

ABSTRACT
Kinase inhibitors targeting the mitogen/extracellular signal-regulated kinase kinase (MEK)- extracellular signal related kinase (ERK) signaling pathway have limited durability in inhibiting growth of triple-negative breast cancer. We defined genome wide enhancer remodeling following MEK inhibition capable of driving adaptive gene transcription. Targeting positive elongation factor (P-TEFb) transcriptional regulatory complex members can block enhancer remodeling making the response to MEK-ERK inhibition durable.
KEYWORDS: Adaptive resistance, BRD4, enhancer remodeling, MEK inhibition, P-TEFb complex
Triple-Negative Breast Cancer (TNBC) is characterized by the loss of expression of the estrogen receptor, progesterone receptor, and human epidermal growth factor receptor 2 (HER2) and there is currently no (Food and Drug Administration) FDA-approved targeted therapy for these patients. TNBC is largely devoid of oncogenic driver mutations, which could otherwise point to therapeutic vulnerabilities. However, frequent elevated expression of epidermal growth factor receptor, KRAS, or BRAF increases the activity of the mitogen/extracellular signal-regulated kinase kinase (MEK)-extracellular signal-related kinase (ERK) pathway making TNBC sensitive to MEK inhibition.1 While TNBC initially responds with strong growth inhibition to MEK inhibitors such as trametinib, the tumor cells rapidly adapt to the drug by transcriptional upregulation and activation of receptor tyrosine kinases (RTKs) which reactivate the growth of the tumor cells.2 This inevitable bypass of MEK-ERK inhibition is a fundamental problem, which prevents kinase inhibitors from having durable growth suppression in the clinic across multiple tumor types. Tumor heterogeneity also makes it difficult to predict which kinases will drive resistance to treatment, calling for a more fundamental understanding of how tumors adapt to pathway inhibition leading to resistance.
In our recent work published in Cancer Discovery,3 we defined the transcriptional response of TNBC patient tumors to trametinib, a FDA-approved MEK inhibitor (MEKi), using paired tumor samples before and after 7 d of treatment with the drug. We observed a large transcriptional response to MEKi in patient tumors involving up to 22% of the transcriptome, effectively changing the phenotypic state of the tumor cell. Analysis of the tyrosine kinome revealed heterogeneity across tumors with multiple RTKs upregulated in treated samples relative to controls. Further quantitative proteomic analysis revealed that these kinases were functionally upregulated in tumor samples. Statistical analyses using the Differential Expression-Seq2 algorithm allowed us to profile kinases transcriptionally induced in both basal-like cell lines and patient tumors including fyn-related Src family tyrosine kinase, erb-b2 receptor tyrosine kinase 2, discoidin domain receptor tyrosine kinase 1 (DDR1), CDC42 binding protein kinase gamma, cyclin-dependent kinase like 5, and cyclin-dependent kinase 19. We also compared subtype-specific responses of RTKs to MEKi between basal-like and claudin-low cell lines with fibroblast growth factor receptor 2 and colony stimulating factor 1 receptor uniquely induced in basal-like cells and platelet-derived growth factor receptorβ and erb-b2 receptor tyrosine kinase 4 uniquely induced in claudin-low cell lines. These subtype differences were confirmed in the SUM-229PE cell line which contains two intrinsic subpopulations, which profiled as either basal-like or claudin-low. Importantly, exome sequencing identified no unique nucleotide variants or copy number alterations between these two populations. Genome-wide DNA methylation analysis further showed no significant difference between the two populations that could account for differential transcriptional responses to trametinib, suggesting epigenetic chromatin modifications beyond that of DNA methylation.
Analysis of enhancer and promoter marks using chromatin immunoprecipitation coupled with deep sequencing (ChIPseq) revealed a significant rearrangement of the genomic landscape following trametinib treatment in TNBC cell lines including the formation of new enhancers proximal to adaptive response genes. Using antibodies to the bromodomain containing 4 (BRD4) protein for ChIPseq to identify enhancers, we observed an increase in the number of BRD4-enriched enhancers starting as early as 1 hour post MEK inhibition that continued to rise across a 72-hour time frame. Many of the enhancers formed in response to trametinib were mapped to genomic loci proximal to genes transcriptionally responsive to trametinib including DDR1, kinase insert domain receptor, and phosphoinositide-3-kinase regulatory subunit 1 (Fig. 1). Clustered, regularly interspaced short palindromic repeats -Cas9 deletion of the DDR1 enhancer confirmed the newly seeded enhancer functioned in regulating the expression of DDR1 following MEKi. Adding the BET bromodomain inhibitor JQ1 in combination with MEKi pharmacologically inhibited the formation of BRD4 enhancers. Illumina deep mRNA sequencing analysis revealed that BRD4 bromodomain inhibition blocked 25% of trametinib-induced gene transcripts, specifically genes associated with enhancer formation in response to drug. The combination treatment translated to sustained growth inhibition in TNBC cell lines and multiple pre-clinical mouse models of TNBC.
Figure 1.
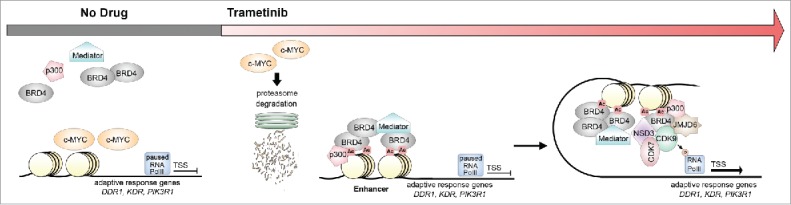
MEK inhibition results in c-MYC degradation and enhancer formation proximal to adaptive response genes. Following treatment with the mitogen/extracellular signal-regulated kinase kinase (MEK) inhibitor trametinib, proteasome degradation and turnover of v-myc avain myelocytomatosis viral oncogene homolog (c-MYC) protein allows for the recruitment of transcriptional regulators proximal to adaptive genes including discoidin domain receptor tyrosine kinase 1 (DDR1), kinase insert domain receptor (KDR), and phosphoinositide-3-kinase regulatory subunit 1 (PIK3R1). Enhancer formation includes the recruitment of the acetyltransferase CREB binding protein/E1A binding protein P300 (p300), the mediator complex, and the positive elongation factor (P-TEFb) complex including bromodomain containing 4 (BRD4), cyclin-dependent kinase 9 (CDK9), cyclin-dependent kinase 7 (CDK7), nuclear receptor binding SET domain protein 3 (NSD3), and jumonjii domain containing 6 (JMJD6). Upon enhancer formation, the P-TEFb complex phosphorylates paused RNA polymerase II at transcriptional start sites (TSSs) leading to productive transcriptional elongation and expression of adaptive response genes.
Inhibiting MEK-ERK signaling results in rapid degradation of v-myc avain myelocytomatosis viral oncogene homolog (c-MYC) protein due to loss of stabilizing serine 62 phosphorylation. Previous studies have characterized c-MYC as a master transcriptional regulator and shown that c-MYC turnover is critical for the recruitment of transcriptional complexes to genomic loci including BRD4 and associated positive elongation factor (P-TEFb) regulatory complexes.4 c-MYC knockdown experiments using an inducible shRNA showed BRD4 recruitment in the 50 highest ranking trametinib-induced enhancer regions. Conversely, the loss of c-MYC turnover during MEK inhibition suppressed enhancer formation across the genome and induction of the adaptive bypass response.
Interest in using BET bromodomain inhibitors to treat cancer has grown in recent years with 10 inhibitors currently in phase 1 and 2 clinical trials. Previous studies have suggested combining BET inhibitors with targeted therapies in various tumor types including luminal and HER2-positive breast cancer to increase the durability of targeted therapies.5-7 Our study provides an epigenetic mechanism by which this combination is effective. We also investigated novel pharmacologic targets to block enhancer seeding in response to MEKi. Targeting additional factors associated with the P-TEFb complex with siRNA including cyclin-dependent kinase 9, cyclin-dependent kinase 7, jumonjii domain containing protein 6, and nuclear receptor binding SET domain protein 3, blocked expression of adaptive RTKs in response to MEKi. Growth assays using small molecule inhibitors against jumonjii methyltransferases and the CREB binding protein (CREBBP)/E1A binding protein P300 (EP300) acetyltransferase also synergized with trametinib. Cumulatively our data show that P-TEFb complex inhibition, and enhancer perturbation, provide a promising avenue for attenuating the adaptive response to targeted inhibition of the MEK-ERK pathway.
Disclosure of potential conflicts of interest
GLJ is a co-founder of KinoDyn.
Funding
This work was supported by the Susan G. Komen Foundation under grant IIR12–225201; the NIH under grants CA058223 and GM116534; the UNC Junior Faculty Development Award; the University Research Council Small Grant Award; and the University Cancer Research Fund.
References
- 1.Cancer Genome Atlas Network Comprehensive molecular portraits of human breast tumours. Nat Nat Res 2012; 490(7418):61-70; PMID:23000897; https://doi.org/ 10.1038/nature11412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Duncan JS, Whittle MC, Nakamura K, Abell AN, Midland AA, Zawistowski JS, Johnson NL, Granger DA, Jordan NV, Darr DB et al.. Dynamic reprogramming of the kinome in response to targeted MEK inhibition in triple-negative breast cancer. Cell 2012; 149(2):307-21; PMID:22500798; https://doi.org/ 10.1016/j.cell.2012.02.053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zawistowski JS, Bevill SM, Goulet DR, Stuhlmiller TJ, Beltran AS, Olivares-Quintero JF, Singh D, Sciaky N, Parker JS, Rashid NU et al.. Enhancer remodeling during adaptive bypass to MEK inhibition is attenuated by pharmacological targeting of the P-TEFb complex. Cancer Discov 2017; 7(3):302-21; PMID:28108460; https://doi.org/ 10.1158/2159-8290.CD-16-0653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jaenicke LA, Eyss von B, Carstensen A, Wolf E, Xu W, Greifenberg AK, Geyer M, Eilers M, Popov N. Ubiquitin-dependent turnover of MYC Antagonizes MYC/PAF1C complex accumulation to drive transcriptional elongation. Mol Cell 2016; 61(1):54-67; PMID:26687678; https://doi.org/ 10.1016/j.molcel.2015.11.007 [DOI] [PubMed] [Google Scholar]
- 5.Stuhlmiller TJ, Miller SM, Zawistowski JS, Nakamura K, Beltran AS, Duncan JS, Angus SP, Collins KA, Granger DA, Reuther RA et al.. Inhibition of lapatinib-induced kinome reprogramming in ERBB2-positive breast cancer by targeting BET family bromodomains. Cell Rep 2015; 11(3):390-404; PMID:25865888; https://doi.org/ 10.1016/j.celrep.2015.03.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stratikopoulos EE, Dendy M, Szabolcs M, Khaykin AJ, Lefebvre C, Zhou M-M, Parsons R. Kinase and BET inhibitors together clamp inhibition of PI3K signaling and overcome resistance to therapy. Cancer Cell 2015; 27(6):837-51; PMID:26058079; https://doi.org/ 10.1016/j.ccell.2015.05.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Matkar S, Sharma P, Gao S, Gurung B, Katona BW, Liao J, Muhammad AB, Kong XC, Wang L, Jin G et al.. An epigenetic pathway regulates sensitivity of breast cancer cells to HER2 inhibition via FOXO/c-Myc Axis. Cancer Cell 2015; 28(4):472-85; PMID:26461093; https://doi.org/ 10.1016/j.ccell.2015.09.005 [DOI] [PMC free article] [PubMed] [Google Scholar]