

Abstract
Regulation of skeletal muscle development and organization is a complex process that is not fully understood. Here, we focused on amphiphysin 2 (BIN1, also known as bridging integrator-1) and dynamin 2 (DNM2), two ubiquitous proteins implicated in membrane remodeling and mutated in centronuclear myopathies (CNMs). We generated Bin1–/– Dnm2+/– mice to decipher the physiological interplay between BIN1 and DNM2. While Bin1–/– mice die perinatally from a skeletal muscle defect, Bin1–/– Dnm2+/– mice survived at least 18 months, and had normal muscle force and intracellular organization of muscle fibers, supporting BIN1 as a negative regulator of DNM2. We next characterized muscle-specific isoforms of BIN1 and DNM2. While BIN1 colocalized with and partially inhibited DNM2 activity during muscle maturation, BIN1 had no effect on the isoform of DNM2 found in adult muscle. Together, these results indicate that BIN1 and DNM2 regulate muscle development and organization, function through a common pathway, and define BIN1 as a negative regulator of DNM2 in vitro and in vivo during muscle maturation. Our data suggest that DNM2 modulation has potential as a therapeutic approach for patients with CNM and BIN1 defects. As BIN1 is implicated in cancers, arrhythmia, and late-onset Alzheimer disease, these findings may trigger research directions and therapeutic development for these common diseases.
Keywords: Muscle Biology
Keywords: Neuromuscular disease, Skeletal muscle
Introduction
Skeletal muscle requires adequate membrane remodeling to achieve and maintain its normal structure and function (1). It is thus not surprising that several human myopathies are due to mutations in proteins implicated in membrane trafficking and remodeling (2). Amphiphysins and dynamins are recognized hubs in the membrane trafficking pathway and are known to physically interact in cells (3, 4). Amphiphysin 2 (BIN1) and dynamin 2 (DNM2) are mutated in rare congenital myopathies called centronuclear myopathies (CNMs) (5–7), characterized by severe muscle weakness and hypotonia associated with mislocalization of nuclei and other organelles and the abnormal orientation and shape of T-tubules in skeletal muscle fibers (8). Despite numerous studies in vitro or in cell culture, the regulation between amphiphysins and dynamins and their physiological importance are barely understood.
BIN1 contains a BAR domain that can sense and induce membrane curvature and a C-terminal SH3 domain that binds to different effectors containing a proline-rich domain (PRD), such as dynamins (9, 10). Based on its membrane tubulation properties and on the muscle phenotype of the amphiphysin Drosophila mutant, BIN1 was proposed to participate in the biogenesis of T-tubules, plasma membrane invaginations forming along with the sarcoplasmic reticulum calcium stores the structural basis of the excitation-contraction coupling machinery in cardiac and skeletal muscles (3, 11). BIN1 recessive mutations in CNMs either impair its membrane tubulation properties, its binding to dynamin 2, or the inclusion of a muscle-specific exon encoding a phosphoinositide-binding (PI-binding) domain (6, 7, 12). DNM2 is a large GTPase involved in membrane fission and endocytosis (13, 14). Biochemical studies indicated that several DNM2 mutations causing CNMs increase dynamin oligomer stability and GTPase activity (15–17). Moreover, overexpression of WT or a DNM2-CNM mutation leads to CNM-like features in mice and perturbation of muscle and T-tubules in Drosophila (17–19). We hypothesize that CNM mutations in BIN1 are loss-of-function while CNM mutations in DNM2 create a gain of function. Here, to study the genetic and functional links between BIN1 and DNM2 and the physiological impact of the BIN1/DNM2 balance in skeletal muscle, we created a Bin1–/– mouse and modulated DNM2 expression in this model, and analyzed the impact of BIN1 on DNM2 activity.
Results
BIN1 is required in skeletal muscle for perinatal survival.
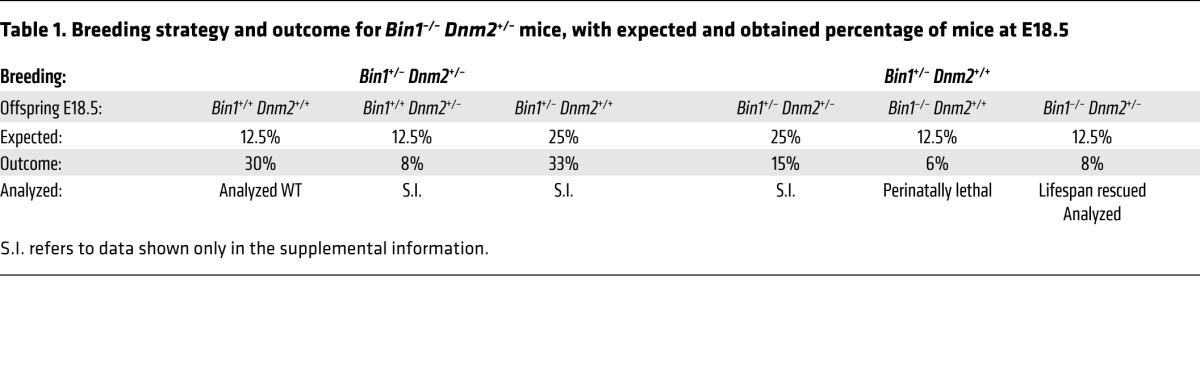
To study the interplay between BIN1 and DNM2 during muscle maturation, Dnm2+/– and Bin1–/– mice were created. The Dnm2+/– mice are viable without obvious phenotypes (20). To develop a mouse model for autosomal recessive BIN1-CNM, we floxed exon 20 that is found in all isoforms of BIN1, is frequently mutated in patients, and encodes for the SH3 domain known to bind to DNM2 (7) (Figure 1A). Following either constitutive or skeletal muscle deletion with CMV or human skeletal actin (HSA) promoters driving the Cre recombinase, Bin1–/– was neonatally lethal in mice, with no pups surviving the first day of postnatal life (Table 1 and Supplemental Table 1; supplemental material available online with this article; https://doi.org/10.1172/JCI90542DS1). A similar perinatal lethality was previously reported for another constitutive Bin1–/– mouse deleted for exon 3 and the phenotype was first attributed to severe ventricular cardiomyopathy (21); however, mice with exon 3 cardiac-specific deletion survived to adulthood (22). At least in our Bin1–/– model, the lethality is due to a primary defect of skeletal muscle, as the HSA-driven Cre recombinase is only expressed in skeletal muscle, and not cardiac muscle (23), and this defines an essential role of BIN1 in perinatal muscle maturation. Similarly, most BIN1-CNM patients display neonatal muscle weakness and several died within the first year (7).
Figure 1. Dnm2 downregulation rescues the neonatal lethality of Bin1–/– mice.

(A) Targeted disruption of mouse Bin1 gene. Exon 20 (blue) and surrounding intronic region (orange). (B) Overview of lifespan and time points used to analyze Bin1–/– Dnm2+/– mice. (C) Mice whole-body weight, and representative photo of mice at 12 months of age. Note that all genotyped Bin1–/– Dnm2+/– mice survived beyond 12 months of age. (D) Immunoblot analysis of DNM2 and BIN1 protein expression from muscle lysates. (E) Relative level of DNM2 protein expression was determined by densitometry of DNM2 signal standardized to GAPDH. n = 4 mice/genotype. (F) Hanging test where 10-week-old mice were required to hang from a cage lid for up to 60 seconds. (G) Four-paw grip test. (H) Rotarod test performed under acceleration mode (4–40 rpm in 5 minutes). n = 3 trials/mouse/day, 6-month-old mice. (I) Specific muscle force (sPo) of the tibialis anterior (TA) muscle (mN force/mg TA muscle). (J) Fatigue of the TA muscle, measured as the time taken to reach 50% of the maximum muscle force (seconds). All graphs depict the mean ± SEM. Statistical analysis was performed using an unpaired 2-tailed Student’s t test for all graphs except H, where a 2-way ANOVA followed by Dunn’s multiple comparison test was used. *P < 0.05, **P < 0.01. n = minimum 5 mice per group for C, F–J.
Table 1. Breeding strategy and outcome for Bin1–/– Dnm2+/– mice, with expected and obtained percentage of mice at E18.5.
Reducing DNM2 expression prevents the neonatal lethality caused by BIN1 defect.
Recessive BIN1-CNM is due to BIN1 loss of function and dominant DNM2-CNM is probably due to DNM2 gain of function (5, 7, 15–17). We hypothesize that these 2 proteins are working antagonistically in the same pathway for muscle maturation and that reducing DNM2 expression might rescue the neonatal lethality observed in Bin1–/– mice. To test this hypothesis, we produced Bin1–/– mice with reduced DNM2 expression (Bin1–/– Dnm2+/–) (Table 1 and Figure 1B). Unlike Bin1–/– mice, Bin1–/– Dnm2+/– mice are viable to at least 18 months (oldest age at sacrifice), with only a slight but significant reduction in body weight from 6 months of age compared with Bin1+/+ Dnm2+/+ (hereafter named WT) littermates (Figure 1C). Furthermore, Bin1–/– Dnm2+/– mice appeared healthy (Figure 1C and Supplemental Videos 1–3). As phenotyping performed on other genotypes (namely, Bin1+/– mice) produced in this cross indicated no obvious phenotypes (Table 1 and Supplemental Figure 1), this study focused on WT and Bin1–/– Dnm2+/– mice.
No difference was observed between WT and Bin1–/– Dnm2+/– mice in blood metabolites, liver function, organ weight, or histology in brain and liver at 12 months (Supplemental Figure 2). No difference in muscle mass or histology of the heart was noted, as ventricle thickness was similar and no increased fibrosis was observed at 12 months (Supplemental Figure 3). Western blot confirmed that BIN1 protein expression was barely detectable in Bin1–/– Dnm2+/– mice and that DNM2 expression was reduced to approximately 50% of WT levels (Figure 1, D and E), therefore validating that reducing DNM2 expression can rescue the neonatal lethality caused by BIN1 defect.
Bin1-KO mice with Dnm2 haploinsufficiency have normal muscle strength.
As Bin1–/– Dnm2+/– and WT mice appeared clinically similar, we focused on muscle-related functions. No difference was observed in hanging test, grip test, and rotarod performance at all different ages analyzed (10 weeks, 6 months, and 12 months) and for both sexes (Figure 1, F–H, Supplemental Figure 4). No difference in weights of gastrocnemius, soleus, extensor digitorum longus (EDL), or tibialis anterior (TA) skeletal muscles was observed in Bin1–/– Dnm2+/– mice compared with WT (Supplemental Figure 4). Specific muscle force and fatigability were measured in situ in the TA muscle at 6 and 12 months of age, and no difference between groups was observed (Figure 1, I and J, and Supplemental Figure 4). Overall, these results indicate that Bin1–/– Dnm2+/– have normal whole-body strength and coordination, and individual muscle strength. As no Bin1–/– mice survived past 24 hours, their strength could not be compared.
Dnm2 reduction rescues CNM histology in Bin1-KO mice.
To determine if the skeletal muscle structure was affected in Bin1–/– Dnm2+/– mice, TA muscle cryosections were stained with hematoxylin and eosin (H&E) and for succinate dehydrogenase (SDH, mitochondrial enzyme) at 10 weeks and 12 months of age. At 10 weeks, muscle fibers were slightly but significantly smaller than WT, while by 12 months this difference was not significant (Figure 2, A, B, D, and E). No difference was observed in SDH staining at either time point. The main sustained difference was a 9-fold increase in centralized nuclei at 10 weeks up to 20% of fibers and to a lesser extent at 12 months (Figure 2, A, C, D, and F). BIN1-CNM histology is characterized by hypotrophic fibers with mislocalized internal nuclei in patient biopsies (24), reminiscent of an important role of BIN1 in nuclear positioning (25). The slight CNM-like histological features in Bin1–/– mice with reduced DNM2 level appear compatible with normal muscle strength and coordination.
Figure 2. Skeletal muscle histology is mildly affected in surviving Bin1–/– mice with reduced DNM2 expression.

Transverse TA sections from 10-week-old (10 wk) (A) or 12-month-old (12 mo) (D) mice were stained with H&E or for SDH. Arrows point to mislocalized nuclei. Scale bars: 100 μm. H&E–stained muscle sections were then analyzed for fiber area for 10 wk (B) and 12 mo (E). Fiber diameter is grouped into 10-μm intervals, and represented as the percentage of total fibers. (C and F) The frequency of fibers with central or internal nuclei were scored. Internal nuclei are defined as neither subsarcolemmal nor central. All graphs depict the mean ± SEM. Statistical analysis was performed using an unpaired 2-tailed Student’s t test. **P < 0.01, ***P < 0.001, n = minimum 5 mice per group.
As Bin1–/– mice did not survive past day 1 for comparison of the nuclei position, we next analyzed skeletal muscles from mice at embryonic day 18.5 (E18.5). In WT mice at this age, muscles are not yet fully structured, and several fibers with centralized nuclei can still be observed by H&E staining (Supplemental Figure 5A). In contrast, nearly all fibers from Bin1–/– mice had centralized nuclei, a feature that was ameliorated by reducing DNM2. Both Bin1–/– and Bin1–/– Dnm2+/– mice displayed an accumulation of SDH staining towards the center of the fibers (Supplemental Figure 5A); however, no difference was observed in SDH staining in adult mice (Figure 2), indicating that this disease feature is corrected during postnatal development in Bin1–/– Dnm2+/– muscles. Overall, these results show that reducing DNM2 expression can improve the histological CNM hallmarks observed in Bin1–/– mice.
Bin1-KO mice with Dnm2 haploinsufficiency have normal muscle ultrastructure and T-tubules.
As BAR proteins can remodel membrane in vitro and as Bin1 downregulation with shRNA in adult muscle led to swollen T-tubules (3, 26, 27), the sarcomere and triad structures were investigated. In skeletal muscles from E18.5 WT mice, DNM2 and α-actinin were detected predominantly in the transverse orientation, consistent with Z-line orientation (Supplemental Figure 5B). This was also observed in both Bin1–/– and Bin1–/– Dnm2+/– muscles, indicating that BIN1 is not required for the transverse localization of DNM2. In adult Bin1–/– Dnm2+/– mice, DNM2 localization was unperturbed and colocalized at the Z-line with α-actinin in 10-week- and 12-month-old mice (Figure 3A and Supplemental Figure 6). By electron microscopy, no obvious structural defects were observed, Z-lines appeared well aligned, myofibrils were of a similar size, and no major differences in organelle size or positioning was noted (Figure 3D). In adult Bin1–/– Dnm2+/– mice, localization of DHPR (T-tubule), RYR1 (junctional sarcoplasmic reticulum), and SERCA (longitudinal sarcoplasmic reticulum) was normal on both longitudinal and transversal sections, indicating no disruption of the triads (Figure 3B and Supplemental Figure 6). Caveolin 3 (CAV3) and dysferlin are localized at the T-tubule during muscle development and regeneration, and at the sarcolemma in nonregenerating mature muscle (28). CAV3 and dysferlin immunolabeling was positive at the sarcolemma in Bin1–/– Dnm2+/– and WT mice, supporting normal development and maintenance of the T-tubules (Figure 3C and Supplemental Figure 6). Triads were well formed, with normal membrane shape, and the number of detectable T-tubules per sarcomere was equivalent to WT (Figure 3, D and E). As triad structures support calcium flux within muscle fibers, we determined if intracellular calcium levels are altered in the absence of BIN1. As Bin1–/– mice did not survive past day 1, we analyzed primary myoblasts derived from E18.5 Bin1–/– mice. At this immature stage of muscle development, no defects in resting cytoplasmic calcium were detected in myoblasts from Bin1–/– mice (Supplemental Figure 5C). In addition, caffeine-stimulated calcium release from the sarcoplasmic reticulum (mediated by RYR1) was not altered (Supplemental Figure 5D), indicating no defect in calcium storage or handling in myoblasts lacking BIN1. Overall, these results show that while BIN1 defect leads to neonatal lethality from a primary skeletal muscle alteration, decreased expression of DNM2 rescues the lifespan of the animals and associates with normal T-tubule maturation and maintenance compatible with normal muscle force and animal behavior.
Figure 3. Normal muscle structure and ultrastructure in Bin1–/– Dnm2+/– mice.

(A) Longitudinal muscle sections were costained for DNM2 (left panel) and α-actinin (middle panel) and imaged by confocal microscopy. Mask image shows areas of colocalization (right panel). Scale bar: 10 μm. (B) Longitudinal (left panel) and transverse (right panel) muscle sections were stained with the T-tubule marker DHPR. Scale bars: 10 μm. (C) Transverse muscle sections stained with CAV3 antibody and viewed by confocal microscopy. Scale bar: 10 μm. (D) Longitudinal muscle sections viewed by transmission electron microscopy. Low-magnification images are in the left panel (scale bar: 0.5 μm); high magnification images are shown in the right panel (scale bar: 200 nm). Arrows point to the triad. All samples are from 12-month-old mice. (E) The number of T-tubules per sarcomere was calculated from TEM images in D. Graph depicts the mean ± SEM. Statistical analysis was performed using an unpaired 2-tailed Student’s t test, n = 3 mice/genotype.
BIN1 and DNM2 achieve tissue-specific isoforms during muscle maturation.
BIN1 and DNM2 are ubiquitously expressed. To tackle the molecular basis of their muscle specificity, their RNA isoforms were identified by RT-PCR, cDNA cloning, and sequencing during muscle cell maturation in culture, at late embryogenesis (E18.5) and in adult muscle. It was shown previously that BIN1 has at least 10 different splicing isoforms, with iso8 containing the PI-binding domain (exon 11, iso8, Figure 4A) that is highly expressed in skeletal muscle (3, 7, 12, 29, 30). BIN1-iso8 is highly expressed in myotubes differentiated for 8 days, and mouse E18.5 and adult muscles (80%–100% of isoforms detected) (Table 2), while it was barely present in undifferentiated myoblasts (7), identifying BIN1-iso8 as the main isoform expressed throughout muscle development. Moreover, mining GTEx human expression data found that BIN1-iso8 was skeletal muscle–specific where it represents 99.6 % of isoforms found in adult (Table 3) (31).
Figure 4. BIN1 and DNM2 in skeletal muscle development.

(A) BIN1 and DNM2 protein domains; corresponding exons and isoforms are shown. N-BAR, N-terminal amphipathic helix and Bin-amphiphysin-Rvs domain; PI, phosphoinositide-binding domain; CBD, clathrin-binding domain; MBD, MYC-binding domain; SH3, Src homology domain; PH, pleckstrin homology domain; GED, GTPase effector domain. Region targeted by exon 20 Bin1–/– mice is indicated. Position of peptides encoded by alternative exons 11 and 12B indicated (not to scale). Predominant BIN1 isoforms are depicted on the right. Iso1 (brain), Iso8 (skeletal muscle), Iso9 (ubiquitous), Iso10 (ubiquitous, cardiac muscle) (adapted from ref. 24). (B) Immunofluorescence staining of primary myotubes (differentiated for 8 days [8d]) and murine muscles at embryonic day 18.5 (E18.5) and in adult (12 weeks) from WT mice. E18.5 muscles have longitudinal (predominant in this image) or transversal triads. DNM2 (upper panel, green in merge) and BIN1 (middle panel, red in merge) immunolabeling is shown. Scale bars: 10 μm. (C) Immunofluorescence staining of semi-thin (200 μm) sections from WT mice, DHPR (upper panel, red in merge), and BIN1 (middle panel, green in merge). Scale bar: 10 μm. (D) Intensity scans spanning 1 complete sarcomere from the above images.
Table 2. Expression of Bin1 and Dnm2 isoforms detected at various stages of muscle differentiation.

Table 3. Number of exon bridging reads over total sequence reads identified in GTEx for BIN1 and DNM2 muscle-specific exons represented in Figure 4A.
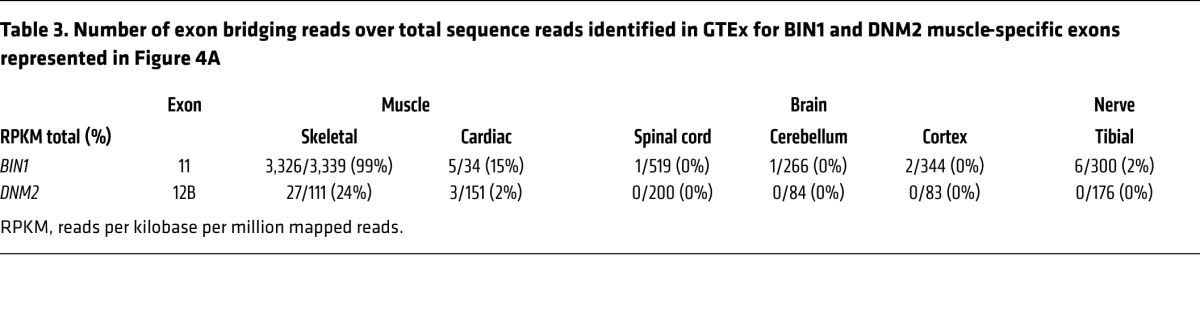
Four transcripts of DNM2 have been previously characterized (32). In addition, we detected the presence of an in-frame exon located between exons 12 and 13, consisting of 30 nucleotides coding for the predicted murine protein sequence CFYTEELVTG, which we named exon 12B (Figure 4A). Inclusion of exon 12B (DNM2+12B) increased from 35% in myoblasts to 44% in differentiated myotubes, and throughout muscle development from 23% (E18.5) to 71% in adult muscle (Table 2). DNM2 exon 12B is specific for skeletal muscle among 43 human tissues (Table 3) (31). Altogether, the data show that while BIN1 and DNM2 are ubiquitously expressed, they achieve specific muscle isoforms during muscle maturation and in adulthood, through alternative splicing.
DNM2 localization correlates with T-tubule maturation.
Inclusion of the PI-binding domain in BIN1 increases its membrane tubulation properties (3), while BIN1 alteration or skipping of this muscle-specific PI-binding domain correlates with T-tubule defects, supporting the notion that BIN1 participates in T-tubule biogenesis (11, 12, 24, 27, 33). During muscle development, forming T-tubules initially have a longitudinal orientation and are then reoriented to their final transverse orientation during the first 3 weeks postnatally in mice (34). To correlate the appearance of BIN1 and DNM2 muscle-specific isoforms with muscle maturation, localization experiments were performed. At E18.5, tubules appear mixed in localization with some fibers exhibiting longitudinal or immature localization (represented in Figure 4B) and others exhibiting transversal or mature orientation (representative image in Supplemental Figure 5). In myotubes and some E18.5 muscle fibers, BIN1 and DNM2 colocalized extensively on longitudinal tubules (Figure 4, B and D). In adult muscle, BIN1 localized on transversal T-tubules with the voltage-dependent calcium channel DHPR (Figure 4C), while DNM2 staining was more intense at the Z-line with α-actinin (Figure 3A) and only partially overlapped with BIN1 at the edge of the I-band (Figure 4, B–D). In the absence of BIN1, DNM2 transversal localization was not perturbed, as a transverse staining pattern was also observed in both Bin1–/– and Bin1–/– Dnm2+/– muscles (Figure 3A and Supplemental Figure 5B), indicating that DNM2 localization at the I-band does not require BIN1. These results indicate that BIN1 and DNM2 may function together in muscle maturation and that increased inclusion of exon 12B in DNM2 correlates with its relocalization away from T-tubules, independent of BIN1 expression.
BIN1 inhibits the GTPase activity of the DNM2 immature muscle isoform.
The expression of the DNM2 skeletal muscle exon 12B is increased throughout skeletal muscle development (Figure 4 and Table 2), and its corresponding peptide is predicted to localize between the pleckstrin homology (PH) lipid-binding domain and the stalk in the dynamin structure (Figure 5A). We first determined if lipid binding was affected by the inclusion of this exon. Full-length purified recombinant DNM2 proteins with or without the exon 12B peptide were produced (Supplemental Figure 7). Cosedimentation assays were performed in the presence of liposomes and confirmed that the ubiquitous isoform of DNM2 (–12B) binds Folch liposomes derived from bovine brain, both in the presence or absence of 5% additional phosphatidylinositol 4,5–bisphosphate (PIP2) (Figure 5B), as previously shown for DNM2 (35). The skeletal muscle–specific isoform of DNM2 (+12B) can also bind liposomes with or without PIP2 (Figure 5B).
Figure 5. BIN1 inhibits DNM2 in an isoform-dependent manner.

(A) Crystal structure model of nucleotide-free human dynamin 1 (adapted from Faeber et al., ref. 52) using PyMOL. The GTPase domain (green), middle domain (light blue), and GTPase effector domain (GED, orange) forming the stalk, and the PH domain (yellow), are depicted, with the sites of DNM2 mutations linked to CNM indicated (red). The peptide encoded by 12B in human (CYYTEQLVTC) spans from position 497 (black 1), followed by 18 unresolved amino acids, and then amino acid 518 (black 2). (B) Cosedimentation assays were performed with recombinant proteins to determine protein binding (in pellet fraction) relative to total protein (pellet+soluble, see Supplemental Figure 7 for raw data), in the presence of liposomes and PIP2. Results are represented as a fold difference versus DNM2 alone. (C) Malachite green assay with DNM2 (±12B) and BIN1 (+exon 11 [Iso8] and –exon 11 [Iso9]) isoforms, with DNM2/BIN1 at a ratio of 1:4. Brain polar lipids with additional 5% PIP2 were used. Results are shown as catalytic rate (mol/mol/min). Results in B and C are an average of 2 (C) or 4 (B) independent experiments. Graph represents the mean + SEM. One-way (B and C) or 2-way (C) ANOVA test was used as described in the Methods section. *P < 0.05, **P < 0.01, ***P < 0.001.
While previous in vitro or in cellulo studies suggested that BIN1 recruits and activates dynamins at membranes for endocytosis, our in vivo rescue data support the hypothesis that BIN1 is rather a negative regulator of DNM2 activity in skeletal muscle. To assess if BIN1 directly inhibits DNM2, we measured GTP hydrolysis by DNM2. Firstly, the GTPase activity of the ubiquitous DNM2-12B was strongly activated by lipids, as previously known (36). Here we show that the DNM2+12B isoform is also activated with lipids (Figure 5C and Supplemental Figure 7). Strikingly, addition of BIN1 with or without exon 11 reduced DNM2-12B activity but did not impact DNM2+12B activity, indicating that BIN1 inhibits GTP hydrolysis by the ubiquitous and predominantly skeletal muscle developmental DNM2 isoform, but not the muscle-specific adult isoform. Thus, muscle maturation correlates with the appearance and predominance of a muscle-specific DNM2 isoform (+12B) with decreased regulation by BIN1. Taken together with the subcellular localization data, it suggests that BIN1 inhibits DNM2 activity during muscle maturation, while the adult DNM2 muscle-specific isoform is uncoupled from BIN1. As BIN1 appears to regulate the embryonic DNM2 isoform but not the adult muscle form, it suggests that adult deletion of Bin1 may not be lethal. Indeed, we found that contrary to embryonic knockout of Bin1, knockout of Bin1 in adult skeletal muscles, using the inducible Cre recombinase expression system, is not lethal (Supplemental Figure 8), supporting the concept that BIN1 is essential for skeletal muscle development but less for muscle maintenance.
Discussion
The ubiquitous BIN1 and DNM2 proteins are well known to participate in membrane remodeling during endocytosis, and in vitro data suggested that amphiphysin proteins recruit and activate dynamins (37). Conversely, we found in this study an antagonistic balance between BIN1 and DNM2, supporting that BIN1 is a negative regulator of DNM2 in vivo, at least in skeletal muscle. This underlines that studying regulatory mechanisms in vivo in relevant tissues where specific isoforms are expressed gives a detailed and somehow unexpected view of these mechanisms. These data call for further analysis of dynamin regulation by amphiphysins in additional physiological contexts. Whether this is also true for dynamin 1, the brain dynamin implicated in synaptic vesicle replenishment (38, 39), remains to be determined.
During muscle development BIN1 and DNM2 strongly colocalize (Figure 4). At this stage the GTPase activity of the major DNM2 isoform is inhibited by BIN1 (Figure 5). The appearance and enrichment of muscle-specific BIN1 and DNM2 isoforms correlate with the maturation of T-tubules, and is accompanied by a decreased sensitivity of DNM2 GTPase activity to BIN1 and the relocalization of DNM2 to the actin-rich I-band partly away from the triad. Thus, for normal muscle formation, BIN1 would partially inhibit DNM2 function on nascent T-tubules. BIN1 and DNM2 muscle-specific exons encode amino acids that are located within or close to regions essential for intramolecular regulation; the PI-binding domain binds the SH3 domain in BIN1 (40) and for DNM2 the PH-stalk domain interface mediates assembly of dimers (37). Thus, BIN1 and DNM2 achieve muscle-specific functions during muscle maturation. These findings indicate for the first time to our knowledge a molecular explanation for the muscle specificity of CNMs. As Bin1–/– Dnm2+/– mice develop normally to adulthood with well-organized triads without BIN1, it is rather the BIN1-DNM2 balance that is important for muscle maturation. Potentially, unregulated DNM2, due either to BIN1 alteration or DNM2 mutations, would compromise this BIN1-DNM2 balance and subsequently muscle maturation/maintenance, leading to CNMs.
Here we show that reducing DNM2 in Bin1–/– mice rescues its neonatal lethality, providing a potential therapeutic approach for patients with BIN1 mutation. We recently discovered that reduction of DNM2 can rescue the lifespan and several pathological features of a murine model of myotubular myopathy, which is the X-linked form of CNM due to mutations in the PI phosphatase MTM1 (20, 41). Altogether, these results indicate that both MTM1 and BIN1 act upstream of DNM2. Indeed, BIN1 can bind to MTM1 and also to DNM2, and BIN1-CNM mutations interfere with its ability to interact with MTM1 and DNM2 (7, 42), indicating the importance of this pathway in muscle and disease. Moreover, DNM2 downregulation becomes a potential strategy to rescue several myopathy forms (linked to either MTM1 or BIN1 mutations). This adds credit to the concept of cross-therapy where phenotypes due to loss of a CNM protein (MTM1 or BIN1) are rescued by downregulating another CNM protein (DNM2). As BIN1 was also implicated in several cancers, arrhythmia, and late-onset Alzheimer disease (43), the epistasis and potential therapeutic approach demonstrated here for myopathies may trigger novel research directions and therapeutic developments for these common diseases.
In conclusion, we show here that reduction of DNM2 can rescue the neonatal lethality due to BIN1 alteration in mice and that the balance and specific regulation between muscle-specific isoforms of BIN1 and DNM2 is necessary for perinatal muscle maturation. These findings highlight that BIN1 is a negative regulator of DNM2 in vitro and in vivo, at least in muscle maturation, and support DNM2 downregulation as a therapeutic strategy to rescue several myopathy forms.
Methods
Materials
Primary antibodies used were mouse anti-DHPRα1 (Cav1.1) subunit (MA3–920; Affinity Bioreagents), α-actinin (EA-53, Sigma-Aldrich), caveolin-3 (clone 26, BD Biosciences), BIN1 (C99D, Upstate), and glyceraldehyde-3-phosphate dehydrogenase (GAPDH, MAB374; Chemicon). Rabbit anti-DNM2 antibodies (R2680 and R2865) (18) and anti-BIN1 (R2405) (7) were made onsite at the polyclonal antibody facility of the IGBMC. Alexa Fluor–conjugated secondary antibodies were purchased from Invitrogen (Alexa Fluor 488 [A11001 and A11008] and Alexa Fluor 594 [A11005 and A11012]). Secondary antibodies against mouse and rabbit IgG, conjugated with horseradish peroxidase (HRP) were purchased from Jackson ImmunoResearch Laboratories (catalog 115-035-146 and 111-036-045). An ECL kit was purchased from Pierce. Plasmids, Sepharose, and PreScission for in vitro enzymatic assays were purchased from GE Healthcare, and lipids were purchased from Avanti Polar Lipids. Indo-1 calcium indicator was purchased from Thermo Fisher Scientific.
Primary cell culture and differentiation
Primary myoblasts from WT newborn mice were prepared using a protocol adapted from De Palma et al. (44). After hind limb muscle isolation, muscles were minced and digested for 1.5 hours in PBS containing 0.5 mg/ml collagenase (Sigma-Aldrich) and 3.5 mg/ml Dispase (Gibco). Cell suspensions were filtered through a 40-μm cell strainer and preplated in Iscove’s Modified Dulbecco’s Medium (IMDM, Gibco) with 10% FCS (Gibco), to discard the majority of fibroblasts and contaminating cells, and then incubated for 3 hours. Nonadherent myogenic cells were collected and plated in IMDM with 20% FCS and 1% Chicken Embryo Extract (MP Biomedical) onto 1:100 Matrigel Reduced Factor (BD) in IMDM-coated fluorodishes. Differentiation was triggered by switching the medium to IMDM with 2% horse serum and 24 hours later a thick layer of matrigel (1:3 in IMDM) was added. For immunofluorescence experiments, myotubes were treated with 80 ng/ml agrin, and the medium was changed every 2 days. Cells in culture were prepared, fixed, and stained with antibodies against BIN1-C99D (1:50) and DNM2-R2680 (1:200).
Primary cell culture and calcium measurements
Primary myoblasts from WT, Bin1+/–, or Bin1–/– mice at E18.5 were prepared using a protocol adapted from De Palma et al. (44) as described above, with minor modifications. Cells were incubated for 1.5 hours. Nonadherent myogenic cells were collected and plated in IMDM with 20% FCS and 1% Chicken Embryo Extract onto 1:100 Matrigel Reduced Factor in IMDM-coated coverslips. Once cells reached 50% confluence, they were incubated for 30 minutes with Indo-1 calcium indicator (5 μM), and then washed and incubated for 30 minutes in medium containing 2 mM calcium. Samples were viewed using a laser scanning confocal microscope (TCS SP2; Leica Microsystems). Baseline measurements were obtained by excitation at 338 nm, and then the ratio of emission wavelengths was measured (405/485 nm, every 2 seconds). Following this, the medium was replaced with calcium-free medium containing EGTA (1 mM) for 5 minutes, and then caffeine was added (25 mM) to activate RYR1-mediated release of calcium from the sarcoplasmic reticulum. The ratio of emission wavelengths was measured (405/485 nm, every 2 seconds).
RT-PCR and isoform detection
Total RNA was extracted from primary myoblasts, differentiated myotubes, and mouse skeletal muscle (E18.5 and adult), using TRIzol (TRI Reagent, Molecular Research Center). Reverse transcription (RT) was performed using Superscript 11 reverse transcriptase (Invitrogen) with 1–2 μg of total RNA, and the presence of BIN1/DNM2 isoforms was detected using our previously described protocol (24). Primers used for PCR amplification were 5′BIN1, ATGGCAGAGATGGGGAGCAAGG; 3′BIN1, TCACTGCACCCGCTCTGTAAAATT; 5′DNM2, CTAGAGTTGTCTGGTGGAGC; and 3′DNM2, GCTTCATGTCCAACAAGCACG. More than 30 cDNA clones were sequenced for each sample. The GTEx database was analyzed for the presence of BIN1 and DNM2 isoforms in various tissues (www.gtexportal.org; latest version V6 dbGaP Accession phs000424.v6.p1) (31).
Immunofluorescence assays
Primary cells were fixed in 4% paraformaldehyde (PFA) for 20 minutes, permeabilized with Triton X-100 (0.2% in PBS), and nonspecific sites were blocked with 10% FCS and 0.1% Triton X-100 in PBS. Primary antibodies were added overnight at 4°C in 0.1% Triton X-100 and 3% FCS in PBS. The following antibodies were used: anti-BIN1 (clone C99D, Sigma-Aldrich) and anti-dynamin 2 (R2680, IGBMC). These antibodies were revealed by employing anti-rabbit Alexa Fluor 594–conjugated or anti-mouse Alexa Fluor 488–conjugated secondary antibodies (Invitrogen). Nuclei were stained with Hoechst 33528 (Sigma-Aldrich) and samples were mounted in FluorSave reagent (Calbiochem).
Protein purification and GTPase activity assay
DNM2+12b was generated by primer-directed PCR mutagenesis from the WT construct using primers (hsDNM12B forward, 5′CTGTTACTATACTGAGCAGCTGGTGACCTGTGCCCAGCAGAGGAGCACGC3′; hsDNM12B reverse, 5′CAGGTCACCAGCTGCTCAGTATAGTAACAGTTGGCAAACCCGATGAAGTC3′) and recombinant DNM2 proteins were produced from pVL1392 plasmids in SF9 cells with the baculovirus system as described previously (45). Briefly, a transfection was performed with DNM2(±12B) plasmids to produce viruses. SF9 cells were infected with viruses and then grown for 3 days at 27°C, and then centrifuged at 2,000 g for 5 minutes. DNM2 recombinant proteins were purified with amphiphysin-SH3 bound to Glutathione-Sepharose 4B beads (GE Healthcare) as previously described (46). BIN1 recombinant protein was produced from pGEX6P1 plasmid in E. coli BL21. E. coli producing BIN1 were induced with IPTG (0.5 mM) for 3 hours at 37°C, centrifuged at 7,500 g, and then proteins were purified using Glutathione Sepharose 4B beads. The GST tag was cut by PreScission enzyme on the beads and the flow through was recovered. DNM2 and BIN1 recombinant proteins were used at a ratio of 1:1 or 1:4 as described (DNM2/BIN1). A malachite green assay for GTP hydrolysis using lipids (95% Brain Polar Lipid Extract and 5% PIP2, both Avanti Polar Lipids) was performed as described previously (47) with a reaction time of 5 minutes at 37°C. Two controls were used for this experiment: one with GTP, liposomes, and buffer (no dynamin); and one with dynamin, liposomes, and buffer (no GTP). Both controls produced zero values.
Cosedimentation assay
This assay was performed as described previously (48). Briefly, 2 μM DNM2 recombinant proteins were mixed with liposomes with or without PIP2 (PE/PC, 80:20, total 1 mg/ml) for 15 minutes at room temperature. High-speed centrifugation was performed to separate pellet and supernatant. The pellet was resuspended and both pellet and supernatant were run in an SDS-PAGE gel. Protein bands were visualized after staining the gel with 2,2,2-trichloroethanol (TCE, Sigma-Aldrich) following activation by UV, allowing labeling of the Trp residues in the DNM2 protein. The ImageLab software from Bio-Rad was used for image acquisition and quantification.
Generation of mouse lines
Bin1 exon 20–KO mice.
The targeting vector was created with LoxP sites flanking exon 20 in a Bin1 exon 20–KO mouse line (referred to here as Bin1–/–) (Figure 1 and Figure 4), linearized, and electroporated into embryonic stem (ES) cells. Recombinant ES cells were injected into C57BL/6 blastocysts that were implanted in pseudopregnant females and germline transmission determined. Recombination was triggered using the CMV promoter. The mice were bred on a B6J background.
Dnm2 heterozygous mice.
The creation of Dnm2+/– mice deleted for exon 8 was described previously (20).
Bin1–/– Dnm2+/– mice.
Female heterozygous Bin1+/– mice were bred with male Dnm2+/– mice to produce 4 possible genotypes in male offspring: Bin1+/+ Dnm2+/+ (WT), Bin1+/+ Dnm2+/–, Bin1+/– Dnm2+/+, and Bin1+/– Dnm2+/–. In the next step, male or female Bin1+/– Dnm2+/– mice were crossed with a female or male Bin1+/– Dnm2+/+. This cross yielded 6 possible genotypes: Bin1+/+ Dnm2+/+ (WT), Bin1+/+ Dnm2+/–, Bin1+/– Dnm2+/+, Bin1+/– Dnm2+/–, Bin1–/– Dnm2+/+, and Bin1–/– Dnm2+/– (Figure 1B). We analyzed female and male Bin1–/– Dnm2+/– mice and compared them to the WT littermates.
Animal experiments
Animals were housed in a temperature-controlled room (19°C–22°C) with a 12-hour light/dark cycle. Mice were weighed weekly until 1 year of age. Mice were humanely killed when required by CO2 inhalation followed by cervical dislocation, according to national and European legislations on animal experimentation. Muscles and other tissues were dissected and frozen in nitrogen-cooled isopentane and liquid nitrogen for histological and immunoblot assays, respectively. For dissection of E18.5 mice, animals were sacrificed by decapitation and pups were dissected. Hind limbs were snap frozen in liquid nitrogen–cooled isopentane and prepared for histology and immunofluorescence as described below.
Phenotyping of Bin1–/– and Dnm2+/– mice
Mice aged 10–15 weeks were phenotyped under the EUMODIC phenotyping program (http://www.eumodic.eu/) with results made publicly available (http://www.europhenome.org/). In both cases male and female mutant mice were comparable to the WT littermates. Blood chemistry, ECG measurements, Dexascan, and qNMR presented here for male mice (n = minimum 10 per group) were performed as part of pipelines 1 and 2 of the EUMODIC phenotyping program, at the Institut Clinique de la Souris (ICS, Illkirch, France; www.ics-mci.fr).
Grip (2 and 4 paws), hang, and rotarod tests
Grip strength tests were performed by placing the 2 front paws or all 4 paws on the grid of a dynamometer (Bioseb) and mice were pulled by the tail in the opposite direction. The maximal strength exerted by the mouse before losing grip was recorded. Three trials per mouse were performed, with 30 seconds of rest between trials (2-paws test, n = minimum 5 mice per group; 4-paws test, n = 5–7 mice per group). For the hanging test, mice were suspended from a cage lid for a maximum of 60 seconds. The time when the mouse fell off the cage was recorded for each trial. Three trials per mouse were performed. In the rotarod test, coordination and whole-body muscle strength were tested using an accelerated rotating rod test (Panlab). Mice were placed on the rod, which accelerated from 4 to 40 rpm during 5 minutes. Three trials per day, with 5 minutes of rest between trials were performed for day 1 (training day), and then 4 days which were recorded. Animals were scored for their latency to fall (in seconds). The mean of the 3 trials was calculated for each experiment listed above (n = 5–7 mice per group).
TA muscle contractile properties
Muscle force measurements were evaluated by measuring in situ muscle contraction in response to nerve and muscle stimulation, as described previously with a force transducer (Aurora Scientific) (18, 49). Results from nerve stimulation are shown (n = 5–11 mice per group). Fatigue was measured as time taken to reach 50% of the maximum force produced. After contractile measurements, the animals were killed by cervical dislocation. TA muscles were then dissected and weighed to determine specific maximal force.
Western blotting
Mouse muscles were minced and homogenized on ice for 3 times 30 seconds (Ultra Turrax homogenizer) in 10 times the weight/volume of 1% NP-40 Tris-Cl buffer, pH 8, then extracted for 30 minutes at 4°C. Protein concentration was determined using a DC Protein Assay kit (Bio-Rad), and lysates analyzed by SDS-PAGE and Western blotting on nitrocellulose membranes. Primary antibodies used were DNM2-R2680 (1:500), DNM2-R2865 (1:500), BIN1-R2405 (1:1,000), and GAPDH (1:10,000); secondary antibodies were anti-rabbit HRP or anti-mouse HRP (1:10,000). Western blot films were scanned and band intensities were determined using ImageJ software (NIH, http://rsb.info.nih.gov/ij/, 1997–2009). Densitometry values were standardized to corresponding total GAPDH values and expressed as a fold difference relative to the indicated control (n = 3–5 mice per group).
Histological and immunofluorescence analysis of skeletal muscle
Longitudinal and transverse cryosections (8 μm) of mouse skeletal muscles were prepared, fixed, and stained with antibodies against DHPRα1 (1:100), α-actinin (1:1,000), caveolin-3 (1:1,000); DNM2-R2680 (1:200), and BIN1-C99D (1:100). Samples were viewed using a laser scanning confocal microscope (TCS SP8; Leica Microsystems). Air-dried transverse sections were fixed and stained with H&E or for SDH, and image acquisition performed with a slide scanner NanoZoomer 2 HT equipped with the fluorescence module L11600-21 (Hamamatsu Photonics) or a DMRXA2 microscope (Leica Microsystems Gmbh). Cross-sectional area (CSA) was analyzed in H&E–stained sections from TA mouse skeletal muscle, using FIJI image analysis software. CSA (μm2) was calculated (>500 fibers per mouse) from 4–7 mice per group. The percentage of TA muscle fibers with centralized or internalized nuclei was counted in more than 500 fibers from 4–6 mice using the cell counter plugin in ImageJ.
Transmission electron microscopy
TA muscle biopsies were fixed with 2.5% glutaraldehyde in 0.1 M cacodylate buffer (pH 7.2) and processed as described previously (18, 50). Triad structures were identified on longitudinal sections of muscle and the number of triads per sarcomere was quantified. The ratio of triads/sarcomere was calculated by dividing number of triads clearly identified by the total number of sarcomeres present in the image, as described previously (51). Forty to 80 triads were counted per mouse, 3 mice/genotype.
Microscopy and analysis
All microscopy was performed at the IGBMC Imaging Centre. All samples for microscopy were mounted in Fluorsave reagent (Merck) and viewed at room temperature. Light microscopy was performed using a fluorescence microscope (DM4000; Leica microsystems) fitted with a color CCD camera (CoolSNAP CF color, Photometrics). Confocal microscopy was performed using a confocal laser scanning microscope (TCS SP2 or SP5; Leica Microsystems). ImageJ and FIJI analysis software were used for image analysis.
Statistics
Statistical analysis was performed using the unpaired 2-tailed Student’s t test unless stated otherwise. When stated, a 1- or 2-way ANOVA test was used with Bonferroni or Dunn’s Multiple Comparison post hoc test. P values less than 0.05 were considered significant.
Study approval
Animal experimentation was approved by the institutional ethical committee Com’Eth IGBMC-ICS (2012-128, 2013-034).
Author contributions
BSC, IP, and JL designed the study and analyzed the data. BSC, IP, HT, ASN, FH, CK, A. Rabai, SF, and BR performed experiments. BSC, JL, and A. Roux supervised the study. BSC, IP, and JL wrote the manuscript.
Supplementary Material
Acknowledgments
We thank Arnaud Ferry (UMRS974, Paris, France) and Xenia Massana, Catherine Koch, Julien Becker, Jean-Luc Weickert, Nadia Messaddeq, Olivia Wendling, Hugues Jacobs, Marc Koch, and Pascal Kessler from the IGBMC for excellent technical assistance; and the animal house, histology platform and Imaging Centre of the IGBMC for support. This study was supported by INSERM, CNRS, University of Strasbourg, the Agence Nationale de la Recherche (14-CE12-0009), SATT Conectus Alsace (2014), Association Francaise contre les Myopathies (grant 15352), Myotubular Trust and Sparks - The Children’s Medical Research Charity, and ANR-10-LABX-0030-INRT, a French State fund managed by the Agence Nationale de la Recherche under the frame program Investissements d’Avenir ANR-10-IDEX-0002-02.
Version 1. 11/13/2017
Electronic publication
Version 2. 12/01/2017
Print issue publication
Footnotes
Conflict of interest: H. Tasfaout, J. Laporte, and B.S. Cowling are inventors on a patent (number Ep13306440.2 - 1412 [EP20130306440], filed 10/28/2013, second file international extension [PCT] PCT/EP2014/072466, 10/20/2014) for targeting DNM2 for the treatment of centronuclear myopathies, and cofounders of Dynacure. J. Laporte and B.S. Cowling are scientific advisors for Dynacure.
Reference information: J Clin Invest. 2017;127(12):4477–4487.https://doi.org/10.1172/JCI90542.
Contributor Information
Ivana Prokic, Email: iprokic@anagenesis-biotech.com.
Hichem Tasfaout, Email: tasfaout@igbmc.fr.
Aymen Rabai, Email: rabai@igbmc.fr.
Frédéric Humbert, Email: Frederic.Humbert@unige.ch.
Bruno Rinaldi, Email: b.rinaldi@unistra.fr.
Anne-Sophie Nicot, Email: anne-sophie.nicot@univ-grenoble-alpes.fr.
Christine Kretz, Email: cricri@igbmc.fr.
Sylvie Friant, Email: s.friant@unistra.fr.
Jocelyn Laporte, Email: jocelyn@igbmc.fr.
References
- 1.Towler MC, Kaufman SJ, Brodsky FM. Membrane traffic in skeletal muscle. Traffic. 2004;5(3):129–139. doi: 10.1111/j.1600-0854.2003.00164.x. [DOI] [PubMed] [Google Scholar]
- 2.Cowling BS, Toussaint A, Muller J, Laporte J. Defective membrane remodeling in neuromuscular diseases: insights from animal models. PLoS Genet. 2012;8(4):e1002595. doi: 10.1371/journal.pgen.1002595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lee E, et al. Amphiphysin 2 (Bin1) and T-tubule biogenesis in muscle. Science. 2002;297(5584):1193–1196. doi: 10.1126/science.1071362. [DOI] [PubMed] [Google Scholar]
- 4.David C, McPherson PS, Mundigl O, de Camilli P. A role of amphiphysin in synaptic vesicle endocytosis suggested by its binding to dynamin in nerve terminals. Proc Natl Acad Sci U S A. 1996;93(1):331–335. doi: 10.1073/pnas.93.1.331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bitoun M, et al. Mutations in dynamin 2 cause dominant centronuclear myopathy. Nat Genet. 2005;37(11):1207–1209. doi: 10.1038/ng1657. [DOI] [PubMed] [Google Scholar]
- 6.Böhm J, et al. Adult-onset autosomal dominant centronuclear myopathy due to BIN1 mutations. Brain. 2014;137(Pt 12):3160–3170. doi: 10.1093/brain/awu272. [DOI] [PubMed] [Google Scholar]
- 7.Nicot AS, et al. Mutations in amphiphysin 2 (BIN1) disrupt interaction with dynamin 2 and cause autosomal recessive centronuclear myopathy. Nat Genet. 2007;39(9):1134–1139. doi: 10.1038/ng2086. [DOI] [PubMed] [Google Scholar]
- 8.Ravenscroft G, Laing NG, Bönnemann CG. Pathophysiological concepts in the congenital myopathies: blurring the boundaries, sharpening the focus. Brain. 2015;138(Pt 2):246–268. doi: 10.1093/brain/awu368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sakamuro D, Elliott KJ, Wechsler-Reya R, Prendergast GC. BIN1 is a novel MYC-interacting protein with features of a tumour suppressor. Nat Genet. 1996;14(1):69–77. doi: 10.1038/ng0996-69. [DOI] [PubMed] [Google Scholar]
- 10.Peter BJ, et al. BAR domains as sensors of membrane curvature: the amphiphysin BAR structure. Science. 2004;303(5657):495–499. doi: 10.1126/science.1092586. [DOI] [PubMed] [Google Scholar]
- 11.Razzaq A, et al. Amphiphysin is necessary for organization of the excitation-contraction coupling machinery of muscles, but not for synaptic vesicle endocytosis in Drosophila. Genes Dev. 2001;15(22):2967–2979. doi: 10.1101/gad.207801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Böhm J, et al. Altered splicing of the BIN1 muscle-specific exon in humans and dogs with highly progressive centronuclear myopathy. PLoS Genet. 2013;9(6):e1003430. doi: 10.1371/journal.pgen.1003430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Faelber K, et al. Oligomerization of dynamin superfamily proteins in health and disease. Prog Mol Biol Transl Sci. 2013;117:411–443. doi: 10.1016/B978-0-12-386931-9.00015-5. [DOI] [PubMed] [Google Scholar]
- 14.Praefcke GJ, McMahon HT. The dynamin superfamily: universal membrane tubulation and fission molecules? Nat Rev Mol Cell Biol. 2004;5(2):133–147. doi: 10.1038/nrm1313. [DOI] [PubMed] [Google Scholar]
- 15.Kenniston JA, Lemmon MA. Dynamin GTPase regulation is altered by PH domain mutations found in centronuclear myopathy patients. EMBO J. 2010;29(18):3054–3067. doi: 10.1038/emboj.2010.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang L, Barylko B, Byers C, Ross JA, Jameson DM, Albanesi JP. Dynamin 2 mutants linked to centronuclear myopathies form abnormally stable polymers. J Biol Chem. 2010;285(30):22753–22757. doi: 10.1074/jbc.C110.130013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chin YH, et al. Dynamin-2 mutations associated with centronuclear myopathy are hypermorphic and lead to T-tubule fragmentation. Hum Mol Genet. 2015;24(19):5542–5554. doi: 10.1093/hmg/ddv285. [DOI] [PubMed] [Google Scholar]
- 18.Cowling BS, et al. Increased expression of wild-type or a centronuclear myopathy mutant of dynamin 2 in skeletal muscle of adult mice leads to structural defects and muscle weakness. Am J Pathol. 2011;178(5):2224–2235. doi: 10.1016/j.ajpath.2011.01.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu N, et al. Mice lacking microRNA 133a develop dynamin 2–dependent centronuclear myopathy. J Clin Invest. 2011;121(8):3258–3268. doi: 10.1172/JCI46267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cowling BS, et al. Reducing dynamin 2 expression rescues X-linked centronuclear myopathy. J Clin Invest. 2014;124(3):1350–1363. doi: 10.1172/JCI71206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Muller AJ, et al. Targeted disruption of the murine Bin1/Amphiphysin II gene does not disable endocytosis but results in embryonic cardiomyopathy with aberrant myofibril formation. Mol Cell Biol. 2003;23(12):4295–4306. doi: 10.1128/MCB.23.12.4295-4306.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hong T, et al. Cardiac BIN1 folds T-tubule membrane, controlling ion flux and limiting arrhythmia. Nat Med. 2014;20(6):624–632. doi: 10.1038/nm.3543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Miniou P, Tiziano D, Frugier T, Roblot N, Le Meur M, Melki J. Gene targeting restricted to mouse striated muscle lineage. Nucleic Acids Res. 1999;27(19):e27. doi: 10.1093/nar/27.19.e27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Toussaint A, et al. Defects in amphiphysin 2 (BIN1) and triads in several forms of centronuclear myopathies. Acta Neuropathol. 2011;121(2):253–266. doi: 10.1007/s00401-010-0754-2. [DOI] [PubMed] [Google Scholar]
- 25.D’Alessandro M, et al. Amphiphysin 2 orchestrates nucleus positioning and shape by linking the nuclear envelope to the actin and microtubule cytoskeleton. Dev Cell. 2015;35(2):186–198. doi: 10.1016/j.devcel.2015.09.018. [DOI] [PubMed] [Google Scholar]
- 26.Rao Y, Haucke V. Membrane shaping by the Bin/amphiphysin/Rvs (BAR) domain protein superfamily. Cell Mol Life Sci. 2011;68(24):3983–3993. doi: 10.1007/s00018-011-0768-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tjondrokoesoemo A, et al. Disrupted membrane structure and intracellular Ca²+ signaling in adult skeletal muscle with acute knockdown of Bin1. PLoS One. 2011;6(9):e25740. doi: 10.1371/journal.pone.0025740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Klinge L, et al. Dysferlin associates with the developing T-tubule system in rodent and human skeletal muscle. Muscle Nerve. 2010;41(2):166–173. doi: 10.1002/mus.21166. [DOI] [PubMed] [Google Scholar]
- 29.Ramjaun AR, McPherson PS. Multiple amphiphysin II splice variants display differential clathrin binding: identification of two distinct clathrin-binding sites. J Neurochem. 1998;70(6):2369–2376. doi: 10.1046/j.1471-4159.1998.70062369.x. [DOI] [PubMed] [Google Scholar]
- 30.Wechsler-Reya RJ, Elliott KJ, Prendergast GC. A role for the putative tumor suppressor Bin1 in muscle cell differentiation. Mol Cell Biol. 1998;18(1):566–575. doi: 10.1128/MCB.18.1.566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.GTEx Consortium. Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science. 2015;348(6235):648–660. doi: 10.1126/science.1262110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cao H, Garcia F, McNiven MA. Differential distribution of dynamin isoforms in mammalian cells. Mol Biol Cell. 1998;9(9):2595–2609. doi: 10.1091/mbc.9.9.2595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fugier C, et al. Misregulated alternative splicing of BIN1 is associated with T tubule alterations and muscle weakness in myotonic dystrophy. Nat Med. 2011;17(6):720–725. doi: 10.1038/nm.2374. [DOI] [PubMed] [Google Scholar]
- 34.Takekura H, Flucher BE, Franzini-Armstrong C. Sequential docking, molecular differentiation, and positioning of T-Tubule/SR junctions in developing mouse skeletal muscle. Dev Biol. 2001;239(2):204–214. doi: 10.1006/dbio.2001.0437. [DOI] [PubMed] [Google Scholar]
- 35.Bagatolli LA, Binns DD, Jameson DM, Albanesi JP. Activation of dynamin II by POPC in giant unilamellar vesicles: a two-photon fluorescence microscopy study. J Protein Chem. 2002;21(6):383–391. doi: 10.1023/A:1021126415320. [DOI] [PubMed] [Google Scholar]
- 36.Lin HC, Barylko B, Achiriloaie M, Albanesi JP. Phosphatidylinositol (4,5)-bisphosphate-dependent activation of dynamins I and II lacking the proline/arginine-rich domains. J Biol Chem. 1997;272(41):25999–26004. doi: 10.1074/jbc.272.41.25999. [DOI] [PubMed] [Google Scholar]
- 37.Daumke O, Roux A, Haucke V. BAR domain scaffolds in dynamin-mediated membrane fission. Cell. 2014;156(5):882–892. doi: 10.1016/j.cell.2014.02.017. [DOI] [PubMed] [Google Scholar]
- 38.Kokotos AC, Cousin MA. Synaptic vesicle generation from central nerve terminal endosomes. Traffic. 2015;16(3):229–240. doi: 10.1111/tra.12235. [DOI] [PubMed] [Google Scholar]
- 39.Farsad K, De Camilli P. Neurotransmission and the synaptic vesicle cycle. Yale J Biol Med. 2002;75(5-6):261–284. [PMC free article] [PubMed] [Google Scholar]
- 40.Kojima C, et al. Regulation of Bin1 SH3 domain binding by phosphoinositides. EMBO J. 2004;23(22):4413–4422. doi: 10.1038/sj.emboj.7600442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tasfaout H, et al. Antisense oligonucleotide-mediated Dnm2 knockdown prevents and reverts myotubular myopathy in mice. Nat Commun. 2017;8:15661. doi: 10.1038/ncomms15661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Royer B, Hnia K, Gavriilidis C, Tronchère H, Tosch V, Laporte J. The myotubularin-amphiphysin 2 complex in membrane tubulation and centronuclear myopathies. EMBO Rep. 2013;14(10):907–915. doi: 10.1038/embor.2013.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Prokic I, Cowling BS, Laporte J. Amphiphysin 2 (BIN1) in physiology and diseases. J Mol Med. 2014;92(5):453–463. doi: 10.1007/s00109-014-1138-1. [DOI] [PubMed] [Google Scholar]
- 44.De Palma C, et al. Nitric oxide inhibition of Drp1-mediated mitochondrial fission is critical for myogenic differentiation. Cell Death Differ. 2010;17(11):1684–1696. doi: 10.1038/cdd.2010.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Quan A, Robinson PJ. Rapid purification of native dynamin I and colorimetric GTPase assay. Meth Enzymol. 2005;404:556–569. doi: 10.1016/S0076-6879(05)04049-8. [DOI] [PubMed] [Google Scholar]
- 46.Kalia R, Talledge N, Frost A. Structural and functional studies of membrane remodeling machines. Methods Cell Biol. 2015;128:165–200. doi: 10.1016/bs.mcb.2015.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Koutsopoulos OS, et al. Dynamin 2 homozygous mutation in humans with a lethal congenital syndrome. Eur J Hum Genet. 2013;21(6):637–642. doi: 10.1038/ejhg.2012.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Itoh T, Erdmann KS, Roux A, Habermann B, Werner H, De Camilli P. Dynamin and the actin cytoskeleton cooperatively regulate plasma membrane invagination by BAR and F-BAR proteins. Dev Cell. 2005;9(6):791–804. doi: 10.1016/j.devcel.2005.11.005. [DOI] [PubMed] [Google Scholar]
- 49.Vignaud A, Hourde C, Medja F, Agbulut O, Butler-Browne G, Ferry A. Impaired skeletal muscle repair after ischemia-reperfusion injury in mice. J Biomed Biotechnol. 2010;2010:724914. doi: 10.1155/2010/724914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Buj-Bello A, et al. The lipid phosphatase myotubularin is essential for skeletal muscle maintenance but not for myogenesis in mice. Proc Natl Acad Sci U S A. 2002;99(23):15060–15065. doi: 10.1073/pnas.212498399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Amoasii L, et al. Phosphatase-dead myotubularin ameliorates X-linked centronuclear myopathy phenotypes in mice. PLoS Genet. 2012;8(10):e1002965. doi: 10.1371/journal.pgen.1002965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Faelber K, et al. Crystal structure of nucleotide-free dynamin. Nature. 2011;477(7366):556–560. doi: 10.1038/nature10369. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.