

Summary
Various factors differentially recognize trimethylated histone H3 lysine 4 (H3K4me3) near promoters, H3K4me2 just downstream, and promoter-distal H3K4me1 to modulate gene expression. This methylation “gradient” is thought to result from preferential binding of the H3K4 methyltransferase Set1/COMPASS to promoter-proximal RNA polymerase II. However, other studies have suggested that location-specific cues allosterically activate Set1. ChIP-Seq experiments show that H3K4 methylation patterns on active genes are not universal or fixed, and change in response to both transcription elongation rate and frequency, as well as reduced COMPASS activity. Fusing Set1 to RNA polymerase II results in H3K4me2 throughout transcribed regions, and similarly extended H3K4me3 on highly transcribed genes. Tethered Set1 still requires histone H2B ubiquitylation for activity. These results show higher-level methylations reflect not only Set1/COMPASS recruitment, but also multiple rounds of transcription. This model provides a simple explanation for non-canonical methylation patterns at some loci or in certain COMPASS mutants.
eTOC blurb
Soares et al. show that the H3K4 methylation gradient, an important chromatin modification at active genes, is determined not only by targeted recruitment of the Set1 methyltransferase, but also transcription frequency and elongation rate. Fusing Set1 to RNA polymerase results in extended H3K4 methylation throughout the gene.
Introduction
A major mechanism for gene regulation is control of DNA accessibility by histone modifications (Cairns, 2009; Venkatesh and Workman, 2015). Nucleosomes comprise two copies each of histones H2A, H2B, H3, and H4 wrapped around DNA. Histone “tails” extend out from the nucleosome core and are subject to a various post-translational modifications. Tail lysines can be mono- (me1), di- (me2), or tri-methylated (me3). Individual histone methyltransferases (HMTs) generally modify a single lysine, often to a specific degree of methylation. Distinct methylations are in turn recognized by specific complexes that mediate transcriptional activation or repression.
Active genes are marked by H3K4 and K36 methylation, and disruptions of these marks or their associated enzymes are linked to several pathologies (Shilatifard, 2012). H3K4me3 is found close to transcription start sites (TSSs), with H3K4me2 and H3K4me1 peaking further downstream on longer transcriptional units (Barski et al., 2007; Liu et al., 2005; Pokholok et al., 2005), creating a 5′ to 3′ gradient of H3K4 methylation. Importantly, different H3K4me states have distinct functions (Buratowski and Kim, 2010). H3K4me3 recruits mostly positive transcription regulators, while H3K4me2 recruits HDACs to suppress cryptic internal transcriptional initiation (reviewed in Buratowski and Kim, 2010; Pinskaya and Morillon, 2009). More complex situations arise when H3K4 methylation from overlapping or antisense non-coding transcription represses mRNA promoters (Kim et al., 2012; Pinskaya et al., 2009). Thus, differential placement of different H3K4me states is essential for proper gene regulation.
Metazoans have multiple H3K4 HMTs (Shilatifard, 2012). While the MLL proteins function at specific genomic loci, H3K4 methylation at active genes is primarily due to Set1. Mammals have two Set1 proteins, while flies and yeast have one. In Saccharomyces cerevisiae, Set1 is the sole H3K4 HMT, making it a good model system for studying co-transcriptional methylation. The yeast COMPlex ASsociated with Set1 (COMPASS) consists of catalytic subunit Set1 and seven additional proteins, all conserved in fly and mammalian Set1 complexes (Shilatifard, 2012). Indeed, all Set1 and MLL complexes share a four subunit subcomplex (Wdr5-Rbbp5-Ash2-Dpy30, or WRAD, in higher eukaryotes: Swd3-Swd1-Bre2-Sdc1 in yeast) intimately associated with the catalytic SET domain (Kim et al., 2013; Lee and Skalnik, 2008). Other subunits involved in enzyme targeting and regulation vary between complexes. In yeast COMPASS, these are the WD40 protein Swd2, the PHD finger protein Spp1, and Shg1. Functions for the non-catalytic subunits remain to be elucidated.
Given the distinct functions of different H3K4me states, it is important to understand how appropriate levels of each are achieved. Two non-exclusive models can be invoked. A “time” model posits the H3K4 methylation level corresponds to the amount of time of Set1 spends at each position along the gene. This idea is supported by the observation that COMPASS preferentially interacts with RNA polymerase II (RNApII) phosphorylated at serine 5 of the Rpb1 C-terminal domain (CTD-Ser5P) (Ng et al., 2003; Lee and Skalnik, 2008). As CTD-Ser5P predominates in early elongation, binding enriches Set1 at promoter-proximal regions to allow higher levels of methylation.
The second model postulates that subunits of COMPASS sense cues, perhaps encoded in histone modifications or other chromatin modifiers, to differentially regulate Set1 catalytic activity along the gene. This “location” model arises from observations that mutations in specific COMPASS subunits appear to block particular levels of methylation. For example, yeast lacking Spp1 have markedly lower H3K4me3 levels, while both H3K4me3 and H3K4me2 levels are severely reduced in the absence of Sdc1 or Bre2 (Dehé et al., 2006; Mersman et al., 2012; Morillon et al., 2005; Schneider et al., 2005; South et al., 2010; Takahashi et al., 2009). These findings led to suggestions that these COMPASS subunits modulate Set1 catalytic activity in a context-specific manner (Thornton et al., 2014).
Interestingly, histone H2B ubiquitylation at lysine 123 (H2Bub) by the Rad6/Bre1 E3 ligase is important for H3K4me3 and H3K4me2, but less so for H3K4me1 (Dehé et al., 2006; Dover et al., 2002; Shahbazian et al., 2005; Sun and Allis, 2002). This histone modification crosstalk could play a part in the “time” or “location” model. One study proposed that H2Bub recruits Swd2 to 5′ ends of active genes, in turn recruiting the rest of COMPASS (Lee et al., 2007). Therefore, H2Bub could help establish the gradient by localizing Set1 to promoter-proximal regions of active genes. Other studies suggest that H2Bub or Rad6/Bre1 can regulate Set1 catalytic activity. One study suggests H2Bub promotes higher methylation states through Spp1, which interacts with the N-SET domain of Set1 to stimulate its activity (Kim et al., 2013). Another argues for an indirect mechanism where Rad6/Bre1 ubiquitylates Swd2 as well as H2B, with ubiquitylated Swd2 recruiting Spp1 to enhance Set1 methylation activity (Vitaliano-Prunier et al., 2008). If H2Bub levels or turnover differ in specific gene regions, this could stimulate COMPASS activity in specific locations to help create a methylation gradient. However, genome-wide ChIP experiments in yeast suggest H2Bub levels are relatively consistent throughout transcribed regions (Bonnet et al., 2014; Schulze et al., 2011).
There are caveats to interpreting the earlier data. First, most COMPASS subunits that affect Set1 methyltransferase activity are also important for Set1 stability in vivo (Dehé et al., 2006; Mersman et al., 2012; Soares et al., 2014), confounding analysis of deletion mutants that decrease Set1 recruitment to active genes. Deletions of SWD1, SWD2, SWD3, or SPP1 decrease both Set1 protein levels and H3K4me. Furthermore, a positive feedback mechanism exists such that catalytic deficient alleles of SET1, or even reduced gene transcription, also reduce Set1 levels (Soares et al., 2014). Another issue is that many studies only monitored H3K4 methylation states by immunoblotting bulk histones. Variations in H3K4me patterns can be missed by not assessing H3K4 methylation distribution genome-wide.
To better understand determinants of H3K4me patterns in S. cerevisiae, we performed ChIP-Seq experiments with normalizing S.pombe “spike-ins” for the different methylation states in wild-type and mutant backgrounds. Our data shows more variability in H3K4 methylation patterns than previously assumed. Both levels and positions of higher methylation states change with transcription frequency. Using mutants that modulate transcription rate, we show that H3K4 methylation patterns are best explained by the “time” model. Indeed, tethering COMPASS to RNApII leads to increased levels of H3K4 methylation throughout transcribed regions, arguing against location-specific cues. Surprisingly, multiple rounds of transcription may be required to attain H3K4me3. Overall, our results provide further insights into the mechanisms that establish H3K4me patterns and open new possibilities for understanding the roles of this modification in gene expression.
Results
H3K4 methylation patterns are not universal
H3K4 methylation distribution are not as homogenous as generally accepted. Certain genes are reported to lack high levels of H3K4me3 in promoter proximal regions (Kim et al., 2012; Orford et al., 2008), while others have extended H3K4me3 zones (Benayoun et al., 2014; Chen et al., 2015). We previously showed that different classes of genes vary in how their methylation states respond to SET1 mutations (Soares et al., 2014). To probe for heterogeneity of H3K4me patterns in the yeast Saccharomyces cerevisiae, we performed whole genome ChIP-Seq analysis of all three states of this histone modification. Previous whole genome analyses of H3K4me in S. cerevisiae (Liu et al., 2005; Pokholok et al., 2005) focused on averaged patterns and often excluded significant numbers of genes with other transcription units in close proximity. Despite potential complications from overlapping or antisense transcripts, we find that including all transcription units allows an unbiased analysis of H3K4me patterns.
Consistent with previous work, when the averaged H3K4me3 level is plotted relative to the TSS, a peak is seen around +200 base pairs (bp), just downstream of the nucleosome depleted region (NDR) and consistent with the +1 nucleosome position (Fig 1A). As expected, due to divergent transcription and the compactness of the yeast genome, a smaller averaged peak of H3K4me3 is also observed upstream of the NDR. Importantly, heat map visualization of individual genes shows a more complex distribution (Fig 1B). While the majority of genes display considerable H3K4me3, roughly 1000 transcription units have little or none (Fig 1B, left panels, with bottom panel showing modification levels normalized to total histone H3). Plotting the H3K4me3 maximum value for each gene confirms this observation, showing a bimodal distribution in a gaussian kernel density estimate fit, with one peak near zero corresponding to genes lacking H3K4me3 (Fig 1C, left panel). Notably, the peak position of H3K4me3 shows little variation (Fig 1B, left panels), occurring just downstream of the NDR seen by total H3 ChIP-Seq (Fig 1B, upper right panel). Interestingly, transcriptional units with strongest H3K4me3 also show increased H3 signal at the +1 nucleosome (Fig 1B, top right panel). These are mostly TFIID-dominant genes with well-defined and stable +1 nucleosomes and stronger transcription, as confirmed by RNApII (Rpb3) Chip-Seq (Fig 1B, bottom right panel).
Figure 1. Distribution of H3K4me states in S.cerevisiae.

A Anchor plot of H3K4me states centered at the transcription start site (TSS) for RNApII transcripts. ChIP-Seq SPMR (sequence tags per million reads) values for nucleotide positions between −1500 and +1500 were averaged and plotted; red, H3K4me3; green, H3K4me2; orange, H3K4me1. B Heat maps of H3K4me states. SPMR values were mapped for individual RNApII transcriptional units and stacked in order of calculated maximum value for H3K4me3 peaks associated with that TSS (see methods). Color code for H3K4me states is as in panel A, with histone H3 in gray, and Rpb3 in purple. Upper panels and Rpb3 show total reads, while lower H3K4 methylation panels show values normalized to total H3. C Kernel density estimation plot and histogram of H3K4me maximum values. Maximum values were calculated for each methylation state at individual genes and plotted both as an histogram representation and a kernel density estimation plot. Color codes as in panel A. D Representative ChIP-Seq tracks. SPMR values for H3K4me3, me2 and me1 are plotted from 200 bp upstream of the TSS to 200 bp downstream of the transcription termination site for YLR249W, YGL008C and YBR085W. Color code as in panel A. See also Fig S1.
Analysis of the average H3K4me2 pattern also agrees with previous studies, with a maximum average peak located approximately 600 bp downstream of the TSS or around 400 bp downstream from the H3K4me3 peak (Fig 1A). However, detailed analysis of individual genes again reveals significant heterogeneity (Fig 1B, middle left panels). Two features are apparent. First, the H3K4me2 peak positions span a wider range than H3K4me3 (Levene test statistic, 3451.8), with a large number of genes having H3K4me2 peaks close to the TSS. Second, the number of genes with little or no H3K4me2 is significantly lower than those lacking H3K4me3 (Fig 1C, compare left and middle left panels).
The distribution of H3K4me1 is similar to that of H3K4me2, but shifted downstream at most genes (Fig 1B, middle right panels). A few genes completely lack a H3K4me1 peak (Fig 1C, right panel), and these have the lowest levels of RNApII (Fig 1B, bottom right panel). After normalizing for lower total H3, the −1 nucleosome also appears enriched for H3K4me1 (Fig 1B, lower middle right panel), perhaps reflecting lower level transcription from divergent promoters.
The distributions of H3K4me marks (Fig 1B) show that genes lacking significant H3K4me3 have H3K4me2 peak positions closer to the TSS, while higher levels of H3K4me3 correlate with more downstream positions of maximum H3K4me2. Similarly, promoters lacking H3K4me2 or me3 often have a peak of promoter-proximal H3K4me1. In addition to the canonical pattern (Fig 1D, top panel), single gene traces often show overlapping H3K4me3 and H3K4me2 peaks near the promoter (Fig 1D, middle panel), or H3K4me2 peaks near the TSS without H3K4me3 (Fig 1D, lower panel).
One possible source of non-canonical methylation patterning is targeted demethylation. In cells lacking Jhd2, the sole H3K4 demethylase, the H3K4me2 and me3 peak positions do not change, but higher levels of these modifications are sometimes seen throughout the gene (Fig S1A–C). Some promoters with little or no H3K4me2 show a slight increase of this modification (Fig S1D–E). However, it is clear that Jhd2 is not the primary cause of variation in methylation positions.
H3K4 methylation patterns respond to transcription frequency
A comparison plot of maximum values for H3K4me2 and me3 at each gene shows an interesting pattern, distinct from a simple linear relationship predicted by a “location” model (Fig 2A). H3K4me3 peak levels increase slowly relative to H3K4me2 levels up to a certain threshold, upon which H3K4me3 increases while H3K4me2 remains high. A subset of genes with high H3K4me3 and low me2 (Fig 2A) are enriched for genes smaller than 1kb, where the expected H3K4me2 peak location would lie outside the transcription unit (Fig S2A).
Figure 2. Effects of transcription rate and COMPASS activity on H3K4me distribution.

A Scatter plot of H3K4me3 versus H3K4me2 maximum SPMR values, with each individual transcription unit represented by a spot. B Scatter plot of relation between H3K4me properties and normalized RNA transcript levels (Xu et al., 2009; http://steinmetzlab.embl.de/NFRsharing/). For each gene, maximum peak value (upper panels) and position (lower panels) for H3K4me3 (left panels, red) and H3K4me2 (right panels, green) were plotted on the y-axis versus RNA levels (log2 scale) on the x-axis. Spearman correlation coefficient is above each plot. C Heat maps of H3K4me3 and H3K4me2 ChIP-Seq signal in spp1Δ. SPMR values were mapped for individual RNApII transcriptional units and stacked in order of H3K4me3 maximum value as in Fig 1B, right panel corresponds to the difference heat map between SPP1 and spp1Δ strains. D Distribution of H3K4me2 peak positions in SPP1 and spp1Δ strains. Position of maximum SPMR value for each gene was calculated and plotted as a kernel density estimate plot using the same number of total bins: red, SPP1; green spp1Δ.E Scatter plot of maximum H3K4me2 value in spp1Δ versus RNA transcript levels (log2 scale). F Scatter plot of maximum H3K4me2 value in spp1Δ versus maximum H3K4me3 in SPP1 cells. See also Fig S2.
Given the apparent correlation between Rpb3 and H3K4me3 levels (Fig 1B), we plotted H3K4me3 and me2 peak values and positions against RNA levels, which are a reasonable proxy for transcription frequency (Xu et al., 2009). H3K4me3 levels, but not peak position, show a moderate correlation with RNA levels (Fig 2B, left panels). Interestingly, H3K4me2 peak position, rather than levels, correlates better with RNA levels (Fig 2B, right panels; Fig S2B). Importantly, using other datasets of transcript levels or RNApII occupancy as a measure of transcription frequency (Pelechano et al., 2010) gave similar correlations with H3K4me3 peak levels and H3K4me2 peak positions (Fig S2B).
Taken together, our results suggest a stepwise transition from H3K4me1 to me2 and then me3. Genes with increased transcription frequency reach the transition threshold more efficiently. These results argue against a “location” model, and suggest that higher levels of methylation are promoted not only by targeted recruitment of COMPASS, but also by repeated passage of elongation complexes.
H3K4 methylation pattern shifts in response to reduced COMPASS activity
Based on immunoblotting, the COMPASS subunit Spp1 was reported to specifically stimulate H3K4me3 (Dehé et al., 2006; Mersman et al., 2012; Schneider et al., 2005). Given the results above, we revisited this effect using ChIP-Seq. As reported, H3K4me3 levels in spp1Δ are strongly decreased compared to SPP1, yet many genes retain significant levels near the promoter (Fig 2Cleft panel, Fig S2C). Interestingly, H3K4me2 shows both an increase in peak levels (Fig S2D) and an upstream shift of peak position to within 450 of the TSS (Fig 2Cright panel, 2D). In the absence of Spp1, there is increased correlation between the H3K4me2 maximum value and transcript levels (Fig 2E), and H3K4me2 maximum value in spp1Δ strongly correlates with that of H3K4me3 in an SPP1 strain (Fig 2F). Lack of Spp1 causes an upstream shift of the H3K4 methylation gradient, as seen by loss of downstream H3K4me2 in a difference heat map comparing SPP1 and spp1Δ (Fig 2C, right panel). These findings are most simply explained by a model where Spp1 contributes to overall COMPASS activity or recruitment, rather than specifically affecting only trimethylation.
H3K4 methylation patterns are sensitive to transcription elongation rate
A “location” model predicts that H3K4me patterns are fixed and should be independent of transcription elongation rate. In contrast, if COMPASS dissociation from elongation complexes occurs with a fixed half-life after initiation, faster elongation should carry COMPASS further and result in increased downstream methylation. To test these predictions we assayed H3K4me in Rpb1 mutant strains with altered transcription elongation rates. The N488D and E1103G alleles respectively decrease (“slow”) and increase (“fast”) polymerization rate (Malagon et al., 2006). Anchor plot representations of average ChIP-Seq results for H3K4me3 and H3K4me2 reveal clear effects (Fig 3A). The average maximum peak position of H3K4me2 is significantly shifted upstream by “slow” polymerase and downstream by the “fast” (Fig 3A, right panel). H3K4me3 peak position shifts are more subtle, but in the same directions (Fig 3A, left panel). These effects are clear in difference plots of individual gene heatmaps (Fig 3B, right panels, Fig S3A). A representative individual gene profile of the H3K4me2 shifts is shown in Fig 3C. The prevalence and magnitude of shifts are also seen in a histogram of maximum signal position differences in each mutant relative to wild type (Fig 3D; for H3K4me3, Pearson 2 skewness coefficient of −0.243 and 0.345 for ‘slow’ and ‘fast’ polymerase respectively; −0.730 and 0.344 for H3K4me2).
Figure 3. H3K4me distribution is sensitive to RNApII elongation rate.

A Anchor plot of H3K4me3 and me2 states in RPB1 (black), rpb1-N488D (“slow” elongation, light brown) and rpb1-E1103G (“fast” elongation, dark brown) strains, averaged and plotted as in Fig 1A. B Heat map of H3K4me3 and me2 states in rpb1-N488D and rpb1-E1103G strains. Heat maps were created as described in Fig 1B, ordered according to H3K4me3 values in RPB1 cells. Upper panels H3K4me3; lower panels H3K4me2; right panels show calculated differences between rpb1-N488D and rpb1-E1103G matrixes. C ChIP-Seq tracks for H3K4me2 on representative gene YLR222C (+/− 200bp from transcribed region) in RPB1, rpb1-N488D, and rpb1-E1103G strains. Color code as in panel A. D Histogram of H3K4me peak position shifts (mutant - wild type) between RPB1 and rpb1-N488D (left panels) or rpb1-E1103G (right panels). H3K4me3 (upper panels), H3K4me2 (lower panels). E Spread of H3K4me3 and H3K4me2 in rpb1-E1103G strain. Distance was calculated between the position of maximum SPMR for each H3K4me state and the position at which the SPMR drops to 10, α; or 5, β (top panel). Paired T-test statistic appears above each plot. See also Fig S3.
In addition to peak shifts, the “fast” polymerase leads to wider methylation spreads as quantitated by calculating the distance between the position of maximum H3K4me SPMR (sequence tags per million reads) and the position at which the SPMR value drops to 10 (spread α) or 5 (spread β) for each gene (Fig 3E). Spreading by “fast” polymerase is clear for both H3K4me3 and me2. Therefore, distributions of H3K4 methylations respond to modulating RNApII elongation rate, more consistent with a “time” rather than “location” model for gradient generation. While this paper was under review, a similar conclusion was reached for H3K36 methylation in mammalian cells (Fong et al., 2017).
Because COMPASS association with RNApII is linked to CTD serine 5 phosphorylation (Ser5P), we used ChIP-seq to test how RPB1 elongation rate mutants affect RNApII occupancy and CTD phosphorylation (Fig S3B). Total RNApII, monitored by Rpb3, was not dramatically affected, although there was slightly less “slow” polymerase in 3′ regions and increased “fast” polymerase signal over the gene body. Consistent with displacement of histones during transcription, H3 occupancy showed an inverse correlation with Rpb3. CTD Ser2P showed very clear shifts, upstream in the “slow” mutant and downstream in the “fast”. In contrast, Ser5P is mostly unaffected in the “fast” mutant, but strongly decreased by the “slow” polymerase (Fig S3B). The observation that H3K4 methylation shifts do not obviously track Ser5P in elongation rate mutants argues that, while CTD-Ser5P promotes initial COMPASS binding, Ser5P dephosphorylation may not determine the downstream extent of H3K4 methylation.
Downstream regions of genes are competent for H3K4me2 and me3
We and others (Kim et al., 2013; Schlichter and Cairns, 2005; Soares et al., 2014; Thornton et al., 2014) showed that truncated Set1 lacking over 700 N-terminal residues produces near wild type levels of total K4 methylation in vivo, despite the fact that smaller deletions cause large decreases. Proposed explanations for how larger deletions paradoxically rescue activity include removal of autoinhibitory regions (Kim et al., 2013; Schlichter and Cairns, 2005), loss of recruitment specificity leading to promiscuous methylation (Thornton et al., 2014), and overexpression of truncated Set1 due to misregulation of protein stability control (Soares et al., 2014). Set1 N-terminal regions undoubtedly play a role in establishing H3K4 methylation patterns, yet most previous studies analyzed methylation levels by immunoblotting extracts or by ChIP metagene analysis, assuming a uniform response of all genes. We previously observed surprisingly large differences in how SAGA-dominant and TFIID-dominant gene classes respond to Set1 truncations or COMPASS depletion (Soares et al., 2014). Given the heterogeneity in H3K4 methylation patterns in wild type cells, and the possible role of Set1 N-terminal domains in establishing these patterns, we used ChIP-Seq to assay H3K4me in a genomically integrated set1Δ700 strain.
Consistent with previous data (Soares et al., 2014; Thornton et al., 2014), set1Δ700 decreases average H3K4me3 levels on genes (Fig 4A, Fig S4A). However, individual gene comparisons between set1Δ700 and SET1 strains show a wide spectrum of effects. Some genes show complete loss of H3K4me3 in set1Δ700, some are relatively unaffected, and others show increased maximum levels or spreading (Fig 4B, C). As previously observed, SAGA-dominated genes are on average more prone to loss or reduction of H3K4me3 upon Set1 truncation (Fig S4B), likely due to faster turnover rates of the +1 nucleosome at SAGA- relative to TFIID-dominated genes (Soares et al., 2014).
Figure 4. The overexpressed Set1Δ700 protein changes H3K4me distribution.

A Anchor plot of H3K4me3 (red), me2 (green), and me1 (orange) states in set1Δ700 cells, averaged and plotted as in Fig 1A. B Heat maps of H3K4me3 and H3K4me2 states in set1Δ700 cells were created as in Fig 1B, ordered according to H3K4me3 SET1 values. Upper panels H3K4me3, lower panels H3K4me2; right panels represent the differences between set1Δ700 and SET1 matrixes. C ChIP-Seq SPMR tracks for H3K4me3 and me2 on representative genes YAL041W (upper panel); YAL035W (middle); and YAL038W (lower). SET1 H3K4me3, red; H3K4me2, green; set1Δ700: H3K4me3, blue; H3K4me2, purple. D Comparison between set1Δ700 and SET1 for H3K4me3 and me2 in different regions of genes, showing decreases near the promoter and increases downstream. Difference between SPMR values (set1Δ700 - SET1) for each gene zone indicated (upper panels) were calculated and plotted as a gaussian kernel density plot. H3K4me3, left; H3K4me2, right. See also Fig S4.
A subset of genes shows significant downstream spreading of H3K4me3 in the Set1Δ700 mutant, generally encompassing the entire transcriptional unit (Fig 4B, upper right panel; Fig 4C, middle panel). This effect can be quantified as the difference in SPMR between SET1 and set1Δ700 backgrounds in different regions of genes (Fig 4D, upper panel). A gaussian kernel density estimate fit plot of the calculated differences clearly shows that in promoter-proximal regions (0 to 500 bp from the TSS), the majority of the values are negative (indicating a decrease in signal in set1Δ700). In contrast, in intervals between 500 to 1000 and 1000 to 1500 bp downstream from the TSS, a large proportion of genes shows increased signal in set1Δ700 (Fig 4D, left lower panel).
H3K4me2 shows a consistent shift upstream in set1Δ700 (Fig 4A; Fig 4B, lower panels). Both average anchor plot representation (Fig 4A) and a histogram of distance between maximum SPMR positions show that peaks of both H3K4me2 and me3 overlap at the +1 nucleosome in the mutant (Fig S4C). Based on our results above (Fig 1 and Fig 2), the upstream “shift” of H3K4me2 and reduction of H3K4me3 likely reflect reduced ability of the truncated COMPASS to reach the highest level of methylation in promoter-proximal regions. However, as seen for H3K4me3, some genes also show increased H3K4me2 in downstream promoter distal regions in set1Δ700 (Fig 4D, right panels; Fig 4C).
Several conclusions can be drawn from this experiment. First, nothing intrinsic to the chromatin environment in downstream gene regions prevents H3K4 di- and tri-methylation. Therefore, any required molecular cues, e.g. H2Bub, are not exclusively localized to promoter-proximal nucleosomes. Second, although overall levels are reduced, H3K4me2 and me3 still show clear promoter-proximal enrichment in the set1Δ700 background, suggesting the mutant COMPASS complex may retain some targeting to 5′ ends. Alternatively, other factors may come into play. In particular, dynamics of nucleosome turnover (Soares et al., 2014) and RNApII elongation can strongly affect H3K4me levels (see Discussion).
Tethering Set1 to RNApII leads to extended H3K4me2 and me3 patterns
In the simplest version of the “time” model, the H3K4me gradient results from increased COMPASS occupancy in promoter-proximal regions (Ng et al., 2003). If true, preventing release of COMPASS should produce high levels of H3K4 methylation throughout the transcribed region. To test this hypothesis, we genetically fused Set1 to an RNApII subunit (Fig S5A). Rpb4 was chosen because the Rpb4-Rpb7 “stalk” protrudes away from the Pol II core, close to the presumed position of the CTD. This should be an acceptable location for CTD binding proteins, as the CTD itself can be transferred from Rpb1 to Rpb4 without loss of function (Suh et al., 2013). Unfortunately, Rpb4 fused to full-length Set1 was unstable (Fig 5A, lane 7), consistent with observations that Set1 levels are kept low through protein degradation signals in the N-terminal domains (Soares et al., 2014).
Figure 5. Fusing Set1 to RNApII extends H3K4 methylation downstream.

A Immunoblot of Rpb4 and H3K4me levels in relevant strains. Protein extracts from the indicated cells were separated by SDS-PAGE and probed with antibodies indicated on the left. H3 and TBP were used as loading controls. Position of Rpb4 proteins is indicated on the right, asterisk indicates a degradation product present in the Rpb4-Set1 (full length) background. FL indicates full length Set1. Lane 1, YSB2723+pRS314; lane 2, YSB2723+pRS414-SET1(1-1080); lane 3, YSB2723+pRS414-SET1Δ500; lane 4, YSB2824+pRS314; lane 5, YSB2827 = YSB2824+pRS314-RPB4NheI; lane 6, YSB2826 = YSB2824+pRS414-SET1(1-1080); lane 7, YSB2828 = YSB2824+pRS314-RPB4-SET1; lane 8, YSB3344 = YSB2824+pRS414-SET1Δ500; lane 9, YSB2829 = YSB2824+pRS314-Rpb4-SET1Δ500. B Growth phenotypes of strains used in panel A (corresponding to lanes 1–9 from top to bottom) were assayed by spotting dilutions on selective media at the indicated temperatures (top). C Average ChIP-seq profiles of Rpb3 (red) and Rpb4 (blue). ChIP-Seq in wild type and RPB4-SET1Δ500 strains were generated using 500 bp windows around the transcription start site (TSS) and transcription termination site (tts), as well as middle (>250 from TSS and <250 from tts) regions, and scaling all genes to 100 data points. Genes shorter than 600 nt were excluded. wild type, solid lines; Rpb4-SetΔ500 fusion, dashed lines. D Scatter plot of maximum H3K4me SPMR values in set1Δ500 or rpb4-set1Δ500 fusion cells (y-axes) versus wild type cells (x-axes). Upper panels H3K4me3, lower panels H3K4me2; left panels, set1Δ500 versus wild type, right panels, rpb4-set1Δ500 versus wild type. E Heat map of H3K4me3 and H3K4me2 states in wild type SET1, set1Δ500, and rpb4-set1Δ500 cells. SPMR maps were vertically ordered according to the maximum value for wild type H3K4me3. Upper plots, H3K4me3, lower plots H3K4me2. Right plots represent the difference matrix from subtraction of wild type from mutant values. See also Figs S5 and S6.
To bypass these effects, we fused the C-terminus of Rpb4 to Set1Δ500, a truncation lacking some of these degradation signals. Set1Δ500 removes the RNA recognition motif and binding site for COMPASS subunit Swd2, but retains the N-SET and SET domains as well as interactions with all other COMPASS subunits, including Spp1 (Kim et al., 2013, Fig S5A). In contrast to Set1Δ700, this truncation retains some degradation signals and so is expressed at levels similar to wild type Set1, yet has strongly reduced H3K4me3 and me2 in vivo (Fig 5A, lane 3 (Soares et al., 2014)). The Rpb4-Set1Δ500 fusion protein is expressed at levels slightly below those of wild type Rpb4 (Fig 5A, lane 9), but complements the growth defects of rpb4Δ (Fig 5B). ChIP-Seq for Rpb3 showed no major changes in RNApII distribution in the fusion strain (Fig 5C). Levels of Rpb4- Set1Δ500 were modestly reduced near promoters, but not further downstream, relative to Rpb4 (Fig 5C). Genome-wide sequencing also revealed that the centromeric Rpb4-Set1Δ500 plasmid was amplified beyond a single copy (Fig S5B), presumably due to the previously described degradation mechanisms controlling Set1 levels (Soares et al., 2014). Nevertheless, immunoblotting shows that Rpb4-Set1Δ500 restores bulk H3K4me2 and me3 to wild type levels or more (Fig 5A).
Because immunoblotting obscures gene-specific effects, we carried out ChIP-Seq for H3K4me3, me2, and me1 in wild type Set1, Set1Δ500, and Rpb4-Set1Δ500 strains. As in all our experiments, S. pombe chromatin was used as an internal normalizing control. Consistent with immunoblotting (Fig 5A), Set1Δ500 shows widespread reduction of H3K4me3 in maximum value scatter plots (Fig 5D, upper left panel) and heatmaps (Fig 5E). Interestingly, H3K4me2 is more resilient, with some genes showing a strong reduction and others none at all (Fig 5D, lower left panel). As with Set1Δ700, H3K4me2 peak location shifts to a promoter-proximal position (Fig 5E). In sum, the Set1Δ500 effects are quite similar to those of Set1Δ700, except Set1Δ500 does not produce downstream spreading of H3K4 methylation, presumably because Set1Δ700 is expressed at several-fold higher levels than Set1Δ500 or wild type Set1 (Soares et al., 2014).
ChIP-Seq confirms that Rpb4-Set1Δ500 fusion restores both H3K4me3 and H3K4me2 to levels comparable with wild type Set1 at essentially all genes (Fig 5D, right panels), even those extreme cases where Set1Δ500 completely abolishes higher states of H3K4me (Fig S5C). At many genes, peak H3K4me3 values actually increase (Fig 5D, right panels). Most importantly, heatmap analysis (Fig 5E) and individual gene traces (Fig S5C) show that tethering Set1 to transcribing polymerase results in extensive downstream spreading of H3K4me2 and me3. Indeed, H3K4me2 almost completely loses defined peaks and instead shows consistently high levels throughout transcribed regions of all genes. These effects are distinct from those of Set1Δ700 (Fig 4B), arguing the changes are not simply due to overexpression of the fusion. Although Rpb4-Set1Δ500 fusion is clearly non-physiological, these results strongly support a “time” model in which COMPASS recruitment is the main factor determining the H3K4me patterns.
Interestingly, although downstream H3K4me3 increases, most genes still show promoter-proximal enrichment of H3K4me3 in Rpb4-Set1Δ500 (Fig 5E). Therefore, while tethered Set1 produces elevated H3K4me2 throughout the gene, some additional feature still produces higher H3K4me3 near many promoters (see Discussion). In wild type cells, H3K4me2 peak position shifts downstream with increased transcription frequency (Fig 1E), suggesting that repeated passage of RNApII leads to higher methylation states. We asked whether this property also contributes to variability of downstream H3K4me3 in Rpb4-Set1Δ500 fusion cells. Genes with lengths of 1.5 kb or more were ordered by their transcript levels as a proxy for transcription frequency. In a heatmap of SPMR level differences between SET1 and rpb4-set1Δ500, higher downstream H3K4me3 clearly correlates with increased expression (Fig 6A, upper panel). In contrast, H3K4me2 is uniformly high, independent of transcription levels (Fig 6A, lower panels).
Figure 6. Effect of transcription frequency on H3K4me spreading in the Rpb4-Set1Δ500 fusion.

A Heatmaps of H3K4me3 and me2 show SPMR values for nucleotide positions 0 to +1500 relative to the TSS for genes longer than 1.5kb, with genes stacked according to RNA levels. Upper panels, H3K4me3; lower panels H3K4me2; left panels wild type SET1; middle panels rpb4-set1Δ500; right panels show difference values (mutant - wild type) between corresponding row matrices. B RNA levels of six gene groups generated by k-means clustering of the H3K4me3 difference matrix in panel A. RNA levels are shown as box plots. Clusters are ordered from largest to smallest H3K4me3 difference. C Averaged anchor plots (TSS at 1) of H3K4me3 and me2 at genes in each cluster from panel B; H3K4me3 (red), H3K4me2 (green), wild type SET1 dashed, rpb4-set1Δ500 solid line. D Histogram of H3K4me1 difference levels in each cluster. Value of maximum SPMR for H3K4me1 in wild type SET1 was subtracted from that in rpb4-set1Δ500 for each gene and differences plotted as histograms. See also Figs S5 and S6.
Correlation between increased transcription and downstream H3K4me3 in the Rpb4-Set1Δ500 fusion strain was further shown using k-means clustering of the difference data for all genes (Fig 6B). Average gene profiles of each cluster reveal that downstream H3K4me3 is strongest in the most highly transcribed genes, less in intermediate genes, and only partially increased in lowly expressed genes (Fig 6C). As seen on long genes in Fig 6A, H3K4me2 is prevalent throughout genes in all clusters, although a downstream shift of the shallow peak correlated with higher levels of H3K4me3 upstream (Fig 6C). Changes in H3K4 monomethylation varied in the Rpb4-Set1Δ500 fusion strain. Maximum H3K4me1 levels increase on lowly expressed genes upon Set1 tethering, but there is a decrease in H3K4me1 at strongly expressed genes; presumably as it is replaced by high levels of H3K4me2 and me3 (Fig 6D). From these results we reach the surprising conclusion that tethering Set1 to RNApII is sufficient to place H3K4me2 throughout the entire gene, but more frequent passage of the RNApII elongation complex is needed to generate H3K4me3, at least in downstream regions.
How H2B ubiquitylation promotes higher levels of H3K4 and H3K79 methylation is still being debated (Soares and Buratowski, 2013), but some proposals invoke H2Bub in recruiting COMPASS. We tested whether the Rpb4-Set1Δ500 fusion bypasses the requirement for Bre1, a component of the H2B ubiquitin ligase, to acheive H3K4me2 and me3. Immunoblotting (Fig S6) shows Bre1 is still required for higher states of H3K4 methylation. Therefore, even if H2Bub helps recruit COMPASS, it must be important for another function needed for efficient H3K4 methylation.
Discussion
H3K4 methylation influences gene expression through recruitment of both positive and negative transcriptional regulators (reviewed in Buratowski and Kim (2010)). Averaged patterns from genome-wide analyses reveal a gradient, with promoter-proximal nucleosomes marked by H3K4me3, followed by H3K4me2 and then H3K4me1 nucleosomes downstream. As Set1/COMPASS interacts with CTD-Ser5-phosphorylated RNApII, it is thought that increased occupancy near the 5′ nucleosomes allows more time for Set1 to reach the higher levels of methylation. However, it has also been suggested that Set1 processivity towards multiple methylations is allosterically regulated by one or more COMPASS subunits responding to location cues in the chromatin.
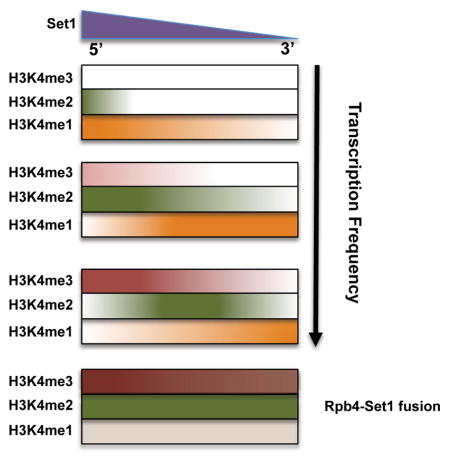
ChIP-Seq shows most genes have the canonical H3K4me pattern, yet a large number deviate (Fig 1). The +1 nucleosome is not universally marked by H3K4me3, and instead can be di- or mono-methylated. Strongly expressed genes show the highest H3K4me3, with a more distal me2 peak. Genes with low transcription often lack H3K4me3 and instead have H3K4me2 or even me1 peaks near the promoter. This correlation between methylation state and transcription frequency argues against the canonical methylation gradient being produced by a single round of transcription. Instead, transitions from H3K4me1 to me2 and me3 likely occur in a stepwise manner over multiple rounds of transcription with nucleosome retention. Our results suggest a more complex “time” model, where H3K4me level is determined by the length of time Set1 is tethered near a nucleosome in each round of transcription, multiplied by the number of transcription events over a unit of time (Fig 7).
Figure 7. Both Set1 localization and transcription frequency determine H3K4 methylation levels.

See Discussion for details of an extended “time” model.
In this “time” model, COMPASS mutants that reduce enzymatic activity or recruitment will affect both levels and location of H3K4me. For example, deletion of SPP1 appears to specifically reduce H3K4me3 by immunoblotting, but ChIP-Seq shows that normally trimethylated regions now have H3K4me2, while dimethylation is reduced at its usual location (Fig 2). These results suggest Spp1 plays a role in overall Set1 activity, not just trimethylation.
While a “location” model restricts higher H3K4me states to promoter proximal regions, a “time” model predicts that H3K4me2 and me3 can occur downstream if Set1 occupancy in those regions increases. We confirmed this prediction in multiple experiments. First, downstream H3K4me2 and me3 are increased by Set1Δ700, a truncation mutant that causes overexpression (Fig 4, (Soares et al., 2014)). This mutant loses Swd2 interaction (Kim et al., 2013), arguing against a requirement for Swd2 to activate COMPASS for higher H3K4me (Vitaliano-Prunier et al., 2008). Interestingly, while downstream H3K4me2 and me3 increased in Set1Δ700, there is still a 5′ bias. Therefore, overexpressed Set1Δ700 may produce “rogue” methylation as proposed for Set1Δ762 (Thornton et al., 2014), but its activity is still targeted to transcribed regions.
Further evidence for the “time” model comes from experiments using RPB1 mutations that affect transcription elongation rate (Fig 3). “Slow” RNApII shifts peaks of methylation upstream, while “fast” polymerase has the opposite effect. “Fast” polymerase also increases the spread of H3K4 methylations. We also created a strain where Set1 is covalently fused to RNApII (Rpb4-Set1Δ500), making recruitment independent of any other factors. Tethered Set1 greatly elevates H3K4me2 levels throughout the entire length of genes, while H3K4me3 is similarly extended at more highly transcribed genes (Figs 5 and 6). These results are most consistent with a “time” model.
An obvious question is why Rpb4-Set1Δ500 doesn’t lead to H3K4me3 along the entire gene. The striking correlation of downstream H3K4me3 with transcription frequency again argues that multiple passes of the RNApII elongation complex are required. However, genes with lower transcript levels still have a 5′ H3K4me3 peak (Fig 6). This observation may support a “location” model, and a recent paper showed that phosphorylated RNApII CTD boosts in vitro trimethylation by mammalian Set1 complex (Ebmaier et al., 2017). However, other explanations are consistent with a “time” model. Histone dynamic studies reveal a phenomenon called “treadmilling” or “passback”, where nucleosomes in transcribed genes gradually move upstream due to repeated passages of the RNApII elongation complex (Radman-Livaja et al., 2011). The “oldest” histones accumulate at 5′ ends of genes, particularly at lowly expressed genes, perhaps producing the H3K4me3 peak in Rpb4-Set1Δ500 cells. Another possible explanation is that occupancy time of the RNApII elongation complex is not uniform during a single transcription cycle. Although yeast does not show large peaks of paused RNApII just downstream of the promoter as in metazoans, both ChIP (Mayer et al., 2010, Fig 1B) and NET-Seq (Churchman and Weissman, 2011) show more RNApII near 5′ ends. This increased RNApII occupancy could extend the time Rpb4-Set1Δ500 spends near upstream nucleosomes.
Several studies proposed that H2Bub stimulates Set1 catalytic activity to produce higher states of H3K4me via the Swd2 or Spp1 subunits of COMPASS, either through recruitment or allosteric activation (Kim et al., 2013; Lee et al., 2007; Vitaliano-Prunier et al., 2008). H2Bub is relatively uniform throughout transcribed regions (Bonnet et al., 2014; Schulze et al., 2011), so this alone would not produce the gradient. We find that higher state methylation by Rpb4-Set1Δ500 still requires the ubiquitin ligase Bre1 (Fig S6), suggesting H2Bub plays an key role beyond simply recruiting Set1 to active genes.
It is exceedingly likely that our mechanistic conclusions in yeast apply to metazoans. Although the six COMPASS-related complexes in mammals have differentiated roles, the Set1A and Set1B complexes most closely resemble yeast COMPASS. These are responsible for the majority of global H3K4 methylation at active promoters, while the more distantly related MLL methyltransferases display gene-specific functions (reviewed in Shilatifard, 2012). As in yeast, mammalian Set1 complex preferentially interacts with Ser5-phosphorylated RNApII (Lee and Skalnik, 2008). Mapping of H3K4 in mammalian cells shows a methylation gradient similar to yeast (Barski et al., 2007), and H2Bub is needed for full methylation (Kim et al., 2009). Therefore, mechanisms for transcription-coupled H3K4 methylation appear conserved.
Interestingly, Benayoun et al. (2014) and Chen et al. (2015) reported extended zones of H3K4me3 over a small set of mammalian genes that also had high levels of elongation-associated marks. Similar broad H3K4me3 zones were claimed to occur in yeast (Benayoun et al., 2014), but our analysis showed these to be multiple promoters from closely spaced genes. Assuming the mammalian annotations are more accurate, these H3K4me3-extended genes fit our model. It was originally concluded these genes are characterized by reduced transcriptional noise, but a subsequent correction (Benayoun et al., 2015) showed extended methylation also correlates with transcription frequency. Our model suggests both increased transcription frequency and consistency would promote H3K4me3 spreading, due to more passages of the RNApII elongation complex per unit time. Thus, increased transcription would be the cause, rather than the result, of methylation spreading.
One mammalian-specific feature is H3K4 monomethylation at enhancers dependent upon MLL3 and MLL4 (Shilatifard, 2012). H3K4me1 is often used as a diagnostic marker of enhancers, but several recent papers suggest enhancers can also have H3K4me3 near TSSs of enhancer RNAs (Core et al., 2014; Hu et al., 2017). It is tempting to speculate that some H3K4me1 at enhancers may result from canonical mechanisms linked to low frequency eRNA transcription, rather than a completely distinct and specialized mechanism.
In summary, our results suggest the H3K4 methylation gradient at active genes is largely explained by the time Set1/COMPASS spends near the nucleosome, without having to invoke location-specific regulation of the enzyme. Surprisingly, the highest level of methylation apparently requires multiple rounds of transcription. As various transcriptional regulators differentially interact with H3K4me2 and me3, the same promoter may respond very differently to these factors depending on whether it is di- or tri-methylated. We believe our simplified model for H3K4 methylation will help explain results looking at these interactions.
STAR METHODS
Detailed methods are provided in the online version of this paper and include the following:
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| α-H3K4me3 | Millipore | 07-473 |
| α-H3K4me2 | Millipore | 07-030 |
| α-H3K4me1 | Millipore | 07-436 |
| α-H3 | Abcam | Ab1791 |
| α-Rpb3 | Neoclone | W0012 |
| α-Rpb4 | Neoclone | W0013 |
| α-CTD-Ser5P | Custom, Warren et al. (1992) J. Cell Sci. | H14 |
| α-CTD-Ser2P | Custom, Warren et al. (1992) J. Cell Sci. | H5 |
| Critical Commercial Assays | ||
| Qubit dsDNA HS Assay kit | ThermoFisher Scientific | Q32851 |
| QIAquick PCR Purification Kit | Qiagen | ID:28104 |
| QIAquick Gel Extraction Kit | Qiagen | ID:28704 |
| MinElute PCR Purification Kit | Qiagen | ID:28004 |
| T4 Polynucleotide Kinase | NEB | M0201S |
| T4 DNA Polymerase | NEB | M0203S |
| DNA Polymerase I, Large (Klenow) Fragment | NEB | M0210S |
| Klenow Fragment (3′->5′ exo-) | NEB | M0212S |
| Quick Ligation Kit | NEB | M2200S |
| Bioanalyzer High Sensitivity DNA kit | Agilent | 5067-4626 |
| Deposited Data | ||
| Raw and analyzed data | This Study | GEO: GSE95356 |
| SAGA versus TFIID dominated gene classification | Huisinga and Pugh (2004) | N/A |
| Transcriptional unit coordinates | Pelechano et al. (2014) | N/A |
| RNA levels | Xu et al. (2009) | http://steinmetzlab.embl.de/NFRsharing/ |
| S.cerevisiae Reference Genome version R64-1-1 | Engel et al., (2013) | https://downloads.yeastgenome.org/sequence/S288C_reference/genome_releases |
| S.pombe Reference Genome version ASM294v2 | Wood et al., (2002) | ftp://ftp.ebi.ac.uk/pub/databases/pombase/pombe/Chromosome_Dumps/ |
| Experimental Models: Organisms/Strains | ||
| S.cerevisiae Strain: MATa, ura3Δ0, leu2Δ0, his3Δ1, met15Δ0 |
http://www-sequence.stanford.edu/group/yeast_deletion_project/deletions3.html/ OpenBiosystems (now Dharmacon) | BY4741 |
| S.cerevisiae Strain: MATa, ura3-52, leu2Δ1, trp1Δ63, his4-912∂, lys2-128δ |
Fred Winston | FY118 |
| S.cerevisiae Strain: MATa, ura3Δ0, leu2Δ0, his3Δ1, met15Δ0, spp1Δ::KanMX |
http://www-sequence.stanford.edu/group/yeast_deletion_project/deletions3.html/ OpenBiosystems (now Dharmacon) | YF523 |
| S.cerevisiae Strain: MATa, ura3Δ0, leu2Δ0, his3Δ1, met15Δ0, jhd2Δ::KanMX |
http://www-sequence.stanford.edu/group/yeast_deletion_project/deletions3.html/ OpenBiosystems (now Dharmacon) | YF1474 |
| S.pombe strain: h+ N |
Fred Winston | 975, FWP9 |
| S.cerevisiae Strain: MATa, URA3::CMV-tTA, leu2Δ, trp1Δ::hisG, his3Δ, met15Δ, lys2Δ |
Malagon et al., (2006) | GRY3020 |
| S.cerevisiae Strain: MATa, URA3::CMV-tTA, leu2Δ, trp1Δ::hisG, his3Δ, met15Δ, lys2Δ, rpb1-E1103G |
Malagon et al., (2006) | GRY3028 |
| S.cerevisiae Strain: MATa, URA3::CMV-tTA, leu2Δ, trp1Δ::hisG, his3Δ, met15Δ, lys2Δ, rpb1-N488D |
Malagon et al., (2006) | GRY3027 |
| S.cerevisiae Strain: MATa, ura3Δ0, leu2Δ0, his3Δ1, met15Δ0, FLAG-Set1(Δ700) |
Soares et al., (2014) | YSB3119 |
| S.cerevisiae Strain: MATa, ura3Δ0, leu2Δ0, trp1Δ::LEU2/KanR, his3Δ1, met15Δ0, set1Δ::KanMX [pRS414] |
This study | YSB3342 |
| S.cerevisiae Strain: MATa, ura3Δ0, leu2Δ0, trp1Δ::LEU2/KanR, his3Δ1, met15Δ0, set1Δ::KanMX, rpb4Δ::NatMX [pRS314-RPB4NheI] |
This study | YSB2827 |
| S.cerevisiae Strain: MATa, ura3Δ0, leu2Δ0, trp1Δ::LEU2/KanR, his3Δ1, met15Δ0, set1Δ::KanMX, rpb4Δ::NatMX [pRS414-SET1 (1-1080)] |
This study | YSB2826 |
| S.cerevisiae Strain: MATa, ura3Δ0, leu2Δ0, trp1Δ::LEU2/KanR, his3Δ1, met15Δ0, set1Δ::KanMX, rpb4Δ::NatMX [pRS414-RPB4-SET1] |
This study | YSB2828 |
| S.cerevisiae Strain: MATa, ura3Δ0, leu2Δ0, trp1Δ::LEU2/KanR, his3Δ1, met15Δ0, set1Δ::KanMX, rpb4Δ::NatMX [pRS414] |
This study | YSB3343 |
| S.cerevisiae Strain: MATa, ura3Δ0, leu2Δ0, trp1Δ::LEU2/KanR, his3Δ1, met15Δ0, set1Δ::KanMX, rpb4Δ::NatMX [pRS414-SET1 Δ500] |
This study | YSB3344 |
| S.cerevisiae Strain: MATa, ura3Δ0, leu2Δ0, trp1Δ::LEU2/KanR, his3Δ1, met15Δ0, set1Δ::KanMX [pRS414-SET1 (1-1080)] |
This study | YSB3345 |
| S.cerevisiae Strain: MATa, ura3Δ0, leu2Δ0, trp1Δ::LEU2/KanR, his3Δ1, met15Δ0, set1Δ::KanMX [pRS414-SET1 Δ500] |
This study | YSB3346 |
| S.cerevisiae Strain: MATa, ura3Δ0, leu2Δ0, trp1Δ::LEU2/KanR, his3Δ1, met15Δ0, set1Δ::KanMX, rpb4Δ::NatMX [pRS314-RPB4-SET1ΔN500] |
This study | YSB3347 |
| S.cerevisiae Strain: MATa, ura3Δ0, leu2Δ0, trp1Δ::LEU2/KanR, his3Δ1, met15Δ0, set1Δ::KanMX, bre1Δ::URA3MX [pRS414-SET1 (1-1080)] |
This study | YSB3318 |
| S.cerevisiae Strain: MATa, ura3Δ0, leu2Δ0, trp1Δ::LEU2/KanR, his3Δ1, met15Δ0, set1Δ::KanMX, rpb4Δ::NatMX, bre1Δ::URA3MX [pRS314-RPB4-SET1ΔN500] |
This study | YSB3319 |
| Oligonucleotides | ||
| Barcoded adapter oligo 1 (BA1): 5′ ACACTCTTTCCCTACACGACGCTCTTCCGATCT-barcode-T 3′ |
Wong et al., (2013) | Reference table 7.11.2 |
| Barcoded adapter oligo 2 (BA2): 5 p-barcode(reverse complement)-AGATCGGAAGAGCGGTTCAGCAGGAATGCCGAG 3a |
Wong et al., (2013) | Reference table 7.11.2 |
| Library amplification PCR primer forward (PCR1): CAAGCAGAAGACGGCATACGAGATCGGTCTCGG CATTCCTGCTGAACCGCTCTTCCGATC*T * Indicates phosphorothioate bond |
Wong et al., (2013) | N/A |
| Library amplification PCR primer reverse (PCR2): AATGATACGGCGACCACCGAGATCTACACTCTT TCCCTACACGACGCTCTTCCGATC*T * Indicates phosphorothioate bond |
Wong et al., (2013) | N/A |
| Recombinant DNA | ||
| Plasmid: pRS414-SET1(1-1080) (ADH1 promoter driving SET1 (1-1080) with N-terminal FLAG tag, TRP1, CEN/ARS, f1+ ori, AmpR) |
Fingerman et al., (2005) | N/A |
| Plasmid: pRS414-SET1Δ500 (ADH1 promoter driving SET1 Δ500 with N-terminal FLAG tag, TRP1, CEN/ARS, f1+ ori, AmpR) |
This study | N/A |
| Plasmid: pRS314-RPB4NheI (RPB4 with NheI site inserted in front of stop codon, TRP1, CEN/ARS, f1+ ori, AmpR) |
Suh et al., (2013) | N/A |
| Plasmid: pRS314-RPB4-SET1 (RPB4-SET1 fusion, 3 glycine residues link the RPB4 and SET1 ORFs, TRP1, CEN/ARS, f1+ori, AmpR) |
This study | N/A |
| Plasmid: pRS314-RPB4-SET1ΔN500 (RPB4-SET1ΔN500 fusion, 3 glycine residues link the RPB4 and SET1ΔN500 ORFs, TRP1, CEN/ARS, f1+ori, AmpR) |
This study | N/A |
| Software and Algorithms | ||
| Sabre | N/A | https://github.com/najoshi/sabre |
| Bowtie 1.1.1 | Langmead et al., (2009) | http://bowtie-bio.sourceforge.net/index.shtml |
| Samtools 1.2 | Li et al., (2009) | http://samtools.sourceforge.net/ |
| MACS 2.1.0 | Zhang et al., (2008) | https://pypi.python.org/pypi/MACS2 |
| Python 3.4 | N/A | https://www.python.org/download/releases/3.4.0/ |
| Python Scripts | This study | https://github.com/LuisSoares/Manuscript |
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Stephen Buratowski, (steveb@hms.harvard.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
S. cerevisiae culture
All S.cerevisiae strains for ChIP-Seq experiments were grown in 300 ml of media at 25°C until OD600 reached 0.5. In Figures 1–4 used YPD (Yeast Extract Peptone Dextrose = 20g Bacto-peptone, 20g Glucose, 10g Yeast Extract 0.15g Tryptophan, dH20 to 1 liter) was used, in Figures 5–6 used MM (Minimal Media = 3g yeast nitrogen base, 10g ammonium sulfate, 4g -4aa powder, 2ml 200mM inositol, dH20 to 1 liter) supplemented with uracil, histidine, and leucine. For protein extracts used for immunoblotting, cells were grown in supplemented MM in the same conditions except using 50 ml cultures.
S. pombe culture
S.pombe strain 975 used for ChIP-Seq experiments was grown at 25°C in 300 ml of YPD media until OD600 reached 0.5.
METHOD DETAILS
Chromatin immunoprecipitation
Cells were grown in appropriate media at 25°C to an OD600 of 0.5, when formaldehyde was added to a final concentration of 1% for cross-linking. Cells were incubated for 20 min at room temperature and the crosslinking reaction quenched for 5 minutes at room temperature with 3M glycine. Cell pellets were recovered and washed with FA buffer (50 mM HEPES-KOH, pH 7.5, 150 mM NaCl, 1mM EDTA,1% Triton X-100, and 0.1% Na Deoxycholate) containing 0.1% SDS. Cells were lysed in FA buffer/0.5% SDS using glass beads and vortexing for 30 cycles of 30 seconds with cooling on ice in between. After spinning out beads and cell debris by microcentrifugation, cross-linked extracts were sonicated using a Misonix 3000 with cup horns, with 10s on/off pulses for a total sonication time of 20 min. Soluble chromatin was recovered after pelleting insoluble material by centrifugation for 10 minutes, 14000 rpm at 4°C. Protein concentration was quantified by Bradford assay. A total of 700 μg of chromatin was used per immunoprecipitation. All ChIP-Seq experiments included S.pombe “spike-in” chromatin prepared similarly and added at 10% relative to S.cerevisiae chromatin. For immunoprecipitation, the indicated antibody and 10 μl of Protein-G sepharose beads were added, followed by overnight incubation at 4°C on a rotator. Beads were washed once with FA buffer, 0.1% SDS, 275 mM NaCl; once with FA buffer, 0.1% SDS, 500 mM NaCl; once with 10 mM Tris-HCl, pH 8.0, 0.25 M LiCl, 1 mM EDTA, 0.5% NP-40, 0.5% Na Deoxycholate; and once with TE (10 mM Tris-HCl, pH 8.0, 1mM EDTA). Immunoprecipitated material was eluted with 50 mM Tris-HCl 7.5, 10 mM EDTA, 1% SDS by incubating at 65°C for 20 min. Beads were washed once with TE, and the supernatant added to eluted material. Recovered immunoprecipitate was de-crosslinked with 40 μg of Pronase for 1 hour at 42°C and overnight at 65°C. Samples were treated with 5 μg RNAse A, phenol-chloroform/chloroform extracted, and precipitated with 400 mM LiCl, 40 μg Glycogen, and 100% EtOH. Precipitated DNA was resuspended in 45 microliters of water and quantified using Qubit assay.
ChIP-Seq Library Preparation
5 ng of immunoprecipitated DNA was used for ChIP-Seq library production as described (Wong et al., 2013). Immunoprecipitated DNA was end repaired using 1 μl T4 DNA polymerase, 1 μl T4 PNK, 0.2 μl DNA Polymerase I Large (Klenow) fragment, 5 μl T4 DNA Ligase buffer and 2μl of 10 mM dNTP Mix in a total volume of 50 μl. The reaction was incubated for 45 minutes at room temperature followed by purification with QIAquick PCR Purification Kit. A single adenosine “tail” was added to the 3′ end of purified repaired fragments using 1 μl Klenow (3′ to 5′ exo minus) in a volume of 50 μl supplemented with 10 μl of 1 mM dATP and 5 μl of NEB buffer 2. The reaction was incubated for 30 minutes at 37°C and purified using QIAquick MinElute column. Adapters containing inline multiplexing barcodes were ligated using 4 μl of T4 Quick DNA ligase, 15 μl of 2xDNA Quick ligase buffer in a total volume of 30 μl for 15 minutes at room temperature. Adapter ligated DNA was run in TBE agarose electrophoresis gel and fragments between 200 and 500 bp were gel purified using QIAGEN Gel Extraction Kit. DNA was PCR amplified using Phusion DNA polymerase with 16 amplification cycles (30 sec at 98°C, [10 sec at 98°C, 30 sec at 65°C, 30 sec at 72° C], repeat 16 cycles, 5 min at 72°C). Library quality was assessed using Agilent 2100 Bioanalyzer. Equal molarity of each library was mixed and 50 bp single-end sequenced in High Output (Standard) v3 Illumina HiSeq 2000 (Harvard Bauer Center Core Facility).
Immunoblotting
For whole cell extracts, 50 ml of selective media was inoculated with 1 ml of an overnight culture and cells were grown until OD[600] reached 0.5. Cells were harvested by centrifugation and resuspended in 500 μl lysis buffer (50 mM Tris-Cl (pH 8.0), 0.1% NP-40, 2 μg/ml Aprotinin, 2 μg/ml Leupeptin, 2 μg/ml Pepstatin A, 2 μg/ml Antipain, 1 mM PMSF) with 150 mM NaCl. 500 μl of glass beads were added and cells were lysed by repeating 20 seconds of vortexing and 20 seconds of cooling on ice for a total of 10 cycles. Lysate was transferred to a 1.5 ml microfuge tube, and insoluble material was precipitated by centrifugation. Supernatant was transferred to a fresh tube, and protein concentration was measured using Coomassie Protein Assay Reagent (Thermo Scientific). For SDS-PAGE and immunoblot analysis, 20–60 μg of protein was loaded per well. Proteins were transferred from the SDS-PAGE gel onto a PVDF membrane and visualized with Ponceau S stain to confirm transfer. Membranes were blocked for 1 hour at room temperature in 5% w/v skim milk or BSA in Tris-buffered Saline with Tween-20 (TBST). Membranes were incubated with primary antibody overnight at 4°C, washed with TBST, and then HRP-conjugated secondary antibody was applied. The membrane was washed again with TBS and blots were visualized on radiography film with Pierce Pico and Femto chemiluminescent substrates.
Bioinformatics Analysis
Sequence reads from ChIP-seq were demultiplexed using SABRE (https://github.com/najoshi/sabre.git) allowing for one mismatch in the barcode. Demultiplexed reads were first aligned to the S.pombe genome (version ASM294v2.31), unaligned reads were subsequently aligned to the S. cerevisiae genome (version R64-1-1). Genome alignment was performed using BOWTIE 1.1.1 (Langmead et al., 2009) excluding the first base and filtering out multiply aligned reads. Alignment files were converted using SAMTOOLS 1.2 (Li et al., 2009) and pileup tracks calculated using MACS2.1.0 (Feng et al., 2012) callpeak function, removing duplicate reads, extending tags to 150 and normalizing values to sequence tags per million reads (SPMR). Coverage tracks were converted to high density wig files and analyzed using custom Python3.4 scripts (https://github.com/LuisSoares/Manuscript).
Annotation Datasets
S. cerevisiae transcriptional unit coordinates were obtained from Pelechano et al. (2014), transcriptional units resulting from duplications that cannot be assigned unique reads were removed from the analysis.
RNA levels were extracted from Xu et al. (2009), considering the RNA levels for each transcriptional unit as the median of the intensity of the entire transcriptional unit (obtained as described above) in the corresponding strand track.
SAGA versus TFIID dominated gene classification data was obtained from Huisinga and Pugh (2004). For assignment of methylation peak position to specific transcription units, the maximum peak had to occur within a window established between the TSS and the calculated 90th percentile value of maximum peak position for all genes. This filter allows exclusion of outlier peak positions that are likely to represent misannotated TSSs or strong antisense promoters.
QUANTIFICATION AND STATISTICAL ANALYSIS
Chip-seq Normalization
For “spike-in” normalization only sequence reads that could be exclusively assigned to each genome were considered for total number of reads. Normalization factors were calculated by first calculating the proportion of reads of S.pombe versus total reads in input reads, and dividing the value by the square root (to account for the SPMR normalization) of the proportion of reads of S.pombe versus total reads in each immunoprecipitation reads.
Statistical analysis
All statistical analysis was performed using Python 3.4 numpy, Pandas, and scipy.stats packages.
Figure 1
The gene list used in Figure 1 (n=6369) was obtained by considering the transcriptional units coordinates described in Pelechano et al. (2014), both transcription units resulting from duplications that cannot be assigned unique reads and units for which the range of −1500 to 1500bp from the transcription start site could not be retrieved (genes close to the edge of chromosomes) were removed from the analysis. To define the position of the peak for each gene, first the distance to the TSS for the maximum SPMR for each transcriptional unit was calculated, followed by defining the 90 percentile of all obtained distances and retrieving for each transcriptional unit the position and value of maximum SPMR located within such percentile distance. This filter allows exclusion of outlier peak positions that are likely to represent misannotated TSSs or strong antisense promoters. The heat maps presented in panel C were created using the filtered gene list and the ordering was established by ranking the values of maximum SPMR for the H3K4me3 track as described above, no averaging or interpolation was used for heat map creation.
Figure 2
The gene list used in Figure 2 (n=6369) was the same used in Figure 1 obtained as described above. The correlation values were calculated using Spearman rank correlation due to the fact that the variables do not have a normal distribution.
Figure 3
The gene list used in Figure 3 (n=6369) was the same used in Figure 1 obtained as described above. The calculation of both peak maximum value and position used was the same as the one described for Figure 1. The heat maps presented were ordered similarly to Figure 1. The spread values depicted were calculated according to the description in the figure legend: the distance was calculated between the position of maximum SPMR for each H3K4me state and the position at which the SPMR drops to 10, alpha; or 5, beta. Genes with maximum SPMR values below 10 and 5 respectively were excluded, as were extreme outlier values beyond 4 standard deviations (generally due to antisense transcripts). For genes that do not drop to 10 and 5 SPMR, the distance from peak to the gene end was used. For comparison paired T-test statistic was used since each transcriptional unit is compared between wild type and mutant conditions. The p-value for all T-tests performed was below 10−3.
Figure 4
The gene list used in Figure 4 (n=6369) was the same used in Figure 1 obtained as described above. The calculation of both peak maximum value and position used was the same as the one described for Figure 1. The heat maps presented were ordered similarly to Figure 1. The values used in panel D were obtained as described in the corresponding figure legend.
Figure 5
The gene list used in Figure 5 panels D and E (n=6369) was the same used in Figure 1 obtained as described above. The calculation of both peak maximum value and position used was the same as that described for Figure 1. The heat maps presented were ordered similarly to Figure 1. For the metagene analysis presented in panel C only transcriptional units longer than 600 bp were considered (n=5152). Each transcriptional unit was split in 3 regions: 5′, from −250 to +250 from the transcription start site; 3′, from −250 to +250 to the transcription termination site; middle, middle region of the each transcriptional unit was split in 100 equal sized regions and each region was averaged. Average of all transcriptional units at base pair resolution was calculated for 5′ and 3′ regions and for each of the 100 middle regions.
Figure 6
The gene list used in Figure 6 was obtained by considering the transcriptional units coordinates described in Pelechano et al. (2014); transcription units resulting from duplications that cannot be assigned unique reads and units were removed and only units longer than 1500 bp were considered (n=2583). The heat maps in panel A were ordered according to RNA levels obtained as described above. K-means clustering was performed using kmeans function from sklearn.cluster Python package with 6 clusters and random state set to 0. The matrix used for cluster calculation is the same used for heat map visualization in Figure 6 panel A, top right image. Clusters were ordered according to intracluster maximum value. Cluster sizes are as follow: 1, 88 genes; 2, 259 genes; 3, 452 genes; 4, 347 genes; 5, 733 genes; 6, 704 genes. Maximum values for H3K4me1 used in panel D were calculated as the maximum SPMR value for each of the transcriptional units.
DATA AND SOFTWARE AVAILABILITY
Sequence data available at GEO database, accession number GSE95356.
Custom Python scripts used in the manuscript available at https://github.com/LuisSoares/Manuscript
Supplementary Material
Highlights.
Many genes do not show the canonical H3K4 methylation gradient pattern.
H3K4 methylation levels are determined by the time Set1 spends near the nucleosome.
Multiple rounds of transcription contribute to H3K4 trimethylation levels.
Set1 fused to RNA pol II places H3K4me2 and me3 throughout transcribed regions.
Acknowledgments
We thank Fred Winston, Scott Briggs, and Jeffrey Strathern for strains. This work was supported by grants GM46498 and GM56663 from the US. National Institutes of Health to SB and 2013S1A2A2035342 from the Korean National Research Foundation to TSK and SB. CH was a Herchel Smith-Harvard Undergraduate Science Research Fellow and received support from the Harvard College Research Program.
Footnotes
Supplemental Information includes six figures and two tables and can be found with this article online at http://dx.doi.org/xxx.
Author Contributions
Conceptualization: L.S., T.K., and S.B.; Software, Formal Analysis, and Visualization: L.S.; Investigation: L.S., P.C.H., Y.C., and H.S.; Writing-Original Draft, L.S., P.C.H. and S.B.; Writing-Review and Editing: L.S. and S.B.; Project Administration, S.B.; Funding Acquisition: S.B. and T.K.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, Wei G, Chepelev I, Zhao K. High-Resolution Profiling of Histone Methylations in the Human Genome. Cell. 2007;129:823–837. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- Benayoun BA, Pollina EA, Ucar D, Mahmoudi S, Karra K, Wong ED, Devarajan K, Daugherty AC, Kundaje AB, Mancini E, Hitz BC, Gupta R, Rando TA, Baker JC, Snyder MP, Cherry JM, Brunet A. H3K4me3 Breadth Is Linked to Cell Identity and Transcriptional Consistency. Cell. 2015;163:1281–1286. doi: 10.1016/j.cell.2015.10.051. [DOI] [PubMed] [Google Scholar]
- Benayoun BA, Pollina EA, Ucar D, Mahmoudi S, Karra K, Wong ED, Devarajan K, Daugherty AC, Kundaje AB, Mancini E, Hitz BC, Gupta R, Rando TA, Baker JC, Snyder MP, Cherry JM, Brunet A. H3K4me3 breadth is linked to cell identity and transcriptional consistency. Cell. 2014;158:673–688. doi: 10.1016/j.cell.2014.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnet J, Wang CY, Baptista T, Vincent SD, Hsiao WC, Stierle M, Kao CF, Tora L, Devys D. The SAGA coactivator complex acts on the whole transcribed genome and is required for RNA polymerase II transcription. Genes Dev. 2014;28:1999–2012. doi: 10.1101/gad.250225.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buratowski S, Kim T. The role of cotranscriptional histone methylations. Cold Spring Harb Symp Quant Biol. 2010;75:95–102. doi: 10.1101/sqb.2010.75.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cairns BR. The logic of chromatin architecture and remodelling at promoters. Nature. 2009;461:193–198. doi: 10.1038/nature08450. [DOI] [PubMed] [Google Scholar]
- Chen K, Chen Z, Wu D, Zhang L, Lin X, Su J, Rodriguez B, Xi Y, Xia Z, Chen X, Shi X, Wang Q, Li W. Broad H3K4me3 is associated with increased transcription elongation and enhancer activity at tumor-suppressor genes. Nat Genet. 2015;47:1149–1157. doi: 10.1038/ng.3385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churchman LS, Weissman JS. Nascent transcript sequencing visualizes transcription at nucleotide resolution. Nature. 2011;469:368–373. doi: 10.1038/nature09652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Core LJ, Martins AL, Danko CG, Waters CT, Siepel A, Lis JT. Analysis of nascent RNA identifies a unified architecture of initiation regions at mammalian promoters and enhancers. Nat Genet. 2014;46:1311–1320. doi: 10.1038/ng.3142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehé PM, Dichtl B, Schaft D, Roguev A, Pamblanco M, Lebrun R, Rodríguez-Gil A, Mkandawire M, Landsberg K, Shevchenko A, Shevchenko A, Shevchenko A, Rosaleny LE, Tordera V, Chávez S, Stewart AF, Géli V. Protein interactions within the Set1 complex and their roles in the regulation of histone 3 lysine 4 methylation. J Biol Chem. 2006;281:35404–35412. doi: 10.1074/jbc.M603099200. [DOI] [PubMed] [Google Scholar]
- Dover J, Schneider J, Tawiah-Boateng MA, Wood A, Dean K, Johnston M, Shilatifard A. Methylation of histone H3 by COMPASS requires ubiquitination of histone H2B by Rad6. J Biol Chem. 2002;277:28368–28371. doi: 10.1074/jbc.C200348200. [DOI] [PubMed] [Google Scholar]
- Ebmaier CC, Erickson B, Allen BL, Allen MA, Kim H, Fong N, Jacobsen JR, Liang K, Shilatifard A, Dowell RD, Old WM, Bentley DL, Taatjes DJ. Human TFIIH kinase CDK7 regulates transcription-associated chromatin modifications. Cell Rep. 2017;20:1173–1186. doi: 10.1016/j.celrep.2017.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel SR, Cherry JM. The new modern era of yeast genomics: community sequencing and the resulting annotation of multiple Saccharomyces cerevisiae strains at the Saccharomyces Genome Database. Database. bat012. 2013 doi: 10.1093/database/bat012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng J, Liu T, Qin B, Zhang Y, Liu XS. Identifying ChIP-seq enrichment using MACS. Nat Protoc. 2012;7:1728–1740. doi: 10.1038/nprot.2012.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fingerman IM, Wu CL, Wilson BD, Briggs SD. Global loss of Set1-mediated H3 Lys4 trimethylation is associated with silencing defects in Saccharomyces cerevisiae. J Biol Chem. 2005;280:28761–28765. doi: 10.1074/jbc.C500097200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong N, Saldi T, Sheridan RM, Cortazar MA, Bentley DL. RNA pol II dynamics modulate co-transcriptional chromatin modification, CTD phosphorylation, and transcriptional direction. Mol Cell. 2017;66:546–560. doi: 10.1016/j.molcel.2017.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu D, Gao X, Cao K, Morgan MA, Mas G, Smith ER, Volk AG, Bartom ET, Crispino JD, Di Croce L, Shilatifard A. Not All H3K4 Methylations Are Created Equal: Mll2/COMPASS Dependency in Primordial Germ Cell Specification. Mol Cell. 2017;65:460–475. e6. doi: 10.1016/j.molcel.2017.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huisinga KL, Pugh BF. A genome-wide housekeeping role for TFIID and a highly regulated stress-related role for SAGA in Saccharomyces cerevisiae. Mol Cell. 2004;13:573–585. doi: 10.1016/s1097-2765(04)00087-5. [DOI] [PubMed] [Google Scholar]
- Kim J, Guermah M, McGinty RK, Lee JS, Tang Z, Milne TA, Shilatifard A, Muir TW, Roeder RG. RAD6-Mediated Transcription-Coupled H2B Ubiquitylation Directly Stimulates H3K4 Methylation in Human Cells. Cell. 2009;137:459–471. doi: 10.1016/j.cell.2009.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Kim JA, McGinty RK, Nguyen UTT, Muir TW, Allis CD, Roeder RG. The n-SET Domain of Set1 Regulates H2B Ubiquitylation-Dependent H3K4 Methylation. Mol Cell. 2013;49:1121–1133. doi: 10.1016/j.molcel.2013.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim T, Buratowski S. Dimethylation of H3K4 by Set1 recruits the Set3 histone deacetylase complex to 5′ transcribed regions. Cell. 2009;137:259–272. doi: 10.1016/j.cell.2009.02.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim T, Xu Z, Clauder-Münster S, Steinmetz LM, Buratowski S. Set3 HDAC mediates effects of overlapping noncoding transcription on gene induction kinetics. Cell. 2012;150:1158–1169. doi: 10.1016/j.cell.2012.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, Skalnik DG. Wdr82 is a C-terminal domain-binding protein that recruits the Setd1A Histone H3-Lys4 methyltransferase complex to transcription start sites of transcribed human genes. Mol Cell Biol. 2008;28:609–618. doi: 10.1128/MCB.01356-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JS, Shukla A, Schneider J, Swanson SK, Washburn MP, Florens L, Bhaumik SR, Shilatifard A. Histone Crosstalk between H2B Monoubiquitination and H3 Methylation Mediated by COMPASS. Cell. 2007;131:1084–1096. doi: 10.1016/j.cell.2007.09.046. [DOI] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. Genome Project Data Processing Subgroup (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics. 1000;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu CL, Kaplan T, Kim M, Buratowski S, Schreiber SL, Friedman N, Rando OJ. Single-nucleosome mapping of histone modifications in S. cerevisiae. PLoS Biol. 2005;3:e328. doi: 10.1371/journal.pbio.0030328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malagon F, Kireeva ML, Shafer BK, Lubkowska L, Kashlev M, Strathern JN. Mutations in the Saccharomyces cerevisiae RPB1 gene conferring hypersensitivity to 6-azauracil. Genetics. 2006;172:2201–2209. doi: 10.1534/genetics.105.052415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer A, Lidschreiber M, Siebert M, Leike K, Söding J, Cramer P. Uniform transitions of the general RNA polymerase II transcription complex. Nat Struct Mol Biol. 2010;17:1272–1278. doi: 10.1038/nsmb.1903. [DOI] [PubMed] [Google Scholar]
- Mersman DP, Du HN, Fingerman IM, South PF, Briggs SD. Charge-based interaction conserved within histone H3 lysine 4 (H3K4) methyltransferase complexes is needed for protein stability, histone methylation, and gene expression. J Biol Chem. 2012;287:2652–2665. doi: 10.1074/jbc.M111.280867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morillon A, Karabetsou N, Nair A, Mellor J. Dynamic Lysine Methylation on Histone H3 Defines the Regulatory Phase of Gene Transcription. Mol Cell. 2005;18:723–734. doi: 10.1016/j.molcel.2005.05.009. [DOI] [PubMed] [Google Scholar]
- Ng HH, Robert F, Young RA, Struhl K. Targeted recruitment of Set1 histone methylase by elongating Pol II provides a localized mark and memory of recent transcriptional activity. Mol Cell. 2003;11:709–719. doi: 10.1016/S1097-2765(03)00092-3. [DOI] [PubMed] [Google Scholar]
- Orford K, Kharchenko P, Lai W, Dao MC, Worhunsky DJ, Ferro A, Janzen V, Park PJ, Scadden DT. Differential H3K4 Methylation Identifies Developmentally Poised Hematopoietic Genes. Dev Cell. 2008;14:798–809. doi: 10.1016/j.devcel.2008.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelechano V, Chávez S, Pérez-Ortín JE. A complete set of nascent transcription rates for yeast genes. PLoS ONE. 2010;5:e15442. doi: 10.1371/journal.pone.0015442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelechano V, Wei W, Jakob P, Steinmetz LM. Genome-wide identification of transcript start and end sites by transcript isoform sequencing. Nat Protoc. 2014;9:1740–1759. doi: 10.1038/nprot.2014.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinskaya M, Gourvennec S, Morillon A. H3 lysine 4 di- and tri-methylation deposited by cryptic transcription attenuates promoter activation. EMBO J. 2009;28:1697–1707. doi: 10.1038/emboj.2009.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinskaya M, Morillon A. Histone H3 lysine 4 di-methylation: A novel mark for transcriptional fidelity? Epigenetics. 2009;4:302–306. doi: 10.4161/epi.4.5.9369. [DOI] [PubMed] [Google Scholar]
- Pokholok DK, Harbison CT, Levine S, Cole M, Hannett NM, Lee TI, Bell GW, Walker K, Rolfe PA, Herbolsheimer E, Zeitlinger J, Lewitter F, Gifford DK, Young RA. Genome-wide map of nucleosome acetylation and methylation in yeast. Cell. 2005;122:517–527. doi: 10.1016/j.cell.2005.06.026. [DOI] [PubMed] [Google Scholar]
- Radman-Livaja M, Verzijlbergen KF, Weiner A, van Welsem T, Friedman N, Rando OJ, van Leeuwen F. Patterns and Mechanisms of Ancestral Histone Protein Inheritance in Budding Yeast. PLoS Biol. 2011;9:e1001075. doi: 10.1371/journal.pbio.1001075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlichter A, Cairns BR. Histone trimethylation by Set1 is coordinated by the RRM, autoinhibitory, and catalytic domains. EMBO J. 2005;24:1222–1231. doi: 10.1038/sj.emboj.7600607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider J, Wood A, Lee JS, Schuster R, Dueker J, Maguire C, Swanson SK, Florens L, Washburn MP, Shilatifard A. Molecular Regulation of Histone H3 Trimethylation by COMPASS and the Regulation of Gene Expression. Mol Cell. 2005;19:849–856. doi: 10.1016/j.molcel.2005.07.024. [DOI] [PubMed] [Google Scholar]
- Schulze JM, Hentrich T, Nakanishi S, Gupta A, Emberly E, Shilatifard A, Kobor MS. Splitting the task: Ubp8 and Ubp10 deubiquitinate different cellular pools of H2BK123. Genes Dev. 2011;25:2242–2247. doi: 10.1101/gad.177220.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahbazian MD, Zhang K, Grunstein M. Histone H2B ubiquitylation controls processive methylation but not monomethylation by Dot1 and Set1. Mol Cell. 2005;19:271–277. doi: 10.1016/j.molcel.2005.06.010. [DOI] [PubMed] [Google Scholar]
- Shilatifard A. The COMPASS family of histone H3K4 methylases: mechanisms of regulation in development and disease pathogenesis. Annu Rev Biochem. 2012;81:65–95. doi: 10.1146/annurev-biochem-051710-134100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soares LM, Buratowski S. Histone Crosstalk: H2Bub and H3K4 Methylation. Mol Cell. 2013;49:1019–1020. doi: 10.1016/j.molcel.2013.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soares LM, Radman-Livaja M, Lin SG, Rando OJ, Buratowski S. Feedback control of Set1 protein levels is important for proper H3K4 methylation patterns. Cell Rep. 2014;6:961–972. doi: 10.1016/j.celrep.2014.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- South PF, Fingerman IM, Mersman DP, Du HN, Briggs SD. A conserved interaction between the SDI domain of Bre2 and the Dpy-30 domain of Sdc1 is required for histone methylation and gene expression. J Biol Chem. 2010;285:595–607. doi: 10.1074/jbc.M109.042697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh H, Hazelbaker DZ, Soares LM, Buratowski S. The C-terminal domain of Rpb1 functions on other RNA polymerase II subunits. Mol Cell. 2013;51:850–858. doi: 10.1016/j.molcel.2013.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun ZW, Allis CD. Ubiquitination of histone H2B regulates H3 methylation and gene silencing in yeast. Nature. 2002;418:104–108. doi: 10.1038/nature00883. [DOI] [PubMed] [Google Scholar]
- Takahashi YH, Lee JS, Swanson SK, Saraf A, Florens L, Washburn MP, Trievel RC, Shilatifard A. Regulation of H3K4 trimethylation via Cps40 (Spp1) of COMPASS is monoubiquitination independent: implication for a Phe/Tyr switch by the catalytic domain of Set1. Mol Cell Biol. 2009;29:3478–3486. doi: 10.1128/MCB.00013-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornton JL, Westfield GH, Takahashi YH, Cook M, Gao X, Woodfin AR, Lee JS, Morgan MA, Jackson J, Smith ER, Couture JF, Skiniotis G, Shilatifard A. Context dependency of Set1/COMPASS-mediated histone H3 Lys4 trimethylation. Genes Dev. 2014;28:115–120. doi: 10.1101/gad.232215.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatesh S, Workman JL. Histone exchange, chromatin structure and the regulation of transcription. Nat Rev Mol Cell Biol. 2015;16:178–189. doi: 10.1038/nrm3941. [DOI] [PubMed] [Google Scholar]
- Vitaliano-Prunier A, Menant A, Hobeika M, Géli V, Gwizdek C, Dargemont C. Ubiquitylation of the COMPASS component Swd2 links H2B ubiquitylation to H3K4 trimethylation. Nat Cell Biol. 2008;10:1365–1371. doi: 10.1038/ncb1796. [DOI] [PubMed] [Google Scholar]
- Warren SL, Landolfi AS, Curtis C, Morrow JS. Cytostellin: a novel, highly conserved protein that undergoes continuous redistribution during the cell cycle. J Cell Sci. 1992;103:381–388. doi: 10.1242/jcs.103.2.381. [DOI] [PubMed] [Google Scholar]
- Wong KH, Jin Y, Moqtaderi Z. Multiplex Illumina sequencing using DNA barcoding. Curr Protoc Mol Biol. 2013;Chapter 7(Unit 7.11.–7.11.11) doi: 10.1002/0471142727.mb0711s101. [DOI] [PubMed] [Google Scholar]
- Wood V, et al. The genome sequence of Schizosaccharomyces pombe. Nature. 2002;415:871–880. doi: 10.1038/nature724. [DOI] [PubMed] [Google Scholar]
- Xu Z, Wei W, Gagneur J, Perocchi F, Clauder-Münster S, Camblong J, Guffanti E, Stutz F, Huber W, Steinmetz LM. Bidirectional promoters generate pervasive transcription in yeast. Nature. 2009;457:1033–1037. doi: 10.1038/nature07728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, Liu XS. Model-based analysis of ChIP-Seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.