

Abstract
The mammalian genome contains thousands of loci that transcribe long noncoding RNAs (lncRNAs)1-3, some of which are known to play critical roles in diverse cellular processes4-7. LncRNA loci can contribute to cellular regulation through a variety of mechanisms: while some encode RNAs that act non-locally (in trans)6,8, emerging evidence indicates that many lncRNA loci act locally (in cis)—for example, through functions of the lncRNA promoter, the process of lncRNA transcription, or the lncRNA transcript itself in regulating the expression of nearby genes7,9-11. Despite their potentially important roles, it remains challenging to identify functional lncRNA loci and distinguish among these and other mechanisms. To address these challenges, we developed a genome-scale CRISPR-Cas9 activation screen targeting more than 10,000 lncRNA transcriptional start sites to identify noncoding loci that influence a phenotype of interest. We found 11 novel lncRNA loci that, upon recruitment of an activator, each mediate BRAF inhibitor resistance in melanoma. We investigated potential local and non-local mechanisms at these candidate loci and found that most appear to regulate nearby genes. Detailed analysis of one candidate, termed EMICERI, revealed that its transcriptional activation results in dosage-dependent activation of four neighboring protein-coding genes, one of which confers the resistance phenotype. Our screening and characterization approach provides a CRISPR toolkit to systematically discover functions of noncoding loci and elucidate their diverse roles in gene regulation and cellular function.
We have previously used the Cas9 Synergistic Activation Mediator (SAM) to screen for protein-coding genes that confer resistance to the BRAF inhibitor vemurafenib in melanoma cells12, making this an ideal phenotype for high-throughput screening of functional lncRNA loci (Supplementary Note 1). We designed a genome-scale sgRNA library targeting 10,504 unique intergenic lncRNA TSSs (>50 bp apart; see Methods, Supplementary Table 1)2,13. We transduced A375 (BRAF(V600E)) melanoma cells with the sgRNA library, cultured them in 2 μM vemurafenib or control (dimethyl sulfoxide, DMSO), and sequenced the distribution of sgRNAs after 14 days of drug treatment (Fig. 1a-b and Extended Data Fig. 1a). RIGER analysis14 identified 16 significantly enriched candidate loci (FDR < 0.05, Fig. 1c,d, Extended Data Fig. 1b,c, and Supplementary Table 2), none of which had been previously functionally characterized.
Figure 1. Genome-scale activation screen identifies lncRNA loci involved in vemurafenib resistance.
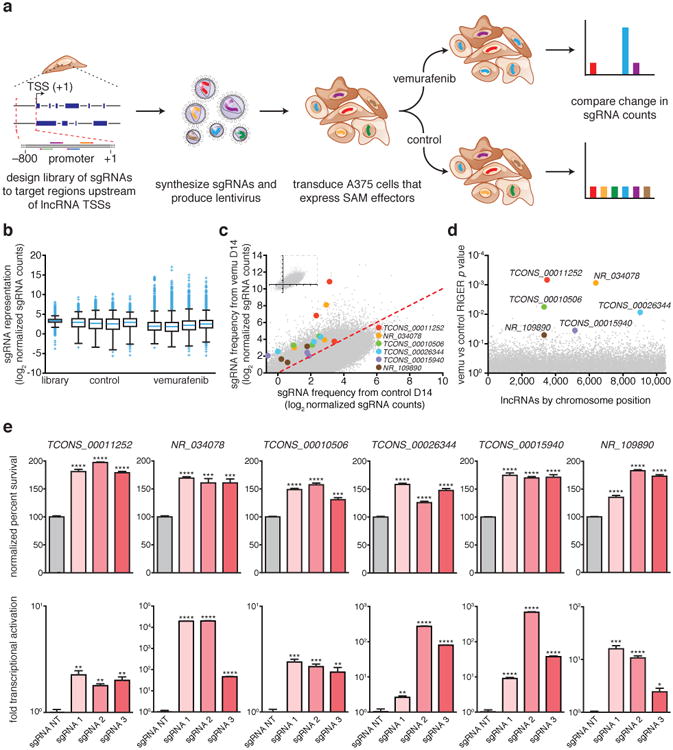
a, A375 cells expressing SAM effectors are transduced with the pooled sgRNA library targeting >10,000 lncRNA TSSs and treated with BRAF inhibitor vemurafenib or DMSO (control) for 14 days. Deep sequencing identified changes in sgRNA distribution. b, Box plot showing the distribution of sgRNA frequencies after vemurafenib or control treatment from n = 4 infection replicates. c, Scatterplot showing enrichment of sgRNAs targeting 6 candidate lncRNA loci. d, RIGER P values of the candidate lncRNA loci. e, Validation of vemurafenib resistance and transcriptional activation in A375 cells expressing individual sgRNAs targeting 6 candidate lncRNA loci or non-targeting (NT) control sgRNA. All values are mean ± SEM with n = 4. ****P < 0.0001; ***P < 0.001; **P < 0.01; *P < 0.05.
To validate the screening results, we individually expressed the 3 most enriched sgRNAs targeting each of the top 16 candidate lncRNA loci in A375 cells. In all 16 cases, the sgRNAs conferred significant vemurafenib resistance (Extended Data Fig. 2), verifying the robustness of our screening approach. We performed RNA sequencing upon activation of each of the 11 loci with the strongest effects (Extended Data Fig. 2, Supplementary Table 3) and found global changes in gene expression consistent with vemurafenib resistance, supporting the functional relevance of these loci to the screening phenotype (Extended Data Fig. 3a).
Next, we turned to classifying the mechanisms by which activation of these loci might lead to resistance, which could include (i) a non-local function of the lncRNA transcript, (ii) a local function of the lncRNA transcript or its transcription; (iii) a local function of a DNA element in the lncRNA locus; and (iv) a local function of SAM, for example activating a nearby promoter (Supplementary Note 2). To focus on loci where the mechanism might require the lncRNA or its transcription (i and ii above), we activated each locus and detected a robust lncRNA transcript upregulation for 6 of these 11 loci (Fig. 1e, Supplementary Table 3). The remaining 5 loci may function through a mechanism other than activation of the lncRNA transcript (e.g., iii and iv above; Supplementary Note 3 and Supplementary Table 4).
We explored whether activating each of these 6 lncRNA loci might affect vemurafenib resistance through non-local (i above) or local (ii and iii above) functions. To test whether candidate lncRNAs contribute to vemurafenib resistance via non-local functions, we overexpressed cDNAs encoding each lncRNA through random lentiviral integration and did not find any that affected drug resistance (Extended Data Fig. 3b), suggesting that these loci likely do not act through non-local functions (Supplementary Note 4 and Supplementary Table 3). To determine if the phenotype might result instead from local functions of the lncRNA loci in regulating a nearby gene7,9,10, we examined the expression of all genes within 1 Mb of the targeted sites. At 5 of the 6 loci, we found that SAM targeting led to differential expression of between 1 and 8 nearby protein-coding genes (Supplementary Table 4; for remaining locus, see Supplementary Note 5). For example, activation of NR_109890 upregulated its neighboring gene EBF1 (Extended Data Fig. 3c), and activation of TCONS_00015940 led to dosage-dependent upregulation of 4 neighboring protein-coding genes (Fig. 2a,b). Together, these analyses indicate that none of the lncRNA loci appear to confer vemurafenib resistance by producing trans-acting RNAs; rather, the loci may regulate the expression of one or more nearby genes.
Figure 2. Activation of the EMICERI promoter produces dosage-dependent upregulation of neighboring genes.

a, Genomic locus of EMICERI contains four neighboring genes (EQTN, MOB3B, IFNK, and C9orf72) and a putative enhancer. b, Expression of EMICERI and its neighboring genes after transduction with non-targeting (NT) or EMICERI-targeting sgRNAs and SAM. ND = not detected. c, Expression of EMICERI and MOB3B after transduction with sgRNAs tiling SAM across the EMICERI locus normalized to a NT sgRNA. All values are mean ± SEM with n = 4. ****P < 0.0001; ***P < 0.001; **P < 0.01.
To further dissect the mechanism for one of these candidate local regulators, we focused on TCONS_00015940, which, when targeted, led to a remarkable dosage-dependent activation of the 4 closest nearby genes (EQTN, MOB3B, IFNK, and C9orf72) (Fig. 2a,b). The targeted site is proximal to the boundary of a topological domain (Fig. 2a and Extended Data Fig. 4). Upon examining this locus, we found that TCONS_00015940 is actually comprised of two separate transcripts (Extended Data Fig. 5a and Supplementary Note 6). We named these transcripts “EQTN MOB3B IFNK C9orf72 enhancer RNA I”, or EMICERI, and EMICERII. The EMICERI promoter, which we targeted in our screen, is actually the promoter for two genes, which are transcribed divergently and initiate ∼66bp apart: EMICERI and MOB3B, a protein-coding gene (Fig. 2a). Tiling SAM across this region indicated that targeting a ∼200 bp region activated both of these genes (Fig. 2a,c). In contrast, targeting SAM to the promoters of the other three nearby genes did not produce coordinated transcriptional activation in the region, although targeting the promoter of C9orf72 led to a slight activation of EMICERI alone (Extended Data Fig. 5b and Supplementary Note 7). Together, these results demonstrate that the EMICERI/MOB3B promoter influences gene expression in a ∼300 kb gene neighborhood.
To determine how coordinated upregulation of the EMICERI gene neighborhood led to vemurafenib resistance, we overexpressed the cDNA for each of the 4 protein-coding genes as well as EMICERI or II lncRNAs from randomly integrated lentivirus. Only MOB3B overexpression led to vemurafenib resistance (Fig. 3a and Extended Data Fig. 6a), indicating that although activation of the EMICERI/MOB3B promoter leads to transcriptional upregulation of 4 protein-coding genes and two lncRNA genes, overexpression of only one of these genes is sufficient for the resistance phenotype. Notably, MOB3B, a novel kinase activator of unknown function, is a paralog of MOB1A/B, known components of the Hippo signaling pathway, whose activation has been shown to confer vemurafenib resistance15-18. We found that MOB3B overexpression downregulates LATS1 to activate the Hippo signaling pathway (Fig. 3b,c, Extended Data Fig. 6f-h, and Supplementary Note 8). We extended our observations beyond the cell line used in our initial screen, by showing that activation of EMICERI and MOB3B conferred vemurafenib resistance in two additional sensitive melanoma cell lines (Fig. 3d,e, Extended Data Fig. 6i) and correlated with a gene-expression signature of vemurafenib resistance in melanoma patients from The Cancer Genome Atlas (Fig. 3f, Extended Data Fig. 3,7, and Supplementary Note 8). Together, these results indicate that activation of the EMICERI locus confers vemurafenib resistance via upregulation of MOB3B and subsequent activation of the Hippo signaling pathway.
Figure 3. MOB3B mediates vemurafenib resistance through the Hippo signaling pathway in melanoma models.
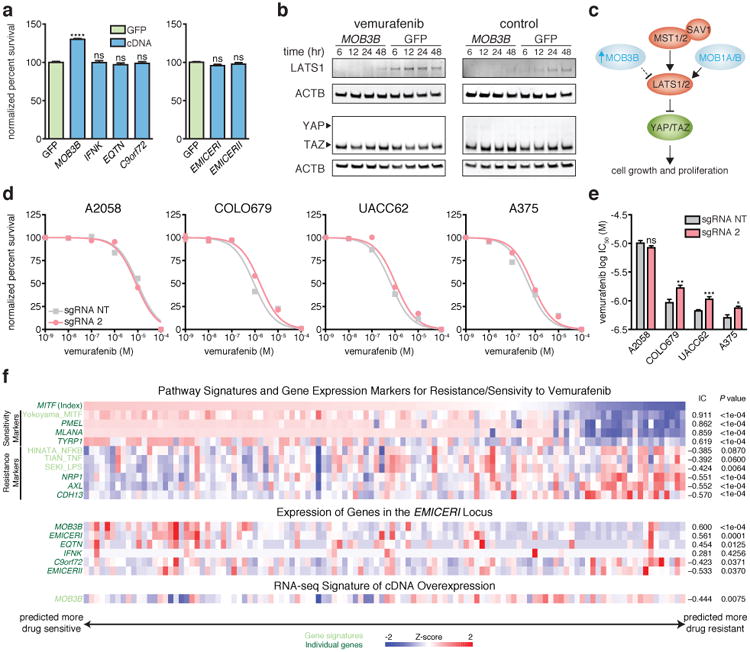
a, Vemurafenib resistance of A375 cells overexpressing each neighboring gene or lncRNA cDNA normalized to GFP. b, Western blots of LATS1, YAP, and TAZ in A375 stably overexpressing MOB3B cDNA or GFP after vemurafenib or control (DMSO) treatment. c, Schematic of MOB3B mechanism in the Hippo signaling pathway. d, Vemurafenib dose response curves for EMICERI activation in different melanoma cell lines. e, Vemurafenib half maximal inhibitory concentration (IC50) for the same conditions in (d). f, Heat map showing expression of gene/signature markers for BRAF inhibitor sensitivity (top), expression of genes in the EMICERI locus (middle), and MOB3B overexpression RNA-seq signature scores (bottom) in 113 different BRAF (V600) patient melanoma samples (primary or metastatic) from The Cancer Genome Atlas. All associations are measured using the information coefficient (IC) between the index and each of the features and P values are determined using a permutation test. Panels show Z scores. All values are mean ± SEM with n = 4. ****P < 0.0001; ***P < 0.001; **P < 0.01. *P < 0.05. ns = not significant.
As an aside, we sought to understand why MOB3B had not been identified in our previous SAM screen for protein-coding genes12. The explanation appears to be that the previous sgRNA library targeted MOB3B upstream of its TSS, whereas the optimal position for activation is downstream (Fig. 2c), and because resistance conferred by MOB3B activation is weaker than for the top candidate genes in the previous screen (Extended Data Fig. 6b-e)12.
We next considered whether transcriptional activation of EMICERI is required for full MOB3B upregulation. Alternatively, it is possible that targeting SAM to the shared EMICERI/MOB3B promoter may confer resistance only through direct activation of MOB3B. Accordingly, we used three perturbation methods to interfere with EMICERI transcription and observed effects on MOB3B:
To block transcription of EMICERI, we targeted dCas9 downstream of the EMICERI TSS. This intervention reduced the expression not only of EMICERI, but also of MOB3B and the other neighboring genes (Fig. 4a,b). We then used a bimodal perturbation system that uses an sgRNA without the SAM-recruitment sequences to target dCas9 to block EMICERI transcription and an sgRNA with the SAM-recruitment sequences to activate the promoter region (Fig. 4a, c). Different combinations of repression and activation sgRNAs targeting the EMICERI locus demonstrated that the transcriptional levels of EMICERI and MOB3B are tightly coupled across several orders of magnitude (correlation coefficient r = 0.98, P < 0.0001) (Fig. 4d).
We generated clonal A375 cell lines carrying insertions of 3 tandem polyadenylation signals (pAS) downstream of the EMICERI TSS, which eliminated production of most of the EMICERI RNA without disrupting the promoter sequence (Fig. 4e, Extended Data Fig. 8a-c, and Supplementary Note 9). Upon SAM activation, the pAS-insertion clones showed significantly reduced expression of EMICERI, MOB3B, and the three other nearby genes compared to wild type clones (Fig. 4f, g and Extended Data Fig. 8d-f), and, as expected, reduced vemurafenib resistance (Fig. 4h and Extended Data Fig. 9). This provides genetic evidence that transcription of EMICERI is involved in MOB3B activation.
We knocked down the EMICERI transcript by transient transfection with antisense oligonucleotides (ASOs), which can lead to RNase H-mediated cleavage of nascent transcripts and transcriptional termination of EMICERI (Fig. 4a and Supplementary Note 10)19. We performed these experiments in the context of activating EMICERI by targeting SAM to the promoter. ASOs targeting EMICERI reduced expression of both EMICERI and MOB3B in a dosage-dependent manner (Fig. 4i and Extended Data Fig. 10a), consistent with the dCas9 and pAS insertion results.
Figure 4. Transcription of EMICERI modulates MOB3B expression.

a, Targeting positions of sgRNAs and antisense oligonucleotides (ASOs) in the EMICERI and MOB3B locus. b, Expression of EMICERI and its neighboring genes in A375 cells transduced with non-targeting (NT) or EMICERI-targeting sgRNAs and dCas9. c, Schematic for bimodal perturbation of EMICERI transcription. sgRNAs 1-3 use MS2 loops to recruit MS2-P65-HSF1 to dCas9 to activate EMICERI, whereas sgRNAs 4-5 recruit only dCas9 to repress EMICERI. d, Correlation between MOB3B and EMICERI expression produced by different combinations of sgRNAs with and without MS2 loops. e, Schematic for inserting polyadenylation signals (pAS) downstream of the EMICERI TSS. SV40, Simian virus 40; PGK, phosphoglycerate kinase. f, EMICERI expression after SAM activation of EMICERI for the wild type and pAS clones. g, MOB3B expression after the same perturbations as (f). h, Vemurafenib resistance after SAM activation of EMICERI. i, Expression of EMICERI and MOB3B after ASO knockdown of EMICERI in the context of SAM activation. j, Expression of MOB3B and EMICERI after ASO knockdown of MOB3B in the context of SAM activation. All values are mean ± SEM with n = 4. ****P < 0.0001; ***P < 0.001; **P < 0.01; *P < 0.05. ns = not significant.
These EMICERI perturbation experiments demonstrate that transcription of EMICERI is required for full activation of MOB3B, confirming that EMICERI is a functional noncoding locus that activates four neighboring protein-coding genes and contributes to the screening phenotype.
Although the experiments above demonstrate that EMICERI transcription is required for MOB3B activation, the precise mechanism may involve either a function of the EMICERI transcript itself or the process of its transcription (e.g., recruitment of transcriptional co-activators)10,20. In the latter case, we might expect that MOB3B transcription would reciprocally regulate EMICERI expression. Indeed, we found that targeting dCas9 downstream of the MOB3B TSS successfully blocked MOB3B transcription and reduced expression of EMICERI and other neighboring genes (Extended Data Fig. 10b); and similarly, in the context of SAM activation, ASOs targeting MOB3B introns reduced the activation of both MOB3B and EMICERI (Fig. 4j and Extended Data Fig. 10c). Together, the EMICERI and MOB3B perturbation experiments suggested that transcription of both the lncRNA (EMICERI) and the mRNA (MOB3B) regulate one another in a positive feedback mechanism that then activates a broader gene neighborhood, potentially through general processes associated with transcription.
A major challenge in understanding the regulatory logic of the genome has been to identify functional lncRNA loci and characterize their mechanisms. Here we demonstrate that genome-scale activation screens enable systematic identification of many lncRNA loci that influence a specific cellular process, facilitating efforts to understand the functions and mechanisms of these key loci. Through a series of functional experiments, we provide a framework for distinguishing categories of regulatory mechanisms, including non-local (trans) functions as well as a diverse array of possible local regulatory mechanisms. Interestingly, the candidate lncRNA loci we identified appear to involve largely local, rather than non-local, regulation of gene expression (Supplementary Table 3), including a remarkable case involving coordinated activation of 4 nearby genes. Further application of this noncoding gain-of-function screening approach in other contexts, together with loss-of-function screening methods21-24 and our characterization strategy, will help elucidate the complex roles of these poorly understood players in development and disease.
Methods
Design and cloning of SAM lncRNA library
RefSeq noncoding RNAs (Release 69) were filtered for lncRNA transcripts that were longer than 200 bp and not overlapping with RefSeq coding gene isoforms 13. The RefSeq lncRNA catalog was combined with the Cabili lncRNA catalog and filtered for unique lncRNA transcriptional start sites (TSSs) defined as TSSs that were >50 bp apart 2. This resulted in 10,504 unique lncRNA TSSs that were targeted with ∼10 single guide RNAs (sgRNAs) each for a total library of 95,958 sgRNAs. sgRNAs were designed to target the first 800 bp upstream of each TSS and subsequently filtered for GC content >25%, minimal overlap of the target sequence, and homopolymer stretch <4 bp. After filtering, the remaining sgRNAs were scored according to predicted off-target matches as described previously 25, and 6 sgRNAs with the best off-target scores were selected in the first 200 bp region upstream of the TSS, 1 in the 200-300 bp region, 1 in the 300-400 bp region, 1 in the 400-600 bp region, and 1 in the 600-800 bp region. In regions with an insufficient number of possible sgRNAs, sgRNAs were selected from the neighboring region closer to the TSS. The ideal location for sgRNA targeting to achieve maximal activation, either upstream or downstream of the TSS, may be unique for each lncRNA locus and dependent on the local regulatory context (e.g., locations of TF binding sites). An additional 500 non-targeting sgRNAs from the GeCKO library 26 were included as controls. Cloning of the SAM sgRNA libraries was performed as previously described with a minimum representation of 100 transformed colonies per sgRNA followed by next-generation sequencing (NGS) validation 27.
Lentivirus production and transduction
For transduction, plasmids were packaged into lentivirus via transfection of library plasmid with appropriate packaging plasmids (psPAX2: Addgene 12260; pMD2.G: Addgene 12259) using Lipofectamine 2000 (Thermo Fisher 11668019) and Plus reagent (Thermo Fisher 11514015) in HEK293FT (Thermo Fisher R70007) as described previously 27. Human melanoma A375 cells (Sigma-Aldrich 88113005) were cultured in R10 media: RPMI 1640 (Thermo Fisher 61870) supplemented with 10% FBS (VWR 97068-085) and 1% penicillin/streptomycin (Thermo Fisher 15140122). Cells were passaged every other day at a 1:5 ratio. Concentrations for selection agents were determined using a kill curve: 300 μg/mL Zeocin (Thermo Fisher R25001), 10 μg/mL Blasticidin (Thermo Fisher A1113903), and 300 μg/mL Hygromycin (Thermo Fisher 10687010). Cells were transduced via spinfection and selected with the appropriate antibiotic as described previously 27. During selection, media was refreshed when cells were passaged every 3 days. The duration of selection was 7 days for Zeocin and 5 days for Hygromycin and Blasticidin. Lentiviral titers were calculated by spinfecting cells with 5 different volumes of lentivirus and determining viability after a complete selection of 3 days 27.
Vemurafenib resistance screen
The vemurafenib resistance screen was conducted similarly to a previously described genome-scale SAM coding gene screen 12. A375 stably integrated with dCas9-VP64 (Addgene 61425) and MS2-P65-HSF1 (Addgene 61426) were transduced with the pooled sgRNA library (Addgene 61427) as described above at an MOI of 0.3 for a total of 4 infection replicates, with a minimal representation of 500 transduced cells per sgRNA in each replicate. Cells were maintained at >500 cells per sgRNA during subsequent passaging. After 7 days of Zeocin selection and 2 days of no antibiotic selection, cells were split into control (DMSO) and vemurafenib (2 μM PLX-4720 dissolved in DMSO, Selleckchem S1152) conditions. Cells were passaged every 2 days for a total of 14 days of control or vemurafenib treatment. The 14-day screening duration was selected based on previous studies 12,23,26. At the end of the screening selection, >500 cells per sgRNA in each condition were harvested for gDNA extraction and amplification of the virally integrated sgRNAs as described previously 27. Resulting libraries were deep-sequenced on Illumina MiSeq or NextSeq platforms with a coverage of >25 million reads passing filter per library.
NGS and screen hits analysis
NGS data was de-multiplexed using unique index reads. sgRNA counts were determined based on perfectly matched sequencing reads only. For each condition, a pseudocount of 1 was added to the sgRNA count and the counts were normalized to the total number of counts in the condition. The sgRNA fold change as a result of screening selection was calculated by dividing the normalized sgRNA counts in the vemurafenib condition by the control and taking the base 2 logarithm. RIGER 14 analysis was performed using GENE-E based on the normalized log2 ratios for each infection replicate. Since a low percentage of functional sgRNAs was expected for each lncRNA loci, the weighted sum method was used. To determine the empirical false discovery rate (FDR) of candidate lncRNA loci, the weighted sum for 10 randomly selected non-targeting sgRNAs in the sgRNA library was used to estimate the P value for each lncRNA locus and a threshold based on a FDR of 0.05 (Benjamini-Hochberg) was selected that corresponded to a P value of 0.031. 7 candidate lncRNA loci were selected based on the average ranking between infection replicates 1 and 2, and 9 candidate lncRNA loci were selected based on the average ranking in all 4 infection replicates. All candidate lncRNA loci had P value < 10-5.
Vemurafenib resistance assay
A375 cells stably integrated with dCas9-VP64 and MS2-P65-HSF1 were transduced with individual sgRNAs targeting the 16 top candidate lncRNA loci from the vemurafenib resistance screen (3 sgRNAs with the highest enrichment per lncRNA locus; Supplementary Table 5) or with control non-targeting sgRNA at an MOI of <0.5 and selected with Zeocin for 5 days as described above. For cDNA overexpression, A375 cells were transduced with cDNA (Supplementary Table 7) or control GFP at an MOI of <0.5 and selected with Hygromycin for 4 days. At 5 days post transduction, cells were replated at low density (3 × 103 cells per well in a 96-well plate). 2 μM vemurafenib or control DMSO was added 3h after plating and refreshed every 2 days for 3-4 days before cell viability was measured using CellTiter-Glo Luminescent Cell Viability Assay (Promega G7571). Significance testing was performed using Student's t-test. For primary patient tumor-derived melanoma cell lines, cells were plated at low density (2 × 103 cells per well in a 96-well plate) and vemurafenib was added 24h after plating. Cells were treated for 3 days before cell viability was measured. For vemurafenib dose response curves, the indicated concentrations of vemurafenib were added and the normalized percent survival values were fitted with a nonlinear curve (log(inhibitor) vs normalized response; Prism 6). Significant differences in logIC50 values was determined using the extra sum-of-squares F test.
qPCR quantification of transcript expression
A375 cells stably integrated with SAM components were transduced with individual sgRNAs targeting top candidate lncRNA loci (Supplementary Table 5), perturbing the EMICERI locus (Supplementary Table 8), or non-targeting control at an MOI of <0.5 and selected with Zeocin for 5 days as described above. For cDNA overexpression, A375 cells were transduced with cDNA (Supplementary Table 7) or control GFP at an MOI of <0.5 and selected with Hygromycin for 4 days. Cells were plated at 5 days post transduction at 70% confluency (3 × 104 cells per well in a 96-well plate), and harvested for RNA 24h after plating as described previously 27. For transcripts that this method could not detect, cells transduced with the respective sgRNAs were plated at 5 days post transduction (1.8 × 105 cells per well in a 24-well plate). RNA was harvested using the RNeasy Plus Mini Kit (Qiagen 74134) and 1 μg of RNA was used for reverse transcription with the qScript Flex cDNA Kit (VWR 95049) and lncRNA-specific primers (Supplementary Table 6). After reverse transcription, TaqMan qPCR was performed with custom or readymade probes as described previously (Supplementary Table 7) 27. Significance testing was performed using Student's t-test.
RNA sequencing and data analysis
A375 cells transduced with individual sgRNAs targeting candidate lncRNA loci or with control non-targeting sgRNAs (Supplementary Table 5) were plated 5 days post transduction at 9 × 104 cells per well or 1.8 × 105 cells per well respectively in a 24-well plate. 3 bioreps per condition were plated. For cDNA overexpression, A375 cells were transduced with cDNA (Supplementary Table 7) or control GFP at an MOI of <0.5 and selected with Hygromycin for 4 days. Cells were treated with 2 μM vemurafenib for 3 days before RNA was harvested as described above. The 6 candidate lncRNA loci with detectable transcript upregulation were prepped with TruSeq Stranded Total RNA Sample Prep Kit with Ribo-Zero Gold (Illumina RS-122-2302) and all other samples were prepped with NEBNext Ultra RNA Library Prep Kit for Illumina (NEB E7530S) and NEBNext PAS mRNA Magnetic Isolation Module (NEB E7490S). Libraries were deep-sequenced on the Illumina NextSeq platform (>9 million reads per condition). Bowtie 28 index was created based on the human hg19 UCSC genome and known gene and lncRNA transcriptome constructed as described above. Paired-end reads were aligned directly to this index using Bowtie with command line options “-q --phred33-quals -n 2 -e 99999999 -l 25 -I 1 -X 1000 --chunkmbs 512 -p 1 -a -m 200 -S”. Next, RSEM v1.2.22 29 was run with default parameters on the alignments created by Bowtie to estimate expression levels.
RSEM's TPM estimates for each transcript were transformed to log-space by taking log2(TPM+1). Transcripts were considered detected if their transformed expression level was equal to or above 1 (in log2(TPM+1) scale). All genes detected in at least one library (out of three libraries per condition) were used to find differentially expressed genes. For lncRNA loci activation, the Student's t-test was performed on each of the 3 replicates for each targeting sgRNA against both non-targeting sgRNAs. For MOB3B cDNA overexpression, the t-test was performed on the cDNA overexpression against GFP control. Only genes that were significant (p-value pass 0.05 FDR correction) were reported. For lncRNA loci activation, the genes overlapping all 3 targeting sgRNAs were reported as differentially expressed as a result of lncRNA loci activation. Power analysis for two-sided t-test were performed on each targeting sgRNA against both non-targeting sgRNAs to determine the probability of correctly identifying a gene as differentially expressed.
For annotating EMICERI, TopHat 30 was used to align RNA-seq reads from A375 transduced with sgRNA 2 or sgRNA 3 (Supplementary Table 8) with command line options “--solexa-quals --num-threads 8 --library-type fr-firststrand --transcriptome-max-hits 1 --prefilter-multihits --keep-fasta-order”. To further investigate the mechanism for MOB3B overexpression, Ingenuity Pathway Analysis was applied to all genes differentially expressed with at least 1.2-fold change or less than 0.7-fold change and the most likely upstream regulator was reported.
Hi-C and chromatin immunoprecipitation with sequencing (ChIP-seq) in GM12878
In situ Hi-C data for GM12878 was obtained and visualized using 2.5kb-resolution KL-normalized observed matrix 31. Hi-C data from 7 cell lines suggested similar topological domain annotations as GM12878 (Rao et al. Cell 2014), suggesting that the TAD present in GM12878 is consistent across cell types. CTCF ChIP-seq for GM12878 and hg19 generated by the ENCODE Project Consortium 32 was downloaded from UCSC Genome Browser. CTCF motifs were identified using FIMO 33 to search for the “V_CTCF_01” and “V_CTCF_02” position weight matrices from TRANSFAC 34 as described previously 21.
Assay for transposable and accessible chromatin sequencing (ATAC-seq)
ATAC-seq samples were prepared as described previously 23. A375 cells were cultured in R10 as described above and 5 × 104 cells in log-phase growth were harvested using an existing ATAC library preparation protocol with minor modifications 35. Library was sequenced using the Illumina NextSeq platform at ∼136 million paired-end reads. Samples were aligned to the human hg19 UCSC genome using Bowtie 28 with command line options “--chunkmbs 256 -p 24 -S -m 1 -X 2000”. For quality control, the duplicate read rate was measured using Picard-Tools Mark Duplicates (10-30%) and the mitochondrial read rate was measured by Bowtie alignment to chrM (<5%) 36.
PhastCons sequence conservation
PhastCons data for primates (n=10 animals), placental mammals (n=33), and vertebrates (n=46) for hg19 were downloaded from UCSC Genome Browser and aligned to the EMICERI locus 37.
ChIP-seq for histone modifications
ChIP samples were prepared as described previously 23. Briefly, A375 cells were plated in T-225 flasks and grown to 70-90% confluence. Formaldehyde was added directly to the growth media for a final concentration of 1% for 10 mins at 37°C to initiate chromatin fixation. The entire two-day ChIP procedure was performed using the EZ-Magna ChIP HiSens Chromatin Immunoprecipitation Kit (Millipore 1710460) according to the manufacturer's protocol. Samples were pulse sonicated with 2 rounds of 10 mins (30s on-off cycles, high frequency) in a rotating water bath sonicator (Diagenode Bioruptor) with 5 mins on ice between each round. To detect histone modifications, antibodies (H3K4me2: Millipore 17-677, H3K4me3: Millipore 04-745, H3K27ac: Millipore 17-683) were optimized individually for each antibody to be 0.5 μL for 1 million cells. 1 μL of IgG (Millipore 12-370) was used for negative control.
After verifying that the IgG ChIP had minimal background, ChIP samples were prepped with NEBNext Ultra II DNA Library Prep Kit for Illumina (NEB E7645S) and deep-sequenced on the Illumina NextSeq platform (>60 million reads per condition). Bowtie 28 was used to align paired-end reads to the human hg19 UCSC genome with command line options “-q -X 500 --sam --chunkmbs 512”. Next, Model-based analysis of ChIP-seq (MACS) 38 was run with command line options “-g hs -B -S --call-subpeaks” to identify histone modifications.
Western blot
A375 cells transduced with MOB3B cDNA or GFP control were plated 5 days post transduction at 1.8 × 105 cells per well in a 24-well plate. Cells were treated with 2 μM vemurafenib for 6, 12, 24, or 48 h before protein lysates were harvested with RIPA lysis buffer (Cell Signaling Technologies 9806S) containing protease inhibitor (Roche 05892791001) and phosphatase inhibitor (Cell Signaling Technologies 5870S) cocktails. Samples standardized for protein concentration with the Pierce BCA protein assay (Thermo Fisher 23227) were incubated at 70°C for 10 mins under reducing conditions. After denaturation, 10 μg of the samples were separated by Bolt 4-12% Bis-Tris Plus Gels (Thermo Fisher NW04120BOX) and transferred onto a polyvinylidene difluoride membrane using iBlot Transfer Stacks (Thermo Fisher IB401001). Blots were blocked with Odyssey Blocking Buffer (TBS; LiCOr 927-50000) and probed with different primary antibodies [anti-pERK (Cell Signaling Technologies 4370, 1:2000 dilution), anti-ERK (Cell Signaling Technologies 4695, 1:1000 dilution), anti-pAKT (Ser473, Cell Signaling Technologies 4060, 1:1000 dilution), anti-AKT (Cell Signaling Technologies 4691, 1:1000 dilution), anti-LATS1 (Cell Signaling Technologies 3477, 1:1000 dilution), anti-YAP/TAZ (Cell Signaling Technologies 8418, 1:1000 dilution), anti-MST1 (Cell Signaling Technologies 3682, 1:1000 dilution), anti-ACTB (Sigma A5441, 1:5000 dilution)] overnight at 4°C. Blots were then incubated with secondary antibodies IRDye 680RD Donkey anti-Mouse IgG (LiCOr 925-68072) and IRDye 800CW Donkey anti-Rabbit IgG (LiCOr 925-32213) at 1:20,000 dilution in Odyssey Blocking Buffer for 1 hr at room temperature. p-ERK and p-AKT blots were stripped with Restore PLUS Western Blot Stripping Buffer (Thermo Fisher 46430) before probing for ERK and AKT respectively. Blots were imaged using the Odyssey CLx (LiCOr).
Primary patient melanoma-derived cell lines
CLF_SKCM_001_T and CLF_SKCM_004_T melanoma tumor tissues were obtained from Dana-Farber Cancer Institute hospital with informed consent and the cancer cell model line generation was approved by the ethical committee. Tumor tissues were dissected into tiny pieces by scalpers around 100 times. Dissected tissues were dissociated in the collagenase/hyaluronidase (STEMCELL technologies 07912) medium for 1 hour. The red blood cells were further depleted by adding the Ammonium Chloride Solution (STEMCELL technologies 07800). The dissociated cells were plated with the smooth muscle growing medium-2 (Lonza CC-3181) in the six well plate and split when the well confluency reached to 80%. Cells were passaged for 5 times with 1:4 splitting ratio for a sequencing verification. The confirmed BRAF V600E melanoma cell models were be propagated for another 7-15 passages and cryovial preserved. We used passage 12 cells for this study. All cells were refed every 3-4 days.
Gene expression and pharmacological validation analysis
Gene expression data (CCLE, TCGA) and pharmacological data (CCLE) were analyzed to better understand the biological relevance of EMICERI and MOB3B. Transcript expression in TCGA and CCLE samples was quantified as follows: 1) FASTQ files were generated from available BAM files using SamToFastq in Picard Tools (https://broadinstitute.github.io/picard/); 2) reads were aligned with STAR v2.5.2b 39 using parameters from the GTEx Consortium pipeline (https://github.com/broadinstitute/gtex-pipeline) and genome indexes generated for read lengths of 48bp (TCGA) and 101bp (CCLE) (--sjdbOverhang option); 3) expression was quantified using RSEM v1.2.22 29. For the alignment and quantification steps, annotations for TCONS_00011252, NR_034078, TCONS_00010506, TCONS_00026344, TCONS_00015940_1, TCONS_00015940_2, and NR_109890 were appended to the GENCODE 19 GTF (https://www.gencodegenes.org/releases/19.html). Gene-level quantifications were also calculated with RNA-SeQC 40 to validate the RSEM results.
Gene expression (RNA-sequencing) were collected from 113 BRAFV600-mutant primary and metastatic patient tumors from The Cancer Genome Atlas (TCGA: https://tcga-data.nci.nih.gov/tcga/). Because pharmacological data was not available for the TCGA melanoma samples, signature gene sets, including some from the Molecular Signature Database (MSigDB) 41, were used to fully map the transcriptional BRAF-inhibitor resistant/sensitive states in TCGA as previously described 42. The TCGA dataset was used for determining the association between resistance and the expression of candidate lncRNA loci or genes in the EMICERI locus. Additionally, we sought a more robust scoring system independent of any single gene. Gene expression signatures were generated based on the genes that were differentially expressed (top 1000 most differentially expressed) as a result of candidate lncRNA loci or MOB3B overexpression identified from RNA-seq. Using single-sample Gene Set Enrichment Analysis (ssGSEA) 43, a score was generated for each sample that represents the enrichment of the gene expression signature in that sample and the extent to which those genes are coordinately up- or down-regulated. Patient tumors were also sorted by EMICERI expression to determine correlation between expression of EMICERI and its neighboring genes.
In the CCLE dataset 44, gene expression data (RNA-sequencing, GCHub: https://cghub.ucsc.edu/datasets/ccle.html) and pharmacological data (activity area for MAPK pathway inhibitors) from BRAFV600 mutant melanoma cell lines were used to compute the association between PLX-4720 resistance and the expression of genes in the EMICERI locus. Similar to the TCGA analysis, the MOB3B overexpression gene signature was determined using ssGSEA 43 projected onto the CCLE RNA-sequencing dataset. Cell lines were also sorted by EMICERI expression to determine correlation between expression of EMICERI and its neighboring genes.
To measure correlations between different features (signature scores, gene expression, or drug-resistance data) in the external cancer datasets, an information-theoretic approach (Information Coefficient; IC) was used and significance was measured using a permutation test (n=10,000), as previously described 42. The IC was calculated between the feature used to sort the samples (columns) in each dataset and each of the features plotted in the heat map (pharmacological data, gene expression, and signature scores).
Polyadenylation signal (pAS) insertion
To truncate EMICERI, the following pAS sequences were inserted consecutively 103, 156, and 198 bp downstream of each copy of EMICERI's TSS:
Synthetic pAS
AATAAAAGATCTTTATTTTCATTAGATCTGTGTGTTGGTTTTTTGTGTG
SV40 pAS
GTTTATTGCAGCTTATAATGGTTACAAATAAAGCAATAGCATCACAAATTTCACAAATAAAGCATTTTTTTCACTGCATTCTAGTTGTGGTTTGTCCAAACTCATCAATGTATCTTATCATGTCT
PGK pAS
AAATTGATGATCTATTAAACAATAAAGATGTCCACTAAAATGGAAGTTTTTTCCTGTCATACTTTGTTAAGAAGGGTGAGAACAGAGTACCTACATTTTGAATGGAAGGATTGGAGCTACGGGGGTGGGGGTGGGGTGGGATTAGATAAATGCCTGCTCTTTACTGAAGGCTCTTTACTATTGCTTTATGATAATGTTTCATAGTTGGATATCATAATTTAAACAAGCAAAACCAAATTAAGGGCCAGCTCATTCCTCCACTCACGATCTATA
PAS clones were generated using CRISPR-Cas9 mediated homology-directed repair (HDR). 3 different sgRNAs targeting 103, 156, and 198 bp downstream of EMICERI (HDR sgRNA 1-3, Supplementary Table 9) and corresponding pAS HDR plasmids were used for inserting pAS into each of the 3 copies of EMICERI in A375. To construct the PAS HDR plasmids, for each sgRNA the HDR templates that consisted of the 850-900 bp genomic regions flanking the sgRNA cleave site were PCR amplified from A375 genomic DNA using KAPA HiFi HotStart Readymix (KAPA Biosystems KK2602). Then 3 pAS sequences (in the order listed above) flanked by the HDR templates were cloned into pUC19 (Addgene 50005). To insert pAS downstream of EMICERI's TSS, 3 rounds of HDR were performed with a different sgRNA and respective pAS HDR plasmid at each round such that selected clones contained pAS sequences in 1 copy of EMICERI in the first round, 2 copies in the second round, and 3 copies in the third round. At each round of HDR, A375 cells were nucleofected with 4 μg of sgRNA and Cas9 plasmid (Addgene 52961) and 2.5 μg of pAS HDR plasmid using SF Cell Line 4D-Nucleofector X Kit L (Lonza V4XC-2024) according to the manufacturer's instructions. Cells were then seeded sparsely (5 × 104 cells per 10-cm Petri dish) to form single-cell clones. After 24h, cells were selected for Cas9 expression with 1 μg/mL Puromycin for 2 days and expanded until colonies can be picked (∼5 days).
To pick colonies, cells were detached by replacing the media with PBS and incubating at room temperature for 15 mins. Each cell colony was removed from the Petri dish using a 200 μL pipette tip and transferred a well in a 96-well plate for expansion. Clones with pAS insertions were identified by 2-round PCR amplification (Supplementary Table 9), first with primers amplifying outside of the HDR template (HDR primer F1 and HDR primer R, 15 cycles) and then with primers amplifying the region of insertion (HDR primer F2 and HDR primer R, 15 cycles) to avoid detecting the HDR template plasmid as a false positive. Products were run on a gel to identify insertions and Sanger sequencing confirmed that the pAS sequences had been inserted at the appropriate site. During each round of HDR, 3 clones with pAS insertions and 1 clone without pAS insertion (wild type) were selected for further expansion and characterization. The wild type clone controls for potential on- and off-target indels.
Antisense oligonucleotide (ASO) knockdown
ASOs targeting EMICERI/II and MOB3B were custom designed using Exiqon's Antisense LNA GapmeR designer (Supplementary Table 10) and a non-targeting ASO (Exiqon 300610) was included for control. ASOs were resuspended in water to a final concentration of 100 μM. A375 stably expressing SAM components dCas9-VP64 and MS2-p65-HSF1 were nucleofected with 500 ng sgRNA (Supplementary Table 8; Addgene 73795) and 100 pmol ASO using the SF Cell Line 4D-Nucleofector X Kit S (Lonza V4XC-2032) according to the manufacturer's instructions. Cells were then seeded at 3 × 104 cells per well in a 96-well plate. 24h after nucleofection, cells were selected for the sgRNA plasmid with 1 μg/mL Puromycin (Thermo Fisher A1113803) for 2 days and changes in transcript expression were determined by qPCR as described above.
Code availability
Code for the analyses described in this paper is available from the authors upon request.
Extended Data
Extended Data Figure 1. Genome-scale activation screen for lncRNA loci involved in BRAF inhibitor resistance.
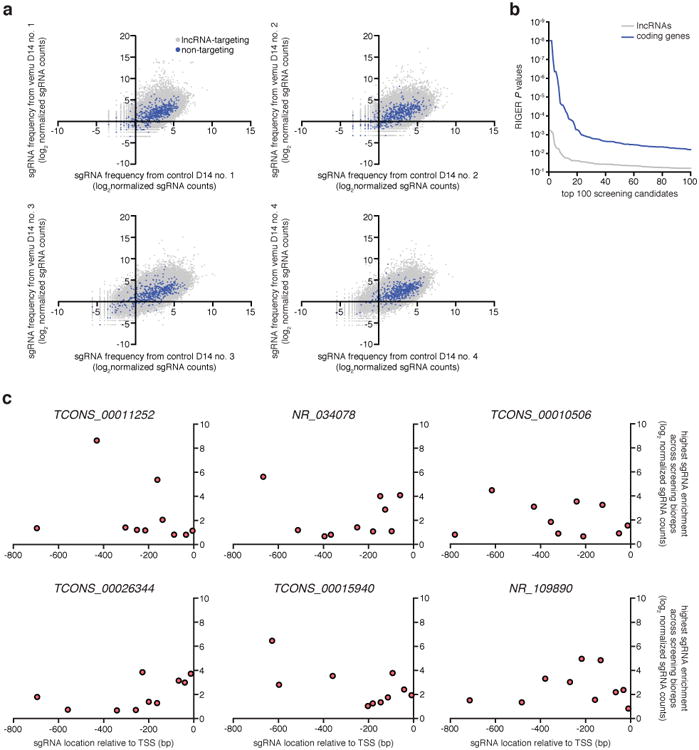
a, Scatterplots showing lncRNA-targeting and non-targeting sgRNA frequencies after vemurafenib (vemu) or control treatment from n = 4 infection replicates. b, RIGER P values for the top 100 hits from the previous SAM protein-coding gene screen 12 compared to the SAM lncRNA loci screen. c, For each candidate lncRNA locus, 10 sgRNAs were designed to target the proximal promoter region (800 bp upstream of the TSS). The relationship between the highest sgRNA enrichment in vemurafenib-treated compared to control condition across screening bioreps (n = 4) and respective spacer position suggests that sgRNAs targeting closer to the annotated TSS are not necessarily more effective, consistent with previous results 12.
Extended Data Figure 2. Validation of candidate lncRNA loci for vemurafenib resistance.

Vemurafenib resistance for A375 transduced with SAM and individual sgRNAs targeting the top 16 candidate lncRNA loci normalized to a non-targeting (NT) sgRNA. All values are mean ± SEM with n = 4. ****P < 0.0001; ***P < 0.001; **P < 0.01.
Extended Data Figure 3. Activation of candidate lncRNA loci mediate vemurafenib resistance by potentially acting locally to regulate expression of nearby genes.

a, Heat map showing expression of gene/signature markers for BRAF inhibitor sensitivity (top), expression of candidate lncRNA loci (middle), and RNA-seq signature of gene expression changes upon activation of candidate lncRNA loci (bottom) in 113 different BRAF (V600) patient melanoma samples (primary or metastatic) from The Cancer Genome Atlas. All associations are measured using the information coefficient (IC) between the index and each of the features and P values are determined using a permutation test. Panels show Z scores. b, Vemurafenib resistance of A375 cells overexpressing each candidate lncRNA cDNA or protein-coding gene normalized to GFP. GPR35 and LPAR1 are positive controls identified previously 12. The same set of sgRNAs targeted TCONS_00012395 and TCONS_00011252; NR_034078 and NR_034079; TCONS_00015940_1 and TCONS_00015940_2. c, Expression of NR_109890 and its neighboring gene EBF1 after SAM activation of NR_109890. All values are mean ± SEM with n = 4. ****P < 0.0001; ***P < 0.001; *P < 0.05. ns = not significant.
Extended Data Figure 4. Topological domain in the EMICERI locus is consistent across cell types.

Hi-C data and topological domain annotations (dotted lines) in the EMICERI locus from 7 cell lines 31. Heat map shows KR-normalized contact matrix at 5-kb resolution. Domain annotations for chromosome 9 were not available in K562, but the same topological domain structure is evident.
Extended Data Figure 5. Dosage-dependent upregulation of the EMICERI locus is specific to activation of EMICERI at its conserved regulatory element.

a, TopHat alignment of RNA-seq paired-end reads suggests that EMICERI is located at chr9:27,529,917-27,531,782 and EMICERII at chr9:27,535,711-27,540,711 (UCSC hg19) (Supplementary Note 6). A375 ATAC-seq and phastCons conservation scores for primates, placental mammals, and vertebrates at the EMICERI locus. Scale bar, 1 kb. b, Expression of EMICERI and its neighboring genes MOB3B, IFNK, EQTN, and C9orf72 after transduction with sgRNAs targeting SAM to the promoters of neighboring genes. All values are mean ± SEM with n = 4. ****P < 0.0001; ***P < 0.001; **P < 0.01; *P < 0.05. ns = not significant. ND = not detected.
Extended Data Figure 6. Activation of EMICERI mediates vemurafenib resistance through MOB3B.
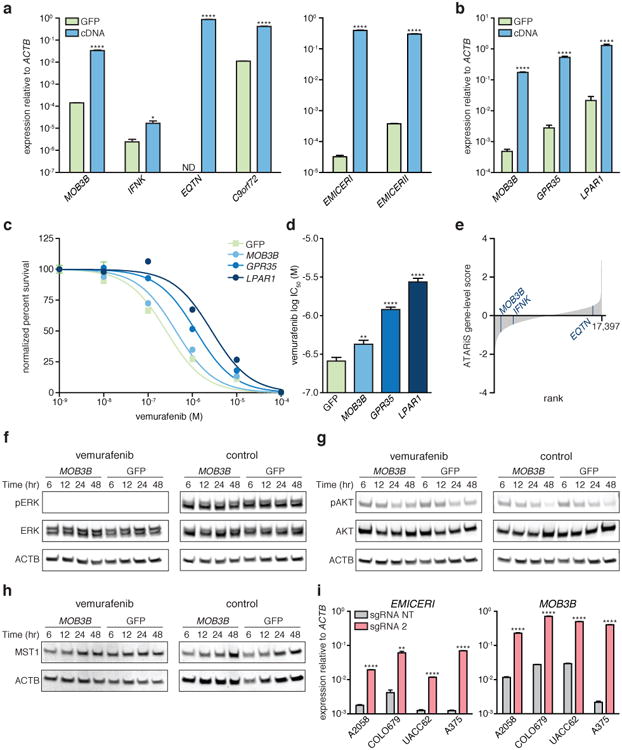
a, Expression of the neighboring genes or EMICERI/II after cDNA overexpression compared to GFP control. b, cDNA overexpression of top hits from the SAM protein-coding gene screen for vemurafenib resistance (GPR35 and LPAR1) 12 or MOB3B compared to GFP control. c, Vemurafenib dose response curves for A375 cells overexpressing cDNA or GFP control. d, Vemurafenib half maximal inhibitory concentration (IC50) for the same conditions in (c). e, ATARiS gene-level scores from the Achilles Project that reflect genetic vulnerabilities of A375. Lower ATARiS gene-level scores indicate stronger dependency on the gene. Rank of MOB3B, 1,084; IFNK, 3,078; EQTN, 15,939. f to h, Western blots of A375 stably overexpressing MOB3B cDNA or GFP control after vemurafenib or control (DMSO) treatment. i, Expression of EMICERI and MOB3B after SAM activation in different melanoma cell lines. All values are mean ± SEM with n = 4. ****P < 0.0001; **P < 0.01; *P < 0.05. ND = not detected.
Extended Data Figure 7. EMICERI expression is strongly correlated with MOB3B expression and vemurafenib sensitivity in melanoma cell lines and patient samples.

a, Heat map showing expression of genes in the EMICERI locus in 113 different BRAF (V600) patient melanoma samples (primary or metastatic) from The Cancer Genome Atlas. Samples are sorted by EMICERI expression. b, Heat map showing expression of genes in the EMICERI locus in melanoma cell lines from the Cancer Cell Line Encyclopedia (CCLE) sorted by EMICERI expression. c, Heat map showing sensitivity to different drugs (top), expression of genes in the EMICERI locus (middle), and MOB3B cDNA overexpression RNA-seq signature (bottom; see Methods for signature generation) in melanoma cell lines from CCLE. Drug sensitivities are measured as Activity Areas. The melanoma cell lines are sorted by PLX-4720 (vemurafenib) drug sensitivity. RAF inhibitors: PLX-4720 and RAF265; MEK inhibitors: AZD6244 and PD-0325901. d, Expression of EMICERI and MOB3B in two primary patient-derived BRAF(V600E) melanoma cell lines. e, Vemurafenib dose response curves for the same cell lines. f, Vemurafenib half maximal inhibitory concentration (IC50) for the same conditions as (e). All associations are measured using the information coefficient (IC) between the index and each of the features and P values are determined using a permutation test. Heat maps show Z scores. All values are mean ± SEM with n = 4. ****P < 0.0001; *P < 0.05.
Extended Data Figure 8. Transcriptional activation of EMICERI modulates expression of neighboring genes.
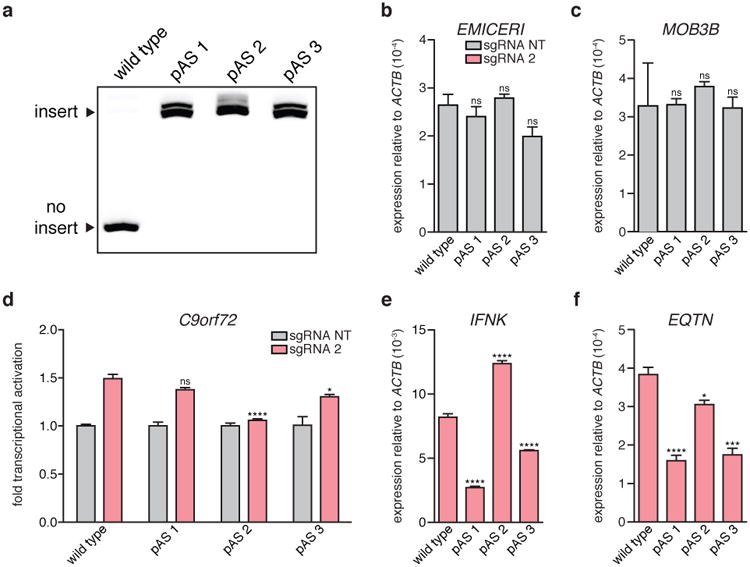
a, Gel confirming polyadenylation signal (pAS) insertion into all 3 copies of EMICERI for each pAS clone. b to c, Basal expression of EMICERI and MOB3B for the wild type and pAS clones. d to f, Expression of C9orf72, IFNK, and EQTN after targeting SAM to EMICERI for the wild type and pAS clones. All values are mean ± SEM with n = 4. ****P < 0.0001; ***P < 0.001; *P < 0.05. ns = not significant.
Extended Data Figure 9. Transcriptional activation of EMICERI confers vemurafenib resistance.

a, Vemurafenib dose response curves for wild type and polyadenylation signal (pAS) clones transduced with SAM and EMICERI-targeting or non-targeting (NT) sgRNAs. b, Vemurafenib half maximal inhibitory concentration (IC50) for the same conditions as (a). All values are mean ± SEM with n = 4. *P < 0.05. ns = not significant.
Extended Data Figure 10. EMICERI and MOB3B act reciprocally to regulate each other through the process of transcription.

a, Expression of EMICERI and MOB3B after antisense oligonucleotide (ASO) knockdown of EMICERI in the context of SAM activation. b, Expression of MOB3B and neighboring genes in A375 cells transduced with non-targeting (NT) or MOB3B-targeting sgRNAs and dCas9. c, Expression of MOB3B and EMICERI after ASO knockdown of MOB3B in the context of SAM activation. c, All values are mean ± SEM with n = 4. ****P < 0.0001; ***P < 0.001; **P < 0.01; *P < 0.05. ns = not significant.
Supplementary Material
Acknowledgments
We would like to thank M. Guttman, C.M. Johannessen, and M. Ghandi for helpful discussions and insights; A. Sayeed, R. Deasy, A. Rotem, and B. Izar for generating the Cancer Cell Line Factory models; R. Belliveau for overall research support; R. Macrae for critical reading of the manuscript; and the entire Zhang laboratory for support and advice. O.O.A. is supported by a Paul and Daisy Soros Fellowship, a Friends of the McGovern Institute Fellowship, the Poitras Center for Affective Disorders, and the National Defense Science and Engineering Graduate Fellowship. J.S.G. is supported by a D.O.E. Computational Science Graduate Fellowship. N.E.S. is supported by the NIH through a Pathway to Independence Award (R00-HG008171) from the National Human Genome Research Institute and a postdoctoral fellowship from the Simons Center for the Social Brain at the Massachusetts Institute of Technology. J.B.W. is supported by the NIH through a Ruth L. Kirschstein National Research Service Award (F32-DK096822). C.P.F. is supported by the National Defense Science and Engineering Graduate Fellowship. J.M.E. is supported by the Fannie and John Hertz Foundation. F.Z. is supported by the NIH through NIMH (5DP1-MH100706 and 1R01-MH110049), the Howard Hughes Medical Institute, the New York Stem Cell, Poitras, Simons, Paul G. Allen Family, and Vallee Foundations; and David R. Cheng, Tom Harriman, and B. Metcalfe. F.Z. is a New York Stem Cell Foundation-Robertson Investigator. Reagents are available through Addgene; support forums and computational tools are available via the Zhang lab website (http://www.genome-engineering.org).
Footnotes
Author contributions: J.J., S.K., and F.Z. conceived and designed the study. J.J., S.K., V.K.V., and J.S.G. performed experiments. J.J. analyzed data. O.O.A. and F.A. performed the analysis of clinical data sets. N.E.S. and J.B.W. performed ATAC-seq and ChIP experiments. J.M.E., C.P.F., and E.S.L. helped with lncRNA experimental design and interpretation. Y.Y.T., C.H.Y., and J.S.B. acquired and generated the Cancer Cell Line Factory models. J.J., J.M.E., E.S.L., and F.Z. wrote the paper with help from all authors.
References
- 1.Guttman M, et al. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature. 2009;458:223–227. doi: 10.1038/nature07672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cabili MN, et al. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev. 2011;25:1915–1927. doi: 10.1101/gad.17446611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Derrien T, et al. The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res. 2012;22:1775–1789. doi: 10.1101/gr.132159.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brown CJ, et al. The human XIST gene: analysis of a 17 kb inactive X-specific RNA that contains conserved repeats and is highly localized within the nucleus. Cell. 1992;71:527–542. doi: 10.1016/0092-8674(92)90520-m. [DOI] [PubMed] [Google Scholar]
- 5.Engreitz JM, et al. The Xist lncRNA exploits three-dimensional genome architecture to spread across the X chromosome. Science. 2013;341:1237973. doi: 10.1126/science.1237973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kretz M, et al. Control of somatic tissue differentiation by the long non-coding RNA TINCR. Nature. 2013;493:231–235. doi: 10.1038/nature11661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang KC, et al. A long noncoding RNA maintains active chromatin to coordinate homeotic gene expression. Nature. 2011;472:120–124. doi: 10.1038/nature09819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Guttman M, Rinn JL. Modular regulatory principles of large non-coding RNAs. Nature. 2012;482:339–346. doi: 10.1038/nature10887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Anderson KM, et al. Transcription of the non-coding RNA upperhand controls Hand2 expression and heart development. Nature. 2016 doi: 10.1038/nature20128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Engreitz JM, et al. Local regulation of gene expression by lncRNA promoters, transcription and splicing. Nature. 2016 doi: 10.1038/nature20149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Paralkar VR, et al. Unlinking an lncRNA from Its Associated cis Element. Mol Cell. 2016;62:104–110. doi: 10.1016/j.molcel.2016.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Konermann S, et al. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature. 2015;517:583–588. doi: 10.1038/nature14136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.O'Leary NA, et al. Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016;44:D733–745. doi: 10.1093/nar/gkv1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Konig R, et al. A probability-based approach for the analysis of large-scale RNAi screens. Nat Methods. 2007;4:847–849. doi: 10.1038/nmeth1089. [DOI] [PubMed] [Google Scholar]
- 15.Johannessen CM, et al. A melanocyte lineage program confers resistance to MAP kinase pathway inhibition. Nature. 2013;504:138–142. doi: 10.1038/nature12688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lei QY, et al. TAZ promotes cell proliferation and epithelial-mesenchymal transition and is inhibited by the hippo pathway. Mol Cell Biol. 2008;28:2426–2436. doi: 10.1128/MCB.01874-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lin L, et al. The Hippo effector YAP promotes resistance to RAF- and MEK-targeted cancer therapies. Nat Genet. 2015;47:250–256. doi: 10.1038/ng.3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Praskova M, Xia F, Avruch J. MOBKL1A/MOBKL1B phosphorylation by MST1 and MST2 inhibits cell proliferation. Curr Biol. 2008;18:311–321. doi: 10.1016/j.cub.2008.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.West S, Proudfoot NJ, Dye MJ. Molecular dissection of mammalian RNA polymerase II transcriptional termination. Mol Cell. 2008;29:600–610. doi: 10.1016/j.molcel.2007.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Skalska L, Beltran-Nebot M, Ule J, Jenner RG. Regulatory feedback from nascent RNA to chromatin and transcription. Nat Rev Mol Cell Biol. 2017 doi: 10.1038/nrm.2017.12. [DOI] [PubMed] [Google Scholar]
- 21.Fulco CP, et al. Systematic mapping of functional enhancer-promoter connections with CRISPR interference. Science. 2016;354:769–773. doi: 10.1126/science.aag2445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu SJ, et al. CRISPRi-based genome-scale identification of functional long noncoding RNA loci in human cells. Science. 2017;355 doi: 10.1126/science.aah7111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sanjana NE, et al. High-resolution interrogation of functional elements in the noncoding genome. Science. 2016;353:1545–1549. doi: 10.1126/science.aaf7613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhu S, et al. Genome-scale deletion screening of human long non-coding RNAs using a paired-guide RNA CRISPR-Cas9 library. Nat Biotechnol. 2016 doi: 10.1038/nbt.3715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hsu PD, et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol. 2013;31:827–832. doi: 10.1038/nbt.2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shalem O, et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science. 2014;343:84–87. doi: 10.1126/science.1247005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Joung J, et al. Genome-scale CRISPR-Cas9 knockout and transcriptional activation screening. Nat Protoc. 2017;12:828–863. doi: 10.1038/nprot.2017.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011;12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rao SS, et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell. 2014;159:1665–1680. doi: 10.1016/j.cell.2014.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Consortium, E. P. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Grant CE, Bailey TL, Noble WS. FIMO: scanning for occurrences of a given motif. Bioinformatics. 2011;27:1017–1018. doi: 10.1093/bioinformatics/btr064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Matys V, et al. TRANSFAC and its module TRANSCompel: transcriptional gene regulation in eukaryotes. Nucleic Acids Res. 2006;34:D108–110. doi: 10.1093/nar/gkj143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Buenrostro JD, Wu B, Chang HY, Greenleaf WJ. ATAC-seq: A Method for Assaying Chromatin Accessibility Genome-Wide. Curr Protoc Mol Biol. 2015;109:21 29 21–29. doi: 10.1002/0471142727.mb2129s109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Van der Auwera GA, et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinformatics. 2013;43:11 10 11–33. doi: 10.1002/0471250953.bi1110s43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Felsenstein J, Churchill GA. A Hidden Markov Model approach to variation among sites in rate of evolution. Mol Biol Evol. 1996;13:93–104. doi: 10.1093/oxfordjournals.molbev.a025575. [DOI] [PubMed] [Google Scholar]
- 38.Feng J, Liu T, Qin B, Zhang Y, Liu XS. Identifying ChIP-seq enrichment using MACS. Nat Protoc. 2012;7:1728–1740. doi: 10.1038/nprot.2012.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dobin A, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.DeLuca DS, et al. RNA-SeQC: RNA-seq metrics for quality control and process optimization. Bioinformatics. 2012;28:1530–1532. doi: 10.1093/bioinformatics/bts196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liberzon A, et al. Molecular signatures database (MSigDB) 3.0. Bioinformatics. 2011;27:1739–1740. doi: 10.1093/bioinformatics/btr260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Konieczkowski DJ, et al. A melanoma cell state distinction influences sensitivity to MAPK pathway inhibitors. Cancer Discov. 2014;4:816–827. doi: 10.1158/2159-8290.CD-13-0424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Barbie DA, et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature. 2009;462:108–112. doi: 10.1038/nature08460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Barretina J, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483:603–607. doi: 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.