

ABSTRACT
We recently reported that depletion of FANCM in ALT cells induces replication stress mainly at their telomeres. Additionally, we found that co-depletion of FANCM and BLM, or FANCM and BRCA1 induces synthetic lethality in the ALT cells. Our new findings could have important implications for cancer prevention and treatment.
KEYWORDS: ALT telomere, FANCM, BLM, BRCA1, replication stress
Recently, Vogelstein and colleagues revealed seemingly surprising source for the majority of human cancers: accidental errors from DNA replication during normal stem cell division.1,2 On the other hand, this makes perfect sense because it has been well-recognized that the DNA replication machinery (i.e., the replisome) as well as the DNA repair systems are not error proof. An old Chinese proverb states that if you often walk by the riverside, you cannot avoid getting your shoes wet. The more a stem cell proliferates, the more likely the replisome makes mistakes. Therefore, Vogelstein and colleagues proposed that, to make a greater impact on reducing the mortality of cancers, prevention and early detection become as important, if not more important, as anti-tumor therapy. At least in theory, the best strategy to prevent the cancer from occurring is to increase the fidelity of DNA replication and DNA repair.
The temporary/transient slowing or stalling of the replication fork is defined as replication stress (RS).3,4 The re-start and/or repair of the stalled or collapsed replication fork could dramatically increase the chances of errors. Certain loci/regions within the human genome are more prone to the RS, for example, centromeres, common fragile sites, subtelomeres, and telomeres.4 Therefore, a better understanding of how these endogenous barriers respond to the RS will be crucial to find a way to lower the errors during DNA replication and DNA repair. Most investigators studying RS response have been using either chemicals or ultra violet (UV) light to induce RS. One of the major issues using these RS inducers (RSI) is that one cannot clearly define where in the genome they induce the RS. Different loci/regions likely respond to the RS differently. Furthermore, in addition to RS, the exogenous RSIs often induce other types of DNA damages. Therefore, the cellular responses to the exogenous RSIs are often mixed. A recent publication from our lab in the Proceedings of the National Academy of Sciences (PNAS) addressed some of these issues.5 We showed that depletion of FANCM, encoded by one of the Fanconi Anemia genes, in cancer cells utilizing the alternative lengthening of telomeres (ALT) as their telomere maintenance mechanism (TMM) induces RS primarily at their telomeres. We thus referred to this endogenous RS system as MR-SAT (FANCM deficiency induced replication stress at ALT telomeres). Similar to the RS induced by the exogenous RSIs, the RS checkpoint, as indicated by the phosphorylation of Chk1 on Serine-345 and RPA32 on Serine-4, Serine-8, and Serine-33, is activated in the MR-SAT system. In addition, we found that homologous recombination (HR) is dramatically upregulated as indicated by the activation of DNA end resection and the recruitment of many important HR proteins, including BLM, BRCA1 and Rad51. Most importantly, we demonstrated that the recruitment of these HR proteins is dependent on both ATR and Chk1, two of the most important kinases regulating the RS response. Our findings indicate that ALT cells not only rely on HR to maintain the length of their telomeres, they also utilize HR to prevent and repair the stalled replication fork at their telomeres. We therefore propose that the MR-SAT system can be used as a robust endogenous RS model at a well-defined genetic locus/region, i.e., the ALT telomere.
At present, we do not know the exact causes of the replication stress in FANCM deficient ALT cells. But we are actively testing the following hypotheses (Fig. 1): (1) FANCM reconfigures and/or overcomes the intermediates of HR during the replication of ALT telomeres; (2) FANCM disrupts the R-loop formed between TERRA and telomeric DNA during the replication of ALT telomeres; and (3) FANCM unfolds the G-quadruplex at the telomeres.
Figure 1.
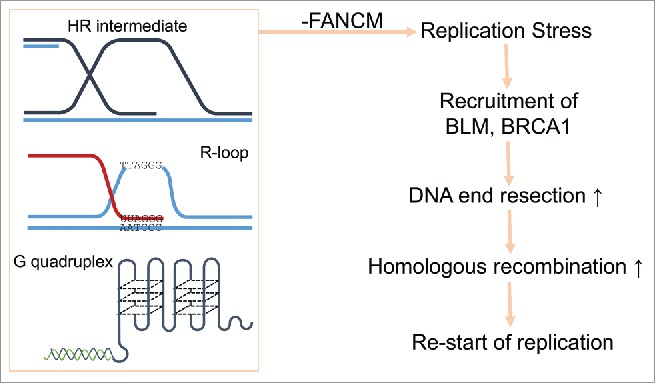
A model of how FANCM, BLM, and BRCA1 cooperatively resolve the replication stress at ALT telomeres.
Synthetic lethality (SL) was put forth sixteen years ago as a revolutionary concept to target cancer cells with lower toxicity compared to the conventional chemotherapy drugs.6 The approval of Lynparza™ (Olaparib) in December 2014 to treat women with advanced ovarian cancers and also carrying the BRCA gene mutations validated SL as a feasible and superior anti-tumor strategy. Another important discovery made in our recent PNAS paper is that we found that deficiency of FANCM and BLM, or FANCM and BRCA1, in ALT cells are synthetically lethal. We think that these SLs are likely due to the failure to repair the DNA damages caused by RS at the ALT telomeres because we observed pronounced increase of micronuclei formation, an indicator of unrepaired DNA damage, in FANCM and BLM, or FANCM and BRCA1 co-depleted ALT cells. These new synthetic lethal interactions could be explored for novel targeted therapies for ALT cancers, which currently can only be treated with conventional chemotherapy drugs. Both BLM and BRCA1 contain domains that have demonstrated enzymatic activities. The N-terminus of BLM has a SF2 helicase domain and can unwind a variety of secondary DNA structures, at least in vitro. The N-terminus of BRCA1 has a RING domain and can function as a E3 ligase to catalyze the conjugation of ubiquitin. In fact, a BLM inhibitor, was discovered recently by Hickson and Colleagues and it showed mild anti-proliferative activity.7 In light of our recent findings, it will be interesting to test whether ML216 preferentially targets ALT cancers.
In summary, our recent findings established the first robust endogenous RS system. A better understanding of the RS response may help either to avoid the errors during the replication or improve the repair efficiency when RS occurs. In addition, the SL interactions uncovered in our study could potentially be explored for developing new targeted therapy for ALT cancers.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgement
we want to thank Dr. Charles Pavia for proofreading this manuscript.
References
- 1.Tomasetti C, Li L, Vogelstein B. Stem cell divisions, somatic mutations, cancer etiology, and cancer prevention. Science. 2017;355(6331):1330-34. doi: 10.1126/science.aaf9011. PMID:28336671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tomasetti C, Vogelstein B. Cancer etiology. Variation in cancer risk among tissues can be explained by the number of stem cell divisions. Science. 2015;347(6217):78-81. doi: 10.1126/science.1260825. PMID:25554788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Berti M, Vindigni A. Replication stress: Getting back on track. Nat Struct Mol Biol. 2016;23(2):103-109. doi: 10.1038/nsmb.3163. PMID:26840898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zeman MK, Cimprich KA. Causes and consequences of replication stress. Nat Cell Biol. 2014;16(1):2-9. doi: 10.1038/ncb2897. PMID:24366029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pan X, Drosopoulos WC, Sethi L, Madireddy A, Schildkraut CL, Zhang D. FANCM, BRCA1, and BLM cooperatively resolve the replication stress at the ALT telomeres. Proc Natl Acad Sci U S A. 2017;114(29):E5940-949. doi: 10.1073/pnas.1708065114. PMID:28673972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hartman JLT, Garvik B, Hartwell L. Principles for the buffering of genetic variation. Science. 2001;291(5506):1001-4. doi: 10.1126/science.291.5506.1001. PMID:11232561 [DOI] [PubMed] [Google Scholar]
- 7.Nguyen GH, Dexheimer TS, Rosenthal AS, Chu WK, Singh DK, Mosedale G, Bachrati CZ, Schultz L, Sakurai M, Savitsky P, et al., A small molecule inhibitor of the BLM helicase modulates chromosome stability in human cells. Chem Biol. 2013;20(1):55-62. doi: 10.1016/j.chembiol.2012.10.016. PMID:23352139 [DOI] [PMC free article] [PubMed] [Google Scholar]