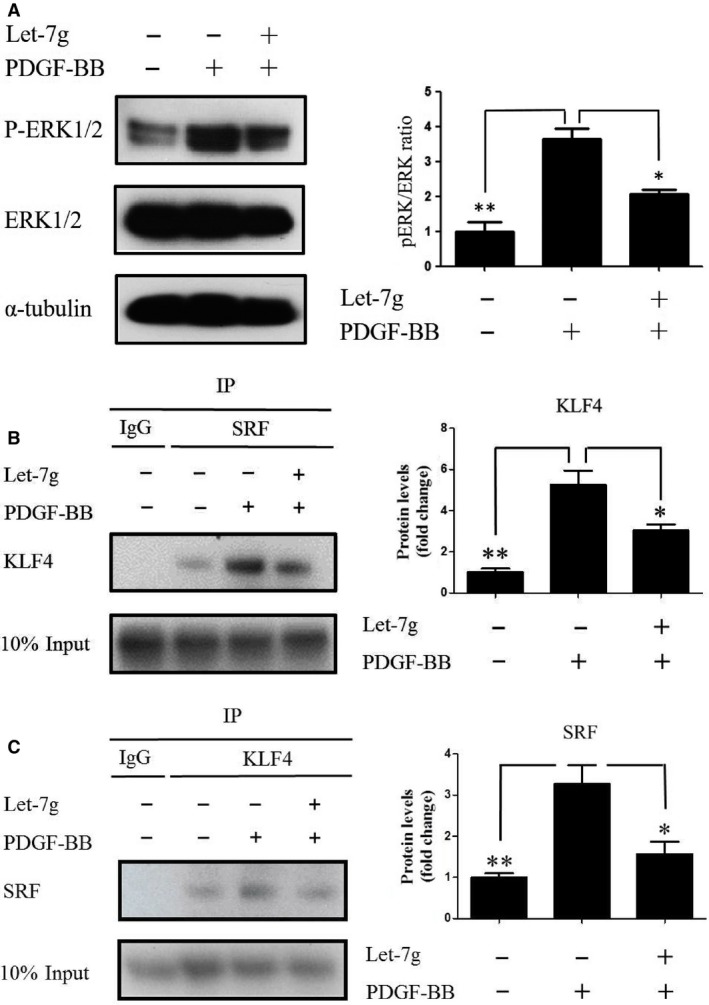
Let‐7g inhibits PDGF‐induced activation of ERK1/2 and KLF4. HASMCs were incubated with PDGF‐BB (5 ng/ml) for 48 hrs before the transfection of let‐7g (5 nM) for 24 hrs. The cell extracts were analysed. (A) Western blot was used to detect phosphorylated ERK1/2 and total ERK1/2. (B and C) Co‐immunoprecipitation assay was used to test for the interaction between KLF4 and SRF. Data represent mean ± S.E.M. of three independent experiments. *P < 0.05; **P < 0.01.
Effects of let‐7g on α‐SMA and calponin expression in PDGF‐BB‐treated HASMCs
We further tested whether let‐7g can maintain VSMC in the contractile status by measuring α‐SMA and calponin levels in the cells. Our data showed that PDGF‐BB decreased α‐SMA and calponin mRNA levels, whereas let‐7g could dose dependently restore their expression (Fig. 3A). The protein data and immunofluorescence imaging analysis also showed the consistent results (Fig. 3B and 3C, Fig. S2). Taken together, these findings indicate that let‐7g can restore PDGF‐BB‐reduced VSMC marker gene expression, and therefore, let‐7g can prevent VSMC from atherosclerotic change.
Figure 3.
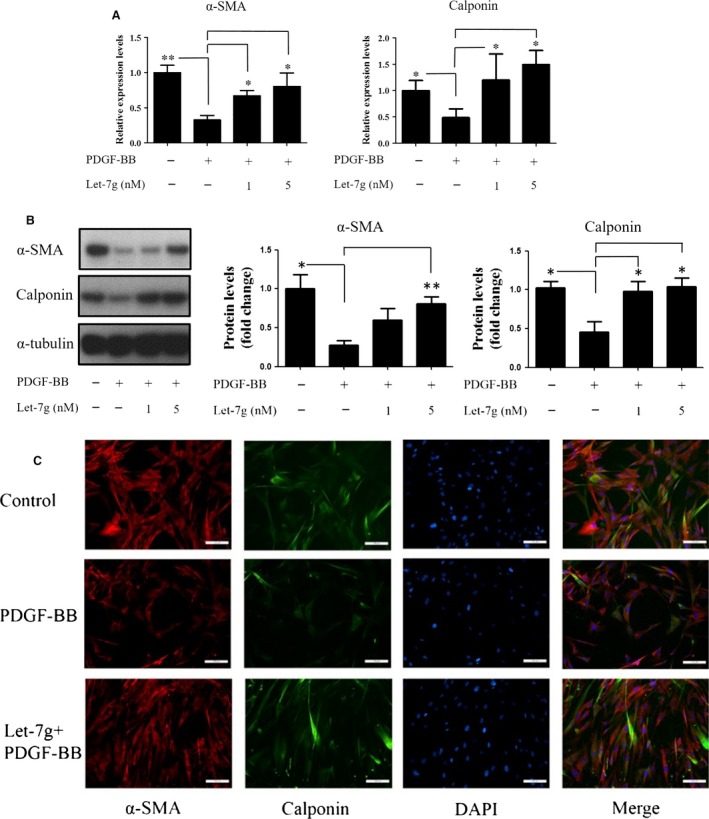
Let‐7g effect on α‐SMA and calponin expression in PDGF‐BB‐treated HASMCs. Cells were incubated with PDGF‐BB (5 ng/ml) for 48 hrs and then transfected with let‐7g (1 or 5 nM) for another 24 hrs. (A) The mRNA levels of α‐SMA and calponin were measured by real‐time PCR. (B) The protein levels of α‐SMA and calponin were determined by Western blot. Data are presented as mean ± S.E.M. from three independent experiments, and each experiment was performed in triplicate. *P < 0.05 and **P < 0.01 versus the negative control group. The entire Western blotting analysis of α‐SMA and calponin is shown in Figure S2 (C) Immunofluorescent stain was used to detect α‐SMA and calponin in HASMCs treated with PDGF‐BB along with let‐7g. Nuclei were stained with DAPI (blue). Scale bars, 40 μm.
Let‐7g reduces atherosclerotic plaque formation
To test for the effect of let‐7g on prevention of atherosclerosis in vivo, apoE−/− mice under a high‐fat diet were injected lentivirus carrying either the let‐7g‐expressing vector or negative control vector for 12 weeks. The atherosclerotic areas in the entire aorta were decreased in mice treated with let‐7g compared to those in the mice treated with placebo (Fig. 4A and B). Let‐7g treatment indeed increased let‐7g levels by 3.6‐fold in the aortas (Fig. 4C). These results confirm that let‐7g suppressed atherosclerotic plaque formation. The mRNAs levels of PDGFB, MEKK1, α‐SMA and calponin from entire aorta were measured. Our in vivo results showed that PDGFB and MEKK1 mRNA levels were suppressed by let‐7g treatment (Fig. 4D), while α‐SMA and calponin mRNA levels were increased by let‐7g treatment (Fig. 4E).
Figure 4.
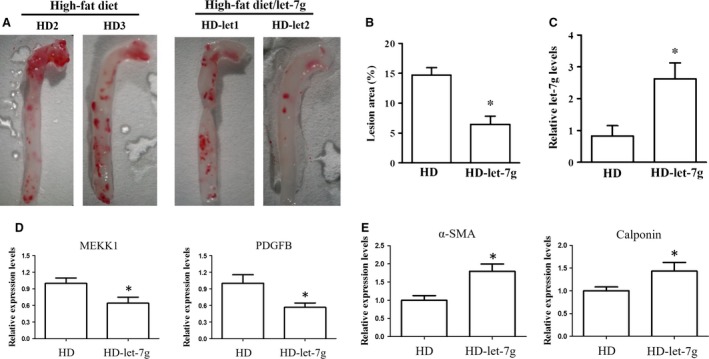
Effect of let‐7g on atherosclerotic plaque formation. (A) The atherosclerotic plaques in the aortas of apoE−/− mice under a high‐fat diet. (B) The quantitative result of atherosclerotic areas and (C) let‐7g levels in the aortas. (D and E) The mRNA levels of MEKK1, PDGFB, α‐SMA and calponin in aortas were determined by qPCR. N = 6 for each group, * means P < 0.05. The data presented as mean ± S.E.
Let‐7g decreases body weight and improves lipid profile
The let‐7g‐treated apoE−/− mice had slower gain of body weight than the placebo‐treated mice when all mice were under a high‐fat diet (Fig. S1A). The placebo‐treated mice had twofold gain of body weight than the let‐7g‐treated mice in the end of 12‐week experiment (Fig. S1B). Fasting blood sugar, the lipid profile and liver function glutamic‐pyruvic transaminase (also known as alanine aminotransferase, ALT) levels were significantly better in the let‐7g‐treated apoE−/− mice than in the placebo‐treated apoE−/− mice in the end of the 12‐week experiment (Fig. [Link], [Link]).
Discussion
The present study demonstrated that let‐7g has multiple inhibitory effects on the PDGF signalling in VSMCs. First, let‐7g could directly down‐regulate the PDGFB and MEKK1 genes leading to reduced interaction between KLF4 and SRF. Secondly, both in vitro and in vivo data showed that let‐7g could keep VSMCs in the contractile status by restoring α‐SMA and calponin expression. The major findings in the present study are schematically summarized in Figure 5. In addition, the present study also showed that let‐7g could reduce the formation of atherosclerotic lesions in apoE−/− mice under a high‐fat diet. Apart from our previous findings regarding let‐7g's effect on LOX‐1 and TGF‐β signalling 18, 19, here we further revealed another mechanism accounting for let‐7g beneficial effects.
Figure 5.
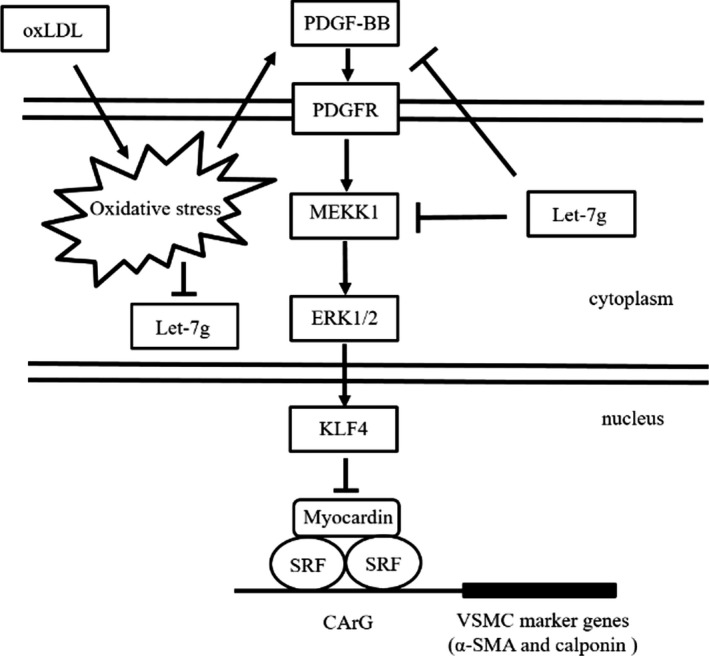
Schematic diagram shows that let‐7g affects PDGF/MEKK1/ERK/KLF4 signalling.
It is known that PDGF‐BB can cause VSMC phenotypic switching from the contractile type to the synthetic type 24. Disruption of PDGF‐BB signalling has been shown to delay or prevent atherogenesis 8. Our data showed that α‐SMA and calponin were down‐regulated in PDGF‐BB‐treated HASMCs, but overexpression of let‐7g could restore their expression. MEKK1 is involved in the PDGF‐BB‐induced VSMC phenotypic switching 25, and we confirmed PDGFB and MEKK1 were let‐7g target genes. A recent study also demonstrated the direct effect of let‐7g on MEKK1 26. The myocardin/SRF complex is a key component to maintain the VSMC contractile status. Activated form of KLF4 can interact with SRF and also repress myocardin 13. Previous studies demonstrated that PDGF‐BB suppresses the promoter activity of VSMC marker genes via regulation of KLF4 21. ERK1/2 are the upstream kinases of KLF4 in the PDGF‐BB‐induced signalling pathway 21, 27. Here, we showed that let‐7g reduced the levels of phosphorylated‐ERK1/2 in PDGF‐BB‐treated HASMCs. Through the direct knockdown of PDGFB and MEKK1, let‐7g eventually suppressed the interaction between KLF4 and myocardin/SRF. These results supported let‐7g as an important regulator to maintain VSMC in the contractile status.
A recent study of PDGF/PDGFRβ signalling in VSMCs revealed that activation of this pathway increased inflammation but not atherosclerosis under a normal cholesterol level 6. However, the same authors further demonstrated that the PDGF signalling amplified hypercholesterolaemia‐induced atherosclerosis 6. The current study showed that let‐7g could simultaneously reduce cholesterol levels and block PDGF/PDGFRβ signalling in VSMCs. Furthermore, we have found that let‐7g decreases PDGF‐BB‐induced inflammatory genes (TNF‐α, IL‐8, IL‐1β and granulocyte‐macrophage colony‐stimulating factor (GM‐CSF)) expression in HASMCs (Fig. S3). Our previous studies also demonstrated that let‐7g could reduce inflammation by suppressing TGF‐β signalling in endothelial cells and knock down LOX‐1, both of which further augment the importance of let‐7g's role in preventing atherosclerosis. Negative feedback regulation plays an important role in homoeostasis. Our results showed that let‐7g can directly suppress PDGFB and inhibit PDGF‐BB‐activated MEKK1/ERK/KLF4 pathway in VSMCs, while PDGF‐BB can reduce let‐7g expression. Accordingly, a negative feedback regulation exists between let‐7g and PDGF‐BB. The possible mechanism of PDGF‐BB reduces let‐7g expression was showed in Figure S4
The present study found that let‐7g could improve the lipid profile in mice under high‐fat diet. Our group previously reported that let‐7g can up‐regulate SIRT‐1 expression 19, and SIRT‐1 has been indicated to play an important role in lipid and glucose metabolism 28, 29, 30, 31. Fasting can increase SIRT‐1 that further interacts with and deacetylates PGC‐1α in liver leading to improve glucose homoeostasis 28. The liver X receptor (LXR) mediates HDL synthesis, and SIRT‐1 plays an important role in cholesterol homoeostasis by regulating LXR 29. Specific knockout of SIRT‐1 in the liver (SIRT1 LKO) decreases fatty acid oxidation, which leads to body weight gain and the development of hepatic steatosis 30. In addition, the serum levels of cholesterol and LDL were increased in SIRT1 LKO mice on a lithogenic diet 31. Accordingly, let‐7g effect on SIRT‐1 may partially explain why let‐7g treatment can improve lipid profile and sugar level.
There are some limitations in the present study. We did not examine the atherosclerotic changes in the carotid artery. However, previous studies have shown that atherosclerotic changes are similar between carotid artery and aorta 32, 33 in the animal experiments. Therefore, we assumed that the results should be comparable between aortic lesion and carotid lesion in our apoE−/− mice. As our present study focuses on VSMC changes, we did not conduct in‐depth studies to investigate the mechanisms for the change of biochemistry data and body weight in the animal study.
In summary, let‐7g inhibits formation of atherosclerotic lesions via regulation of multiple mechanisms. Our in vivo study showed that let‐7g improves lipid profile and sugar level in mice under a high‐fat diet. Our in vitro study demonstrated that let‐7g substantially inhibits the phenotypic switching of VSMCs, which is mediated through targeting PDGFB and MEKK1 genes. As contractile VSMCs are required to maintain normal functions arterioles and to prevent atherosclerosis, maintaining a sufficient level of let‐7g may reduce a risk for cardiovascular diseases.
Conflicts of interest
The authors confirm that there are no conflicts of interest.
Supporting information
Table S1 PCR primer sequences.
Figure S2 Entire Western blotting analysis of α‐SMA and calponin.
Figure S3 Let‐7g inhibits inflammatory genes expression in PDGF‐BB‐treated HASMCs.
Figure S4 Schematic diagram showing the possible mechanism of PDGF‐BB reduces let‐7g expression.
Acknowledgements
This work was supported by the following funding: Ministry of Science and Technology (Taiwan, R.O.C. MOST103‐2314‐B‐037‐026‐MY3, MOST103‐2314‐B‐037‐027‐MY2, MOST103‐2314‐B‐075‐076‐MY3, MOST104‐2745‐B‐037‐001, MOST105‐2314‐B‐039‐050), National Health Research Institutes (Taiwan, R.O.C. NHRI‐EX106‐10605PI). In this manuscript, Wang TM and Chen KC designed the research study, conducted experiments and prepared manuscript. Hsu PY and Chen CY helped experiments. Wang YS designed study and prepared the manuscript. Lin HF designed study and interpreted the data. Liao YC interpreted the data interpretation and prepared manuscript. Juo SH designed the study, supervised the progress, coordinated the experiments, interpreted the data and finalized manuscript.
References
- 1. Silvestre‐Roig C, de Winther MP, Weber C, et al Atherosclerotic plaque destabilization: mechanisms, models, and therapeutic strategies. Circ Res. 2014; 114: 214–26. [DOI] [PubMed] [Google Scholar]
- 2. Liu S, Yang P, Kang H, et al NDRG2 induced by oxidized LDL in macrophages antagonizes growth factor productions via selectively inhibiting ERK activation. Biochim Biophys Acta. 2010; 1801: 106–13. [DOI] [PubMed] [Google Scholar]
- 3. Akiba S, Kumazawa S, Yamaguchi H, et al Acceleration of matrix metalloproteinase‐1 production and activation of platelet‐derived growth factor receptor beta in human coronary smooth muscle cells by oxidized LDL and 4‐hydroxynonenal. Biochim Biophys Acta. 2006; 1763: 797–804. [DOI] [PubMed] [Google Scholar]
- 4. Evanko SP, Raines EW, Ross R, et al Proteoglycan distribution in lesions of atherosclerosis depends on lesion severity, structural characteristics, and the proximity of platelet‐derived growth factor and transforming growth factor‐beta. Am J Pathol. 1998; 152: 533–46. [PMC free article] [PubMed] [Google Scholar]
- 5. Heldin CH, Westermark B. Mechanism of action and in vivo role of platelet‐derived growth factor. Physiol Rev. 1999; 79: 1283–316. [DOI] [PubMed] [Google Scholar]
- 6. He C, Medley SC, Hu T, et al PDGFRbeta signalling regulates local inflammation and synergizes with hypercholesterolaemia to promote atherosclerosis. Nat Commun. 2015; 6: 7770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ricci C, Ferri N. Naturally occurring PDGF receptor inhibitors with potential anti‐atherosclerotic properties. Vascul Pharmacol. 2015; 70: 1–7. [DOI] [PubMed] [Google Scholar]
- 8. Rutherford C, Martin W, Carrier M, et al Endogenously elicited antibodies to platelet derived growth factor‐BB and platelet cytosolic protein inhibit aortic lesion development in the cholesterol‐fed rabbit. Int J Exp Pathol. 1997; 78: 21–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sano H, Sudo T, Yokode M, et al Functional blockade of platelet‐derived growth factor receptor‐beta but not of receptor‐alpha prevents vascular smooth muscle cell accumulation in fibrous cap lesions in apolipoprotein E‐deficient mice. Circulation. 2001; 103: 2955–60. [DOI] [PubMed] [Google Scholar]
- 10. Tallquist M, Kazlauskas A. PDGF signaling in cells and mice. Cytokine Growth Factor Rev. 2004; 15: 205–13. [DOI] [PubMed] [Google Scholar]
- 11. Salabei JK, Cummins TD, Singh M, et al PDGF‐mediated autophagy regulates vascular smooth muscle cell phenotype and resistance to oxidative stress. Biochem J. 2013; 451: 375–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Johnson JL. Emerging regulators of vascular smooth muscle cell function in the development and progression of atherosclerosis. Cardiovasc Res. 2014; 103: 452–60. [DOI] [PubMed] [Google Scholar]
- 13. Liu Y, Sinha S, McDonald OG, et al Kruppel‐like factor 4 abrogates myocardin‐induced activation of smooth muscle gene expression. J Biol Chem. 2005; 280: 9719–27. [DOI] [PubMed] [Google Scholar]
- 14. Doi H, Iso T, Yamazaki M, et al HERP1 inhibits myocardin‐induced vascular smooth muscle cell differentiation by interfering with SRF binding to CArG box. Arterioscler Thromb Vasc Biol. 2005; 25: 2328–34. [DOI] [PubMed] [Google Scholar]
- 15. Deaton RA, Gan Q, Owens GK. Sp1‐dependent activation of KLF4 is required for PDGF‐BB‐induced phenotypic modulation of smooth muscle. Am J Physiol Heart Circ Physiol. 2009; 296: H1027–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Torella D, Iaconetti C, Catalucci D, et al MicroRNA‐133 controls vascular smooth muscle cell phenotypic switch in vitro and vascular remodeling in vivo . Circ Res. 2011; 109: 880–93. [DOI] [PubMed] [Google Scholar]
- 17. Romaine SP, Tomaszewski M, Condorelli G, et al MicroRNAs in cardiovascular disease: an introduction for clinicians. Heart. 2015; 101: 921–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chen KC, Hsieh IC, Hsi E, et al Negative feedback regulation between microRNA let‐7g and the oxLDL receptor LOX‐1. J Cell Sci. 2011; 124: 4115–24. [DOI] [PubMed] [Google Scholar]
- 19. Liao YC, Wang YS, Guo YC, et al Let‐7g improves multiple endothelial functions through targeting transforming growth factor‐beta and SIRT‐1 signaling. J Am Coll Cardiol. 2014; 63: 1685–94. [DOI] [PubMed] [Google Scholar]
- 20. McDonald OG, Wamhoff BR, Hoofnagle MH, et al Control of SRF binding to CArG box chromatin regulates smooth muscle gene expression in vivo . J Clin Invest. 2006; 116: 36–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kawai‐Kowase K, Owens GK. Multiple repressor pathways contribute to phenotypic switching of vascular smooth muscle cells. Am J Physiol Cell Physiol. 2007; 292: C59–69. [DOI] [PubMed] [Google Scholar]
- 22. Shao M, Rossi S, Chelladurai B, et al PDGF induced microRNA alterations in cancer cells. Nucleic Acids Res. 2011; 39: 4035–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kolch W. Coordinating ERK/MAPK signalling through scaffolds and inhibitors. Nat Rev Mol Cell Biol. 2005; 6: 827–37. [DOI] [PubMed] [Google Scholar]
- 24. Yu X, Li Z. MicroRNAs regulate vascular smooth muscle cell functions in atherosclerosis (review). Int J Mol Med. 2014; 34: 923–33. [DOI] [PubMed] [Google Scholar]
- 25. Zhao Y, Biswas SK, McNulty PH, et al PDGF‐induced vascular smooth muscle cell proliferation is associated with dysregulation of insulin receptor substrates. Am J Physiol Cell Physiol. 2011; 300: C1375–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cao R, Wu W, Zhou X, et al Let‐7g induces granulosa cell apoptosis by targeting MAP3K1 in the porcine ovary. Int J Biochem Cell Biol. 2015; 68: 148–57. [DOI] [PubMed] [Google Scholar]
- 27. Kim MO, Kim SH, Cho YY, et al ERK1 and ERK2 regulate embryonic stem cell self‐renewal through phosphorylation of Klf4. Nat Struct Mol Biol. 2012; 19: 283–90. [DOI] [PubMed] [Google Scholar]
- 28. Rodgers JT, Lerin C, Haas W, et al Nutrient control of glucose homeostasis through a complex of PGC‐1alpha and SIRT1. Nature. 2005; 434: 113–8. [DOI] [PubMed] [Google Scholar]
- 29. Li X, Zhang S, Blander G, et al SIRT1 deacetylates and positively regulates the nuclear receptor LXR. Mol Cell. 2007; 28: 91–106. [DOI] [PubMed] [Google Scholar]
- 30. Purushotham A, Schug TT, Xu Q, et al Hepatocyte‐specific deletion of SIRT1 alters fatty acid metabolism and results in hepatic steatosis and inflammation. Cell Metab. 2009; 9: 327–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Purushotham A, Xu Q, Lu J, et al Hepatic deletion of SIRT1 decreases hepatocyte nuclear factor 1alpha/farnesoid X receptor signaling and induces formation of cholesterol gallstones in mice. Mol Cell Biol. 2012; 32: 1226–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Eitzman DT, Westrick RJ, Xu Z, et al Plasminogen activator inhibitor‐1 deficiency protects against atherosclerosis progression in the mouse carotid artery. Blood. 2000; 96: 4212–5. [PubMed] [Google Scholar]
- 33. Kumar S, Chen M, Li Y, et al Loss of ADAMTS4 reduces high fat diet‐induced atherosclerosis and enhances plaque stability in ApoE(‐/‐) mice. Sci Rep. 2016; 6: 31130. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 PCR primer sequences.
Figure S2 Entire Western blotting analysis of α‐SMA and calponin.
Figure S3 Let‐7g inhibits inflammatory genes expression in PDGF‐BB‐treated HASMCs.
Figure S4 Schematic diagram showing the possible mechanism of PDGF‐BB reduces let‐7g expression.