

10.1 Myocarditis
Development of autoimmune diseases is a consequence of lack of self-tolerance, which can arise due to defective function of any of the features contributing to the unresponsive status of the immune system toward self-antigens, i.e., anergy, regulation, and clonal deletion [1, 2]. Autoimmunity requires a self-specific response, despite the fact that innate response might be involved. That is the main difference with the rare autoinflammatory diseases, in which an uncontrolled but unspecific response occurs, with minimal involvement of the adaptive response [3].
Myocarditis can be defined as the inflammatory process affecting the muscular tissues of the heart (myocardium). Myocarditis is considered an important cause of sudden death as has been shown by autopsy-based studies in the general population [4–7]. Regardless of its etiology, the acute inflammation may progress to subacute and chronic stages and finally to tissue remodeling, fibrosis, and loss of myocardium architecture and contractile function [4, 6, 8]. The latter chronic damage corresponds to development of dilated cardiomyopathy (DCM). Its overall incidence among myocarditis cases is not accurately known, but retrospective studies report that 9–16% of unexplained nonischemic DCM cases have histological evidence of myocarditis, thus suggesting a persistent/self-sustained autoimmune inflammatory progression as a requirement [4, 7, 9, 10]. Other studies estimate a wider range of 10–50% of DCM cases having evidence of myocarditis as could be determined by endomyocardial biopsy [11].
Thus, DCM is the most severe complication of myocarditis. DCM implies the dilatation of heart chambers leading to impairment of systolic function [5, 12]. The consequence of such systolic dys-function is heart failure, for which the only definitive treatment is transplantation, since the fiber damage and fibrosis are, so far, irreversible processes [4, 5, 9]. Other complications might occur as consequence of tissue damage, like valve insufficiency due to rupture of papillary muscles, arrhythmias due to damage of conduction system, reentry phenomenon associated with myocardial fibrosis, and adrenergic stimulation by autoantibodies [4, 5].
10.1.1 Epidemiology
Myocarditis is a disease with an extreme wide range of clinical presentations, from asymptomatic to life-threatening, importantly including sudden death. This fact, in conjunction to the lack of noninvasive highly specific diagnostic methods and its overlapping symptoms with other more prevalent/incident cardiovascular diseases, makes it an underdiagnosed entity [8]. Myocarditis is the cause of sudden death in around 10% of cases [7, 13]. Important efforts had been made to develop noninvasive imaging diagnostic methods. Cardiac magnetic resonance (CMR) has been proposed as a reliable approach based on magnetic relaxation times suggesting regional myocardial edema and fluid leak. Lake Louise criteria seem to be an accurate diagnostic method based on evidence of myocardial edema on T2-weighted images plus capillary leak in T1 [4, 14–16].
Consequence of such diagnostic conundrum, retrospective studies based on autopsies have reported an extremely wide range of association of myocarditis with sudden death, from 2 to 42% [17, 18]. Importantly all of them are relatively small studies with lack of multicentric approach, thus probably biased by environmental and genetic factors.
On the other hand, prospective studies analyzing progression of myocarditis diagnosed by gold-standard biopsy parameters, an ideal clinical-epidemiologic design, report a consistent progression of myocarditis to DCM of about 30% [5, 19–21]. Post-myocarditis DCM patients have a poor prognosis, and when American Heart Association (AHA) heart failure functional classes III–IV are reached, only heart transplantation provides a definitive resolution [5].
Viral myocarditis is thought to be the most frequent type, mostly affecting children and young adults. Studies report that 60% of children diagnosed with acute myocarditis have a transplant-free survival of 10 years [21]. Also, 25–40% of DCM patients have viral genome detected in the endomyocardial biopsy [22].
10.1.2 Clinical Presentation
The presentation of acute myocarditis is extremely variable and frequently subclinical. Acute myocarditis can either lead to acute complications or progress relatively asymptomatically to chronic heart failure, then debuting clinically with chronic late-stage complications like heart failure due to DCM. As a consequence, a high level of suspicion is required by the clinician for deciding to rule out myocarditis with specific tests. The gold standard for myocarditis diagnosis is still a positive endomyocardial biopsy, an invasive procedure which requires a high clinical pretest likelihood and low risk/benefit ratio to be performed [10]. The main clinical presentation features of myocarditis are:
10.1.2.1 Acute Coronary Syndrome-Like Symptoms
Considering the overwhelmingly higher incidence of “real” acute coronary syndrome (ACS, consequence of coronary artery disease or vasculopathy) as compared with myocarditis, this is probably the presentation needing higher clinical suspicion index. In that regard, consideration of classic risk factors for ACS like age, sex, family history, and metabolic disturbances is of extreme importance in clinical care. Angiographic studies lacking evidence of coronary atherosclerosis or vasculopathy, the latter typical of transplant chronic allorejection, strongly suggest myocarditis rather than ACS [10, 23]. Functional cardiac tests (echocardiography and cardiac magnetic resonance (CMR)) or indexes of myocardial damage (like cardiac troponin T or I, cTnT or cTnI) may show results equivalent to ACS, as unspecifically reflect cardiac tissue injury. Even more important is the fact that electrocardiogram (EKG), the first-line diagnostic test for ACS in clinical practice, may show similar findings in myocarditis as compared with ACS, mainly ST segment elevation/depression and T-wave inversion. Inflammation-associated coronary vasospasm had been identified as one of the main mechanisms leading to ACS-like symptoms in myocarditis, thus also involving transient ischemia [24].
10.1.2.2 New Onset or Worsening of Heart Failure Symptoms in the Absence of Coronary Artery Disease (CAD) or Other Typical Causes
In this regard, it must be considered that CAD, long-term high blood pressure and metabolic disturbances (like Diabetes Mellitus, which turns out to be pathophysiologically related to CAD and high blood pressure) are epidemiologically the main factor predisposing to heart failure. Acute decompensation of heart failure functional status without paralleling changes in underlying disease or, even more, the unexplained new onset of heart failure signs must lead to rule out myocarditis of any etiology [4–6].
10.1.2.3 Chronic and Persistent Symptoms Compatible with Heart Failure, but Not Implying Acute Worsening nor Typical Risk Factors
Patients with stable symptoms of heart failure as the ones enounced above, but lacking typical risk factors, must be considered potential myocarditis patients, despite the absence of acute signs of worsening or previous history/symptoms of acute myocarditis [4].
10.1.2.4 Acute Life-Threatening Cardiac Conditions
This category includes arrhythmias, aborted sudden death, and cardiogenic shock [6]. Evidently, in these cases, the priority is the resolution of the acute event, but afterward proper studies are needed to rule out myocarditis.
10.1.3 Histopathology
Myocarditis can be classified based on several parameters, mainly histopathology, time progression, and etiology. It is important to consider that once the acute inflammatory phase is surpassed, the long-term fibrosis and loss of myocardial architecture features are usually similar regardless of original type of myocarditis. The Dallas classification was defined in order to standardize the pathology reports and account for the technical issues associated with endomyocardial biopsy sampling process. It is based on conventional staining procedures (hematoxylin-eosin) and not immunohistochemistry. It categorizes myocarditis in (a) myocarditis with/without fibrosis, (b) borderline myocarditis, and (c) no myocarditis (subsequent biopsies should be classified in persistent, healing, or healed myocarditis) [25, 26]. Nevertheless, it is not particularly useful in pathophysiologic and immunologic grounds, and its usefulness has decayed over time [25]. Based on the immunopathology findings, myocarditis is classified in:
10.1.3.1 Acute Lymphocytic Myocarditis
Acute lymphocytic myocarditis is characterized by a predominant myocardial patchy infiltration of T lymphocytes, typically identified in immunohistochemistry [IHC] by CD3 expression, with minimal fibrosis. As expected, areas of lymphocyte infiltration co-localize with CD68+ macrophages. This is the most common pathologic type of myocarditis and is most frequently of viral etiology, mainly Coxsackievirus B and adenoviruses [5, 8].
10.1.3.2 Chronic Lymphocytic Myocarditis (CLM)
CLM is thought to be a chronic stage of acute lymphocytic myocarditis; this entity is pathologically characterized by existence of myocardial fibrosis but still accompanied by leukocyte infiltration. The timing for such progression from acute to chronic inflammation is variable and currently unpredictable. In this case, the areas of fibrosis are consequence of the persistent evolution of the inflammatory processes [4].
10.1.3.3 Giant Cell Myocarditis
Giant cell myocarditis is characterized by unique histological features and particular aggressiveness and capacity to progress to chronic and subacute life-threatening complications like DCM. The prevalence of progression to DCM is considered to be up to 80% [8]. In fact, despite immunosuppressive therapy, two studies have shown that only around 10% of giant cell myocarditis patients survive 4 years without transplantation, compared with 44% of lymphocytic myocarditis [10, 27]. Although other studies reported improved transplant-free survival between 40 and over 70%, some of these studies might have not included the most severe cases with early complications and mortality [28, 29]. Giant cell myocarditis is thought to be a consequence of autoimmunity. That autoimmune origin is also supported by association of giant cell myocarditis with numerous autoimmune diseases and post-transplantation appearance of giant cells in heart grafts [30–35]. Histologically, giant cell myocarditis is characterized also by a prominent leukocyte infiltration. In this case, the inflammatory areas are more extensive, but with myeloid cell (mainly CD68+ macrophages) predominance as compared with T cell infiltration. Interestingly, eosinophils are also often present in the cardiac infiltrate; however, eosinophils are not an independent predictor of mortality [9]. It is important to notice that the murine model of autoimmune myocarditis, experimental autoimmune myocarditis (EAM), resembles the features of this pathologic subtype [8].
10.1.3.4 Sarcoidosis
This is a systemic “idiopathic” disease in the frontier between autoimmunity and autoinflammatory disease, characterized by an antigen presenting cells (APC) dys-function, generating chronic tissue inflammation and granulomatous lesions in organs like the lung, kidney, and heart. Sarcoidotic myocarditis display extensive infiltration by activated macrophages, leading to chronic inflammation and tissue damage [4, 23].
10.1.3.5 Eosinophilic Myocarditis
This form of myocarditis is observed in entities associated with peripheral eosinophilia (like primary idiopathic hypereosinophilia or chronic eosinophilia due to infectious causes). Eosinophilic myocarditis may also appear as a primary disease, most likely of autoimmune origin. Its landmark is the presence of eosinophils in significant proportions in myocardial infiltrates. This entity, like the giant cell myocarditis, shows also a poor long-term prognosis despite broad immunosuppressive treatment [4, 8, 10]. A murine model of this human type of myocarditis has been developed by our laboratory and will be described later in this chapter [36].
Different etiologic agents might lead to similar histological characteristics. Toxic (drug-induced), viral, radiation-associated, and autoimmune myocarditis might generate acute lymphocytic myocarditis with similar pathologic findings. Similarly, most cases of myocarditis associated with systemic autoimmune diseases (Sjögren’s disease, systemic lupus erythematous, vasculitis) also have lymphocytic or giant cells as features [37, 38].
10.1.4 Etiology
Myocarditis is a very broad pathologic definition, as it does not account for the trigger or specific immunologic features involved in the disease. As a consequence, myocarditis can also be classified based on its etiology. Several factors might lead to development of myocarditis, including viral (believed to have the higher incidence and prevalence, being Coxsackievirus B and adenovirus the main causes), physical noxa (radiation), pharmacologic (like anthracyclines, 5-fluorouracil, alcohol, tricyclic antidepressants), hematologic (essentially the eosinophilic myocarditis, either associated with hypereosinophilic conditions or as primary Th2-skewed autoimmune response), and autoimmune, existing mechanistic overlap between those categories as will be described below. Other significantly less frequent infectious causes have been identified: Mycobacterium species, Mycoplasma pneumoniae, Cryptococcus species, and Trypanosoma cruzi [6]. It is controversial if HIV itself can trigger the myocardial inflammation or if the myocarditis occurs secondarily to AIDS-related complications like autoimmunity and/or opportunistic infections [39, 40]. Interestingly, the Smallpox Vaccination Program of the Department of Defense (targeting adults) was stopped for significant increase of myocarditis cases close to the vaccination time [41].
Autoimmune myocarditis may occur as an isolated entity, in which the primary (and usually only) targeted organ is the heart. Typically, that is the case of giant cell myocarditis and certain cases of eosinophilic myocarditis not associated with peripheral hypereosinophilia. Also, several systemic autoimmune diseases may affect heart tissues, generating myocarditis in the context of a broader autoimmune phenomenon. The disease most strongly associated with development of myocarditis is systemic lupus erythematosus (SLE), but it might also occur in association with Sjögren’s syndrome (SS), vasculitis, and polymyositis [8, 37, 38].
In the enounced cases, autoimmune diseases lead mainly to lymphocytic acute/chronic myocarditis, strongly suggesting a T cell-mediated process. Accessory diagnostic tests are needed to determine the actual etiology/trigger of myocarditis and differentiate viral vs primary autoimmune causes. As myocarditis-associated viruses are epidemiologically common, a positive serology (IgG or IgM) should not be considered enough evidence to establish a viral etiology. Simultaneous evidence of active infection in myocardial tissues along with endomyocardial biopsy proof of myocarditis is the most accurate etiologic diagnosis [5, 6].
It is important to notice that the progression rate of myocarditis to irreversible tissue damage varies depending on the etiology. On the other hand, despite the causes, triggers, and initial immunologic driving forces may be different, the clinical and histological characteristics of DCM stage are similar for all the types and causes of myocarditis [42]. In that regard, it is possible that at certain point of the progression of the myocarditis, several etiologic types confluence in a common autoimmune pathogenic process leading to chronic inflammation, tissue remodeling, fibrosis, muscle fiber damage, systolic dysfunction, and finally DCM [43].
10.2 Murine Models of Myocarditis
Significant efforts have been made to study mechanistically the immunologic features associated with the trigger/initiation, acute phase, and progression of myocarditis to DCM. As with other several pathologies, animal models are extremely important to carry out those types of studies.
10.2.1 EAM and Eosinophilic EAM Models
Experimental autoimmune myocarditis (EAM) murine model was developed allowing to address the immunobiology of acute and chronic myocarditis. EAM has immunologic features paralleling and resembling the human entity, making it suitable to pursue pathophysiologic insights on myocarditis. Importantly, EAM mimics the most severe clinical course of giant cell myocarditis, allowing to study the entire temporal spectrum and progression of the disease to DCM. Another important feature of EAM models is a gender bias resembling the atypical pattern of human autoimmune myocarditis, in such a way that male mice are more susceptible to the induction of heart-specific autoimmunity [44].
EAM can be induced by immunization with cardiac myosin or with a myocarditogenic peptide derived from the α-cardiac myosin heavy chain emulsified in complete Freund’s adjuvant (CFA) injected twice in the first 8 days to susceptible mice strains [45, 46]. The mice strain susceptibility to EAM seems to be partially related to MHC haplotypes. The main susceptible strains are cogenic A/J background (A/J H2a, A.BY H2b, A.CA H2f, and A.SW H2s) and Balb/c (H2d). Susceptibility of certain C57BL/10 J background strain had also been described (B10.A H2a, B10.S H2s, and B10.PL H2u) [47]. In the section dedicated to viral etiology and influence of HLA haplotypes, further discussion will be made regarding the influence of MHC and non-MHC genes.
In susceptible strains, the self-peptide presentation is mainly IA restricted, altogether pointing out the importance of that specific class II chain, equivalent to human HLA-DQ. Furthermore, as will be described below, transgenic nonobese diabetic mice expressing human HLA-DQ8 instead of IAb spontaneously develop autoimmune myocarditis.
As might be expected according to the MHC molecular biology, the H2-restricted self-peptides associated with optimal autoimmunization are strain and haplotype-specific, also demonstrating a MHC bias with potential translational implication. Thus, Balb/c mice are susceptible to immunization with α-myosin heavy chain peptide MyHCα614–629, SWXJ to MyHCα406–425, and MyHCα1631–1650, whereas A/Js are susceptible to MyHCα334–352 as well as cTnI105–122 [48].
The rationale of the induction process is to elicit a cellular immune response simultaneously with an antigenic challenge with a cardiac-specific self-peptide in order to generate an adaptive self- and myocardium-specific immune response capable to overcome the regulatory mechanisms, thought to be suppressed in human auto-immune diseases. Myocardium-specific inflammatory process occurs peaking at day 21 after first immunization and progressing to late-stage DCM between day 40 and 60 [44, 48]. Cardiac inflammation at the peak of EAM on day 21 is characterized by a significant leukocyte infiltration in the myocardium, including innate cells like neutrophils, eosinophils, monocytes/macrophages, as well as lymphocytes, representing adaptive cellularity. The adaptive T cell-mediated response is the driving force in the development of this inflammatory process [48].
EAM models also employ the use of adjuvants and Toll-like receptor ligands. In the case of the widely studied Balb/c EAM, the protocol includes subcutaneous injection of MyHCα peptide emulsified in complete Freund’s adjuvant (CFA) and supplemented with heat-inactivated Mycobacterium tuberculosis strain H37Ra to 5 mg/mL on day 0 and 7 of the induction protocol, plus 500 ng of intraperitoneal Pertussis toxin on day 0 [44]. The requirement of coadjuvants underlines the importance of innate immunity activation, presumably via TLRs, in the generation of a robust adaptive autoimmune response [49].
We have developed recently a murine model of eosinophilic experimental autoimmune myocarditis (EoEAM) using mice deficient in IL-17A and IFnγ and IL17A−/−IFNγ−/− double knockout (DKO) mice, immunized similarly to the general EAM model [36]. EAM in IL17A−/−IFNγ−/− DKO results in a condition in which the immune response is preferentially Th2, leading to a lethal eosinophilic myocarditis.
10.2.2 Other Murine Models of Autoimmune Myocarditis
In addition to EAM, troponin-induced myocarditis was developed, in which also susceptible mice are immunized in a similar manner than described above, but using a troponin-derived peptide instead of myosin peptides. It is unclear how closely this model resembles the real pathogenic events occurring in humans. It seems that in human myocarditis the main target of autoimmunity is the myosin heavy chain. Nevertheless, the rationale of the troponin-induced model is to have the possibility to use troponin levels and anti-troponin antibodies as an alternative readout [50].
Nonobese diabetic (NOD) mice which lack expression of the murine MHC-II molecule IA (IAb−/−) but express the human class II haplotype HLA-DQ8 develop spontaneous myocarditis. It is important to clarify that those mice are not a humanized murine model but rather a transgenic strain expressing one human HLA haplotype in substitution of the murine one [51]. Despite the xenogenic differences, the mouse antigen-presenting cells can effectively present peptides in an HLA-restricted manner to mouse T cells, then allowing certain mechanistic studies regarding HLA-biased presentation during autoimmunity. Those studies are potentially translatable to human physiology, mainly in the case of presentation of peptides derived from phylogenetically preserved self-antigens.
Murine models have also focused on the involvement of deficient PD-1 signal on T cells in the development of myocarditis [52, 53]. PD-1 is a member of the CD28 family. Its ligation with PDL1/PDL2 expressed on antigen-presenting cells during the formation of the immune synapse generates a downregulation of effector T cell activation, via anergy, apoptosis, and/or induction of regulatory properties [54]. PD-1-deficient mice (Pd1−/−) develop more severe EAM [52]. That enhanced susceptibility is associated with increased CD4 and CD8 myocardial infiltration. Similar findings were observed using the MRL-Faslpr/lpr, a murine model of autoimmunity with lupus-like features as consequence of a loss-of-function mutation in the Fas gene. If a PD-1 KO condition is introduced in the MLR-Faslpr/lpr strain, then a severe lethal myocarditis occurs spontaneously at 4–8 weeks after birth. The incidence of myocarditis was up to 96% in this murine model. The features were an increased T cell infiltration, characteristically with a Th1 bias. The latter has been proposed as a useful murine model of PD-1 influence in myocarditis development/progression [53]. This is of clinical relevance since cases of acute myocarditis have been reported in human patients as side effect of anti-PD-1 checkpoint treatment for cancer (melanoma and non-small cell carcinoma) [55–57].
10.2.3 Experimental Viral Myocarditis
The most widely used viral models imply the infection of the mouse strain with Coxsackievirus B3 (CVB3), an enterovirus of the Picornaviridae family which is one of the primary pathogens associated with human viral myocarditis [43, 58–60]. Two main models of CVB3-induced myocarditis have been described. First one induces severe acute myocarditis with significant tissue damage and sudden death occurring within the first week of direct intraperitoneal infection [59, 60]. In the second CVB3 myocarditis model, heart-passaged CVB3 viruses are used to induce milder acute myocarditis, which progresses to chronicity and DCM [43]. In this CVB3 model, myocarditis is induced by intraperitoneal injection of heartpassaged Coxsackievirus B3 in a susceptible mice strain. Inflammatory phase is developed 7–14 days postinfection, and progression to DCM occurs 28–56 postinfection [43]. Thus, the model using heart-passaged CVB3 viruses allows to study the whole spectrum of immune processes involved in the progression to DCM, including the post-viral autoimmune phenomena [43]. A similar gender bias than EAM and human viral myocarditis exists in this model, with greater male susceptibility [61]. The fact that CVB is also the main myocarditis-associated virus in human disease, as well as the immunopathogenic similarities between the model and the patients, makes it a powerful research tool.
It has been shown that mice with an A/J background and C57BL/10 J background can develop the inflammatory phase of CVB3-induced myocarditis but only A/J mice progress to a chronic stage and DCM [43, 62]. These facts strongly suggest that progression to sustained inflammation and chronic complications of viral myocarditis might follow a common immunopathogenic pathway with autoimmune myocarditis [8, 61, 63]. Cardiac-tropic viruses might act as initial triggers and “natural adjuvants” of autoimmune processes [61].
10.3 Specific Factors Triggering/Predisposing Myocarditis Development
As most of the autoimmune diseases, myocarditis is considered a multifactorial entity, in which several immunologic mechanisms are involved on its development and progression. Regarding the trigger and initiation of its pathogenesis, so far, no unique sufficient factor has been identified but rather multiple endogenous and environmental confluent factors, in such a way that the myocardium-specific autoimmune process is triggered and sustained (Fig. 10.1). The balance and relative influence of those factors is still unclear but seems to be variable and host-dependent.
Fig. 10.1.

Proposed model for myocarditis development. Coexistence of predisposing factors along with specific triggers of myocardium damage leads to exposure of cryptic self-antigens and a consequent inflammatory process. At this point, both the innate and the myocardium-specific adaptive response generate a self-sustained autoimmune phenomenon independent of the original trigger factor. That autoimmune process is responsible for development of myocarditis and progression to DCM
10.3.1 Gender
In the case of myocarditis, the gender bias is unusual as myocarditis is more likely to occur in males, as compared with females, a feature which is mirrored in the murine models [4, 5, 8, 44, 64]. The reason for that bias is still a conundrum, but certain factors have been found as a possible explanation, importantly differences in TLR4 activation susceptibilities between sexes [4, 5, 8, 64, 65].
Using the CVB3 myocarditis murine model, it was found that myocarditis severity is significantly stronger in males, as well as the likelihood of progression to DCM [43, 66]. This occurs despite a similar viral load and replication rate among genders, suggesting the existence of sex-specific immunologic features not related to virus clearance. Males produce higher amounts of pro-inflammatory cytokines IL1β, IL18, and IFNγ during myocarditis. TLR4, an important inducer of IL1β and IL18 by monocytes/macrophages and mast cells upon ligation, is more strongly expressed in male APCs than in female hosts, in a IFNγ-independent manner. Furthermore, Tim-3, a key peripheral inducer of T regulatory cells (Tregs), is cross regulated in an inverse manner by TLR4 with estrogens as cofactor [49]. Finally, Treg development, which is proportional to Tim-3 expression/functionality, is stronger in females, even during viral myocarditis, in an estrogen-dependent manner. This fact provides not only a plausible explanation for the myocarditis gender bias but also a link between innate and adaptive immunity and the requirement for coadjuvants in EAMs, which typically involves TLR ligands [49].
10.3.2 Environment
An important factor that observationally and mechanistically has been associated with development of human autoimmunity, with some successful animal model correlates, is the environmental influence [67].
Drugs such as digoxin, cephalosporins, diuretics (like furosemide), and tricyclic antidepressants are relatively weakly associated with myocarditis, as the incidence among patient taking those drugs is low. On the other hand, anthracyclines (like doxorubicin) are strongly associated with myocardial inflammation, even in a dose-dependent manner [64, 68, 69]. The mechanisms of drug-induced myocarditis are considered consequence of pleiotropic pharmacologic properties [64, 69]. There are also case reports of myocarditis associated with drug-induced eosinophilia. Despite the fact that no extensive mechanistic studies had been performed in those cases and most of the data is based on clinical reports, it is believed that in the latter cases myocardial inflammation is a consequence of eosinophilic infiltration rather than direct drug-induced damage [70].
Physical factors like radiation are also strongly associated with myocarditis in a dose-dependent manner. The risk of actinic myocarditis increases exponentially above 2Gy [71]. Radiation induces an acute immune response mediated by TNFα and IL1β, which seems to be the initiation mechanism if radiation noxa affects heart tissues [71].
The current concept is that pleiotropic properties of those pharmacologic and physical agents induce a direct myocardial damage. Nevertheless, this direct effect is not the only cause of myocarditis but rather its trigger. After that initial damage, an innate immune response occurs associated with exposure of intracellular self-antigens and eventually modification of those proteins in the inflammatory milieu generating neo-epitopes, altogether priming a myocardial-specific autoimmune adaptive response [64, 68].
10.3.3 Viruses
Viral myocarditis is considered sometimes an independent entity. Nevertheless, it seems that once the trigger infectious noxa exerts its effect, the final effector mechanisms are similar to the ones leading to autoimmune myocarditis progression and chronic complications. Myocardium-tropic viral infection acts as a trigger and “coadjuvant” generating a sustained myocardium-specific autoimmune response. The main viruses associated with myocarditis are the CVB3, adenovirus, influenza (A, B), parvovirus B19, cytomegalovirus, and Epstein-Barr virus. All these viruses cause myocarditis with similar inflammatory features, and all could lead to DCM. The real prevalence of progression to DCM for each specific virus is not known [6, 64, 72].
The current consensus is that both immune-mediated and viral cytotoxic mechanisms play an important role. In vitro experiments have demonstrated that low-level/low-rate enterovirus replication in myocytes generates viral proteins able to produce filament disruption of myocyte structure changes resembling the features observed in vivo. This filament disruption is induced by enteroviral protease [73–75]. Thus, beyond virus cytotoxicity, a low-level viral replication can induce cardiac cell damage even in the absence of production of fully mature and infectious viral particles.
During acute infection, one of the first infiltrating leukocytes to the myocardium are Natural Killer (NK) cells, which seem to play a short-term protective role by limiting the viral infection [76]. In support of that, NK-deficient mice develop a more severe inflammatory viral myocarditis (CVB3 model) [77]. Similar to CVB3 myocarditis, NK cells are protective in EAM model since NK cell depletion led to exacerbated inflammation, fibrosis, and loss of cardiac function [78]. We have shown that the mechanism of NK cell protection is mediated by antagonizing eosinophils trafficking to the heart [78]. Eosinophils, classically considered to exert an innate response, play a final effector role in myocarditis even in parallel to a robust adaptive T cell-mediated response [36].
A “second wave” of infiltrating leukocytes during viral myocarditis is mainly comprised of T cells, peaking 7–14 days after viral infection in murine models. T cells play an important role in the clearance of viruses. Specific T cell clones have the capacity to destroy and lysate infected cardiomyocytes, according to in vitro data [72, 76]. Overall, this antiviral response has a dual effect: (a) it controls viral infection and limits its cytotoxic effects, and (b) the tissue damage associated with the T cell response leads to exposure of cryptic antigens like myosin-derived peptides, which could eventually generate an autoimmune cardiac-specific response [79].
Several facts support the involvement of autoimmunity in the progression of viral myocarditis to a chronic stage. Mice develop cardiac-specific autoantibodies in both CVB3 myocarditis and in EAM. Furthermore, the mice susceptible or resistant to chronic CVB3 myocarditis are also susceptible or resistant to EAM, respectively (see above). C57BL/10 J mice expressing H2b haplotype can develop acute inflammatory viral myocarditis, equivalent to A/J background strains, but are protected from chronic disease and progression to DCM [62]. A similar phenomenon has been described in mice sharing MHC haplotypes but no other non-MHC antigens (A/J B10.A H2a vs. B10.A H2a, resistant and susceptible to virus-associated DCM, respectively) [47]. Thus, both MHC and non-MHC genes are involved in the susceptibility to autoimmune myocarditis.
10.3.4 Mimicry
Recognition of antigens by T cell receptors (TCR), B cell receptors (BCR), and antibodies relies on the existence of specific complementarity of molecular regions of proteins, called epitopes, and the antigen-binding regions of the enounced receptors and antibodies (in a MHC-restricted manner in the case of TCR). Mimicry implies molecular similarity between non-self (microbial or alloepitopes) and self-epitopes, and the consequent cross-reactivity with T cell clones and/or antibodies (BCR are antibody-like surface receptors) [80, 81]. A paradigmatic example is the association of infections of Campylobacter jejuni with the development of Guillain-Barré syndrome [82].
It is still unclear if the molecular mimicry phenomenon is critical in the pathogenic process of myocarditis, but some studies suggest at least a partial contribution. The main proposed target of autoimmune response in myocarditis is the heavy chain of the myosin, specifically the isoform expressed in myocytes αMyHC [83, 84]. Multiple myosin isotypes had been identified, but myosin sequence is relatively phylogenetically preserved [85]. Using an A/J background murine model (H2k haplotype), it was found that autoreactive T cell clones recognizing αMyHC peptides (specifically 334–352) in an IAk-restricted manner have cross-reactivity with microbial epitopes derived from Bacillus spp., Magnetospirillum gryphiswaldense, Zea mays, and, even more important for its human clinical implications, Cryptococcus neoformans [84]. The latter fungi had shown a breakout within the last decades in association with the AIDS pandemic. That might have implication on the pathogenesis of myocarditis, mostly in patients susceptible to C. neoformans infections under chemotherapy or carrying clinical HIV infection.
Another particular kind of molecular mimicry was described in the context of myocarditis as similarity between self-antigens, myosin epitopes, and β-adrenergic receptors. Therefore, the autoantibodies generated during myocarditis against myosin peptides may cross-react with adrenergic receptors and exert an activating adrenergic effect upon ligation, thus potentially contributing to chronic sympathetic-mediated cardiac damage [86].
10.3.5 Exposure of Encrypted Self-Antigens
Another concept demonstrated to play a role in the development of autoimmunity is the exposure of self-antigens which are encrypted and unavailable to the immune system under physiologic conditions [87, 88]. Cardiac myosin (specifically αMyHC) is one of the most important self-targets in myocarditis. Importantly, myosin, which belongs to the contractile apparatus of the myocytes, is not significantly exported to the extracellular matrix.
Myh6, the gene encoding αMyHC, has been shown to not be expressed in medullary thymic epithelial cells (mTECs), which are the main responsible for T cell thymic negative selection, via promiscuous self-antigen presentation. αMyHC is not expressed neither in peripheral lymphoid stromal cells. As consequence, in physiologic conditions, specific self-reactive anti-αMyHC T cell clones escape from negative selection, reaching periphery as autoreactive cardiac-specific clones. That is an important feature predisposing development of myocarditis in mice and humans as long as other events or risk factors take place leading to release of αMyHC, a cryptic self-antigen. In fact, it was shown that transgenic mice, expressing Myh6 in mTECs, are protected from EAM induction in association with clonal deletion of those autoreactive clones [89].
Either myocardial infection, toxins, ischemia or other insults can lead to myocardial damage and subsequent exposure of intracellular proteins (cryptic epitopes). That exposure, in the context of a proper pro-inflammatory microenvironment, triggers the adaptive immune response leading to myocarditis in genetically susceptible individuals [6, 72, 76]. Thus, different triggers could lead to myocarditis and eventually chronic autoimmune myocarditis.
10.3.6 MHC and Non-MHC Bias
Human Leukocyte Antigen (HLA) genes (human form of Major Histocompatibility Complex, MHC) are one of the most polymorphic genes in humans, as well as the coresponding MHC genes are in most mammals. This genetic variability has functional consequences in terms of the affinity of MHC molecules for specific peptides. Despite MHC molecules being promiscuous in terms of peptide presentation, certain biases exist as consequence of the avidity and affinity of MHC molecules for specific peptides [2].
The association of certain autoimmune diseases with specific HLA haplotypes is considered to be consequence of a higher affinity of certain haplotypes for preserved protein self-products.
HLA haplotypes are not sufficient, and may not be main determinants, in myocarditis initiation and progression. Nevertheless, certain associations between DCM of any cause and HLA haplotypes had been found. Specifically, HLA-DR4 is statistically associated with DCM based on retrospective studies [90–92]. Other DCM-HLA-positive associations had been reported, mainly HLA-DR12, DR15, and DRB*0601, as well as negative associations (HLA-DR11, DQB1*0301) [93–95].
Experimental facts observed in murine models of EAM and CVB3 myocarditis also support the influence of MHC haplotypes in the development of myocarditis and its progression to DCM. Mice with b haplotypes in A/J background are less susceptible than other non-H2b A/J strains to EAM and CVB3 myocarditis including the acute inflammatory phase and DCM [96]. Furthermore, that b-associated protection on A/J background parallels development of lower titers of cardiac-specific autoantibodies [62, 96].
Similarly, transgenic nonobese diabetic mouse strain expressing human HLA-DQ8 instead of IAb develops spontaneous myocarditis which progresses to DCM even with electrophysiologic disturbances like heart block (see Sect. 10.2) [51, 97]. Nevertheless, it is of notice that HLA-DQ8 is not within the haplotypes associated with DCM in human studies, which might limit the translational findings of this murine model and/or underscore the importance of other non-MHC genes.
There is a lack of myocarditis-specific association of MHC haplotypes in certain murine models of myocarditis as was shown by using specific strains sharing MHC haplotypes (MHC-full matched), but differing in non-MHC background-associated genes has been performed. A.SW (A/J background) and B10.S (B10 background), both H2s, have differences in myosin-induced EAM susceptibility, being B10.S protected as compared with susceptible A.SW [98]. Similarly, mice with b haplotypes differ in susceptibility to EAM. C57BL/10 J are resistant, while A/J are very susceptible to EAM. That strongly suggests the influence of other non-MHC features in the development of myocardial-specific autoimmunity. Using simple sequence length polymorphism (SSLP) markers in the murine genome, two non-MHC loci were found to be associated with the development of EAM in H2s equivalent strains. Those genes, named in mice Eam1 and Eam2 (located in proximal chromosome 1 and distal chromosome 6, respectively), are linked to myocarditis development. Interestingly, those loci and their human equivalents had been associated with SLE and diabetes, as well as autoimmune experimental encephalitis and orchitis in mouse [98]. Other studies using the CVB3 murine model and also SSLP tracking system found other non-MHC loci associated with viral myocarditis: Vms1 (chromosome 1), Vms2 (chromosome 4), and Vms3 (chromosome 3) [99].
Finally, a specific 14 bp deletion in a region of the HLA-G gene (a human non-classical MHC I molecule) was reported to be associated with DCM. Nevertheless, as linkage disequilibrium exists between HLA-G and HLA-DR and DQ and considering that the reported HLA-G 14 bp deletion occurs in the 3′-untranslated region of the gene, it remains unclear if the HLA-G bias corresponds to a real mechanistic association, or just a correlation whose mechanism relies on class II-linked haplotypes [100, 101].
Other polymorphisms have been identified in association with autoimmunity, mainly on the CTLA-4, PD-1, and ICOS genes. All these genes encode for functional proteins involved in the regulation of T cell activation mainly via induction of apoptosis and/or anergy. Specifically, mutations in PD-1 and ICOS are associated with the development of myocarditis in murine models but in the context of broader systemic autoimmune features [52, 53, 102, 103]. The case of PD-1 deserves particular attention considering the outbreak of the checkpoint blockade in cancer therapy. PD-1 blockade has been shown to provide benefit in certain cancers like non-small cell lung carcinoma and melanoma [55, 56]. Myocarditis has been found to be an infrequent side effect of PD-1 blockade. The histological features were that of acute lymphocytic myocarditis [57]. This validates the importance of the disruption of the PD1/PDL1 axis in the development of myocarditis observed in mice.
10.3.7 Autoimmune Regulator (AIRE)
Autoimmune regulator (AIRE) allows a promiscuous expression of self-antigens by medullary thymic epithelial cells (mTECs) [104, 105]. AIRE is involved in central deletion of effector T cells (Teff) and thymic Treg (tTreg) induction but also in peripheral Teff anergization and pTreg differentiation [104–106].
The absence of AIRE expression in murine models (AIRE−/− mice) leads to an increase of autoreactive effector T cells in the periphery [107]. AIRE also influence self-tolerance by directing autoreactive bone marrow-derived T cell clones into regulatory functions [107].
Taking these concepts to the field of myocardial autoimmunity, some experimental data have demonstrated that defective central (thymicdependent) tolerance and increase of autoreactive Teff (specific for myosin-derived epitopes) as well as decreased thymic Tregs (tTregs, formerly nTregs) with the same specificity are involved in the inflammatory phase of EAM [106, 108]. HLA-DQ8+ IAb−/− NOD mice develop spontaneous myocarditis with cellular and humoral autoimmune responses directed toward epitopes of the α isoform of myosin heavy chain (αMyHC). The same model showed that EAM is dependent on the T cell-mediated anti-MyHC response [109]. Anti-myosin-specific T cells represent the majority of the myocardium-infiltrating Teff [89]. The development of myocarditis mediated by MyHC-specific Teff was associated with lack of expression of αMyHC by mTECs. With more complex genetic manipulations of that murine strain, it was shown that susceptibility to EAM was abrogated by expression of MyHC by mTECs, paralleling a decrease in that specific Teff autoreactive clones in the periphery. Further studies about the functional impact of AIRE in the expression of myosin by human mTECs as a factor determining development of autoimmune myocarditis are needed [89].
10.3.8 Fefz2
It was discovered recently that nuclear factor Fezf2, similarly to AIRE, directs tissue-restricted antigen expression in thymic medulla stromal cells. [110]. Fefz2-deficient mice (Fefz2−/−) develop systemic disease distinct from AIRE−/− mice [110]. Discovery of the importance of Fezf2 in the development of central tolerance is relatively recent. So far, no evidence exists associating autoimmune myocarditis with functional or genetic modifications of Fezf2. Nevertheless, it is a topic that probably will be under scrutiny in the near future.
10.4 Immunopathogenesis of Myocarditis
Studies performed in murine models and observational data from humans have demonstrated that CD4 T cell response is the main driving force of autoimmune myocarditis.
Autoimmunity is characterized by a highly antigen-specific immune response, yet the activation of innate response is required to prime the adaptive response [1]. Its importance in the context of autoimmune myocarditis has been demonstrated by the fact that EAM murine models require the use of coadjuvants (like complete Freund’s adjuvant (CFA)) and unspecific stimulation by pathogen-associated molecular patterns (PAMPs) provided by Mycobacterium antigens. Importantly, the coadjuvant challenge must be provided simultaneously to the exposure of cardiac self-antigens in order to generate the cardiac-specific sustained autoimmunity. If timing is not properly set, then the self-specific immune response is not mounted, even if the innate response is successfully stimulated by the adjuvants [8]. Innate cellular response is required not only for initiation of autoimmunity but also for the maintenance of the adaptive T cell-mediated response and progression to chronicity. Myocardial infection by specific microbes, beyond a direct virus-dependent cytolytic effect, seems to play the role of “coadjuvant” to the autoimmune adaptive response (Fig. 10.1) [61, 81]. In this section, the specific features of this complex cross talk will be described.
10.4.1 Adaptive Cellular Response
The adaptive cellular immune response is characterized by its antigen specificity and high inflammatory efficiency. Its major mediators are the T cells, which specificity relies in the molecular structure of clonal TCRs, able to “recognize” characteristic peptide/MHC complexes.
The classic dichotomist Th1/Th2 model does not provide an accurate representation of the normal immune response [111, 112]. That became evident in the last decades after discovery of other important pro-inflammatory Teff subsets, like Th17 (but also including Th9 and Th22), as well as anti-inflammatory subpopulations, mainly Tregs and Tr1. Furthermore, a high plasticity of T cell subsets has been described, involving not only central (thymic) but also peripheral T cell fate induction [112]. Dysfunction of Th17 and Treg has been widely found to be a key factor in autoimmunity, including myocarditis. Importantly Treg and Th17 activation/differentiation shares certain cytokine requirements, remarkably TGFβ, needed for both subsets but in a concentration-dependent manner. In the presence of TGFβ, IL6/IL-1β vs IL10 balance is responsible for the Th17 vs Treg differentiation [112].
10.4.1.1 Th1 Response
As in other autoimmune diseases, the overall activity of self-specific Th1 response is one of the main mediators of the inflammatory phase of myocarditis [111]. Supporting the importance of Th1 response in myocarditis, several studies had focused on IL12 as primary mediator. It was shown that mice lacking IL12Rβ1 and STAT4 signal (thus unresponsive to IL12) are resistant to myocarditis, whereas exogenous IL12 exacerbates EAM [113]. Notwithstanding, the same study demonstrated that blockade of IFNγ worsen the disease, starting to draw the picture of a counterbalance of IFNγ-IL12 axis and a potential dual role of IFNγ [113].
IL12 family main members and its subunits are IL12 (p35 p40), IL23 (p19 p40), IL27(p28 EBI3), and IL35 (p35 EBI3) [114]. The existence of shared subunits between the cytokines makes difficult the determination of the specific role of each member. Furthermore, IL12Rβ is a receptor chain shared by IL12 and IL23 [114]. Several attempts have been made to elucidate the differential role of IL12 family cytokines. The first studies analyzed the differential EAM phenotype between IL12p40 −/− and IL12p35−/− [115]. It was found that IL23 and IL-12 dual-deficient mice (IL12p40 −/−) were protected from EAM in contrast to IL12-deficient mice (IL12p35−/−), suggesting predominant pathogenic role of IL23 over IL12 [115]. Importantly, IL23 is key in the development and stabilization of the Th17 response [112]. Also, it must be considered that IL12p35 is a subunit shared with the recently discovered IL35, a potent anti-inflammatory cytokine [116]. Furthermore, that study was not able to induce specific knockout of IL23, and the conclusions are rather inferred from the differences between IL12p40 −/− and IL12p35−/−.
In later studies, our laboratory was able to determine the specific role of IL23 in myocarditis using an IL12p19−/− strain (IL23 KO), which is deficient only in IL23, because as far as is known, IL12p19 subunit is not shared with other members of the family [117]. We found that IL23 is transiently required during the early stages to induce CD4 T cell pathogenicity. That process was dependent on GM-CFS. IL23 was required in the early stages but dispensable once the GM-CSF-dependent T cell-mediated autoimmunity is established; thus, IL23 is a required “switch” for the initiation process of EAM [117].
In congruence with the complex regulatory network of the immune system, results observed in IFNγ-deficient models showed paradoxical effects as compared with IL12 family-deficient studies. IFNγ KO (Ifng−/−), IFNγ receptor KO (Ifngr−/−), and mice receiving anti-IFNγ antibody treatment developed more severe inflammatory myocarditis than WT strains [113, 118, 119]. In addition, mice deficient in Tbet (Tbx21−/−), nuclear factor required for Th1 T cell differentiation/development, develop more severe EAM than WT [120]. Similar findings regarding the effect of depletion of IFNγ were observed in CVB3 myocarditis model [121]. The severe CVB3 myocarditis in the absence of IFNγ was not associated with lack of antiviral response but with an enhanced IL1β, IL4, and TGFβ production [121].
An important feature observed in DCM on IFNγ-deficient mice is the severity of the fibrosis and even the development of constrictive hemodynamic complications [122]. The latter might be associated with the regulatory influence of IFNγ on the mediators of tissue remodeling and fibrosis, including monocytes and fibroblasts. IFNγ has multiple potential sources in the context of myocarditis, including T cells, monocytes, macrophages, dendritic cells, and innate lymphoid cells.
As final consequence, Th1 response occurs during the development of myocarditis, with IL12 and TNFα playing essentially a pro-inflammatory role but with IFNγ being dual, thus modulating and homeostatically dampening the immune response and limiting sustained inflammation and fibrosis.
10.4.1.2 Th2 Response
In the murine model of Th2-mediated eosinophilic myocarditis, EoEAM, a massive and severe inflammatory eosinophilic myocarditis progressing to DCM, is observed in a transgenic strain lacking the main Th1 and Th17 final effector cytokines, i.e., IL17A−/−IFNγ−/−, after EAM induction. The lack of Th1 and Th17 responses generates a Th2-biased inflammation leading to eosinophilic myocarditis, resembling the features and severity of the eosinophilic human disease. The main final effectors of the myocardial damage are eosinophils, despite the T cell requirement [36].
Pointing out the complexity of the regulatory process, IL4 and IL13, both classic pro-inflammatory Th2 cytokines, seem to have an opposite effect in the development of EAM- and CVB3-induced myocarditis. Those Th2 cytokines are under the same promoter and are typically dependent on the activation of the nuclear factor GATA3 in T cells [48]. Mice lacking IL4 expression (Il4−/−) develop a milder inflammatory myocarditis and a less severe systolic dysfunction and progression to chronic stages when EAM is induced in an A/J background. However, Balb/c IL4 KO mice develop a disease similar to WTs [123, 124]. This unveils strain-specific immunologic differences but, on the other hand, shows that IL4 pro-inflammatory capacity is variable during EAM but certainly is not a protective mediator. Oppositely, we described that IL13−/− mice on Balb/c background develop more severe EAM- and CVB3-induced myocarditis and progress to severe DCM and the associated heart failure [123].
The latter observation in IL13−/− mice takes us back to the cross talk between adaptive T cell response and the innate cellular response. In fact, IL13 exert a protective role in the development and progression of myocarditis by regulating the macrophage differentiation. Aggressive acute inflammatory features and the important chronic impairment in systolic function during DCM phase were associated with a typical upregulation of a macrophage-derived cytokine cluster: IL1β, IL18, IFNγ, and TGFβ [123]. Aside from an increase in macrophage-derived cytokines, a specific upregulation of CD204+ CD206+ pro-inflammatory/activated macrophages was observed in IL13 KO mice, strongly suggesting a suppressive effect of IL13 in the macrophage side of the innate-adaptive cross talk.
10.4.1.3 Th17 Response
IL17A, the main pro-inflammatory Th17-derived mediator, unexpectedly is not necessary for development of acute inflammatory stages of myocarditis in EAM model [125]. On the other hand, we have found that IL-17A is strictly required for progression to DCM, cardiac fibrosis, and loss of cardiac function. In other words, the acute inflammation can take place in the absence of IL17A; however without IL17A, myocarditis does not progress to DCM [125].
Importantly, the Th17 response required for progression from myocarditis to DCM is not only dependent on leukocytes. We demonstrated that beyond inflammatory Ly6Chigh monocytes, also cardiac fibroblasts play an important role in the inflammatory process. IL17A−/− mice, protected from DCM but not acute EAM, have a diminished myocardial infiltration of neutrophils and Ly6Chigh inflammatory monocytes [126]. Interestingly, a conversion of Ly6Chigh monocytes to Ly6Clo monocytes protects WT host from DCM, strongly suggesting that Ly6Chigh monocytes are associated with the pro-fibrotic IL17A effect. We have found that granulocyte-monocyte colony-stimulating factor (GM-CSF) is required, in conjunction with IL17A, for Ly6Chigh monocyte infiltration in myocardium during EAM. We showed that IL-17A is able to induce high amount of GM-CSF and other myeloid cytokines and chemokines from cardiac fibroblasts. Thus, we have discovered an active Th17-fibroblast-monocyte cross talk in the pathogenesis of myocarditis. To summarize, T cell-derived IL17A plus fibroblast-derived GM-CSF contributes to Ly6Chigh monocyte chemotaxis/activation, which plays an active role in fibrosis and sustains inflammatory process (Fig. 10.2) [126].
Fig. 10.2.
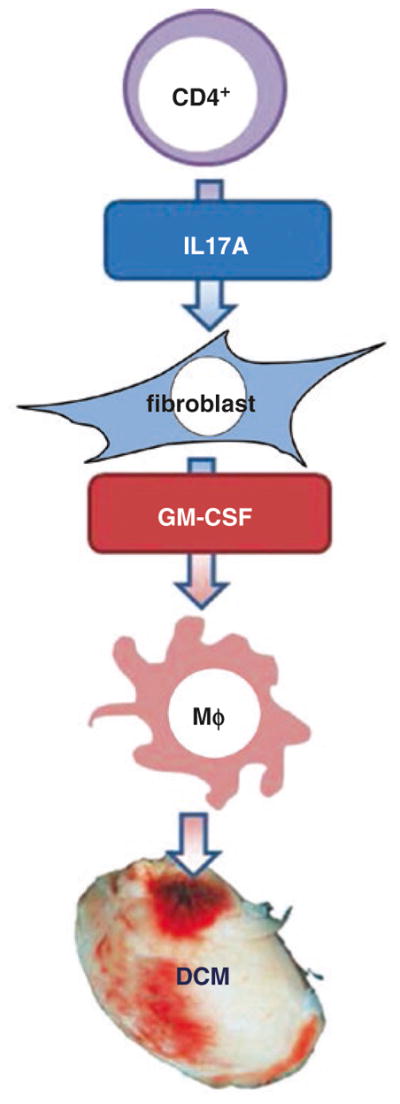
Schematic immunopathogenesis of post-myocarditis DCM development. CD4 T cell autoimmune response induces a Th17-dependent pro-inflammatory process associated with DCM development. T cell-derived IL17A induces cardiac fibroblasts to produce GM-CSF, which in turns activates monocytes toward a highly inflammatory function. Those activated monocytes are required final effectors in the progression to chronic tissue damage and DCM
As described above, a very specific milieu of cytokines is needed for Th17 cell differentiation and establishment of the so-called Th17 environment. That milieu includes IL6, IL23, and TGFβ. Importantly, IL23 is needed for Th17 terminal differentiation and Th17 “stabilization” [111, 112]. We demonstrated the requirement of IL23 (p19) during EAM development [117]. Specifically, CD4 T cell stimulation by IL23 is required during the acute phase of autoimmunity for an effective myocarditis (EAM) and consequent DCM development. The key effect of such IL23 stimulation was proven to be the GM-CSF production by T cells. Furthermore, IL23 effect is only transiently required during the acute phase (early after EAM induction), as later influence cannot restore the EAM development [117].
An important translational study supporting the latter concepts was published recently [127]. That study supports the influence of Th17 in human myocarditis and its progression to DCM. Th17 CD4 T cells were found to be associated with myocarditis, influencing inflammation via IL6, TGFβ, and IL23. Also, an association exists with GM-CSF producing monocytes (CD14+). This interaction was related with TLR2 expression on those inflammatory monocytes, an interestingly finding unveiling the role of this particular TLR type in human myocarditis. Consistently, a decreased classic Treg population (Foxp3+) was observed in association with the enounced features [127].
Those seminal studies made evident potential Th17 response-focused therapeutic targets, not only to treat acute inflammatory myocarditis but also its late-stage complications.
10.4.1.4 T Regulatory Cells
Treg deficiency is associated with autoimmune severity, including in myocarditis models. A mechanism, already described above, is associated with the cross regulation of TLR4-Tim3, which is preferentially associated with Treg development in females. It is a mechanism explaining the constitutive protection of females to myocarditis, both autoimmune and viral associated, as compared with male hosts [49].
In the same way, a possible explanation for the preferential susceptibility of A.SW H2s to EAM as compared with B10.S H2s, despite an identical H2 genotype, is a constitutive strain-specific lower frequency of classic Tregs (CD4 + CD25 + Foxp3+) in A. SW. Interestingly, this Treg profile seems to be really constitutive and not associated with specific myosin-targeted responses, as demonstrated by ovalbumin immunization. Importantly, Tregs showed an opposite trend in terms of frequency with respect to Th17 cells in those strains in the context of EAM [128].
Interestingly, Tregs also provide protection to viral myocarditis, as demonstrated in the CVB3 myocarditis model. Importantly, Tregs not only diminished tissue damage but also were associated with improved viral clearance. Those phenomena were associated with secretion of TGFβ, again demonstrating the multiple roles of that cytokine-like factor [129].
Recently, the severity of EAM was found to be attenuated by cannabidiol (a non-psychoactive constituent of marijuana), in relation with a decrease of CD4 T cells. The mechanism was not entirely elucidated, but the possibility of a change in the Treg/Teff balance is a plausible hypothesis [130]. Overall, the current concept is that a defective Treg function is associated with development of autoimmune diseases, including autoimmune myocarditis, providing a potential therapeutic target, which so far had been elusive.
10.4.2 Adaptive Humoral Response
Despite the existence of clearly humoral-mediated autoimmune diseases (like post-streptococcal glomerulonephritis, Berger disease, and Goodpasture disease), the influence of those autoantibodies on the pathogenesis of most autoimmune diseases is still controversial, at least on its initiation phase. Myocarditis is a cellular-mediated process, and several myocarditis-associated autoantibodies are specific to encrypted antigens that are not available to antibody ligation without tissue damage and cell disruption [87]. As consequence, the generation of autoantibodies by clonal selection of self-reactive B cell clones is secondary to T cell activation, T-B cross talk, and release of intracellular or matrix-encrypted proteins after tissue damage [131, 132].
In myocarditis, this lack of pivotal role during initiation is supported by the fact that up to two-thirds of acute myocarditis patients have no detectable cardiac-specific antibodies at the moment of diagnosis or clinical onset [83, 132]. On the other hand, its correlation with disease progression and detection in symptom-free relatives of DCM patients suggest that they occasionally may be early markers paralleling myocardial damage after its initiation, with minimal to null causative influence in the initial phase [132].
Eighty percent of patients with DCM-related myocarditis develop cardiac-specific antibodies, but also up to 60% of patients with heart failure of any cause have circulating antibodies targeting heart-specific epitopes. The latter supports the idea of the necessity of tissue damage of any cause for development of humoral autoimmunity, as well as the paucity of its inflammatory influence [83, 132–134].
A plethora of autoantibodies has been described in myocarditis, including anti-αMyHC, troponin I and T, and β1-adrenergic receptor. Also, non-cardiac-specific autoantibodies are frequently found in myocarditis patients, including anti-actin, tropomyosin, laminin, and anti-muscarinic receptor [4]. Of those, the ones receiving most attention are the anti-αMyHC and the anti-adrenergic receptor. Anti-αMyHC antibodies have been described to correlate with disease progression (including diminished titers after adequate clinical response) and are also found in murine models [132]. Anti-adrenergic receptor antibodies are also correlated with disease progression, and interestingly a significant proportion of the clones are activating antibodies, so those antibodies might contribute to hemodynamic alterations via autonomic modulation [4, 83, 132, 133, 135, 137].
Antibodies targeting specific receptors might induce persistent activation of such receptors, i.e., induce a sustained positive intracellular signaling, similar to the one generated by physiologic ligands, but abnormally sustained over time. This is not the case of all receptor-specific antibodies but seems to be the case of several clones involved in myocarditis targeting adrenergic receptors [133]. The case of cross-reaction of anti-myosin antibodies with adrenergic receptors was already described above in the section dedicated to mimicry [84].
In the case of activating β1-adrenergic antibodies (with a prevalence of 70–80% among patients with dilated cardiomyopathy of any cause and 60% on patients with myocarditis), they seem to exert their persistent adrenergic effect by stabilizing an activated molecular conformation of the receptor upon ligation. Also, the cross-link of two receptors, generating a stable dimerization, contributes to that effect [132, 133].
A pathogenic sustained β-adrenergic activation has been demonstrated to be able to induce cardiomyocyte apoptosis [137, 138]. Furthermore, once systolic dysfunction begins, the increased adrenergic tone (sympathetic) has deleterious hemodynamic effects [139, 140]. It is believed that beneficial effect of β blocker drugs in patients with heart failure of any cause is related both with the improvement of hemodynamic status and the decrease of the proapoptotic effect. Nevertheless, theoretically, such drugs cannot reverse per se the stabilization of the receptors induced by the activating autoantibodies. In that regard, the turnover rate and upregulation of those receptors must be considered [140].
Therapeutic approaches are focused on the depletion or neutralization of anti-adrenergic activating autoantibodies. Plasmapheresis and immunoadsorption of those antibodies were tested in case-control clinical studies in patients with dilated cardiomyopathy [141]. Interestingly, also benefit from the removal of cardio-depressant autoantibodies by immunoadsorption was also found [142]. Patients receiving immunoadsorption have improved cardiac function. After a 12-month follow-up, anti-β-adrenergic receptor autoantibodies did not return to the original titers [143].
Another treatment strategy with a similar goal is the neutralization of those antibodies using aptamers in the apheresis technology. As aptamers are synthetic oligonucleotide ligands with high specificity to targets like the Fab region of antibodies, its use is proposed as a mean to optimize the removal of autoantibodies by apheresis and decrease toxic and immunogenic complications described for immunoadsorption [144, 145]. The overall utility of this approach has not been fully elucidated.
10.4.3 Innate Immune Response
Innate leukocytes are those ones defined for lacking an antigen-specific receptors, thus responding in an unspecific manner via cytokine chemoattraction and/or nonspecific receptors such as Toll-like receptors (TLRs) and responding with phagocytosis and non-specific cytotoxic chemical species. The main innate immune cells involved in myocarditis are natural killers (NKs), neutrophils, eosinophils, and monocyte/macrophages [8]. It is important to notice that innate lymphoid cells (ILCs), a specific subset of leukocytes having lymphoid properties but lacking antigen-specific receptor, were described recently. ILCs are involved in mucosae patrol and in several immune responses, including antihelminthic responses and immune-mediated diseases like Crohn disease, psoriasis, dermatitis, and airway hyperreactivity [146]. Little is known about its biology in the heart. Nevertheless, NK cells, a particular TCR-lacking subset of lymphocytes with cytolytic capacity, are currently considered ILCs type 1 [147]. Regarding NK cells, its role in the immune response during autoimmune myocarditis (EAM model) and viral myocarditis (CVB3 models) has been described above.
Eosinophils are not only involved in the development of the severely Th2-skewed EoEAM but also in the classic EAM [36]. Eosinophils behave as final effectors and infiltrate and produce myocardial inflammation even once the adaptive T cell response is established.
Neutrophils also infiltrate myocardium during EAM, exerting an inflammatory role, with long-term impact even in the impairment of the cardiac function [48]. Neutrophil activation and recruitment are dependent on the Th17 response via IL17A and GM-CSF [125]. Also, in an OVA-TCR transgenic system of heart inflammation, it was found that neutrophil cross talk with heart-infiltrating CD8 T cells sustains its T cytotoxic response [148].
Finally, one of the most important heart-infiltrating innate cells during myocarditis are the macrophages [48, 149]. Some of its interactions with specific cytokines and T cell responses were described above.
Several attempts have been made to classify macrophages. For instance, depending on its pattern of activation and inflammatory capacities, they are divided into M1 and M2. M1 macrophages (or “classic”) are activated by IFNγ in association with the Th1 response and have strong antigen-presenting and pro-inflammatory capacities. On the other hand, M2 “alternative” differentiation occurs in anti-inflammatory environments and is induced by IL10 and TGFβ. It is proposed that M2 macrophages have a significantly weaker antigen presentation capacity [149]. However, M1-M2 dichotomy is difficult to reproduce and detect in vivo. Also, based on its monocyte precursors and final inflammatory capacities, mouse macrophages are classified in vivo as Ly6Chigh inflammatory and Ly6Clow regulatory subsets. Human CD14high monocyte/macrophages correspond to mouse Ly6Chigh, whereas CD14int are equivalent to Ly6Clow. M1/M2 and Ly6Chigh/low classifications do not completely overlap. A significant plasticity exists in the monocyte/macrophage populations [149]. As consequence, it is considered that such a rigid categorization does not correspond to the real immunobiology of these cells.
Specifically in EAM, monocytes and macrophages are early myocardial-infiltrating cells. Not all myocardial macrophages are strictly “infiltrating cells” but rather derived from resident monocyte differentiation [149]. Even before that massive infiltration, the TLR ligation (mainly 2 and 4) in monocytes/macrophages is crucial in the initiation process of myocarditis and also the targets of coadjuvants in the EAM model [8, 61, 149].
The pro-inflammatory molecule called high-mobility group box 1 (HMGB1) has been found to be important during the initiation process of EAM in studies performed with TnT-EAM model [150]. This is a ligand of a macrophage receptor called receptor for advanced glycation end products (RAGE). This interaction induces a M1-like phenotype/functionality of macrophages, boosting its inflammatory impact on myocarditis. Importantly, TLRs are redundant receptors of HMGB1, which activation is independent of RAGE [150, 151].
It has been found that once in an inflammatory Ly6Chigh status, macrophages express IL17 receptor. Heart-infiltrating macrophages during EAM have these characteristics. In that way, macrophages, along with neutrophils, belong to the innate cells associated with Th17 responses during myocarditis. After IL17A stimulation, Ly6Chigh macrophages produce a particular cluster of cytokines, GM-CSF, already described as an important mediator of EAM, IL3, IL9, CCL4, and CCL5 [152].
Overall, macrophages are potent inflammatory cells during myocarditis and potentially the most abundant in endomyocardial biopsies. These cells are critical in the initiation process. Nevertheless, they are also responsible for the progression of the disease. Macrophages are orchestrated with the T cell response, in such a way that several compensatory and regulatory mechanisms which dampen the severity of EAM (like IFNy) can target macrophages, as described above.
10.5 Therapeutics
Unfortunately, this is one of the less developed areas in the myocarditis field. To begin with, some clinical studies lack a clear diagnostic and etiologic definition, and several times, the term “virus-positive” or “virus-negative” myocarditis is based only on peripheral blood serology [153, 154].
That generates a huge conundrum on how to analyze the results of the therapeutic trials. The trigger is unclear in most of the cases of the “virus-negative” patients. Even more important is the fact that it is unknown at which point of the temporal evolution of the autoimmune process is the patients when enrolled in the therapeutic trials. That makes us hypothesize that case and control groups had been heterogeneous in immunopathogenic terms, in such a way that it is hard to analyze the data from a strict evidence-based medicine perspective.
Notwithstanding, it is understandable that the potential severity and the relatively low incidence of myocarditis, along with the immunologic and diagnostic complexities/limitations enounced above, make almost impossible to carry out optimally designed clinical trials as the ones performed for other high prevalent/incident entities as high blood pressure and diabetes.
Fortunately, there is a growing body of therapeutic knowledge which, despite frequently based on case reports and uncontrolled trials, suggests that immune modulation/suppression is a useful approach in myocarditis, even in viral-triggered cases if it is used together with proper antiviral treatment [72, 153].
10.5.1 Immunosuppression
The rationale of this therapeutic approach is to target the pro-inflammatory mediators of the disease. Immunosuppression is used in treatment of myocarditis types considered to be autoimmune such as giant cell myocarditis. So far, broad immunosuppressive drugs rather than targeted treatments have been used. The main drugs tested for myocarditis are steroids (prednisone, prednisolone), cyclosporine, and azathioprine [9, 30]. These drugs are mostly used in combination; however, the benefits for individual subtypes of myocarditis need to be evaluated in proper clinical comparative studies. Some studies showed significant or at least some improvement in terms of ejection fraction [155], but another study reported no benefit on ejection fraction or survival as compared with placebo [156]. Notwithstanding, differences in the study design exist, and importantly the study reporting benefit was made on nonviral myocarditis cases, whereas in the one reporting no benefit no viral status discrimination was made.
Another interesting study analyzed the effect of muromonab-CD3 (a monoclonal antibody targeting CD3) plus cyclosporine and steroids in a prospective cohort of 12 patients. This study included specifically patients with giant cell myocarditis. Despite there was no control group (which is understandable due to the high severity and lethality of this type of myocarditis), an improved survival was reported [28–30, 157].
10.5.2 Immunomodulation
As described in the section of humoral response, plasmapheresis/immunoadsorption has been studied by several groups. A consistent benefit was observed in hemodynamic terms, mainly improvement of ejection fraction. Those protocols include the coadministration of immunoglobulin. Interestingly, those studies enrolled patients with DCM, which supports the hemodynamic deleterious effect of cardiac autoantibodies. Importantly, DCM patients of any cause were included, which expands the usefulness of this approach to non-myocarditis cases [141, 159–161].
High doses of immunoglobulin exert a beneficial effect (improved survival and ejection fraction) in acute myocarditis patients. Several data come from small case studies [153], but a randomized controlled study on acute myocarditis patients supports the observation [161]. It is of notice that one controlled study reports conflicting data by not finding improvements of the ejection fraction [162]. Also, in those studies, no clear etiologic definition was made.
10.5.3 Antiviral
The main antiviral drugs studied in myocarditis have been acyclovir, peramivir, ganciclovir, ribavirin, and artesunate, in well-defined viral myocarditis patients (etiology was specifically determined: parvovirus B19, influenza, cytomegalovirus, parainfluenza, herpesvirus, respectively) [6, 72, 153]. All of them have been either small studies or case reports, but overall the benefit of antiviral treatment seems to be consistent in terms of ejection fraction, survival, and decreased viral load. All protocols had included immunosuppressive and/or immunomodulatory treatment, as steroids and/or immunoglobulin [153]. The latter is consistent with the described importance of the immune-mediated damage in conjunction with viral cytotoxic effects in the development of viral myocarditis.
Also, interferon α and β were analyzed in viral myocarditis. Interferon α was found beneficial in enterovirus-confirmed DCM, based on hemodynamic parameters and viral clearance. Interferon β significantly improved ejection fraction and viral load in a cohort of acute viral myocarditis [163], whereas no benefit was observed in a cohort of patients treated at DCM stage [164].
10.5.4 Future Therapeutic Challenges
In general, the future directions should focus on a proper diagnostic, not only on the etiologic aspect but also on the specific stage of evolution of the immune/inflammatory process. That would allow to determine specific treatments depending on the underlying cause and also on the predominant immune process taking place in the patient at the moment of the intervention. Targeted treatments should be generated in the near future, addressing the specific immunopathogenic process predominating in the patient, i.e., more effective antiviral treatments, induction of self-tolerance, boost of Treg response, dampening Teff response, and blockade of innate response-associated receptors, among others, including modulation of the immunologic role of stromal cells like fibroblast and endothelial cells. The positive side of the story is the exponentially growing knowledge about the immunopathogenesis of myocarditis, which certainly will end up in development of effective diagnostic, prognostic, and therapeutic strategies.
10.6 Concluding Remarks
Myocarditis is an extremely complex immune-mediated process. Several risk factors and biologic issues predisposing its trigger and progression have been identified. Those include gender-associated immunologic properties, HLA and non-HLA genetic characteristics, exposure of cryptic antigens, mimicry, systemic autoimmune diseases, drugs, and viral infections. Also, the current concept is that once the initial myocardial inflammation is established, then a T cell-dependent autoimmune process takes place despite differences in the specific etiologic factor (Fig. 10.1). That autoimmune process is responsible for the self-sustained inflammation and progression to tissue damage leading to DCM. In general, it has been found that IFNγ play a paradoxical protective role, in opposition to other Th1 cytokines like IL12. Similar counterbalancing system exists between Th2 cytokines like IL4 and IL13. Notwithstanding, the Th17 response, and importantly its timing, is key in the progression of the acute disease to chronic damage and DCM, clinically the most severe complication of myocarditis. IL17A is not required during the acute phase, but IL23 is required to induce a pathogenic priming of self-reactive T cell clones in a GM-CSF-dependent manner. Importantly, the Th17 system is strictly required for the progression to DCM (Fig. 10.2). Despite the growing body of knowledge about the immunopathogenesis of myocarditis, the specific triggers and factors leading to progression in patients are still a conundrum. Also, since no risk or etiologic factor seems to be sufficient for the initiation and progression processes, post-myocarditis progression to DCM is still unpredictable in clinical practice. Advances exist in therapeutics, but still relying in global immunosuppression and unspecific immunomodulation, with is still suboptimal results. The expectation is that future basic and translational studies might provide even deeper insights in the pathogenesis of myocarditis. That would lead to development of better diagnostic tools allowing characterization and stratification of stages of myocarditis progression in each patient. Finally, that translational knowledge would make possible the development of individualized targeted treatments.
Contributor Information
William Bracamonte-Baran, Department of Pathology, Division of Immunology, Johns Hopkins University School of Medicine, 720 Rutland Ave., Baltimore, MD 21205, USA.
Daniela Čiháková, Division of Immunology, Department of Pathology, Johns Hopkins University School of Medicine, 720 Rutland Ave., Baltimore, MD 21205, USA. W. Harry Feinstone Department of Molecular Microbiology and Immunology, Johns Hopkins University Bloomberg School of Public Health, Baltimore, MD 21205, USA.
References
- 1.Mackay IR, Leskovsek NV, Rose NR. Cell damage and autoimmunity: a critical appraisal. J Autoimmun. 2008;30(1–2):5–11. doi: 10.1016/j.jaut.2007.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bracamonte-Baran W, Burlingham W. Non-inherited maternal antigens, pregnancy, and allotolerance. Biom J. 2015;38(1):39–51. doi: 10.4103/2319-4170.143498. [DOI] [PubMed] [Google Scholar]
- 3.Ozen S, Bilginer Y. A clinical guide to autoinflammatory diseases: familial Mediterranean fever and next-of-kin. Nat Rev Rheumatol. 2014;10(3):135–47. doi: 10.1038/nrrheum.2013.174. [DOI] [PubMed] [Google Scholar]
- 4.Caforio AL, et al. Current state of knowledge on aetiology, diagnosis, management, and therapy of myocarditis: a position statement of the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J. 2013;34(33):2636–48. 2648a–2648d. doi: 10.1093/eurheartj/eht210. [DOI] [PubMed] [Google Scholar]
- 5.Kindermann I, et al. Update on myocarditis. J Am Coll Cardiol. 2012;59(9):779–92. doi: 10.1016/j.jacc.2011.09.074. [DOI] [PubMed] [Google Scholar]
- 6.Pollack A, et al. Viral myocarditis-diagnosis, treatment options, and current controversies. Nat Rev Cardiol. 2015;12(11):670–80. doi: 10.1038/nrcardio.2015.108. [DOI] [PubMed] [Google Scholar]
- 7.Fabre A, Sheppard MN. Sudden adult death syndrome and other non-ischaemic causes of sudden cardiac death. Heart. 2006;92(3):316–20. doi: 10.1136/hrt.2004.045518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cihakova D, Rose NR. Pathogenesis of myocarditis and dilated cardiomyopathy. Adv Immunol. 2008;99:95–114. doi: 10.1016/S0065-2776(08)00604-4. [DOI] [PubMed] [Google Scholar]
- 9.Ekstrom K, et al. Long-term outcome and its predictors in giant cell myocarditis. Eur J Heart Fail. 2016;18(12):1452–8. doi: 10.1002/ejhf.606. [DOI] [PubMed] [Google Scholar]
- 10.Cooper LT, et al. The role of endomyocardial biopsy in the management of cardiovascular disease: a scientific statement from the American Heart Association, the American College of Cardiology, and the European Society of Cardiology. Endorsed by the Heart Failure Society of America and the Heart Failure Association of the European Society of Cardiology. J Am Coll Cardiol. 2007;50(19):1914–31. doi: 10.1016/j.jacc.2007.09.008. [DOI] [PubMed] [Google Scholar]
- 11.Kindermann I, et al. Predictors of outcome in patients with suspected myocarditis. Circulation. 2008;118(6):639–48. doi: 10.1161/CIRCULATIONAHA.108.769489. [DOI] [PubMed] [Google Scholar]
- 12.Daubeney PE, et al. Clinical features and outcomes of childhood dilated cardiomyopathy: results from a national population-based study. Circulation. 2006;114(24):2671–8. doi: 10.1161/CIRCULATIONAHA.106.635128. [DOI] [PubMed] [Google Scholar]
- 13.Doolan A, Langlois N, Semsarian C. Causes of sudden cardiac death in young Australians. Med J Aust. 2004;180(3):110–2. doi: 10.5694/j.1326-5377.2004.tb05830.x. [DOI] [PubMed] [Google Scholar]
- 14.Baccouche H, et al. Diagnostic synergy of non-invasive cardiovascular magnetic resonance and invasive endomyocardial biopsy in troponin-positive patients without coronary artery disease. Eur Heart J. 2009;30(23):2869–79. doi: 10.1093/eurheartj/ehp328. [DOI] [PubMed] [Google Scholar]
- 15.Abdel-Aty H, et al. Diagnostic performance of cardiovascular magnetic resonance in patients with suspected acute myocarditis: comparison of different approaches. J Am Coll Cardiol. 2005;45(11):1815–22. doi: 10.1016/j.jacc.2004.11.069. [DOI] [PubMed] [Google Scholar]
- 16.Aletras AH, et al. ACUT2E TSE-SSFP: a hybrid method for T2-weighted imaging of edema in the heart. Magn Reson Med. 2008;59(2):229–35. doi: 10.1002/mrm.21490. [DOI] [PubMed] [Google Scholar]
- 17.Basso C, et al. Postmortem diagnosis in sudden cardiac death victims: macroscopic, microscopic and molecular findings. Cardiovasc Res. 2001;50(2):290–300. doi: 10.1016/S0008-6363(01)00261-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gore I, Saphir O. Myocarditis; a classification of 1402 cases. Am Heart J. 1947;34(6):827–30. doi: 10.1016/0002-8703(47)90147-6. [DOI] [PubMed] [Google Scholar]
- 19.Richardson P, et al. Report of the 1995 World Health Organization/International Society and Federation of Cardiology Task Force on the Definition and Classification of cardiomyopathies. Circulation. 1996;93(5):841–2. doi: 10.1161/01.cir.93.5.841. [DOI] [PubMed] [Google Scholar]
- 20.Felker GM, et al. The spectrum of dilated cardiomyopathy. The Johns Hopkins experience with 1,278 patients. Medicine (Baltimore) 1999;78(4):270–83. doi: 10.1097/00005792-199907000-00005. [DOI] [PubMed] [Google Scholar]
- 21.Towbin JA, et al. Incidence, causes, and outcomes of dilated cardiomyopathy in children. JAMA. 2006;296(15):1867–76. doi: 10.1001/jama.296.15.1867. [DOI] [PubMed] [Google Scholar]
- 22.Cooper LT. Molecular biologic detection of virus infection in myocarditis and dilated cardiomyopathy. In: Cooper LT, editor. Myocarditis: from bench to bedside. Totowa: Humana Press; 2002. pp. 295–324. [Google Scholar]
- 23.Leone O, et al. 2011 consensus statement on endomyocardial biopsy from the Association for European Cardiovascular Pathology and the Society for Cardiovascular Pathology. Cardiovasc Pathol. 2012;21(4):245–74. doi: 10.1016/j.carpath.2011.10.001. [DOI] [PubMed] [Google Scholar]
- 24.Yilmaz A, et al. Coronary vasospasm as the underlying cause for chest pain in patients with PVB19 myocarditis. Heart. 2008;94(11):1456–63. doi: 10.1136/hrt.2007.131383. [DOI] [PubMed] [Google Scholar]
- 25.Baughman KL. Diagnosis of myocarditis: death of Dallas criteria. Circulation. 2006;113(4):593–5. doi: 10.1161/CIRCULATIONAHA.105.589663. [DOI] [PubMed] [Google Scholar]
- 26.Aretz HT. Myocarditis: the Dallas criteria. Hum Pathol. 1987;18(6):619–24. doi: 10.1016/s0046-8177(87)80363-5. [DOI] [PubMed] [Google Scholar]
- 27.Okura Y, et al. A clinical and histopathologic comparison of cardiac sarcoidosis and idiopathic giant cell myocarditis. J Am Coll Cardiol. 2003;41(2):322–9. doi: 10.1016/s0735-1097(02)02715-8. [DOI] [PubMed] [Google Scholar]
- 28.Cooper LT, Jr, et al. Usefulness of immunosuppression for giant cell myocarditis. Am J Cardiol. 2008;102(11):1535–9. doi: 10.1016/j.amjcard.2008.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Maleszewski JJ, et al. Long-term risk of recurrence, morbidity and mortality in giant cell myocarditis. Am J Cardiol. 2015;115(12):1733–8. doi: 10.1016/j.amjcard.2015.03.023. [DOI] [PubMed] [Google Scholar]
- 30.Cooper LT, Jr, Berry GJ, Shabetai R. Idiopathic giant-cell myocarditis – natural history and treatment. Multicenter Giant Cell Myocarditis Study Group Investigators. N Engl J Med. 1997;336(26):1860–6. doi: 10.1056/NEJM199706263362603. [DOI] [PubMed] [Google Scholar]
- 31.Cooper LT, Jr, et al. Giant cell myocarditis. J Heart Lung Transplant. 1995;14(2):394–401. [PubMed] [Google Scholar]
- 32.Kong G, et al. Response of recurrent giant cell myocarditis in a transplanted heart to intensive immunosuppression. Eur Heart J. 1991;12(4):554–7. doi: 10.1093/oxfordjournals.eurheartj.a059938. [DOI] [PubMed] [Google Scholar]
- 33.Gries W, et al. Giant cell myocarditis: first report of disease recurrence in the transplanted heart. J Heart Lung Transplant. 1992;11(2 Pt 1):370–4. [PubMed] [Google Scholar]
- 34.Grant SC. Giant cell myocarditis in a transplanted heart. Eur Heart J. 1993;14(10):1437. doi: 10.1093/eurheartj/14.10.1437. [DOI] [PubMed] [Google Scholar]
- 35.Grant SC. Recurrent giant cell myocarditis after transplantation. J Heart Lung Transplant. 1993;12(1 Pt 1):155–6. [PubMed] [Google Scholar]
- 36.Barin JG, et al. Fatal eosinophilic myocarditis develops in the absence of IFN-gamma and IL-17A. J Immunol. 2013;191(8):4038–47. doi: 10.4049/jimmunol.1301282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Busteed S, et al. Myocarditis as a prognostic indicator in systemic lupus erythematosus. Postgrad Med J. 2004;80(944):366–7. doi: 10.1136/pgmj.2003.012450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Levin MD, Zoet-Nugteren SK, Markusse HM. Myocarditis and primary Sjogren’s syndrome. Lancet. 1999;354(9173):128–9. doi: 10.1016/s0140-6736(99)02251-5. [DOI] [PubMed] [Google Scholar]
- 39.Barbaro G. Cardiovascular manifestations of HIV infection. Circulation. 2002;106(11):1420–5. doi: 10.1161/01.cir.0000031704.78200.59. [DOI] [PubMed] [Google Scholar]
- 40.Frustaci A, et al. Biopsy-proven autoimmune myocarditis in HIV-associated dilated cardiomyopathy. BMC Infect Dis. 2014;14:729. doi: 10.1186/s12879-014-0729-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Halsell JS, et al. Myopericarditis following smallpox vaccination among vaccinia-naive US military personnel. JAMA. 2003;289(24):3283–9. doi: 10.1001/jama.289.24.3283. [DOI] [PubMed] [Google Scholar]
- 42.Root-Bernstein R, Fairweather D. Unresolved issues in theories of autoimmune disease using myocarditis as a framework. J Theor Biol. 2015;375:101–23. doi: 10.1016/j.jtbi.2014.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fairweather D, Rose NR. Coxsackievirus-induced myocarditis in mice: a model of autoimmune disease for studying immunotoxicity. Methods. 2007;41(1):118–22. doi: 10.1016/j.ymeth.2006.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cihakova D, et al. Animal models for autoimmune myocarditis and autoimmune thyroiditis. Methods Mol Med. 2004;102:175–93. doi: 10.1385/1-59259-805-6:175. [DOI] [PubMed] [Google Scholar]
- 45.Donermeyer DL, et al. Myocarditis-inducing epitope of myosin binds constitutively and stably to I-Ak on antigen-presenting cells in the heart. J Exp Med. 1995;182(5):1291–300. doi: 10.1084/jem.182.5.1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pummerer CL, et al. Identification of cardiac myosin peptides capable of inducing autoimmune myocarditis in BALB/c mice. J Clin Invest. 1996;97(9):2057–62. doi: 10.1172/JCI118642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li HS, Ligons DL, Rose NR. Genetic complexity of autoimmune myocarditis. Autoimmun Rev. 2008;7(3):168–73. doi: 10.1016/j.autrev.2007.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Barin JG, Cihakova D. Control of inflammatory heart disease by CD4+ T cells. Ann N Y Acad Sci. 2013;1285:80–96. doi: 10.1111/nyas.12134. [DOI] [PubMed] [Google Scholar]
- 49.Frisancho-Kiss S, et al. Cutting edge: cross-regulation by TLR4 and T cell Ig mucin-3 determines sex differences in inflammatory heart disease. J Immunol. 2007;178(11):6710–4. doi: 10.4049/jimmunol.178.11.6710. [DOI] [PubMed] [Google Scholar]
- 50.Goser S, et al. Cardiac troponin I but not cardiac troponin T induces severe autoimmune inflammation in the myocardium. Circulation. 2006;114(16):1693–702. doi: 10.1161/CIRCULATIONAHA.106.635664. [DOI] [PubMed] [Google Scholar]
- 51.Elliott JF, et al. Autoimmune cardiomyopathy and heart block develop spontaneously in HLA-DQ8 transgenic IAbeta knockout NOD mice. Proc Natl Acad Sci U S A. 2003;100(23):13447–52. doi: 10.1073/pnas.2235552100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tarrio ML, et al. PD-1 protects against inflammation and myocyte damage in T cell-mediated myocarditis. J Immunol. 2012;188(10):4876–84. doi: 10.4049/jimmunol.1200389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang J, et al. PD-1 deficiency results in the development of fatal myocarditis in MRL mice. Int Immunol. 2010;22(6):443–52. doi: 10.1093/intimm/dxq026. [DOI] [PubMed] [Google Scholar]
- 54.Keir ME, et al. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol. 2008;26:677–704. doi: 10.1146/annurev.immunol.26.021607.090331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Naidoo J, Page DB, Wolchok JD. Immune modulation for cancer therapy. Br J Cancer. 2014;111(12):2214–9. doi: 10.1038/bjc.2014.348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Page DB, et al. Immune modulation in cancer with antibodies. Annu Rev Med. 2014;65:185–202. doi: 10.1146/annurev-med-092012-112807. [DOI] [PubMed] [Google Scholar]
- 57.Laubli H, et al. Acute heart failure due to autoimmune myocarditis under pembrolizumab treatment for metastatic melanoma. J Immunother Cancer. 2015;3:11. doi: 10.1186/s40425-015-0057-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Henke A, et al. The role of CD8+ T lymphocytes in coxsackievirus B3-induced myocarditis. J Virol. 1995;69(11):6720–8. doi: 10.1128/jvi.69.11.6720-6728.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Huber SA, Gauntt CJ, Sakkinen P. Enteroviruses and myocarditis: viral pathogenesis through replication, cytokine induction, and immunopathogenicity. Adv Virus Res. 1998;51:35–80. doi: 10.1016/s0065-3527(08)60783-6. [DOI] [PubMed] [Google Scholar]
- 60.Fuse K, et al. Myeloid differentiation factor-88 plays a crucial role in the pathogenesis of Coxsackievirus B3-induced myocarditis and influences type I interferon production. Circulation. 2005;112(15):2276–85. doi: 10.1161/CIRCULATIONAHA.105.536433. [DOI] [PubMed] [Google Scholar]
- 61.Fairweather D, Frisancho-Kiss S, Rose NR. Viruses as adjuvants for autoimmunity: evidence from Coxsackievirus-induced myocarditis. Rev Med Virol. 2005;15(1):17–27. doi: 10.1002/rmv.445. [DOI] [PubMed] [Google Scholar]
- 62.Wolfgram LJ, et al. Variations in the susceptibility to Coxsackievirus B3-induced myocarditis among different strains of mice. J Immunol. 1986;136(5):1846–52. [PubMed] [Google Scholar]
- 63.Fairweather D, et al. IL-12 protects against coxsackievirus B3-induced myocarditis by increasing IFN-gamma and macrophage and neutrophil populations in the heart. J Immunol. 2005;174(1):261–9. doi: 10.4049/jimmunol.174.1.261. [DOI] [PubMed] [Google Scholar]
- 64.Feldman AM, McNamara D. Myocarditis. N Engl J Med. 2000;343(19):1388–98. doi: 10.1056/NEJM200011093431908. [DOI] [PubMed] [Google Scholar]
- 65.Mahfoud F, et al. Blood pressure and heart rate predict outcome in patients acutely admitted with suspected myocarditis without previous heart failure. J Hypertens. 2012;30(6):1217–24. doi: 10.1097/HJH.0b013e328352b9ca. [DOI] [PubMed] [Google Scholar]
- 66.Fairweather D, Frisancho-Kiss S, Rose NR. Sex differences in autoimmune disease from a pathological perspective. Am J Pathol. 2008;173(3):600–9. doi: 10.2353/ajpath.2008.071008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Parks CG, et al. Expert panel workshop consensus statement on the role of the environment in the development of autoimmune disease. Int J Mol Sci. 2014;15(8):14269–97. doi: 10.3390/ijms150814269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ansari A, Maron BJ, Berntson DG. Drug-induced toxic myocarditis. Tex Heart Inst J. 2003;30(1):76–9. [PMC free article] [PubMed] [Google Scholar]
- 69.Feenstra J, et al. Drug-induced heart failure. J Am Coll Cardiol. 1999;33(5):1152–62. doi: 10.1016/s0735-1097(99)00006-6. [DOI] [PubMed] [Google Scholar]
- 70.Lo MH, et al. Drug reaction with eosinophilia and systemic symptoms syndrome associated myocarditis: a survival experience after extracorporeal membrane oxygenation support. J Clin Pharm Ther. 2013;38(2):172–4. doi: 10.1111/jcpt.12025. [DOI] [PubMed] [Google Scholar]
- 71.Madan R, et al. Radiation induced heart disease: pathogenesis, management and review literature. J Egypt Natl Canc Inst. 2015;27(4):187–93. doi: 10.1016/j.jnci.2015.07.005. [DOI] [PubMed] [Google Scholar]
- 72.Rose NR. Viral myocarditis. Curr Opin Rheumatol. 2016;28(4):383–9. doi: 10.1097/BOR.0000000000000303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wessely R, et al. Transgenic expression of replication-restricted enteroviral genomes in heart muscle induces defective excitation-contraction coupling and dilated cardiomyopathy. J Clin Invest. 1998;102(7):1444–53. doi: 10.1172/JCI1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wessely R, et al. Low-level expression of a mutant coxsackieviral cDNA induces a myocytopathic effect in culture: an approach to the study of enteroviral persistence in cardiac myocytes. Circulation. 1998;98(5):450–7. doi: 10.1161/01.cir.98.5.450. [DOI] [PubMed] [Google Scholar]
- 75.Badorff C, et al. Enteroviral protease 2A cleaves dystrophin: evidence of cytoskeletal disruption in an acquired cardiomyopathy. Nat Med. 1999;5(3):320–6. doi: 10.1038/6543. [DOI] [PubMed] [Google Scholar]
- 76.Kearney MT, et al. Viral myocarditis and dilated cardiomyopathy: mechanisms, manifestations, and management. Postgrad Med J. 2001;77(903):4–10. doi: 10.1136/pmj.77.903.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lodge PA, et al. Coxsackievirus B-3 myocarditis. Acute and chronic forms of the disease caused by different immunopathogenic mechanisms. Am J Pathol. 1987;128(3):455–63. [PMC free article] [PubMed] [Google Scholar]
- 78.Ong S, et al. Natural killer cells limit cardiac inflammation and fibrosis by halting eosinophil infiltration. Am J Pathol. 2015;185(3):847–61. doi: 10.1016/j.ajpath.2014.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yao HL, et al. Gene expression analysis during recovery process indicates the mechanism for innate immune injury and repair from Coxsackievirus B3-induced myocarditis. Virus Res. 2016;213:314–21. doi: 10.1016/j.virusres.2016.01.003. [DOI] [PubMed] [Google Scholar]
- 80.Benvenga S, Guarneri F. Molecular mimicry and autoimmune thyroid disease. Rev Endocr Metab Disord. 2016;17(4):485–98. doi: 10.1007/s11154-016-9363-2. [DOI] [PubMed] [Google Scholar]
- 81.Fairweather D, et al. From infection to autoimmunity. J Autoimmun. 2001;16(3):175–86. doi: 10.1006/jaut.2000.0492. [DOI] [PubMed] [Google Scholar]
- 82.Phongsisay V. The immunobiology of Campylobacter jejuni: innate immunity and autoimmune diseases. Immunobiology. 2016;221(4):535–43. doi: 10.1016/j.imbio.2015.12.005. [DOI] [PubMed] [Google Scholar]
- 83.Caforio AL, Mahon NJ, McKenna WJ. Cardiac autoantibodies to myosin and other heart-specific autoantigens in myocarditis and dilated cardiomyopathy. Autoimmunity. 2001;34(3):199–204. doi: 10.3109/08916930109007385. [DOI] [PubMed] [Google Scholar]
- 84.Massilamany C, et al. Identification of novel mimicry epitopes for cardiac myosin heavy chain-alpha that induce autoimmune myocarditis in A/J mice. Cell Immunol. 2011;271(2):438–49. doi: 10.1016/j.cellimm.2011.08.013. [DOI] [PubMed] [Google Scholar]
- 85.Tzolovsky G, et al. Identification and phylogenetic analysis of Drosophila melanogaster myosins. Mol Biol Evol. 2002;19(7):1041–52. doi: 10.1093/oxfordjournals.molbev.a004163. [DOI] [PubMed] [Google Scholar]
- 86.Li Y, et al. Mimicry and antibody-mediated cell signaling in autoimmune myocarditis. J Immunol. 2006;177(11):8234–40. doi: 10.4049/jimmunol.177.11.8234. [DOI] [PubMed] [Google Scholar]
- 87.Moudgil KD, Sercarz EE. Crypticity of self antigenic determinants is the cornerstone of a theory of autoimmunity. Discov Med. 2005;5(28):378–82. [PubMed] [Google Scholar]
- 88.Park AC, et al. Mucosal administration of collagen V ameliorates the atherosclerotic plaque burden by inducing interleukin 35-dependent tolerance. J Biol Chem. 2016;291(7):3359–70. doi: 10.1074/jbc.M115.681882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lv H, et al. Impaired thymic tolerance to alpha-myosin directs autoimmunity to the heart in mice and humans. J Clin Invest. 2011;121(4):1561–73. doi: 10.1172/JCI44583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Carlquist JF, et al. HLA class II (DR and DQ) antigen associations in idiopathic dilated cardiomyopathy. Validation study and meta-analysis of published HLA association studies. Circulation. 1991;83(2):515–22. doi: 10.1161/01.cir.83.2.515. [DOI] [PubMed] [Google Scholar]
- 91.Limas CJ, Limas C. HLA antigens in idiopathic dilated cardiomyopathy. Br Heart J. 1989;62(5):379–83. doi: 10.1136/hrt.62.5.379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Martinetti M, et al. HLA and immunoglobulin polymorphisms in idiopathic dilated cardiomyopathy. Hum Immunol. 1992;35(3):193–9. doi: 10.1016/0198-8859(92)90105-v. [DOI] [PubMed] [Google Scholar]
- 93.Liu W, et al. Association of HLA class II DRB1, DPA1 and DPB1 polymorphism with genetic susceptibility to idiopathic dilated cardiomyopathy in Chinese Han nationality. Autoimmunity. 2006;39(6):461–7. doi: 10.1080/08916930600893709. [DOI] [PubMed] [Google Scholar]
- 94.Rodriguez-Perez JM, et al. MHC class II genes in Mexican patients with idiopathic dilated cardiomyopathy. Exp Mol Pathol. 2007;82(1):49–52. doi: 10.1016/j.yexmp.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 95.Lozano MD, et al. Human leukocyte antigen class II associations in patients with idiopathic dilated cardiomyopathy. Myocarditis Treatment Trial Investigators. J Card Fail. 1997;3(2):97–103. doi: 10.1016/s1071-9164(97)90041-5. [DOI] [PubMed] [Google Scholar]
- 96.Neu N, et al. Cardiac myosin induces myocarditis in genetically predisposed mice. J Immunol. 1987;139(11):3630–6. [PubMed] [Google Scholar]
- 97.Taneja V, et al. Spontaneous myocarditis mimicking human disease occurs in the presence of an appropriate MHC and non-MHC background in transgenic mice. J Mol Cell Cardiol. 2007;42(6):1054–64. doi: 10.1016/j.yjmcc.2007.03.898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Guler ML, et al. Two autoimmune diabetes loci influencing T cell apoptosis control susceptibility to experimental autoimmune myocarditis. J Immunol. 2005;174(4):2167–73. doi: 10.4049/jimmunol.174.4.2167. [DOI] [PubMed] [Google Scholar]
- 99.Aly M, et al. Complex genetic control of host susceptibility to coxsackievirus B3-induced myocarditis. Genes Immun. 2007;8(3):193–204. doi: 10.1038/sj.gene.6364374. [DOI] [PubMed] [Google Scholar]
- 100.Lin A, et al. 14 bp deletion polymorphism in the HLA-G gene is a risk factor for idiopathic dilated cardiomyopathy in a Chinese Han population. Tissue Antigens. 2007;70(5):427–31. doi: 10.1111/j.1399-0039.2007.00926.x. [DOI] [PubMed] [Google Scholar]
- 101.Hviid TV, Christiansen OB. Linkage disequilibrium between human leukocyte antigen (HLA) class II and HLA-G – possible implications for human reproduction and autoimmune disease. Hum Immunol. 2005;66(6):688–99. doi: 10.1016/j.humimm.2005.03.003. [DOI] [PubMed] [Google Scholar]
- 102.Seko Y, et al. Roles of programmed death-1 (PD-1)/PD-1 ligands pathway in the development of murine acute myocarditis caused by coxsackievirus B3. Cardiovasc Res. 2007;75(1):158–67. doi: 10.1016/j.cardiores.2007.03.012. [DOI] [PubMed] [Google Scholar]
- 103.Futamatsu H, et al. Attenuation of experimental autoimmune myocarditis by blocking activated T cells through inducible costimulatory molecule pathway. Cardiovasc Res. 2003;59(1):95–104. doi: 10.1016/s0008-6363(03)00334-1. [DOI] [PubMed] [Google Scholar]
- 104.Abramson J, Goldfarb Y. AIRE: From promiscuous molecular partnerships to promiscuous gene expression. Eur J Immunol. 2016;46(1):22–33. doi: 10.1002/eji.201545792. [DOI] [PubMed] [Google Scholar]
- 105.Abramson J, Husebye ES. Autoimmune regulator and self-tolerance – molecular and clinical aspects. Immunol Rev. 2016;271(1):127–40. doi: 10.1111/imr.12419. [DOI] [PubMed] [Google Scholar]
- 106.Metzger TC, Anderson MS. Control of central and peripheral tolerance by Aire. Immunol Rev. 2011;241(1):89–103. doi: 10.1111/j.1600-065X.2011.01008.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Malchow S, et al. Aire enforces immune tolerance by directing autoreactive T cells into the regulatory T cell lineage. Immunity. 2016;44(5):1102–13. doi: 10.1016/j.immuni.2016.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Metzger TC, Anderson MS. Myocarditis: a defect in central immune tolerance? J Clin Invest. 2011;121(4):1251–3. doi: 10.1172/JCI57211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Smith SC, Allen PM. Myosin-induced acute myocarditis is a T cell-mediated disease. J Immunol. 1991;147(7):2141–7. [PubMed] [Google Scholar]
- 110.Takaba H, et al. Fezf2 orchestrates a thymic program of self-antigen expression for immune tolerance. Cell. 2015;163(4):975–87. doi: 10.1016/j.cell.2015.10.013. [DOI] [PubMed] [Google Scholar]
- 111.Damsker JM, Hansen AM, Caspi RR. Th1 and Th17 cells: adversaries and collaborators. Ann N Y Acad Sci. 2010;1183:211–21. doi: 10.1111/j.1749-6632.2009.05133.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kleinewietfeld M, Hafler DA. The plasticity of human Treg and Th17 cells and its role in autoimmunity. Semin Immunol. 2013;25(4):305–12. doi: 10.1016/j.smim.2013.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Afanasyeva M, et al. Interleukin-12 receptor/STAT4 signaling is required for the development of autoimmune myocarditis in mice by an interferon-gamma-independent pathway. Circulation. 2001;104(25):3145–51. doi: 10.1161/hc5001.100629. [DOI] [PubMed] [Google Scholar]
- 114.Pope RM, Shahrara S. Possible roles of IL-12-family cytokines in rheumatoid arthritis. Nat Rev Rheumatol. 2013;9(4):252–6. doi: 10.1038/nrrheum.2012.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Sonderegger I, et al. Neutralization of IL-17 by active vaccination inhibits IL-23-dependent autoimmune myocarditis. Eur J Immunol. 2006;36(11):2849–56. doi: 10.1002/eji.200636484. [DOI] [PubMed] [Google Scholar]
- 116.Olson BM, Sullivan JA, Burlingham WJ. Interleukin 35: a key mediator of suppression and the propagation of infectious tolerance. Front Immunol. 2013;4:315. doi: 10.3389/fimmu.2013.00315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Wu L, et al. Pathogenic IL-23 signaling is required to initiate GM-CSF-driven autoimmune myocarditis in mice. Eur J Immunol. 2016;46(3):582–92. doi: 10.1002/eji.201545924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Eriksson U, et al. Dual role of the IL-12/IFN-gamma axis in the development of autoimmune myocarditis: induction by IL-12 and protection by IFN-gamma. J Immunol. 2001;167(9):5464–9. doi: 10.4049/jimmunol.167.9.5464. [DOI] [PubMed] [Google Scholar]
- 119.Eriksson U, et al. Lethal autoimmune myocarditis in interferon-gamma receptor-deficient mice: enhanced disease severity by impaired inducible nitric oxide synthase induction. Circulation. 2001;103(1):18–21. doi: 10.1161/01.cir.103.1.18. [DOI] [PubMed] [Google Scholar]
- 120.Rangachari M, et al. T-bet negatively regulates autoimmune myocarditis by suppressing local production of interleukin 17. J Exp Med. 2006;203(8):2009–19. doi: 10.1084/jem.20052222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Fairweather D, et al. Interferon-gamma protects against chronic viral myocarditis by reducing mast cell degranulation, fibrosis, and the profibrotic cytokines transforming growth factor-beta 1, interleukin-1 beta, and interleukin-4 in the heart. Am J Pathol. 2004;165(6):1883–94. doi: 10.1016/s0002-9440(10)63241-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Fairweather D, et al. Complement receptor 1 and 2 deficiency increases coxsackievirus B3-induced myocarditis, dilated cardiomyopathy, and heart failure by increasing macrophages, IL-1beta, and immune complex deposition in the heart. J Immunol. 2006;176(6):3516–24. doi: 10.4049/jimmunol.176.6.3516. [DOI] [PubMed] [Google Scholar]
- 123.Cihakova D, et al. Interleukin-13 protects against experimental autoimmune myocarditis by regulating macrophage differentiation. Am J Pathol. 2008;172(5):1195–208. doi: 10.2353/ajpath.2008.070207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Afanasyeva M, et al. Experimental autoimmune myocarditis in A/J mice is an interleukin-4-dependent disease with a Th2 phenotype. Am J Pathol. 2001;159(1):193–203. doi: 10.1016/S0002-9440(10)61685-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Baldeviano GC, et al. Interleukin-17A is dispensable for myocarditis but essential for the progression to dilated cardiomyopathy. Circ Res. 2010;106(10):1646–55. doi: 10.1161/CIRCRESAHA.109.213157. [DOI] [PubMed] [Google Scholar]
- 126.Wu L, et al. Cardiac fibroblasts mediate IL-17A-driven inflammatory dilated cardiomyopathy. J Exp Med. 2014;211(7):1449–64. doi: 10.1084/jem.20132126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Myers JM, et al. Cardiac myosin-Th17 responses promote heart failure in human myocarditis. JCI Insight. 2016;1(9) doi: 10.1172/jci.insight.85851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Chen P, et al. Susceptibility to autoimmune myocarditis is associated with intrinsic differences in CD4(+) T cells. Clin Exp Immunol. 2012;169(2):79–88. doi: 10.1111/j.1365-2249.2012.04598.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Shi Y, et al. Regulatory T cells protect mice against coxsackievirus-induced myocarditis through the transforming growth factor beta-coxsackie-adenovirus receptor pathway. Circulation. 2010;121(24):2624–34. doi: 10.1161/CIRCULATIONAHA.109.893248. [DOI] [PubMed] [Google Scholar]
- 130.Brusko TM, Putnam AL, Bluestone JA. Human regulatory T cells: role in autoimmune disease and therapeutic opportunities. Immunol Rev. 2008;223:371–90. doi: 10.1111/j.1600-065X.2008.00637.x. [DOI] [PubMed] [Google Scholar]
- 131.Cheng HM, Chamley L. Cryptic natural autoantibodies and co-potentiators. Autoimmun Rev. 2008;7(6):431–4. doi: 10.1016/j.autrev.2008.03.011. [DOI] [PubMed] [Google Scholar]
- 132.Caforio AL, et al. Circulating cardiac autoantibodies in dilated cardiomyopathy and myocarditis: pathogenetic and clinical significance. Eur J Heart Fail. 2002;4(4):411–7. doi: 10.1016/s1388-9842(02)00010-7. [DOI] [PubMed] [Google Scholar]
- 133.Wallukat G, Schimke I. Agonistic autoantibodies directed against G-protein-coupled receptors and their relationship to cardiovascular diseases. Semin Immunopathol. 2014;36(3):351–63. doi: 10.1007/s00281-014-0425-9. [DOI] [PubMed] [Google Scholar]
- 134.Neumann DA, et al. Circulating heart-reactive antibodies in patients with myocarditis or cardiomyopathy. J Am Coll Cardiol. 1990;16(6):839–46. doi: 10.1016/s0735-1097(10)80331-6. [DOI] [PubMed] [Google Scholar]
- 135.Caforio AL, et al. Identification of alpha- and beta-cardiac myosin heavy chain isoforms as major autoantigens in dilated cardiomyopathy. Circulation. 1992;85(5):1734–42. doi: 10.1161/01.cir.85.5.1734. [DOI] [PubMed] [Google Scholar]
- 136.Caforio AL, et al. Idiopathic dilated cardiomyopathy: lack of association between circulating organ-specific cardiac antibodies and HLA-DR antigens. Tissue Antigens. 1992;39(5):236–40. doi: 10.1111/j.1399-0039.1992.tb01941.x. [DOI] [PubMed] [Google Scholar]
- 137.Gu C, et al. Apoptotic signaling through the beta-adrenergic receptor. A new Gs effector pathway. J Biol Chem. 2000;275(27):20726–33. doi: 10.1074/jbc.M000152200. [DOI] [PubMed] [Google Scholar]
- 138.Staudt Y, et al. Beta1-adrenoceptor antibodies induce apoptosis in adult isolated cardiomyocytes. Eur J Pharmacol. 2003;466(1–2):1–6. doi: 10.1016/s0014-2999(03)01431-6. [DOI] [PubMed] [Google Scholar]
- 139.Christ T, et al. Autoantibodies against the beta1 adrenoceptor from patients with dilated cardiomyopathy prolong action potential duration and enhance contractility in isolated cardiomyocytes. J Mol Cell Cardiol. 2001;33(8):1515–25. doi: 10.1006/jmcc.2001.1414. [DOI] [PubMed] [Google Scholar]
- 140.Lymperopoulos A, Rengo G, Koch WJ. Adrenergic nervous system in heart failure: pathophysiology and therapy. Circ Res. 2013;113(6):739–53. doi: 10.1161/CIRCRESAHA.113.300308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Herda LR, et al. Effects of immunoadsorption and subsequent immunoglobulin G substitution on cardiopulmonary exercise capacity in patients with dilated cardiomyopathy. Am Heart J. 2010;159(5):809–16. doi: 10.1016/j.ahj.2010.01.012. [DOI] [PubMed] [Google Scholar]
- 142.Trimpert C, et al. Immunoadsorption in dilated cardiomyopathy: long-term reduction of cardiodepressant antibodies. Eur J Clin Investig. 2010;40(8):685–91. doi: 10.1111/j.1365-2362.2010.02314.x. [DOI] [PubMed] [Google Scholar]
- 143.Muller J, et al. Immunoglobulin adsorption in patients with idiopathic dilated cardiomyopathy. Circulation. 2000;101(4):385–91. doi: 10.1161/01.cir.101.4.385. [DOI] [PubMed] [Google Scholar]
- 144.Haberland A, et al. Aptamer neutralization of beta1-adrenoceptor autoantibodies isolated from patients with cardiomyopathies. Circ Res. 2011;109(9):986–92. doi: 10.1161/CIRCRESAHA.111.253849. [DOI] [PubMed] [Google Scholar]
- 145.Marquis JK, Grindel JM. Toxicological evaluation of oligonucleotide therapeutics. Curr Opin Mol Ther. 2000;2(3):258–63. [PubMed] [Google Scholar]
- 146.Artis D, Spits H. The biology of innate lymphoid cells. Nature. 2015;517(7534):293–301. doi: 10.1038/nature14189. [DOI] [PubMed] [Google Scholar]
- 147.Eberl G, Di Santo JP, Vivier E. The brave new world of innate lymphoid cells. Nat Immunol. 2015;16(1):1–5. doi: 10.1038/ni.3059. [DOI] [PubMed] [Google Scholar]
- 148.Grabie N, et al. Neutrophils sustain pathogenic CD8+ T cell responses in the heart. Am J Pathol. 2003;163(6):2413–20. doi: 10.1016/S0002-9440(10)63596-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Barin JG, Rose NR, Cihakova D. Macrophage diversity in cardiac inflammation: a review. Immunobiology. 2012;217(5):468–75. doi: 10.1016/j.imbio.2011.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Bangert A, et al. Critical role of RAGE and HMGB1 in inflammatory heart disease. Proc Natl Acad Sci U S A. 2016;113(2):E155–64. doi: 10.1073/pnas.1522288113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Su Z, et al. HMGB1 facilitated macrophage reprogramming towards a proinflammatory M1-like phenotype in experimental autoimmune myocarditis development. Sci Rep. 2016;6:21884. doi: 10.1038/srep21884. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 152.Barin JG, et al. Macrophages participate in IL-17-mediated inflammation. Eur J Immunol. 2012;42(3):726–36. doi: 10.1002/eji.201141737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Jensen LD, Marchant DJ. Emerging pharmacologic targets and treatments for myocarditis. Pharmacol Ther. 2016;161:40–51. doi: 10.1016/j.pharmthera.2016.03.006. [DOI] [PubMed] [Google Scholar]
- 154.Escher F, et al. Long-term outcome of patients with virus-negative chronic myocarditis or inflammatory cardiomyopathy after immunosuppressive therapy. Clin Res Cardiol. 2016;105(12):1011–20. doi: 10.1007/s00392-016-1011-z. [DOI] [PubMed] [Google Scholar]
- 155.Frustaci A, Russo MA, Chimenti C. Randomized study on the efficacy of immunosuppressive therapy in patients with virus-negative inflammatory cardiomyopathy: the TIMIC study. Eur Heart J. 2009;30(16):1995–2002. doi: 10.1093/eurheartj/ehp249. [DOI] [PubMed] [Google Scholar]
- 156.Mason JW, et al. A clinical trial of immunosuppressive therapy for myocarditis. The Myocarditis Treatment Trial Investigators. N Engl J Med. 1995;333(5):269–75. doi: 10.1056/NEJM199508033330501. [DOI] [PubMed] [Google Scholar]
- 157.Menghini VV, et al. Combined immunosuppression for the treatment of idiopathic giant cell myocarditis. Mayo Clin Proc. 1999;74(12):1221–6. doi: 10.4065/74.12.1221. [DOI] [PubMed] [Google Scholar]
- 158.Felix SB, et al. Hemodynamic effects of immunoadsorption and subsequent immunoglobulin substitution in dilated cardiomyopathy: three-month results from a randomized study. J Am Coll Cardiol. 2000;35(6):1590–8. doi: 10.1016/s0735-1097(00)00568-4. [DOI] [PubMed] [Google Scholar]
- 159.Knebel F, et al. Reduction of morbidity by immunoadsorption therapy in patients with dilated cardiomyopathy. Int J Cardiol. 2004;97(3):517–20. doi: 10.1016/j.ijcard.2003.12.003. [DOI] [PubMed] [Google Scholar]
- 160.Staudt A, et al. Effects of immunoadsorption on the nt-BNP and nt-ANP plasma levels of patients suffering from dilated cardiomyopathy. Ther Apher Dial. 2006;10(1):42–8. doi: 10.1111/j.1744-9987.2006.00343.x. [DOI] [PubMed] [Google Scholar]
- 161.Kishimoto C, et al. Therapy with immunoglobulin in patients with acute myocarditis and cardiomyopathy: analysis of leukocyte balance. Heart Vessel. 2014;29(3):336–42. doi: 10.1007/s00380-013-0368-4. [DOI] [PubMed] [Google Scholar]
- 162.McNamara DM, et al. Controlled trial of intravenous immune globulin in recent-onset dilated cardiomyopathy. Circulation. 2001;103(18):2254–9. doi: 10.1161/01.cir.103.18.2254. [DOI] [PubMed] [Google Scholar]
- 163.Kuhl U, et al. Interferon-beta treatment eliminates cardiotropic viruses and improves left ventricular function in patients with myocardial persistence of viral genomes and left ventricular dysfunction. Circulation. 2003;107(22):2793–8. doi: 10.1161/01.CIR.0000072766.67150.51. [DOI] [PubMed] [Google Scholar]
- 164.Zimmermann O, et al. Interferon beta-1b therapy in chronic viral dilated cardiomyopathy – is there a role for specific therapy? J Card Fail. 2010;16(4):348–56. doi: 10.1016/j.cardfail.2009.12.016. [DOI] [PubMed] [Google Scholar]