

Abstract
Ebolaviruses and marburgviruses belong to the family Filoviridae and cause high lethality in infected patients. There are currently no licensed filovirus vaccines or antiviral therapies. The development of broad-spectrum therapies against members of the Marburgvirus genus, including Marburg virus (MARV) and Ravn virus (RAVV), is difficult because of substantial sequence variability. RNAi therapeutics offer a potential solution, as identification of conserved target nucleotide sequences may confer activity across marburgvirus variants. Here, we assessed the therapeutic efficacy of lipid nanoparticle (LNP) delivery of a single nucleoprotein–targeting (NP-targeting) siRNA in nonhuman primates at advanced stages of MARV or RAVV disease to mimic cases in which patients begin treatment for fulminant disease. Sixteen rhesus monkeys were lethally infected with MARV or RAVV and treated with NP siRNA-LNP, with MARV-infected animals beginning treatment four or five days after infection and RAVV-infected animals starting treatment three or six days after infection. While all untreated animals succumbed to disease, NP siRNA-LNP treatment conferred 100% survival of RAVV-infected macaques, even when treatment began just 1 day prior to the death of the control animals. In MARV-infected animals, day-4 treatment initiation resulted in 100% survival, and day-5 treatment resulted in 50% survival. These results identify a single siRNA therapeutic that provides broad-spectrum protection against both MARV and RAVV.
Keywords: Infectious disease, Virology
Keywords: Drug therapy
Introduction
From 1967 until late 2013, members of the 2 genera of filoviruses, Marburgvirus and Ebolavirus, have caused periodic occurrences of hemorrhagic fever (HF) in Central Africa, resulting in severe and often lethal disease in infected patients (1). Prior to 2013, these outbreaks were relatively small in size, ranging from a few cases to several hundred cases. However, the recent unprecedented epidemic of Ebola virus disease caused by the Ebola virus (EBOV, species Zaire ebolavirus) in West Africa that resulted in 28,616 cases and 11,310 deaths from December 2013 to January 2016 underscores the public health threat posed by filoviruses. This concern is further heightened by the lack of licensed vaccines and antiviral therapies. While substantial progress has been made in advancing promising candidate EBOV vaccines and antivirals as a result of the 2013–2016 epidemic, less work has been done on developing effective countermeasures against the marburgviruses Marburg virus (MARV) and Ravn virus (RAVV) (2). In addition, the significant sequence variability (up to 21%) (2) between the 2 virus variants presents an obstacle to the development of therapies that are effective across all members of the Marburgvirus genus.
RNAi therapy is a promising drug modality that offers exquisite target specificity and a well-defined mechanism of action. A significant barrier to in vivo application of RNAi has been the development of efficient delivery vehicles that promote cellular uptake (3); however, use of a lipid nanoparticle (LNP) platform not only protects siRNAs from nuclease degradation in the bloodstream but also mediates effective delivery to hepatocytes, a major replication cell for MARV and RAVV. This technology has been used successfully to protect nonhuman primates (NHPs) against EBOV when given before or at the onset of illness (4, 5) and against Sudan virus (SUDV, species Sudan ebolavirus) when given at the late stage of disease (6). Although anti-EBOV siRNA-LNP was recently tested in a very small phase II clinical trial, in which it appeared safe but did not show clear benefit, the extremely high viral loads (>9 log10 copies/ml for all 12 cases) and existing organ injury in the enrolled end-stage patients probably obscured this trial’s ability to detect statistically significant treatment efficacy (7). In addition, LNP technology continues to advance beyond the formulations used in the preceding TKM-Ebola clinical trials, and novel LNP formulations with increased potency have been developed and shown to have utility in filovirus-infected NHPs (5).
In previous marburgvirus siRNA studies, we have (a) identified 2 siRNAs targeting the MARV NP (designated NP-718m and NP-143m) that, when encapsulated in a LNP, inhibited the replication of MARV and RAVV in vitro and provided complete protection to guinea pigs infected with several different MARV and RAVV isolates (8); and (b) showed that NP-718m–LNP conferred complete protection to MARV-infected rhesus macaques when administered at the onset of clinical illness, 3 days after MARV exposure (9). The primary criticism of this earlier MARV macaque work was that treatment was initiated too early in the disease course and had debatable practical utility, with the possible exception of accidental laboratory exposures. Here, we assessed the utility of using NP-718m–LNP as a therapeutic intervention in uniformly lethal rhesus macaque models of MARV and RAVV infection when treatment was delayed until the animals were at advanced stages of illness, as evidenced by fevers and high viral loads. In doing so, importantly, we demonstrated the broad-spectrum activity of NP-718m–LNP as a therapeutic intervention against these sequence-divergent members of the Marburgvirus genus.
Results
NP-718m–LNP treatment results in the survival of MARV-infected NHPs when treatment is initiated at late stages of the disease.
A total of 10 rhesus macaques were infected with a uniformly lethal dose of MARV. Initiation of NP-718m–LNP treatment occurred 4 days or 5 days post infection (p.i.) and consisted of 7 daily i.v. bolus doses. The control animals were not treated with NP-718m–LNP. All 4 animals given NP-718m–LNP beginning on day 4 p.i. survived MARV infection, while 2 of 4 animals given NP-718m–LNP beginning on day 5 p.i. survived infection. Both untreated control animals succumbed on day 8 p.i. (Figure 1A), which was consistent with a mean time to death of 7.86 days across 14 historical control animals (T.W. Geisbert, unpublished observations). To assess whether the 2 treated animals succumbed as a result of the emergence of viral escape mutants to the siRNA, virus was isolated from these animals and subjected to an in vitro infection assay in HepG2 cells. NP-718m–LNP treatment on infected HepG2 cells showed comparable antiviral activity between virus isolated from treated animals that had succumbed and the original virus inoculum (Supplemental Figure 1; supplemental material available online with this article; https://doi.org/10.1172/JCI96185DS1).
Figure 1. NP-718m mediates effective silencing of both MARV and RAVV and confers survival benefit to lethally infected NHPs.

(A) Despite a single nucleotide mismatch present in NP-718m against RAVV target sequences, NP-718m showed activity similar to that of NP-718m RAVV in a dual-luciferase reporter assay assessment of siRNA activity (NP-718m RAVV had perfect complementarity to RAVV sequences after mismatch correction). The target sequence for NP-143m was conserved against both MARV and RAVV. Six doses of each siRNA at 0.0032 nM, 0.016 nM, 0.008 nM, 0.4 nM, 2 nM, and 10 nM, respectively, were assessed in HepG2 cells transfected with a luciferase reporter plasmid encoding MARV or RAVV NP mRNA sequences. Cell lysates were quantitated for Renilla luciferase (fused to MARV or RAVV target transgene expression) and firefly luciferase signals. The Renilla luciferase signal was normalized to the firefly luciferase signal and expressed as the percentage of gene expression relative to a plasmid-only control (pDNA) assigned a value of 100%. Data represent the mean of technical triplicates ± SD of 1 experiment. NP-718m–LNP treatment conferred survival benefit to rhesus macaques lethally infected with (B) MARV or (C) RAVV. *P = 0.0357, by 1-tailed Fisher’s exact test.
NP-718m–LNP treatment results in the survival of RAVV-infected NHPs when treatment is initiated at early onset and late stages of the disease.
A single nucleotide mismatch is present in the NP-718m target site in RAVV NP mRNA (Figure 1B). In vitro assessment of siRNA activity against MARV or RAVV NP transcript sequences in a dual-luciferase reporter assay predicted that this mismatch would have minimal impact on NP-718m anti-RAVV activity (Figure 1B). Previous results assessing the efficacy of NP-718m in lethally infected guinea pig models suggested a decreased efficacy against RAVV (treatment resulted in 60% survival), whereas NP-718m–LNP treatment of MARV-infected animals produced complete protection (8). Interestingly, in the previous study, treatment of RAVV-infected guinea pigs with a siRNA cocktail containing both NP-718m and a siRNA with perfect target site complementarity to RAVV (NP-143m) conferred complete protection. To determine whether these results translated into the more stringent NHP model of RAVV infection, we assessed NP-718m and the cocktail treatment consisting of NP-718m and NP-143m in 5 rhesus macaques lethally infected with RAVV, with treatment beginning 3 days p.i. In contrast to the previous results obtained in the guinea pig model, we found that treatment with NP-781m–LNP alone was sufficient to confer 100% survival to the lethally infected animals. Because no additional advantage was observed in survival or in the control of viral load in animals receiving the NP-718m plus NP-143m cocktail when compared with animals treated with NP-718m alone, we conducted a subsequent study in 5 RAVV-infected macaques to examine the efficacy of the single siRNA NP-718m when treatment was initiated 6 days p.i., only 1 day prior to when the untreated control animal in the study succumbed to disease (mean time to death across 3 historical control animals of 9 days; T.W. Geisbert, unpublished observations). Notably, even when treatment was initiated at this later stage of disease, all animals treated with NP-718m–LNP survived infection (Figure 1C), and treatment-associated survival was found to be statistically significant (P = 0.0357, Fisher’s exact test).
Viremia and viral RNA load are effectively reduced upon NP-718m–LNP treatment.
NP-718m–LNP treatment on day 4 p.i. and day 5 p.i. with MARV was able to reduce peak viremia by up to 4 log10 PFU/ml, as measured by plaque assay, when compared with untreated control animals (Figure 2A). We observed similar reductions with NP-718m–LNP treatment of RAVV-infected animals, which was statistically significant for day 6 (*P < 0.0001, 1-way ANOVA with Dunnett’s correction for multiple comparisons) (Figure 2B). Viral control was rapid, with reductions observed by the next sampling time point, 3 days after treatment initiation. Treatment with NP-718m–LNP reduced the viral RNA load in the blood by a range of 2 to 4 log10 units by day 8 p.i. in MARV-infected macaques and by day 9 p.i. in RAVV-infected animals when compared with viral loads in the untreated animals (Figure 2, C and D). Animals administered NP-718m–LNP treatment also had reduced viral RNA loads in tissues at the study’s end when compared with viral loads in the untreated animals euthanized during peak viremia (Figure 2, E and F).
Figure 2. NP-718m–LNP treatment effectively controls MARV or RAVV viral replication in blood and tissues.

(A) NP-718m–LNP treatment mediates up to 4 log reductions in viremia in MARV-infected NHPs when compared with untreated controls. Protection was greatest when treatment was initiated at earlier p.i. time points. n = 4 treated animals and n = 1 untreated control each for 1 study 4 days p.i. and 1 study 5 days p.i. *P = 0.0159, by 1-tailed, unpaired t test (day 8). (B) Similar reductions in viremia were also observed in RAVV-infected animals treated with NP-718m alone or in a cocktail with NP-143m. n = 2 for NP-718m–LNP and NP-718m plus NP-143m–LNP treatment groups 3 days p.i.. n = 4 treated animals for the day-6 p.i. study. n = 1 untreated control for each study. NP-718m treatment resulted in reductions in viral RNA present in the blood for (C) MARV (*P = 0.0033, by 1-tailed, unpaired t test; day 8) and (D) RAVV, as assessed by viral RNA qRT-PCR. n = 4 treated animals and n = 1 untreated control each for the MARV day-4 p.i. study and the MARV day-5 p.i. study. n = 2 for NP-718m–LNP and NP-718m plus NP-143m–LNP treatment groups for the RAVV day-3 p.i. study and n = 4 treated animals for the RAVV day-6 p.i. study. n = 1 untreated control for each study. Treatment was also able to control RAVV (E) and MARV (F) viral replication in tissues, including those in immune-privileged sites such as the reproductive organs, brain, and eye. The lower limit of quantification (LLOQ) for viral RNA qRT-PCR was 1 × 105 GEq/ml or GEq/g. LN, lymph node.
NP-718m–LNP treatment ameliorates clinical and pathological disease associated with MARV and RAVV HF.
Animals treated with NP-718m–LNP exhibited less severe signs of clinical illness, such as depression, anorexia, petechial rash, and hemorrhage, which are associated with MARV or RAVV infection (Figure 3, A and B; see Tables 1 and 2 for individual animal data). We assessed serum samples from all infected animals for liver- and kidney-associated function markers such as alanine aminotransferase (ALT) and blood urea nitrogen (BUN), respectively, which dramatically increase during MARV or RAVV disease–mediated organ decompensation. We found that NP-718m–LNP treatment was able to protect against liver (Figure 3, C and D, and Figure 4, A–D) and renal damage (Figure 3, E and F, and Figure 4, E and F) induced by MARV or RAVV. All infected animals, independent of treatment, showed thrombocytopenia, lymphopenia, and/or granulocytosis during the peak viremia period, which is typical of MARV and RAVV HF (Figure 5, A–F). Examination of tissue sections showed various degrees of lesions and MARV (Figure 6, A–D) or RAVV (Figure 7, A–D) antigen in the untreated control animals, consistent with historical controls (10). Tissue lesions in NP-718m–LNP–treated animals that did not survive MARV infection were comparable to those in untreated control animals (Figure 6, I–L). None of the tissue sections from the NP-718m–LNP–treated animals that survived out to day 28 p.i. showed any lesions or clear immunoreactivity for MARV or RAVV antigen (Figure 6, E–H and Figure 7, E–H). This ability of NP-718m–LNP to protect against disease is also supported by the relative lack of change in the clinical scores of the treated animals from the baseline scores (Figure 3, A and B). These data collectively suggest that LNP delivery of NP-718m siRNA mediates effective and potent broad-spectrum therapeutic treatment of MARV or RAVV infection.
Figure 3. NP-718m–LNP treatment ameliorates disease symptoms in MARV or RAVV HF.

(A) Clinical scores of untreated or NP-718m–LNP–treated MARV-infected animals. n = 4 treated animals and n = 1 untreated control each for the MARV day-4 p.i. study and the MARV day-5 p.i. study. (B) Clinical scores of untreated or NP-718m–LNP–treated animals lethally infected with RAVV. n = 2 for NP-718m–LNP and NP-718m plus NP-143m–LNP treatment groups for the RAVV day-3 p.i. study and n = 4 treated animals for the RAVV day-6 p.i. study. n = 1 untreated control for each study. Scoring changes measured from baseline included posture and activity level, attitude and behavior, food and water intake, weight, respiration, and disease manifestations such as visible rash, hemorrhage, ecchymosis, or flushed skin. A score of ≥ 9 indicated that an animal met the criteria for euthanasia. (C) Animals treated with NP-718m showed amelioration of liver dysfunction typical of MARV infection. (D) NP-718m–LNP treatment protected against liver dysfunction in macaques lethally infected with RAVV. (E) NP-718m–LNP treatment ameliorated kidney dysfunction resulting from MARV disease. (F) Both NP-718m alone or in a siRNA cocktail with NP-143m conferred similar protection against kidney dysfunction in RAVV-infected NHPs.
Table 1. Clinical description and outcome of MARV-infected NHPs.

Table 2. Clinical description and outcome of RAVV-infected NHPs.

Figure 4. NP-718m–LNP treatment mitigates liver and kidney dysfunction in MARV or RAVV HF.

(A and C) Reductions observed in AST and GGT elevations typical of MARV disease. (B and D) Similar reductions were observed in RAVV-infected animals treated with NP-718m–LNP alone or with the NP-718m plus NP-143m siRNA cocktail, with the greatest amelioration observed in animals in which treatment was initiated within an earlier p.i. time frame. NP-718m treatment was also able to maintain kidney function against (E) MARV or (F) RAVV infection. n = 4 treated animals and n = 1 untreated control each for the MARV day-4 p.i. study and the MARV day-5 p.i. study. n = 2 for NP-718m–LNP and NP-718m plus NP-143m–LNP treatment groups for the RAVV day-3 p.i. study, and n = 4 treated animals for the RAVV day-6 p.i. study. n = 1 untreated control for each study.
Figure 5. MARV- and RAVV-infected rhesus macaques experienced hematological changes after infection.

Changes were observed independent of whether the animals were treated with NP-718m–LNP. (A and B) Thrombocytopenia after MARV and RAVV infection, respectively. (C and D) Lymphopenia and (E and F) granulocytosis. n = 4 treated animals and n = 1 untreated control each for the MARV day-4 p.i. study and the MARV day-5 p.i. study. n = 2 for NP-718m–LNP and NP-718m plus NP-143m–LNP treatment groups for the RAVV day-3 p.i. study, and n = 4 treated animals for the RAVV day-6 p.i. study. n = 1 untreated control for each study.
Figure 6. Comparison of MARV pathology and antigen in representative tissues of rhesus monkeys either treated or not with NP-718m–LNP.
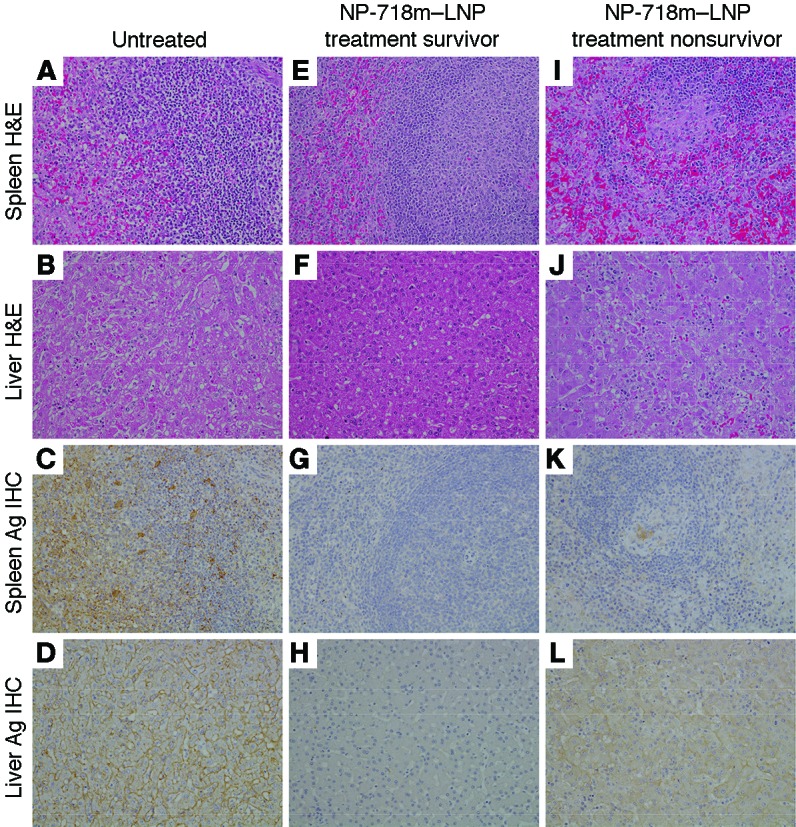
(A and I) Spleen: diffuse lymphoid depletion of the white pulp and fibrin deposition in the red pulp in a MARV-infected control animal and a nonsurviving NP-718m–LNP–treated animal. (C and K) Spleen: diffuse cytoplasmic immunolabeling of dendriform mononuclear cells in the red and white pulp of a MARV-infected control animal and a nonsurviving NP-718m–LNP–treated animal. No overt lesion (E) or immunolabeling (G) was observed in the spleen of a surviving NP-718m–LNP–treated animal. (B and J) Liver: multifocal necrotizing hepatitis, sinusoidal leukocytosis, and eosinophilic cytoplasmic inclusion bodies in a MARV-infected control animal and a nonsurviving NP-718m–LNP–treated animal. (D and L) Liver: diffuse cytoplasmic immunolabeling (brown color) of sinusoidal lining cells, Kupffer cells, and hepatocytes in a nonsurviving NP-718m–LNP–treated animal. No overt lesion (F) or immunolabeling (H) was observed in liver from a surviving NP-718m–LNP–treated animal. All representative images (original magnification, ×20) are from surviving NP-718m-7–treated animal, nonsurviving NP-718m-8–treated animal, and an untreated control animal 5 days p.i. Ag, antigen.
Figure 7. Comparison of RAVV pathology and antigen in representative tissues of rhesus monkeys either treated or not with NP-718m–LNP.

(A) Spleen: diffuse lymphoid depletion of the white pulp and fibrin deposition in the red pulp in a RAVV-infected control animal. (C) Spleen: diffuse cytoplasmic immunolabeling of dendriform mononuclear cells in the red and white pulp of a RAVV-infected control animal. No overt lesion (E) or immunolabeling (G) was observed in spleen of an NP-718m–LNP–treated animal. (B) Liver: multifocal necrotizing hepatitis, sinusoidal leukocytosis, and eosinophilic cytoplasmic inclusion bodies in a RAVV-infected control animal. (D) Liver: diffuse cytoplasmic immunolabeling (brown color) of sinusoidal lining cells, kupffer cells, and hepatocytes. No overt lesion (F) or immunolabeling (H) was observed in the liver of a NP-718m–LNP–treated animal. All representative images were taken (original magnification, ×20) from the NP-718m-11–treated animal or an untreated control animal 6 days p.i.
Discussion
Human fatality rates have ranged from 23% for some isolates of MARV and/or RAVV to as high as 90% for the Angola strain of MARV (1, 2). As evidenced by the recent Ebola virus disease epidemic of unprecedented size in West Africa (11, 12), there is a real and urgent need to develop effective countermeasures against these Tier 1 Select Agents (United States Federal Select Agent Program). Given the rapid disease onset associated with filoviral HF, the ability of a medical countermeasure to provide therapeutic treatment (upon or after initial clinical signs) and possess broad-spectrum activity against multiple species of virus would confer significant benefit in acute situations when the time to treatment initiation is at its most critical.
Historically, most studies in NHPs examined interventions against MARV infection at time points before the animals were viremic or showed any evidence of clinical illness. Furthermore, prior to this year, to our knowledge, there have been no studies of therapeutics against RAVV. Early studies using recombinant vesicular stomatitis virus–based vaccines (13, 14), polyclonal NHP antibodies (15), antisense phosphorodiamidate morpholino oligomers (16), and a broad-spectrum nucleoside analog (17) all demonstrated potential in protecting NHPs against lethal MARV infection when treatment was initiated within 48 hours of exposure, before the onset of viremia or clinical illness. Subsequent work showed that antisense phosphorodiamidate morpholino oligomers could provide partial protection for NHPs when treatment was initiated as late as day 4 p.i. in a macaque model of MARV infection. However, an important consideration in these previous studies is the strain of MARV used. Specifically, 1 MARV postexposure treatment study in NHPs (15) evaluated efficacy against the Ci67 strain associated with the initial MARV outbreak in Europe in 1967 that resulted in 7 fatalities among 31 patients (18). All but 1 other MARV postexposure treatment study in NHPs evaluated efficacy against the Musoke strain. The Musoke strain was isolated from a nonfatal human case (19) and has been associated with a much more protracted disease course in NHPs, in which the animals typically succumb 10–13 days after exposure (13, 14, 16, 17). In contrast, the Angola strain is the causative agent of the largest outbreak of MARV HF thus far, with a mortality rate of 90% in over 200 confirmed cases (2). It would be highly desirable for advanced development of any MARV countermeasure to include the demonstration of efficacy against MARV-Angola, the most pathogenic MARV strain, which also manifests as a more rapid disease course (7–9 days) in NHPs (10). Very recent studies are beginning to address the Angola strain of MARV as well as RAVV. Notably, it was reported that a single monoclonal antibody against the MARV glycoprotein provided complete protection against the Angola strain of MARV when treatment was initiated on day 4 and near-complete protection when treatment was initiated on day 5 (20). Importantly, the same antibody provided complete protection against RAVV when treatment of NHPs was delayed until day 5 after exposure. Prediction of broad-spectrum activity for monoclonal antibodies is difficult, and empirical testing is required to confirm activity against multiple virus species. In contrast, the sequence-dependent mechanism of action of RNAi therapeutics is amenable to design features that can incorporate broad-spectrum activity through the identification of conserved target nucleotide sequences present in multiple species.
The goal of the current study was to determine whether a single siRNA, NP-718m, was able to confer broad-spectrum protection against the sequence-divergent MARV and RAVV variants in NHPs, when treatment was started at considerably advanced disease stages. We have now demonstrated that NP-718m–LNP treatment completely protects rhesus monkeys against lethal MARV and RAVV HF when treatment begins on days 4 and 6 after exposure, respectively. These studies are also the first to our knowledge to report 100% postexposure protection of NHPs from RAVV when treatment was delayed until day 6 after exposure, just 1 day prior to when the control animals in this study began to succumb to disease. This ability to confer protection against both MARV and RAVV is important, since all strains of MARV and RAVV are endemic to the same regions of Africa and have even been identified during the same outbreaks (21).
NP-718m–LNP treatment resulted in 50% survival when administration began 5 days p.i. in animals infected with the MARV-Angola variant. We found no significant delay in the time to death of the treated animals that succumbed and no evidence of viral escape mutants. It is possible that at the start of treatment, these animals may already have been experiencing notable pathology attributable to the disease, which limited the benefit derived from treatment. Similar observations have been made in animals infected with Sudan virus that were treated with siRNA-LNP 5 days p.i. (6) and also reflect findings in the TKM-Ebola-Guinea phase II clinical trial, which suggested that high viral loads and existing organ injury in subjects probably obscured the ability to detect treatment benefit (7). In the surviving treated animals, viral RNA was detected in most of the tissues assayed, such as the axillary and inguinal lymph nodes and spleen (Figure 2, E and F). We detected no lesions or antigen expression in these tissues by histological analysis, suggesting that these are not sites of active viral replication. The presence of viral RNA may reflect retention in immune sites during the development of immunological memory or remnants of a productive adaptive antiviral immune response, as was recently noted for Lassa virus infection (22).
Our results in the stringent NHP model of lethal infection with clinical virus corroborates earlier work demonstrating the ability of NP-718m to effectively protect against all major MARV strains and RAVV in guinea pigs (8). Interestingly, in the NHP model, which more closely approximates human disease, NP-718m–LNP treatment conferred better protection than what was observed previously in infected RAVV guinea pigs, despite the presence of a 1-nucleotide mismatch in the siRNA target sequence against RAVV. The location of this nucleotide change at position 9 in the antisense strand of NP-718m is within the previously predicted low-tolerance region (positions 5–11), in which mismatches have a higher probability of negatively impacting siRNA activity (23–26). However, mismatch tolerance is also dependent on the identity of the nucleotides involved, and the target:siRNA mismatch described here (a G:U wobble pair) is particularly well tolerated (26), a finding that is also consistent with our siRNA activity assessment (Figure 1B). As the target site for NP-718m was not found to differ between the clinical and rodent-adapted MARV and RAVV virus used to infect NHPs and guinea pigs, respectively, the different survival outcomes with treatment in these 2 animal models may reflect differences in disease course (27), such as the more profound weight loss observed in guinea pigs during peak viremia or subtle changes potentially attributable to the genetic changes observed in the guinea pig–adapted virus strains. These observations further emphasizes the need to evaluate medical countermeasures in NHP models that recapitulate multiple aspects of human filovirus HF.
In this study, siRNA encapsulated in a LNP was administered through daily i.v. injection, a route of administration that may present challenges in resource-scarce settings such as Central Africa. The development of alternate routes such as intradermal (28) or subcutaneous (29) routes for the delivery of siRNA may enable simpler and easier methods of administration for antifiloviral siRNA therapeutics in the future.
Together, the results presented here strongly support the further development of NP-718m–LNP as a broad-spectrum therapeutic treatment for MARV and RAVV infection in humans. Its success against the strain of MARV responsible for the largest and most lethal outbreak in history represents a substantial advance in countermeasure development, particularly when coupled with the complete protection seen when treatment is administered late in the disease course. In addition, the ability of a single siRNA to protect against multiple sequence-divergent virus variants, as demonstrated here, represents a proof of principle of the utility of rational design in identifying siRNAs with broad-spectrum activity and a key milestone in progress toward RNAi therapeutics.
Methods
LNP encapsulation of siRNA.
The design, chemical modification, and abrogated immunostimulation testing of NP-718m and NP143m siRNA have been previously described (8). NP-718m or NP143m siRNA (8) (synthesized by Integrated DNA Technologies) was encapsulated in LNP by the process of spontaneous vesicle formation as previously reported (30). The resulting LNPs were dialyzed against PBS and filter sterilized through a 0.2-μm filter before use. Particle sizes were highly consistent, ranging from 78 to 79 nm for all studies, with low polydispersity values (0.04–0.09). High encapsulation efficiencies (97%–98%) were obtained for material prepared for each study.
Dual-luciferase reporter assay.
siRNA activity against MARV and RAVV NP mRNAs was confirmed in vitro using a dual-luciferase reporter assay assessing reductions in the expression of MARV or RAVV NP transgenes cloned into the psiCHECK2 vector (Promega) in HepG2 cells (ATCC; HB-8065) grown in MEM-supplemented media (Gibco, Thermo Fisher Scientific). Gene synthesis and subcloning into psiCHECK2 was conducted by GenScript. HepG2 cells were reverse-transfected with Lipofectamine 2000 (Invitrogen, Thermo Fisher Scientific) complexes containing 0.75 μg psiCHECK2 plasmid and LNP (20–0.8 nM) in 96-well plates. LNPs containing siRNA targeting Renilla luciferase (Rluc) or a nonspecific target (ApoB) were used as positive and negative controls, respectively. Cells were lysed 48 hours after transfection. Luminescence was detected with the Dual Luciferase Reporter Assay kit (Promega) according to the manufacturer’s protocol using a Berthold Luminometer (Berthold Detection Systems). The Renilla luciferase signal (reflecting target transgene expression) was normalized to the firefly luciferase signal and expressed as the percentage of gene expression relative to a plasmid-only control.
Animal infection.
Twenty healthy adult rhesus macaques (Macaca mulatta) of Chinese origin (4–6 kg) (PrimGen) were used to conduct four separate studies (4 treated animals and 1 control per study; control animals were untreated, as previous studies treating filovirus-infected NHPs with nonspecific siRNAs encapsulated in LNP showed no evidence of nonspecific protection or delay in death [refs. 4, 6, 9]). Animals were randomized using Microsoft Excel into treatment or control groups. Ten animals were inoculated i.m. with a target dose of 1,000 PFU MARV-Angola H.sapiens-tc/ANG/2005/Angola-200501379 (Vero E6, passage 2 (ATCC; CRL-1586) (the actual dose of MARV was determined to be 1,063 PFU for the 96-hour delay to treatment study and 1,138 PFU for the 120-hour delay to treatment study). In addition, 10 animals were inoculated i.m. with a target dose of 1,000 PFU RAVV H.sapiens-tc/KEN/1987/Kitum Cave-810040 (Vero E6, passage 3) (the actual dose of RAVV was determined to be 1,125 PFU for the 72-hour delay to treatment study and 1,163 PFU for the 120-hour delay to treatment study). In the first study, NP-718m–LNP (0.5 mg/kg total siRNA dose) was administered to 4 macaques by i.v. bolus infusion 96 hours after MARV infection, while 1 control animal received no treatment. All treated animals were given a total of 7 daily doses of NP-718m–LNP after MARV infection. In the second study using 5 macaques, NP-718m–LNP (0.5 mg/kg) was administered to 4 macaques by i.v. bolus infusion 120 hours after MARV infection, while the control animal received no treatment. The 4 animals received additional treatments of NP-718m–LNP on days 6, 7, 8, 9, 10, and 11 after MARV infection. In the third study involving 5 macaques, NP-718m–LNP (0.5 mg/kg) was administered to 2 macaques, and an equal mixture of NP-718m–LNP and NP-143m–LNP (0.5 mg/kg total dose) was administered to 2 animals by i.v. bolus infusion 72 hours after RAVV infection, while the control animal received no treatment. Treated animals received additional doses of the NP siRNAs on days 4, 5, 6, 7, 8, and 9 after RAVV challenge. In the final study with 5 macaques, NP-718m–LNP (0.5 mg/kg) was administered to 4 macaques by i.v. bolus infusion 144 hours after RAVV infection, while the control animal was not treated. These animals received additional treatments of NP-718m–LNP on days 7, 8, 9, 10, 11, and 12 after RAVV challenge. All 20 animals underwent physical examination, and blood was collected at the time of infection and at various times after MARV or RAVV infection. In addition, all animals were monitored daily and scored for disease progression with an internal filovirus scoring protocol approved by the IACUC of the University of Texas Medical Branch. The scoring changes measured from baseline included posture and activity level, attitude and behavior, food and water intake, weight, respiration, and disease manifestations such as visible rash, hemorrhage, ecchymosis, or flushed skin. A score of 9 or higher indicated that an animal met the criteria for euthanasia. These studies were not conducted in a blinded fashion.
Detection of viremia.
RNA was isolated from whole blood using the Viral RNA Mini Kit (QIAGEN), with 100 μl of blood added to 600 μl of buffer AVL. The primers and probe targeting the NP gene of MARV were used for quantitative real-time PCR (qRT-PCR). The probe used here was 6-carboxyfluorescein (6FAM)-5′5′CCCATAAGGTCACCCTCTT-3′-6 carboxytetramethylrhodamine (TAMRA) (Life Technologies, Thermo Fisher Scientific) (8). MARV RNA was detected using the CFX96 Detection System (Bio-Rad Laboratories) and One-Step Probe qRT-PCR kits (QIAGEN) with the following cycle conditions: 50°C for 10 minutes, 95°C for 10 seconds, and 40 cycles of 95°C for 10 seconds and 59°C for 30 seconds. The Ct values representing MARV genomes were analyzed with CFX Manager Software, and data are shown as genome equivalents (GEq). To create the GEq standard, RNA from MARV stocks was extracted, and the number of MARV genomes was calculated using Avogadro’s number and the molecular weight of the MARV genome.
Virus titration was performed by plaque assay with Vero E6 cells (ATCC; CRL-586) from all serum samples as previously described (10, 13, 14, 31). Briefly, 10-fold serial dilutions of the samples were adsorbed to Vero E6 monolayers in duplicate wells (200 μl); the limit of detection was 15 PFU/ml.
Hematology and serum biochemistry.
Total white blood cell counts, white blood cell differentials, red blood cell counts, platelet counts, hematocrit values, total hemoglobin concentrations, mean cell volumes, mean corpuscular volumes, and mean corpuscular hemoglobin concentrations were analyzed from blood collected in tubes containing EDTA using a laser-based hematologic analyzer (Beckman Coulter). Serum samples were tested for concentrations of albumin, amylase, ALT, aspartate aminotransferase (AST), alkaline phosphatase (ALP), γ-glutamyltransferase (GGT), glucose, cholesterol, total protein, total bilirubin (TBIL), BUN, creatine (CRE), and C-reactive protein (CRP) by using a Piccolo Point-of-Care Analyzer and Biochemistry Panel Plus Analyzer Discs (Abaxis).
Histopathology and IHC.
Necropsy was performed on all subjects. Tissue samples from all major organs were collected for histopathologic and immunohistochemical examination, immersion-fixed in 10% neutral-buffered formalin, and processed for histopathology as previously described (9). For IHC, anti-Marburgvirus genus immunoreactivity was detected using an anti-MARV VP40 protein rabbit primary antibody (Integrated BioTherapeutics; 0303-001) at a 1:4,000 dilution. In brief, tissue sections were processed for IHC using the Dako Autostainer. The secondary antibody used was biotinylated goat anti-rabbit IgG (VECTOR Laboratories) at 1:200, followed by Dako LSAB2 streptavidin-HRP. Slides were developed with Dako DAB chromagen and counterstained with hematoxylin. Nonimmune rabbit IgG was used as a negative control.
Statistics.
Statistical analyses were conducted using GraphPad Prism 6.0 (GraphPad Software). A 1-tailed, unpaired Student’s t test was used to determine whether differences in the means were present. A Fisher’s exact test was used to compare differences in survival between the treated and control groups. A P value of less than 0.05 was considered significant.
Study approval.
The animal studies were performed at the Galveston National Laboratory of the University of Texas Medical Branch at Galveston and approved by the IACUC of the University of Texas Medical Branch.
Author contributions
RUB and MR designed the siRNA, and RUB conducted the dual luciferase reporter studies. EPT, CEM, ACHL, IM and TWG conceived and designed the NHP studies. CEM, JBG, DJD, RWC, and TWG performed the NHP infection and treatment experiments and conducted clinical observations of the animals. JBG, KNA, and DJD performed the clinical pathology assays. JBG performed the MARV and RAVV infectivity assays. CEM and KNA performed the PCR assays. EPT, CEM, JBG, KNA, DJD, RWC, KAF, ASK, ACHL, and TWG analyzed the data. KAF performed histological and immunohistochemical analyses. EPT, CEM, ACHL, and TWG wrote the manuscript. All authors had access to all of the data and approved the final version of the manuscript.
Supplementary Material
Acknowledgments
We thank Viktoriya Borisevich (University of Texas Medical Branch, Galveston, Texas, USA) for assistance with clinical pathology assays performed in the GNL BSL-4 laboratory and Shaun Klassen and Helen Yuen (Arbutus Biopharma Corporation, Burnaby, British Columbia, Canada) for their assistance with formulating material. This study was supported by the Department of Health and Human Services, NIH grant U19AI109711 (to TWG and EPT) and grant UC7AI094660 for BSL-4 operations support of the Galveston National Laboratory. The opinions, interpretations, conclusions, and recommendations are those of the authors and are not necessarily endorsed by the University of Texas Medical Branch.
Version 1. 11/06/2017
Electronic publication
Version 2. 12/01/2017
Print issue publication
Footnotes
Conflict of interest: A.C.H. Lee, I. MacLachlan, and T.W. Geisbert claim intellectual property rights regarding RNAi for the treatment of filovirus infections. I. MacLachlan and T.W. Geisbert are co-inventors on US Patent 7,838,658, “siRNA silencing of filovirus gene expression,” and A.C.H. Lee, I. MacLachlan, and T.W. Geisbert are co-inventors on US Patent 8,716,464, “Compositions and methods for silencing Ebola virus gene expression.”
Reference information: J Clin Invest. 2017;127(12):4437–4448.https://doi.org/10.1172/JCI96185.
Contributor Information
Chad E. Mire, Email: chmire@UTMB.EDU.
Raul Ursic-Bedoya, Email: raursic@hotmail.com.
Marjorie Robbins, Email: marjorierobbins@gmail.com.
Robert W. Cross, Email: rwcross@utmb.edu.
Ian MacLachlan, Email: ian272@shaw.ca.
References
- 1.Rougeron V, Feldmann H, Grard G, Becker S, Leroy EM. Ebola and Marburg haemorrhagic fever. J Clin Virol. 2015;64:111–119. doi: 10.1016/j.jcv.2015.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Towner JS, et al. Marburgvirus genomics and association with a large hemorrhagic fever outbreak in Angola. J Virol. 2006;80(13):6497–6516. doi: 10.1128/JVI.00069-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zatsepin TS, Kotelevtsev YV, Koteliansky V. Lipid nanoparticles for targeted siRNA delivery - going from bench to bedside. Int J Nanomedicine. 2016;11:3077–3086. doi: 10.2147/IJN.S106625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Geisbert TW, et al. Postexposure protection of non-human primates against a lethal Ebola virus challenge with RNA interference: a proof-of-concept study. Lancet. 2010;375(9729):1896–1905. doi: 10.1016/S0140-6736(10)60357-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thi EP, et al. Lipid nanoparticle siRNA treatment of Ebola-virus-Makona-infected nonhuman primates. Nature. 2015;521(7552):362–365. doi: 10.1038/nature14442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Thi EP, et al. Rescue of non-human primates from advanced Sudan ebolavirus infection with lipid encapsulated siRNA. Nat Microbiol. 2016;1(10):16142. doi: 10.1038/nmicrobiol.2016.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dunning J, et al. Experimental treatment of Ebola virus disease with TKM-130803: a single-arm phase 2 clinical trial. PLoS Med. 2016;13(4):e1001997. doi: 10.1371/journal.pmed.1001997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ursic-Bedoya R, et al. Protection against lethal Marburg virus infection mediated by lipid encapsulated small interfering RNA. J Infect Dis. 2014;209(4):562–570. doi: 10.1093/infdis/jit465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thi EP, et al. Marburg virus infection in nonhuman primates: Therapeutic treatment by lipid-encapsulated siRNA. Sci Transl Med. 2014;6(250):250ra116. doi: 10.1126/scitranslmed.3009706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Geisbert TW, et al. Marburg virus Angola infection of rhesus macaques: pathogenesis and treatment with recombinant nematode anticoagulant protein c2. J Infect Dis. 2007;196 Suppl 2:S372–S381. doi: 10.1086/520608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Baize S, et al. Emergence of Zaire Ebola virus disease in Guinea. N Engl J Med. 2014;371(15):1418–1425. doi: 10.1056/NEJMoa1404505. [DOI] [PubMed] [Google Scholar]
- 12.Bagcchi S. Ebola haemorrhagic fever in west Africa. Lancet Infect Dis. 2014;14(5):375. doi: 10.1016/S1473-3099(14)70034-9. [DOI] [PubMed] [Google Scholar]
- 13.Daddario-DiCaprio KM, et al. Postexposure protection against Marburg haemorrhagic fever with recombinant vesicular stomatitis virus vectors in non-human primates: an efficacy assessment. Lancet. 2006;367(9520):1399–1404. doi: 10.1016/S0140-6736(06)68546-2. [DOI] [PubMed] [Google Scholar]
- 14.Geisbert TW, Bausch DG, Feldmann H. Prospects for immunisation against Marburg and Ebola viruses. Rev Med Virol. 2010;20(6):344–357. doi: 10.1002/rmv.661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dye JM, et al. Postexposure antibody prophylaxis protects nonhuman primates from filovirus disease. Proc Natl Acad Sci U S A. 2012;109(13):5034–5039. doi: 10.1073/pnas.1200409109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Warren TK, et al. Advanced antisense therapies for postexposure protection against lethal filovirus infections. Nat Med. 2010;16(9):991–994. doi: 10.1038/nm.2202. [DOI] [PubMed] [Google Scholar]
- 17.Warren TK, et al. Protection against filovirus diseases by a novel broad-spectrum nucleoside analogue BCX4430. Nature. 2014;508(7496):402–405. doi: 10.1038/nature13027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Martini GA, Knauff HG, Schmidt HA, Mayer G, Baltzer G. A hitherto unknown infectious disease contracted from monkeys. “Marburg-virus” disease. Ger Med Mon. 1968;13(10):457–470. [PubMed] [Google Scholar]
- 19.Smith DH, et al. Marburg-virus disease in Kenya. Lancet. 1982;1(8276):816–820. doi: 10.1016/s0140-6736(82)91871-2. [DOI] [PubMed] [Google Scholar]
- 20.Mire CE, et al. Therapeutic treatment of Marburg and Ravn virus infection in nonhuman primates with a human monoclonal antibody. Sci Transl Med. 2017;9(384):eaai8711. doi: 10.1126/scitranslmed.aai8711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bausch DG, et al. Marburg hemorrhagic fever associated with multiple genetic lineages of virus. N Engl J Med. 2006;355(9):909–919. doi: 10.1056/NEJMoa051465. [DOI] [PubMed] [Google Scholar]
- 22.McElroy AK, et al. A case of human Lassa virus infection with robust acute T-cell activation and long-term virus-specific T-cell responses. J Infect Dis. 2017;215(12):1862–1872. doi: 10.1093/infdis/jix201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Elbashir SM, Harborth J, Lendeckel W, Yalcin A, Weber K, Tuschl T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature. 2001;411(6836):494–498. doi: 10.1038/35078107. [DOI] [PubMed] [Google Scholar]
- 24.Jackson AL, et al. Expression profiling reveals off-target gene regulation by RNAi. Nat Biotechnol. 2003;21(6):635–637. doi: 10.1038/nbt831. [DOI] [PubMed] [Google Scholar]
- 25.Amarzguioui M, Holen T, Babaie E, Prydz H. Tolerance for mutations and chemical modifications in a siRNA. Nucleic Acids Res. 2003;31(2):589–595. doi: 10.1093/nar/gkg147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Du Q, Thonberg H, Wang J, Wahlestedt C, Liang Z. A systematic analysis of the silencing effects of an active siRNA at all single-nucleotide mismatched target sites. Nucleic Acids Res. 2005;33(5):1671–1677. doi: 10.1093/nar/gki312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cross RW, Fenton KA, Geisbert JB, Ebihara H, Mire CE, Geisbert TW. Comparison of the pathogenesis of the Angola and Ravn strains of Marburg virus in the outbred guinea pig model. J Infect Dis. 2015;212 Suppl 2:S258–S270. doi: 10.1093/infdis/jiv182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Deng Y, et al. Transdermal delivery of siRNA through microneedle array. Sci Rep. 2016;6:21422. doi: 10.1038/srep21422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nair JK, et al. Multivalent N-acetylgalactosamine-conjugated siRNA localizes in hepatocytes and elicits robust RNAi-mediated gene silencing. J Am Chem Soc. 2014;136(49):16958–16961. doi: 10.1021/ja505986a. [DOI] [PubMed] [Google Scholar]
- 30.Ma H, et al. Formulated minimal-length synthetic small hairpin RNAs are potent inhibitors of hepatitis C virus in mice with humanized livers. Gastroenterology. 2014;146(1):63–66.e5. doi: 10.1053/j.gastro.2013.09.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hensley LE, et al. Pathogenesis of Marburg hemorrhagic fever in cynomolgus macaques. J Infect Dis. 2011;204 Suppl 3:S1021–S1031. doi: 10.1093/infdis/jir339. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.