

Abstract
p120-Catenin (p120) functions as a tumor suppressor in intestinal cancer, but the mechanism is unclear. Here, using conditional p120 knockout in Apc-sensitized mouse models of intestinal cancer, we have identified p120 as an “obligatory” haploinsufficient tumor suppressor. Whereas monoallelic loss of p120 was associated with a significant increase in tumor multiplicity, loss of both alleles was never observed in tumors from these mice. Moreover, forced ablation of the second allele did not further enhance tumorigenesis, but instead induced synthetic lethality in combination with Apc loss of heterozygosity. In tumor-derived organoid cultures, elimination of both p120 alleles resulted in caspase-3–dependent apoptosis that was blocked by inhibition of Rho kinase (ROCK). With ROCK inhibition, however, p120-ablated organoids exhibited a branching phenotype and a substantial increase in cell proliferation. Access to data from Sleeping Beauty mutagenesis screens afforded an opportunity to directly assess the tumorigenic impact of p120 haploinsufficiency relative to other candidate drivers. Remarkably, p120 ranked third among the 919 drivers identified. Cofactors α-catenin and epithelial cadherin (E-cadherin) were also among the highest scoring candidates, indicating a mechanism at the level of the intact complex that may play an important role at very early stages of of intestinal tumorigenesis while simultaneously restricting outright loss via synthetic lethality.
Keywords: Cell Biology, Oncology
Keywords: Cancer, Cell migration/adhesion, Colorectal cancer
Introduction
The vast majority of human colorectal cancer (CRC) is triggered by inactivation of the tumor suppressor adenomatous polyposis coli (APC) in one of several intestinal stem cells (ISCs) present in each intestinal crypt. The result is unscheduled translocation of β-catenin to the nucleus where tumorigenesis is initiated via interaction with the transcription factor TCF4 and unregulated transactivation of Wnt pathway target genes (e.g., AXIN2, MYC) (1–4). The ISC contributes several stem cell–restricted properties (e.g., self-renewal, mesenchymal characteristics) that are hijacked by the constitutively activated Wnt pathway and are essential for tumor initiation. Progression to malignancy involves subsequent genetic and epigenetic alterations, including mutations in genes such as KRAS, SMAD4, and TP53, which are frequently targeted in CRC, and a much larger number of less frequently mutated genes uncovered recently by whole genome sequencing. The transition to malignancy is often accompanied by the onset of metastasis, a phenomenon responsible for most cancer-related mortality and frequently linked to genetic and/or epigenetic dysregulation of the cell-cell adhesion receptor epithelial cadherin (E-cadherin) (5–8).
E-cadherin is widely regarded as the principle organizer of the epithelial phenotype (9, 10). It functions as a classic tumor suppressor in diffuse gastric cancer (11) and lobular carcinoma of the breast (12), where germline mutations in the E-cadherin gene (CDH1) are responsible for familial inheritance of one mutant allele. Early loss of the second allele (i.e., loss of heterozygosity [LOH]) then plays a causal role in tumorigenesis (11–13). In the vast majority of epithelial cancers, however, E-cadherin is considered a metastasis suppressor because it is downregulated in advanced tumors and plays a causal role in the transition to metastasis (5, 7, 14–16). Although factors underpinning the distinct differences in timing of E-cadherin loss between these groups are not understood, the former is frequently irreversible (e.g., gene mutation) while the latter often occurs by epithelial-to-mesenchymal transition (EMT) and is later reversed during colonization of distant sites (17).
As with all classical cadherins, E-cadherin mediates specific adhesion of adjacent cells via homophilic interaction between extracellular domains (9). Strong E-cadherin–mediated adhesion, however, is absolutely dependent upon formation of a multimeric complex with 3 cytoplasmic binding partners, namely α-, β-, and p120-catenins (p120) (9, 18). All members of the classical cadherin family, in fact, use these same core components to organize the actin cytoskeleton and coordinate signaling (9, 19). β-Catenin functionally links the complex to the actin cytoskeleton via recruitment of α-catenin (20–23), a vinculin-like actin-binding protein. On the other hand, p120 directly stabilizes and retains E-cadherin at the cell surface by suppressing E-cadherin endocytosis (19, 24–26) and coordinates local interactions with the cytoskeleton through recruitment of various RhoGTPase modulators (e.g., RhoGEFs, RhoGAPs, Rho kinase 1 [ROCK1], FRMD5) (27–30). Other important signaling systems that interface physically and/or functionally with the E-cadherin complex include the Wnt pathway (e.g., APC, β-catenin) (31), the HIPPO pathway (including downstream effectors YAP and TAZ) (32, 33), receptor tyrosine kinase (RTK) pathways (e.g., EGFR and downstream effectors) (34–36), the TGFβRII pathway (37), and NF-κB signaling (38, 39). Most of these pathways have oncogenic potential when dysregulated and function in opposition to the growth-suppressive effects of the E-cadherin complex in differentiated epithelia. Indeed, E-cadherin and its binding partners are extensively described in the pathology literature (based on immunohistochemical [IHC] staining) as mislocalized, downregulated, and/or lost altogether in a wide variety of carcinoma types, including CRC (40).
To evaluate the potential role of p120 (also known as CTNND1) in tumorigenesis, we previously generated a p120 conditional KO (cKO) mouse (p120fl/fl) that has been widely distributed and used to examine consequences of p120 ablation in many organ systems (39, 41–46). Although effects vary depending on the system and context, phenotypes usually involve adhesion defects and are often severe. The developing mammary gland, for example, does not form in the absence of p120 (46). In both epidermal and esophageal epithelia, p120 KO results in activation of NF-κB, leading to inflammation and tumorigenesis (38, 39), indicating an apparent cadherin-independent tumor suppressor role for p120 in these tissues. Similar experiments targeting p120 KO to the intestinal epithelium, however, led to adhesive defects, intestinal bleeding, and death by 21 days of age (42), precluding studies on tumorigenesis. To enable long-term experiments, a second-generation model was constructed using a tamoxifen-inducible (TAM-inducible) VillinCreERT2 driver and “limited” p120 KO (i.e., limited to approximately 10% of the intestinal epithelium by optimizing TAM dose) (45). In this model, nearly half of the animals developed one or more intestinal tumors by 18 months, consistent with a tumor-suppressor role for p120 in the intestine.
In the present study, we have used a combination of (i) “limited” p120 KO in Apc-sensitized mice and (ii) mutation frequencies derived by Sleeping Beauty (SB) insertional mutagenesis to elucidate the role of p120 as a tumor suppressor in CRC. Interestingly, loss of a single p120 allele induced a large (10-fold) increase in tumor multiplicity. Loss of both p120 alleles, however, was never observed in tumors and was found to be synthetic lethal with Apc LOH (i.e., cells lacking both p120 and APC were not viable). In vitro studies using organoid cultures derived from these tumors identified ROCK-dependent apoptosis as the mechanism for the observed synthetic lethality. Thus, our results reveal an “obligate” haploinsufficient tumor-suppressor role for p120 in the context of intestinal APC LOH. Moreover, in unbiased quantitative analyses based on SB-derived mutation frequencies, p120 ranked third in tumorigenic potency out of more than 919 statistically validated drivers. Notably, α-catenin and E-cadherin were also among the highest scoring candidates and evidently function similarly to p120 as obligate haploinsufficient tumor suppressors. Thus, in combination with Apc LOH, loss of any 1 of the 6 alleles encoding p120, α-catenin, or E-cadherin was sufficient to markedly accelerate tumorigenesis very early in the process. Conversely, biallelic loss at these loci was restricted at the onset of tumorigenesis by a synthetic lethal interaction with Apc LOH, providing a potential mechanistic explanation for why complete loss of E-cadherin is rarely observed prior to late-stage cancer.
Results
p120 Functions as an obligatory haploinsufficient tumor suppressor following loss of Apc.
To ascertain long-term effects of p120 loss in the intestine, we previously developed a TAM regime in p120fl/fl; VillinCreERT2 mice that reproducibly limits the extent of p120 ablation to discreet clonal pockets comprising approximately 10% of the total epithelium (hereafter termed limited p120 KO/ablation) (45). Under conditions of limited p120 ablation, these pockets of p120-null epithelium are well tolerated and maintained for the life of the animal. Almost half of these animals developed one or more intestinal tumors, but surprisingly, none of the tumors exhibited outright loss of p120 immunoreactivity. There was evidence of Wnt pathway upregulation (i.e., elevated β-catenin levels) in the majority of the tumors, but further analyses were complicated by limited amounts of tumor sample.
Here, to address the problem in a more tractable model, we crossed p120fl/fl; VillinCreERT2 mice onto an Apc1638N/+ background to generate Apc1638N/+; p120fl/fl; VillinCreERT2 mice. Experimental (+TAM) and control (corn oil vehicle alone) mice were generated as before using limited p120 ablation (45) and sacrificed 8 months later for analysis. Remarkably, the TAM-treated p120-KO cohort showed a 10-fold increase in tumor number (Figure 1A). The tumors were distributed throughout the small and large intestine (Figure 1B), but again, none of the tumors from this group exhibited complete loss of p120 (Figure 1, C and D, and Table 1) despite the presence of clonal pockets of p120-null cells distributed ubiquitously throughout the surrounding WT epithelium.
Figure 1. p120 Functions as a tumor suppressor in the intestine.

(A) Apc1638N/+; VillinCreERT2; p120fl/fl mice were treated with corn oil alone or with TAM dissolved in corn oil (n = 6 per group). At 8 months after injection, mice were sacrificed and total number of tumors throughout the small and large intestine was determined using a dissecting microscope. Tumor multiplicity was increased 10-fold in TAM-treated mice (49 ± 6 vs. 4.5 ± 1, ***P < 0.001, unpaired t test with Welch’s correction). Error bars represent SEM. (B) Tumor number was increased in TAM-treated mice in all 3 regions of the small intestine, cecum, and colon. Error bars represent SEM. PSI, proximal; MSI, middle; DSI, distal. (C) Representative small intestinal tumors from oil- and TAM-treated mice stained for p120 and β-catenin. p120 Immunoreactivity was reduced and cytosolic β-catenin enhanced in tumors (dotted lines) from both oil- (top panel) and TAM-treated (middle and bottom panels) mice as compared with adjacent normal tissue (arrowhead). Despite its genetic ablation, complete (biallelic) p120 KO was never observed in tumor tissue (e.g., area encircled by dotted line, bottom panel, n = 52 tumors), but occurred frequently in adjacent WT tissue (encircled by solid line). Scale bars: 100 μm. (D) Tumor (T) and adjacent normal (N) tissue were isolated by LCM. DNA was isolated and amplified by PCR and then separated by electrophoresis to confirm retention of 1 allele in tumor tissue. (E) ApcMin/+; VillinCreERT2; p120fl/fl mice were injected with TAM and sacrificed 3 months later. Pockets of p120-null cells (solid line) were frequently observed embedded in and around adenomatous tissue (dotted lines), yet these areas invariably proved to be WT upon closer inspection (as evidenced by absence of β-catenin and WT H&E staining, n = 191 tumors). Adjacent normal staining marked by arrowhead. Scale bars: 100 μm.
Table 1. Biallelic p120 loss is not observed in Apc-dependent tumor models.

Representative examples from immunofluorescence analyses are shown in Figure 1C. Tumors from Apc1638N/+ mice treated with vehicle alone (oil), or TAM were coimmunostained with p120- and β-catenin–specific antibodies, as indicated. Staining with p120 was noticeably reduced by varying degrees in all tumors from the Apc1638N/+ control group as compared with WT tissue (e.g., Figure 1C) and further reduced in tumors from the TAM-treated cohort (Figure 1C and Supplemental Table 1; supplemental material available online with this article; https://doi.org/10.1172/JCI77217DS1). Complete loss of p120, on the other hand, was never observed in these tumors. Additionally, all samples were costained for β-catenin to discriminate clearly between tumor and surrounding normal tissue in areas where p120 staining was lost completely. Retention of β-catenin (despite p120 loss) indicates tumor tissue, whereas β-catenin staining is absent upon loss of p120 from WT tissue (as shown in Figure 1, C and E). Finally, retention of 1 p120 floxed allele was further confirmed by DNA analysis, in which areas corresponding to tumor and putative adjacent normal tissue were isolated by laser capture microdissection (LCM) and analyzed by PCR (Figure 1D). Together, the results show that loss of a single p120 allele in the context of Apc LOH is sufficient to drive a substantial increase in tumor multiplicity. Loss of the second p120 allele, on the other hand, is restricted by synthetic lethal interaction with Apc LOH.
To further validate and extend these observations, we repeated a few key experiments in a different Apc model (i.e., ApcMin/+) and refined the experimental approach so as to isolate p120 heterozygosity as the only remaining variable. First, we asked whether the substantially stronger ApcMin allele would override the synthetic lethal phenotype observed in the Apc1638N/+ system. However, as per the Apc1638N/+ cohort, none of the 191 ApcMin/+ tumors examined exhibited complete p120 loss (Figure 1E and Table 1). For independent confirmation of tumor versus WT tissue, serial sections were examined by H&E staining (Figure 1E). As anticipated, tumor tissue was distinctly basophilic and β-catenin positive, whereas normal tissue was eosinophilic (Figure 1E) and β-catenin negative. Next, the p120 limited KO protocol was bypassed altogether by crossing inducible (p120fl/+) or crossing germline p120 heterozygous (p120+/–) mice onto the ApcMin/+ background. Although many measures of intestinal homeostasis were not obviously impacted by p120 heterozygosity (Supplemental Figure 1), there was nonetheless an appreciable increase in tumor multiplicity comparable to that induced by limited p120 knockout (Supplemental Figures 2 and 3). Thus, the increased tumor multiplicity is predominantly caused by p120 haploinsufficiency and is not dependent on the generation of p120-null foci or secondary effects emanating from the ablation protocol itself.
p120 Staining intensity is transiently reduced in intestinal progenitors during mitotic clonal expansion and permanently reduced upon Apc LOH.
The lining of the small intestine comprises a monolayer of columnar epithelium organized into a contiguous system of alternating crypts and villi, as illustrated schematically in Figure 2A. Multipotent ISCs residing at the base of each crypt give rise to “progenitors,” which in turn undergo 5 or 6 uninterrupted cycles of mitotic clonal expansion as they migrate upward through the “transit-amplifying” (TA) compartment and out onto the villi as terminally differentiated cells. Approximately 48 hours later, they undergo apoptosis as they are extruded from the villus tip into the lumen. This developmentally programmed sequence of proliferation, lineage allocation, terminal differentiation, and apoptotic extrusion is repeated every 4 to 5 days in the normally functioning organ.
Figure 2. Reduced p120 immunoreactivity at intestinal crypt base and in Apc-driven adenomas.

(A) Epithelial architecture and function in the small intestine (modified from Reya and Clevers, Nature 2005) (B) Immunolocalization of p120 and β-catenin and (C) β-catenin and Kaiso in the small intestine. Immunoreactivity in the crypt (bracket) is reduced for p120 and elevated for Kaiso as compared with villous compartment. Boxes represent area enlarged in Merge + Hoechst. Scale bars: 100 μm. (D) p120 Immunoreactivity is reduced in ApcMin/+ adenomatous tissue marked by upregulated β-catenin staining (arrowheads) relative to adjacent normal tissue (asterisks). Scale bars: 100 μm. (E) p120 and β-catenin immunoreactivity in representative Apc1638N/+ adenoma. Weak β-catenin signal is typical of Apc1638N/+. Adjacent normal tissue (asterisk). Scale bar: 100 μm. (F) Reduced p120 IHC staining in human familial adenomatous polyposis (FAP) samples. Images depict a single crypt microadenoma (first panel) and small macroadenoma (second panel, arrowheads). Normal tissue designated by arrows. Scale bar: 100 μm. (G) p120, β-catenin, and Kaiso immunoreactivity in a representative Lrig1CreERT2; Ctnnb1ΔEx3/+–derived adenoma (arrowhead). Scale bar: 50 μm.
To understand the role of p120 in this process, we initially used immunofluorescence and/or IHC staining to characterize expression of p120 and some of its binding partners in WT intestinal epithelium (Figure 2, B and C) and in the context of Apc-associated tumorigenesis (Figure 2, D and E). The staining intensity of p120 in the crypts was invariably reduced relative to that in adjacent villi, where staining was obviously enhanced along basolateral membranes (Figure 2B). In contrast, the transcription factor and p120-binding partner Kaiso was expressed at high levels in the nuclei of all cells present in the crypt and then abruptly downregulated as the cells moved upward onto the villus (Figure 2C). β-Catenin levels are known to be selectively elevated in ISC nuclei due to high levels of local Wnt signaling. However, the absolute levels of this nuclear β-catenin pool are quantitatively insignificant compared with those present in cadherin complexes and are not readily apparent at the exposure levels used here to visualize cadherin complexes. Thus, for this study, we have used Kaiso as a marker for crypt cells and as an indicator of the crypt-progenitor phenotype.
Interestingly, the lower p120 staining intensity observed in normal intestinal crypts was also evident in adenomas arising spontaneously in our Apc haploinsufficient mouse models (Figure 2, D and E). ApcMin/+ mice typically develop 30 to 100 tumors per animal, whereas mice carrying the substantially weaker Apc1638N allele consistently present with just 2 to 7 tumors (47, 48). The tumorigenic strength of the ApcMin allele typically manifests visually in the form of high levels of cytoplasmic (and nuclear) β-catenin (Figure 2D). Whereas tumorigenesis initiated by the Apc1638N allele is also associated with β-catenin stabilization, the phenomenon is substantially less pronounced and often not readily evident by standard immunofluorescent staining (Figure 2E). Notably, p120 staining is reduced in both models, irrespective of β-catenin (Figure 2, D and E). Moreover, p120 IHC staining was similarly reduced (relative to surrounding WT tissue) in early human adenomas from patients with familial adenomatous polyposis (Figure 2F), indicating that the phenomenon is not unique to the mouse, but rather a characteristic common to early intestinal adenomas from both species. Although β-catenin upregulation is often used as a marker for intestinal tumorigenesis, reduced p120 staining and/or elevated Kaiso staining were substantially more robust in essentially all of our Wnt pathway models, including Ctnnb1ΔEx3, which expresses an oncogenic β-catenin allele (Figure 2G). Not surprisingly, immunofluorescent localization of E-cadherin and α-catenin mirrored that of p120 in all models tested (Supplemental Figure 4).
Reduced p120 staining intensity reflects a change in p120 status, not levels.
We next asked whether reduced p120 staining was, in fact, synonymous with reduced p120 levels. Since immunofluorescence staining is unreliable as a means of protein quantification, we considered alternative approaches and settled on a combination of genetic manipulation and Western blotting (Figure 3). The TA compartment, which occupies the upper two-thirds of the crypt, comprises a large progenitor cell niche (under the influence of canonical Wnt signaling) that is largely dedicated to the rapid expansion of intestinal precursors (49). Normally, crypt progenitors undergo terminal differentiation during transit amplification and emerge onto the villus as mature cell types. Upon Apc LOH, however, this developmental program is short-circuited by elevated Wnt signaling, resulting in permanent retention of an undifferentiated crypt-progenitor phenotype and tumor outgrowth (50, 51).
Figure 3. Crypt-villus changes in p120 staining reflect altered status, not levels, of cadherin complex components.

(A) Homozygous Apc floxed (ApcCKO/CKO) mice were crossed with the Lrig1CreERT2 line so that addition of TAM resulted in elimination of Apc in the intestinal crypt epithelium (designated Apc cKO). p120 (green) and Kaiso (red) immunostaining in the ileum at days 0, 3, 4, and 5 after TAM administration. Kaiso staining marks upward progress of crypt progenitor phenotype (dotted line). Images are representative of 5 mice. Scale bars: 100 μm. (B) Sections of ileum from WT (top) and Apc cKO (bottom) at day 5 after TAM administration immunolabeled with antibodies to p120 (green) and Kaiso (red). Kaiso staining confirms conversion to crypt-progenitor phenotype (bottom right). Scale bars: 100 μm. (C) Quantitative protein analysis by SDS-PAGE and immunoblot. Whole cell lysates of samples from B were blotted with p120 mAbs or pAbs as indicated. Tubulin was used as loading control. Blots are representative of 3 independent experiments. (D) No change in p120 mRNA levels from WT and day 5 Apc cKO ileal samples (n = 5 mice per group) by quantitative reverse-transcriptase PCR (qRT-PCR), while Kaiso (Zbtb33) expression increased. Positive controls, cMyc and Axin2. *P < 0.05; **P < 0.001, unpaired t test with Welch’s correction. (E) Representative colonic tissue from VillinCreERT2; p120fl/fl mice immunolabeled for p120 allows discrimination of side-by-side allelic differences in p120 expression (dotted line, 0; arrowhead, 1; arrow, 2 alleles) Scale bar: 100 μm. (F) Protein levels of p120 in p120+/+ and p120+/– mice as determined by p120 immunostaining or (G) immunoblots. Scale bars: 100 μm. Tubulin was used as loading control. Blots are representative of 3 independent experiments.
In the present study, we used ApcCKO/CKO; Lrig1CreERT2/+ mice to simultaneously inactivate Apc in stem cells across the entire intestinal epithelium (Figure 3, A–D). Mice were injected with TAM, sacrificed on days 0, 3, 4, and 5, as indicated, and the intestines processed for coimmunoflourescence staining with p120-specific and Kaiso-specific antibodies (Figure 3A). As reported previously by others (49), Apc-deleted progenitor cells originating in the crypt failed to differentiate and instead continued to migrate upward onto the villus while maintaining a crypt-progenitor phenotype. By 5 days after TAM administration, the entire epithelium comprised undifferentiated crypt-progenitor cells, all of which showed strong immunoreactivity for Kaiso and reduced immunoreactivity for p120 (Figure 3B). To quantify protein levels associated with the crypt-progenitor phenotype, the transformed epithelium generated by Apc ablation was physically isolated on day 5 after TAM administration, extracted in Laemmli sample buffer, and directly measured by SDS PAGE and Western blotting (Figure 3C). Notably, although immunofluorescent staining for p120 was considerably less than that of control WT tissue (e.g., Figure 3B), Western blotting revealed that p120 levels were unchanged while β-catenin levels increased (Figure 3C). Kaiso levels also increased as expected, but E-cadherin levels remained unchanged, consistent with the unchanged levels of p120 and the well-established observation that E-cadherin stability is directly controlled by p120 (19, 25). Positive controls show that p120 allelic differences in intestines of p120 heterozygous mice were readily detected by both immunofluorescence (Figure 3, E and F) and Western blotting (Figure 3G). Thus, in spite of the sharp drop in p120 staining, the reduced signal reflects a qualitative, but not quantitative, change in p120 status.
Synthetic lethality occurs rapidly following forced ablation of p120 in Apc-null tumors.
Throughout these studies, outright p120 loss was never observed in adenomas. As shown in Figure 1, in the few instances where pockets of p120 ablation were initially indistinguishable from the surrounding tumor mass, H&E staining and/or immunostaining for β-catenin revealed clearly that these p120-null areas were in fact derived from WT tissue. Indeed, none of the 52 Apc1638N/+ or 191 ApcMin/+ tumors analyzed in long-term experiments showed any sign of p120-null content.
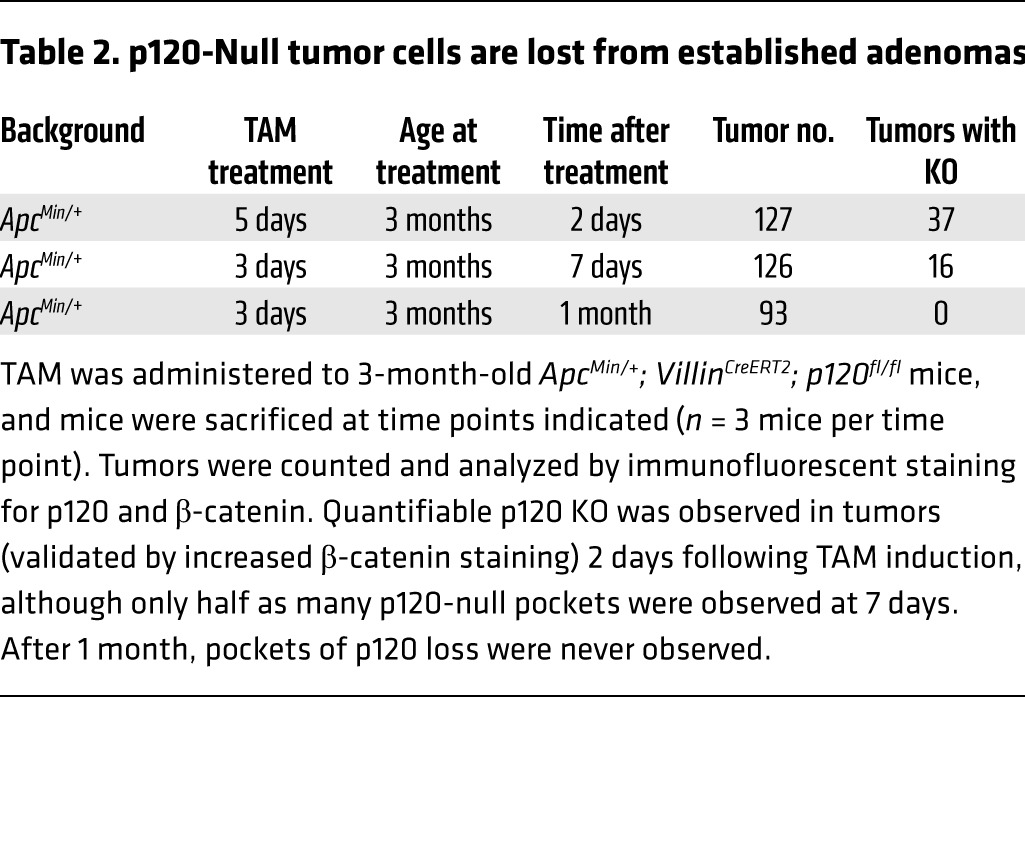
To ascertain the ultimate fate of p120-ablated tumor cells, mice with already well-formed tumors were sacrificed for analysis at 2 days, 1 week, or 1 month following treatment with TAM (Figure 4). Unlike what occurred in the long-term tumor studies, tumors examined within days after TAM administration displayed easily discernible foci of p120-null cells (Figure 4A). p120-Null foci detected at days 2 and 7 after TAM administration, however, were never present at 1 month (Table 2), indicating that p120-null tumor tissue is lost altogether by a few weeks after TAM administration. At day 2 after TAM administration, ApcMin/+ p120–KO tumors stained for the apoptotic marker cleaved caspase 3 (CC3) showed no change in apoptosis while phospho-histone H3 (p-H3) staining revealed a 2-fold increase in cell proliferation (Figure 4B). Nevertheless, it is clear that the synthetic lethal interaction between p120 loss and Apc LOH is absolute and that all tumor cells lacking p120 were eliminated within 1 month of p120 loss.
Figure 4. Obligatory tumor-specific retention of 1 p120 allele.

(A) TAM was administered to ApcMin/+;VillinCreERT2;p120fl/fl mice, and mice were sacrificed at varying times after TAM as indicated in Table 2 (n = 3 mice per group). Representative immunofluorescent image showing p120 KO within adenoma tissue (white dashed boxes), as confirmed by maintained staining for β-catenin (second panel) and Kaiso (third panel). Scale bar: 50 μm. (B) ApcMin/+ p120–KO tumors were costained for p120 (green) and apoptotic marker CC3 (red, n = 23 high-powered fields) or p-H3 (red, n = 19 high-powered fields). Analysis of p120-KO cells as compared with adjacent p120-positive tumor tissue indicates no change in apoptosis, yet a 2-fold increase in cell proliferation. Representative images are shown on the left. Scale bars: 100 μm. Quantification is shown at right. Error bars represent SEM. **P < 0.01, unpaired t test with Welch’s correction.
Table 2. p120-Null tumor cells are lost from established adenomas.
Synthetic lethality following combined loss of APC and p120 results from ROCK-dependent apoptosis.
To clarify the mechanism underlying obligate haploinsufficiency for p120, we reconstructed our model in vitro in the form of organoid cultures derived from p120fl/fl; VillinCreERT2; ApcMin/+ adenomas. Because 4-hydroxytamoxifen induction was relatively inefficient for removing the retained p120 allele, complete p120 ablation was conducted instead with an adenoviral vector expressing Cre-recombinase (Ad-Cre), using the same vector expressing GFP (Ad-GFP) as a negative control. Complete p120 ablation induced a dramatic switch from cyst-like to extensively branched organoid morphology, as revealed by bright field imaging (Figure 5A) and confocal immunofluorescence microscopy (Figure 5B). Although loss of p120 did not always preclude cystic morphology (Supplemental Figure 5), it was clearly prerequisite for branching (Figure 5C). A similar increase in branching was seen in organoids from Apc WT counterparts following loss of one p120 allele (Supplemental Figure 6). In a previous study, we showed that p120 depletion efficiently blocked anchorage-independent growth of MDCK cells transformed with either Src or Rac1, but not H-Ras (52). This effect of p120 depletion for both Src and Rac1 was completely reversed by Y27632-mediated inhibition of ROCK1. Similarly, in the present study, ROCK inhibition had little effect on the survival of cystic organoids, but markedly improved the survival of branched structures, all of which were p120 null (Figure 5D). Under these conditions, expression of CC3 was selectively elevated in p120-depleted cultures upon withdrawal of Y27632. These experiments suggest that p120 ablation in Apc-deficient organoids renders them sensitive to apoptotic cell death (Figure 5E, see also Supplemental Figure 5). In contrast, p120 ablation dramatically accelerated cell proliferation in Apc-deficient organoids (>10-fold) provided the ROCK inhibitor was added to suppress apoptosis (Figure 5F). Collectively, these experiments suggest (i) that cells acquiring combined loss of p120 and Apc in vitro are ultimately eliminated by a cell-autonomous synthetic lethal mechanism involving caspase-3–mediated apoptosis, (ii) that the effect is reversed by inhibition of ROCK, and (iii) that when enabled by inclusion of the ROCK inhibitor, p120 depletion dramatically accelerates cell proliferation in Apc-deficient organoids.
Figure 5. Biallelic p120 loss induces branching and cell death in ApcMin/+ adenoma-derived p120-KO organoids.

(A) Bright field images of ApcMin/+; VillinCreERT2; p120fl/fl small intestinal tumor organoids infected with either adeno-GFP (Ad-GFP, control) or adeno-CRE (Ad-CRE) viral vectors. Images are representative of cultures at day 4 after passage. Scale bars: 200 μm. (B) Confocal images of Ad-CRE–infected ApcMin/+; VillinCreERT2; p120fl/fl small intestinal tumor organoids stained with p120 antibody (green) and DAPI (blue) show mosaic loss of p120. Scale bars: 50 μm. Loss of p120 invariably segregated with branched morphology (arrows), while p120 retention was more commonly associated with cystic morphology (arrowheads). (C) Quantification of cystic and branched structure of Ad-CRE organoids by p120 status and morphology. Organoids were classified as largely p120 negative (with p120–) when half or more of the organoid content was p120 deficient. Error bars represent SEM (n = 3 cultures, approximately 50 organoids per culture). (D) Quantified viability of cystic and branched organoids in the presence or absence of ROCK1 inhibitor Y27632 (Y). Viability was calculated by counting intact organoids before and after 6 days of culture with (Y+) or without (Y-) Y27632. Error bars represent SEM (n = 8–10 cultures for each condition, 10–30 organoids per culture). (E) Immunoblot quantification of apoptosis marker CC3 (cl-CASP3) from infected tumor organoids cultured with or without 10 μM Y27632 for 4 days. β-Actin (ACTB) was used as loading control. (F) Tumor cells infected with indicated virus and noninfected parental cells were seeded at 4 × 104 per well. Total cell number in each well was counted after 5 days of culture with Y27632. Data shown as mean ± SEM of 3 to 4 wells for each condition. ****P < 0.0001, 1-way ANOVA pairwise comparisons with Tukey’s correction. Parental vs. Ad-GFP was not significant.
Insertional mutagenesis identifies p120 haploinsufficiency as a potent driver of intestinal tumorigenesis.
In view of p120’s newly identified activity, we sought an objective means of quantifying the tumorigenic potency of p120 haploinsufficiency relative to other potential drivers of tumorigenesis. SB transposon–mediated insertional mutagenesis has recently emerged as a powerful means of identifying novel drivers of tumorigenesis (53, 54). The system mobilizes modified transposable elements, which then reinsert themselves at random locations across the genome, thereby accelerating the accumulation of stochastic mutations. Depending on orientation and location, the insertions generate gain-of-function or loss-of-function mutations (illustrated in Figure 6A). The inserted transposons enable subsequent genome-wide recovery and quantification of all mutations in each tumor. Optimized versions of the SB system have been used to initiate and accelerate tumor development in WT mice or to accelerate tumorigenesis in mice already predisposed (i.e., sensitized) by selected gatekeeper mutations (e.g., ApcMin/+) (54). Importantly, although SB accelerates the rate of mutagenesis, subsequent forces driving tumor progression are largely unaffected. Most of the mutations have no effect on tumorigenesis (i.e., passengers) and are therefore essentially irrelevant. Driver mutations, on the other hand, confer distinct growth and/or survival advantages that selectively promote tumorigenesis. The relative tumorigenic potency of a given mutation is reflected by the frequency in which it shows up in tumors. Thus, relative hit frequencies provide a means of keeping score, the final tally amounting to a global ranking of the identified drivers in order of relative tumorigenic potency.
Figure 6. Dysregulation of the cadherin complex is frequently observed across multiple drivers of intestinal tumorigenesis.

(A) Schematic of the T2Onc2 SB transposon. The transposon yields gain-of-function mutations by driving transcription of nearby genes through an MSCV-driven splice donor (gene activation). Insertion within a gene leads to loss of function by termination of transcription through a birectional-polyA signal (gene inactivation). (B) Mapping of individual insertion sites (insertions, blue, separate alleles represented above and below gene; gene structure, green, introns represented by thicker line). Raw data for Apc (top) compiled from all 4 genetic backgrounds (ApcMin/+ [n = 446 tumors], KrasG12D [n = 65 tumors], Smad4fl/+ [n = 100 tumors], and p53R175H [n = 55 tumors]) show a mutation pattern similar to that observed in human CRC. Insertion sites for p120 (middle) and Ctnna1 (α-catenin, bottom) in the Smad4 SB background (n = 100 tumors) are consistent with gene inactivation. (C) CIS analysis shows a high percentage of tumors with mutations in p120, E-cadherin (Cdh1), and α-catenin (Ctnna1) in all genetic backgrounds. Collectively, mutations that target the cadherin complex (p120, E-cadherin, and α-catenin combined) were observed in close to 60% of all intestinal tumors. (D) Venn diagram of Smad4 SB results indicates multiple tumors with overlapping cadherin complex mutations, suggesting these mutations are not mutually exclusive. However, SB technology cannot discriminate cooccurring mutations in a given cell from mutations occurring separately in distinct clonal populations within a given tumor. (E) Data from TCGA COAD data set comparing P120 mRNA levels in colon adenocarcinomas (n = 264) and normal colon (n = 39). Data are shown from compiled COAD data set (left) and individual patient-matched COAD samples (right, n = 24). Data were log2 transformed and median centered at 1 (dotted line) for visualization. ****P < 0.0001, unpaired Student’s t test.
In the present study, we analyzed a series of in vivo intestinal SB insertional mutagenesis screens designed originally for unbiased high-throughput driver discovery in genetic backgrounds relevant to human intestinal cancer (i.e., ApcMin/+, K-RasG12D/+, Smad4f/+, p53R172H/+) (53, 55). Breeding schemes and genotypes for each model are shown in Supplemental Figure 7. Global analyses of results compiled from these 4 screens are now published (53, 55). Figure 6B shows collective raw hit data for Apc, p120, and Ctnna1 (α-catenin). The genomic structures of these genes are shown in green across the center, while aqua-blue bars represent individual hits showing the location of mutations across the gene and on each allele (above and below). Notably, the insertion patterns for p120 and Ctnna1 (Figure 6B) and Cdh1 (E-cadherin, not shown) are consistent with gene inactivation, as hits are distributed randomly across both alleles.
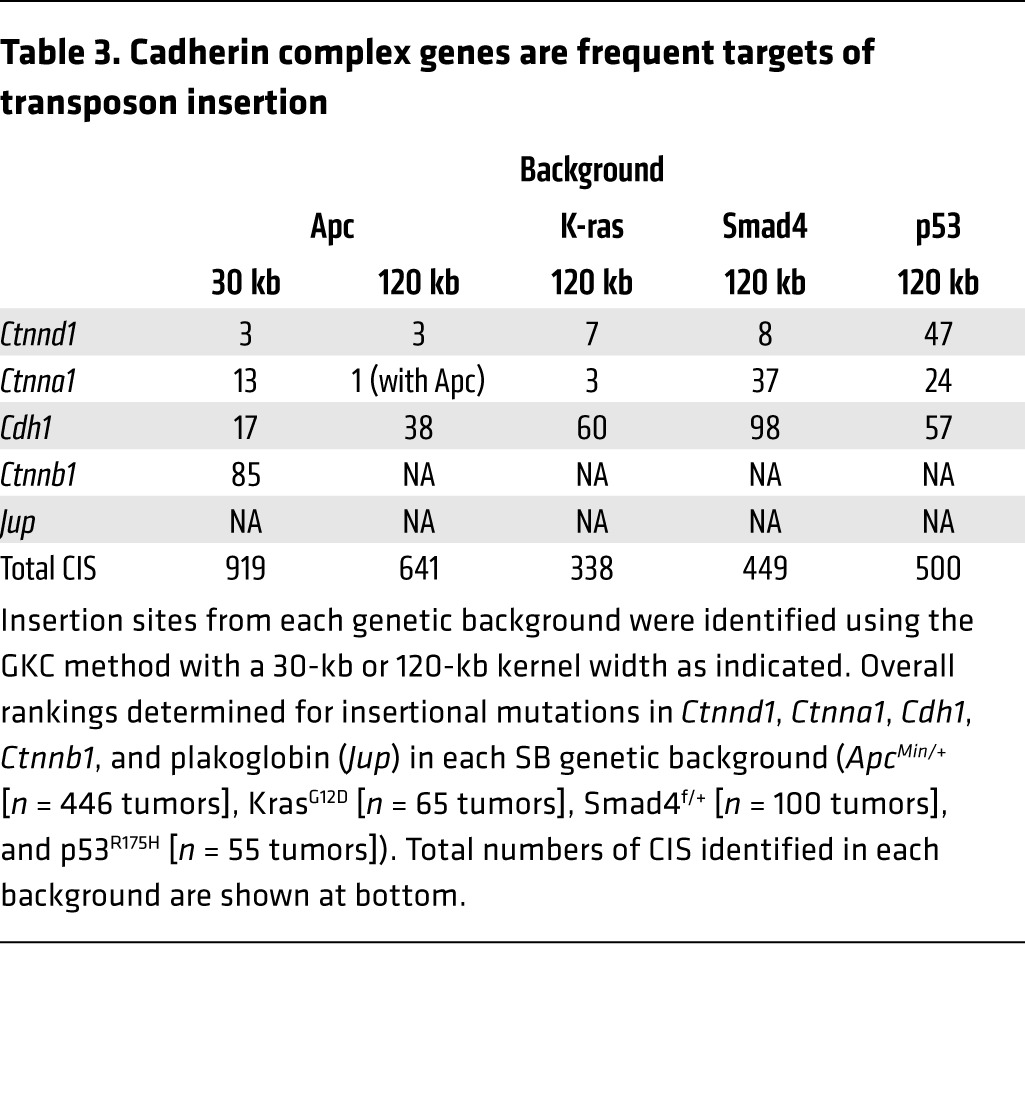
Key quantitative findings from the SB data are summarized in Figure 6C and Table 3. Data from the 4 screens are organized by genetic background, and the proportion of tumors containing mutations (also known as hits) in the indicated genes is shown for each background. Apc was by far the most frequently hit gene in 3 of 4 backgrounds, the unexpected exception being Smad4 (Figure 6C), which is addressed separately in the global analyses of these screens (55). Here, we note that among the 919 drivers identified in the ApcMin/+ background, p120 haploinsufficiency ranked third in overall tumorigenic potency, superseded only by the gatekeeper itself, Apc, and one other gene (Table 3). Moreover, the scores were largely independent of background (i.e., p120 ranked 3rd, 7th, 8th and 47th, respectively, in the Apc, K-Ras, Smad4, and p53 mutant backgrounds) with the possible exception of p53. Notably, similar results posted for α-catenin and E-cadherin such that the scores for all 3 core components of the complex were among the top scores overall in tumorigenic potency.
Table 3. Cadherin complex genes are frequent targets of transposon insertion.
Haploinsufficiency at the level of the cadherin complex.
Among the more surprising results from the SB analyses was that, in addition to exceptionally high scores on the tumorigenic potency scale, both p120 and α-catenin consistently outscored E-cadherin (Table 3). On the other hand, E-cadherin scores were also exceptional, ranging from a high in the top third percentile (Apc cohort) to a low that was still among the top 22nd percentile (Smad4 cohort). In the final analysis, the hit frequencies for p120, α-catenin, and E-cadherin could have differed markedly from one another, but instead clustered together at the extreme high end of the range. The similar scores point to an obvious common denominator, the intact E-cadherin complex, as the functional unit of tumorigenic selection, with loss of any 1 of the 6 alleles encoding these proteins being sufficient to markedly accelerate tumor progression. The individual results for p120, Ctnna1 (α-catenin), and Cdh1 (E-cadherin) were therefore combined to most accurately represent the overall impact of cadherin complex inactivation in intestinal tumorigenesis (Figure 6C). Notably, the data reveal that roughly half of all SB-induced tumors, irrespective of genetic background, sustained an inactivating mutation in at least 1 of these 3 core components.
A number of tumors contained mutations in more than 1 core subunit (Figure 6D), suggesting a possible lack of mutual exclusivity. However, SB does not show whether mutations within a particular tumor exist together in the same cell or separately as distinct clonal populations. Thus, mutual exclusivity cannot be determined from these data alone. In fact, many tumors contained more than 1 mutation in p120, but our finding that complete loss of p120 is synthetic lethal in the context of Apc LOH argues strongly that these mutations represent separate monoclonal events in the same tumor. Thus, these results are in line with the concept that inactivation of any of the core subunits (p120, α-catenin, or E-cadherin) by SB insertion effectively inactivates the entire cadherin complex, resulting in markedly accelerated tumorigenesis.
Finally, given the high tumorigenic potency attributed to p120 haploinsufficiency by SB, we searched the publicly available databases for evidence of physiologically relevant counterparts in human CRC. Analysis of CRC genomes in The Cancer Genome Atlas (TCGA) (https://cancergenome.nih.gov/) revealed that the gene encoding human P120 is not frequently mutated, as is also the case for α-catenin (CTNNA1) and E-cadherin (CDH1). On the other hand, direct measurements of P120 mRNA expression in normal and tumor samples from individual CRC patients revealed a highly significant reduction in P120 mRNA levels in a large proportion of tumors (Figure 6E). Though beyond the scope of the present report, these results may provide insight into physiologically relevant alterations in P120 levels that might affect human tumor progression in a fashion consistent with our SB data.
Discussion
Reduced p120 expression is well documented in many solid tumors, including CRC, but the significance of this observation is incompletely understood (40). Here, we definitively identify p120 as an “obligate” haploinsufficient tumor suppressor in early intestinal neoplasia. Ablation of a single p120 allele in Apc1638N/+ mice resulted in a 10-fold increase in tumor multiplicity. Removal of the second p120 allele, however, did not further enhance tumorigenesis, but instead induced synthetic lethality in combination with Apc LOH. As a result, outright loss of p120 was never observed in tumors from these mice. The unusual behavior of the second p120 allele in these experiments is a hallmark of obligate haploinsufficiency (56).
Whereas classical tumor suppressors such as APC are genetically recessive, haploinsufficiency is a manifestation of genetic dominance that arises when only 1 allele of a normally diploid locus is present (57). The term obligate haploinsufficiency was coined originally to explain a counterintuitive variation on this theme exhibited by PTEN, the prototypical and best-understood member of this unconventional group. As per all haploinsufficient tumor suppressors, loss of just 1 allele is sufficient to actively promote tumorigenesis. In conventional haploinsufficiency, the second allele is also under positive selection and is therefore also frequently lost at some later point in progression. TP53 behaves this way. In contrast, a feature of obligate haploinsufficiency is that LOH is rarely observed because loss of the second allele is either immaterial (i.e., offers no selective advantage) or outright incompatible with survival, as illustrated in prostate cancer by the behavior of PTEN (56). Up to 70% of primary prostate cancer is PTEN haploinsufficient at presentation (56). Loss of the second allele is not evident because PTEN LOH triggers a p53-dependent fail-safe mechanism, resulting in irreversible senescence and removal of the incapacitated cell. This mechanism is operative in prostate cancer over long periods of time and may contribute to the often-indolent behavior of this cancer. However, the fail-safe mechanism itself can fail in late-stage prostate cancer: now, upon loss of p53, PTEN LOH emerges unopposed and actively facilitates tumor progression (56). In our mouse models of intestinal cancer, the mechanism underlying the behavior of p120 is apparently p53 independent, but still obviously reminiscent of the PTEN paradigm: in the wake of Apc LOH, p120 LOH elicits a lethal fail-safe response. Presumably, this arrangement means that in the early stages of the approximately 85% of human CRC that is initiated by APC LOH, complete loss of p120 should rarely, if ever, be observed.
The SB mutagenesis screens afforded an opportunity to directly assess the tumorigenic impact of p120 haploinsufficiency (relative to other candidate drivers) in 4 intestinal tumor–predisposed genetic backgrounds (i.e., ApcMin/+, K-RasG12D/+, Smad4fl/+, p53R172H/+) (53, 55). Importantly, among the candidates identified and ranked, α-catenin and E-cadherin were not merely present, but rather closely aligned with p120 at the very high end of the tumorigenic scale. Cosegregation of these 3 factors is not unexpected, given that they are each indispensable for E-cadherin–mediated adhesion (and suppression of tumorigenicity), but this strong recurring theme nonetheless validates the intact cadherin complex as the functional unit under selection by SB. In the final analysis, 42%–60% of all SB-generated tumors were found to contain at least 1 inactivated allele of p120, α-catenin, or E-cadherin, irrespective of genetic background.
Interestingly, this theme of codependence extends also to obligate haploinsufficiency. Indeed, α-catenin and E-cadherin are known to be haploinsufficient tumor suppressors from previous KO studies in mice (58, 59). Although E-cadherin has not been formally tested for the obligate condition (i.e., synthetic lethality with Apc LOH), the point is essentially proven already for α-catenin in an elegant study originally designed to identify suppressors of Apc-initiated intestinal tumorigenesis (59). Following random mutagenesis, a mutant mouse strain was identified in which Apc-induced tumorigenesis is potently suppressed. The so-called modifier locus was mapped to a mutant (essentially null) α-catenin gene located in cis (on the same allele) approximately 1 MB downstream of Apc on chromosome 18. In most Apc mutant mice, loss of the remaining WT Apc allele occurs by LOH (following somatic recombination). Notably, α-catenin is linked by proximity and generally undergoes LOH at the same time. Thus, Apc LOH in the modifier strain is essentially coupled to complete loss of functional α-catenin, the latter accounting for suppression of tumorigenesis. This scenario is exactly analogous to the synthetic lethal combination we describe herein for Apc LOH and complete loss of p120. Thus, for p120 and α-catenin, it is clear that biallelic loss is potently restricted by Apc LOH. We suggest that this restriction mechanism reflects an APC-associated checkpoint/fail-safe mechanism that is not limited to p120, but rather the working core of E-cadherin partners required for strong intercellular adhesion (minimally p120, α-catenin, and E-cadherin). Presumably, the emergence of E-cadherin loss in late-stage cancer reflects the undoing of the fail-safe mechanism, analogous to the aforementioned loss of p53 followed by subsequent complete loss of PTEN in advanced prostate cancer.
Based on the SB rankings, our analysis suggests that haploinsufficiency at the level of the E-cadherin complex is a serious event in the progression of Apc-initiated intestinal cancer. In that context, the notion of genetic linkage of Apc to the nearby α-catenin locus on mouse chromosome 18 suggests that Apc-LOH could be routinely coupled to monoallelic loss of α-catenin, a potential 2-in-1 hit scenario. Of note, the 120-kb Gaussian kernel analysis of the SB common insertion site (CIS) data ranks α-catenin first with Apc, which makes little sense in any context other than the coupled LOH described above. Interestingly, the human APC and α-catenin genes are also located in relative proximity on 5q21 and 5q32, respectively, and allelic loss of chromosome 5q is present in almost half of sporadic colorectal adenomas and carcinomas. These observations raise the possibility that APC LOH, the event believed to initiate the vast majority of human intestinal cancer, is in fact also frequently coupled to monoallelic loss of α-catenin, in which case the synthetic lethal arrangement as a fail-safe mechanism may actually come into play relatively early and perhaps more often than currently anticipated.
We were initially surprised that, at the molecular level, p53 does not seem to have an obvious role in either the haploinsufficient or synthetic lethal activities observed upon ablation of both p120 alleles. Synthetic lethality, on the other hand, was efficiently blocked in tumor organoids by specific Y27632-mediated inhibition of ROCK. We showed previously that in NIH3T3 fibroblasts, cadherin-linked p120 is required for Rac1-mediated inhibition of RhoA, a signaling module that coordinates the antagonism between these factors. Disruption of the pathway by removing p120 leads to constitutive activation of RhoA and various actomyosin contractile phenotypes that include tension-dependent apoptosis (27, 52, 60, 61). In MDCK cells, Src- and Rac1-mediated (but not Hras mediated) cell transformation is dependent on this pathway and blocked upon removal of p120 (52), and the inverted polarity phenotype described originally by Mostov and colleagues is similarly controlled (52, 62). Interestingly, the mechanistic elements common to all of these examples appear to be shared by APC. For example, Apc LOH is known to induce upregulation of several RacGEFs (e.g., TIAM1, VAV3). Apc-associated tumorigenesis is dependent on Rac1 activity (63), and we show here that the synthetic lethal combination of Apc LOH and cadherin loss of function is rescued by inhibition of ROCK.
We also show that p120 ablation in cultured Apc-deficient organoids causes a dramatic switch from cyst-like to highly branched morphology. However, combined loss of Apc and p120 decidedly sensitized the cells to caspase-3-dependent apoptosis, which was, in fact, efficiently rescued by the ROCK inhibitor. Cell proliferation increased 10-fold upon removal of p120 (provided the ROCK inhibitor was present to block apoptosis). Together, the data reveal that combined loss of Apc and p120 gives rise to Wnt-activated, highly proliferative, E-cadherin–deficient cells that nonetheless are efficiently eliminated due to synthetic lethality. Importantly, the in vitro phenotypes exhibited by p120 heterozygous organoids were intermediate relative to their WT and p120-null counterparts; reducing the dose by 1 allele was sufficient to circumvent the synthetic lethal consequence of complete loss while retaining sufficient oncogenic activity to account for the increase in tumor multiplicity observed in vivo. Though not immediately intuitive, this obligatory haploinsufficient behavior probably confers a significant long-term advantage similar to that of the PTEN phenotype in prostate cancer. In effect, the ability (in the monoallelic state) to efficiently block a worst-case scenario (e.g., malignancy) for years, if not decades, appears to outweigh the increased incidence of tumors associated with haploinsufficiency.
The remarkable efficacy of this apparent fail-safe mechanism over many unsuccessful attempts to generate p120 ablated tumors in Apc mutant mice speaks to the robustness of this synthetic lethal mechanism over time. Indeed, it turns out that it is not actually difficult to generate tumors with easily discernible foci of p120 ablated cells if one simply waits until tumors are already formed before giving TAM. The foci are nonetheless transient, as they are neve r present at 3 weeks after TAM administration or any time thereafter. Given the efficient clearance of thousands if not millions of these cells in a relatively short period of time, we propose that synthetic lethality could easily account for the fact that complete loss of E-cadherin and/or its essential cofactors is rarely if ever observed in the early stages of intestinal tumor progression. It is now well established that the ISC is also the cell of origin for intestinal cancer. Upon loss of APC, the ISC is forced to transition from a transient state of physiologically appropriate dependence on Wnt to a pathological state dominated by autonomous Wnt signaling. Disruption of the Wnt pathway is thus an obvious therapeutic strategy, but one that is intrinsically dose limited, as intestinal homeostasis is dependent on maintained Wnt signaling. Notably, loss of p120 by itself is normally well tolerated in the intestine, provided that the extent of p120 KO does not exceed 15% of the epithelial lining (45). In contrast, the pathologic condition initiated by APC LOH might be selectively targetable on the basis of its synthetic lethal interaction with p120. Though it seems like a very bad idea to target the E-cadherin complex directly, synthetic lethality notably broadens the scope of available candidates. Moreover, reduced, intermittent, or conditional dosing might lead to ways of exploiting synthetic lethality while minimizing single target effects.
Methods
Animals.
Mice containing the floxed p120 allele (fl) were generated as described previously by our lab and backcrossed onto a C57BL/6 background (42). p120fl/fl mice were crossed with VillinCreERT2 (Sylvie Robine, Institut Curie, Paris, France) (64) mice to target TAM-inducible p120 ablation to the small intestine and colon. p120fl/fl; VillinCreERT2 mice were then crossed with Apc1638N/+ (Mouse Models of Human Cancer Consortium Repository, National Cancer Institute [NCI]) or ApcMin/+ (Jackson Laboratories) lines to generate the experimental populations for tumor studies (Apc1638N/+ n = 6 corn oil and n = 6 plus TAM; ApcMin/+ n = 16 p120+/+, n = 12 p120fl/+, and n = 12 p120fl/fl). After genotyping, p120 KO was induced by i.p. injection of 1 mg/20 g mouse weight of TAM (Sigma-Aldrich) dissolved in corn oil for 3 consecutive days. Mice were sacrificed when moribund (~8 months and 3 months after TAM administration for Apc1638N/+ and ApcMin/+, respectively) and tumors counted on a Zeiss Stemi 2000-C dissecting microscope. For analysis of p120 KO in established tumors, TAM treatment was not initiated until ApcMin/+; p120fl/fl; VillinCreERT2 mice were 3 months old.
To generate p120 heterozygous mice, p120fl/fl mice were crossed with transgenic mice expressing Cre-recombinase gene under the control of the adenovirus EIIA promoter. The resulting p120fl/ΔCre pups were verified as expressing 1 p120-null allele (primer sequences: CAGACACATCAGAAGAAGGCG and CACATTCCCGAAGTTTCCTCC) and then crossed into the C57BL6/J strain to yield the p120+/– line. These mice were crossed with ApcMin/+ to yield ApcMin/+; p120+/+ and ApcMin/+; p120+/– littermate sets. Matched mice were sacrificed when one reached experimental end point for tumor burden analysis (n = 7 mice per genotype).
For conditional Apc ablation, ApcCKO/CKO mice (Mouse Models of Human Cancer Consortium Repository, NCI) were crossed with Lrig1CreERT2/+ mice (Robert Coffey).
Homozygous Apc flox mice expressing Lrig1CreERT2 were injected i.p. with 1 mg TAM dissolved in corn oil to induce Apc loss. Littermate control mice were treated with corn oil alone. For β-catenin activation, Lrig1CreERT2/+; Ctnnb1ΔEx3/+ mice obtained from Robert Coffey were treated with 2 mg TAM and were sacrificed 3 weeks later.
Mice were maintained under a strict 12-hour light/12-hour dark cycle and with free access to chow and water. Genotyping was done as described previously (45).
Organoid culture.
Tumor organoid culture were derived from small intestinal tumors from 3-month-old ApcMin/+; p120fl/fl;VillinCreERT2 mice. Tumors were harvested and cultured as described elsewhere (65). Briefly, mice were sacrificed and intestines were splayed to isolate adenomas. Isolated adenomas were rocked in chelation buffer (PBS with 2 mM EDTA) for 1 hour prior to shaking to release surrounding normal epithelium. Digestion buffer was then added as follows: DMEM (Gibco; Thermo Fisher Scientific), 2.5% FBS, 1% antibiotic-antimycotic (Corning), 100 μg/ml collagenase XI (Sigma-Aldrich), and 125 μg/ml Dispase II (Roche). Adenomas were incubated for 30 minutes at 37°C. Digested adenoma fragments were then collected and cultured in growth factor–reduced Matrigel (Corning) overlaid with advanced DMEM/F12 supplemented with 1× N2 (Gibco, Thermo Fisher Scientific), 1× B27 (Gibco, Thermo Fisher Scientific), and 1% antibiotic-antimycotic (Corning). 10 μM Y27632 (TOCRIS) and 50 μM Q-VD-OPh (Sigma-Aldrich) were added to culture medium where described.
For small intestinal organoid culture, 10 cm of duodenum was harvested from 8-week-old p120+/+ and p120+/– mice. Tissue was minced and incubated in chelation buffer (PBS with 2 mM EDTA) for 30 minutes at 4°C. Following chelation, tissue fragments were shaken in buffer (PBS with 43.4 mM sucrose and 54.9 mM D-sorbitol) to release crypts. Supernatants containing crypts were collected and filtered; the number of crypts was counted. Crypts were then plated in growth factor–reduced Matrigel at a concentration of 500 crypts per 50 μl Matrigel. After polymerization, Matrigel was overlaid with culture medium as described above, additionally supplemented with 1× Glutamax (Gibco, Thermo Fisher Scientific), 10 μM HEPES (Gibco, Thermo Fisher Scientific), 50 ng/ml EGF (R&D), 100 ng/ml Noggin (R&D), and 500 ng/ml R-spondin (R&D).
For adenoviral infection, organoids were collected from Matrigel by washing with ice-cold PBS with 0.5% serum and dissociated by TrypLE express (Gibco, Thermo Fisher Scientific). Dissociated cells were incubated with 2 × 105 PFU/μl Adeno-GFP or Adeno-CRE virus in culture medium supplemented with 10 μM Y27632 for 30 minutes at 37°C. Infected cells were then pelleted and resuspended in Matrigel containing another 2 × 105 PFU/μl Adeno-GFP or Adeno-CRE virus and overlaid with culture medium supplemented with 10 μM Y27632.
Immunohistochemistry.
Swiss-rolled intestinal tissue was harvested on ice and fixed in 10% neutral buffered formalin overnight at 4°C prior to embedding in paraffin. Tissue processing and H&E staining were performed by the Vanderbilt Translational Pathology Shared Resource using standard techniques. For fluorescent IHC, antigen retrieval consisted of 10 minutes boiling in 10 mM sodium citrate (pH 6) for all antibodies. After blocking, samples were incubated with primary and secondary antibodies overnight at 4°C and for 2 hours at room temperature, respectively.
The following primary antibodies were utilized: anti-p120 (8D11, custom monoclonal to mouse p120 catenin, A.B. Reynolds) (66); anti-p120/pp120 (BD Biosciences, 610133); anti-Kaiso (custom polyclonal, A.B. Reynolds); anti–E-cadherin (BD Biosciences, 610181); anti–β-catenin (BD Biosciences, 610154); anti–α-catenin (Sigma-Aldrich, C2081); anti–p-H3 (EMD Millipore, 06-570); and anti-CC3 (Cell Signaling Technology, 9661). Secondary antibodies were conjugated to either Alexa Fluor 488 or Alexa Fluor 594 dyes (Invitrogen). Nuclei were visualized by brief incubation in 1 μg/ml Hoechst dye. Fluorescent images were taken on an Axioplan 2 microscope (Carl Zeiss Inc.) with a Hamamatsu Orca-ER digital camera. Images were analyzed and processed with MetaMorph software (MDS Analytical Technologies).
For whole-mount immunostaining of organoids, Matrigel droplets were fixed with 4% PFA for 20 minutes on ice, followed by permeabilization/blocking in PBS supplemented with 2.5% donkey serum and 0.2% Triton X-100 at 4°C overnight. Organoid samples were then probed with anti-p120 antibody at 4°C overnight followed by Alexa Fluor 488–conjugated secondary antibodies and DAPI for 2 hours at room temperature. Stained organoids were imaged using an inverted (IX-71, Olympus) or confocal microscope (Eclipse Ti, Nikon).
Western blot analysis.
Intestines were harvested and flushed with ice-cold PBS. Sections of mouse distal ileum (2 cm) were lysed in RIPA buffer (50 mM Tris pH 7.4, 150 mM NaCl, 1% Nonidet P40, 0.5% deoxycholic acid, 0.1% SDS) containing protease and phosphatase inhibitor cocktails (Sigma-Aldrich) on ice. Samples were homogenized with a motor fitted with a Kontes Microtube Pellet Pestle Rod and cleared by spinning at 14,000 g at 4°C for 10 minutes. Total protein was quantified using a bicinchoninic acid assay (Pierce). Protein (10 μg per sample) was boiled in 2× Laemmli sample buffer and separated by SDS-PAGE. Proteins were transferred to nitrocellulose (PerkinElmer), and nonspecific binding was blocked using Odyssey blocking buffer (LI-COR). Primary antibodies used included the following: anti-p120 mAb (BD Biosciences, 610133); anti-p120 polyclonal Ab (pAb) (F1αSH, custom polyclonal, A.B. Reynolds); anti-Kaiso (custom polyclonal, A.B. Reynolds); anti–E-cadherin (BD Biosciences, 610181); anti–β-catenin (Sigma-Aldrich, C2206); anti–α-tubulin (Vanderbilt Antibody and Protein Resource, clone E7); CC3 (Cell Signaling Technology, 9661); and β-actin (Sigma-Aldrich, A1978). The Odyssey system was used for detection of secondary goat anti-mouse IgG and goat anti-rabbit IRDye antibodies (LI-COR), or anti-mouse and rabbit TrueBlot (Rockland Immunochemicals).
P120 mRNA expression.
P120 mRNA expression was determined with Illumina HiSeq and Illumina GA RNASeqV2 data from TCGA colon adenocarcinoma (COAD) data set (n = 264 CRC, 39 normal colon, and 24 patient matched sets). Normalized RSEM expression data were log2 transformed and median centered at 1 for visualization.
Quantitative reverse-transcriptase PCR.
Sections of mouse ileum (2 cm) were homogenized in TRIzol reagent. Total RNA was purified with RNeasy Mini Kit with on-column DNase treatment (QIAGEN) according to the manufacturer’s instructions. cDNA synthesis was performed using a VILO cDNA Kit (Life Technologies). Real-time PCR was performed on Bio-Rad CFX96 real-time cyclers using LuminoCT SYBR Green Master Mix (Sigma-Aldrich). After preincubation at 95°C, 2-step cycling was performed from 95°C (10 seconds) to 60°C (30 seconds) for 40 cycles. Gapdh was used to calculate normalized fold change. Melting curves of the PCR products were analyzed using CFX Manager software (Bio-Rad) to exclude amplification of nonspecific products. See Supplemental Table 2 for primer list.
LCM.
Tissue was collected into adhesive caps using the Veritas Laser Capture Microdissection System and processed for DNA using the Pico Pure DNA Extraction Kit (Arcturus, KIT0103).Proteinase K solution (10–30 μl) was used for each sample. Extracted DNA was PCR amplified for the p120 null or floxed alleles and run on 2% agarose gels for viewing.
SB insertional mutagenesis screens.
The following alleles were used to generate SB intestinal tumor models: LSL-KrasG12D, p53R175H, Smadfl/+, VillinCreERT2, T2Onc2, and Rosa26LSL–SB11 (See Supplemental Figure 7 for crosses). The resulting cohorts and analysis have been previously described (55). ApcMin/+ cohort and tissue isolation have been described previously (53). Briefly, to identify drivers of intestinal tumorigenesis, isolation of the transposon insertion sites was performed using splinkerette PCR to produce barcoded PCR products that were pooled and sequenced on the 454 GS-Titanium sequencer (Roche) platform. Reads from sequenced tumors were mapped to the mouse genome assembly National Center for Biotechnology Information m37 and merged together to identify nonredundant SB insertion sites. Nonredundant SB insertion sites were then used to identify CISs using a Gaussian kernel convolution (GKC) statistical framework as described elsewhere (53, 55). Kernel sizes used for ApcMin/+ study were 30 kb and 120 kb. For K-ras, Smad4, and p53 screens, static kernel widths of 15, 30, 50, 75, 120, and 240 kb were used to identify CISs, the results of which were then merged according to P values for final insertion tally.
Statistics.
Statistical analyses were performed using GraphPad Prism software (GraphPad). For assays with 2 groups with equal or unequal variance, unpaired 2-tailed Student’s t tests or t test with Welch’s correction was performed, respectively. In instances with 3 or more groups, 1-way ANOVA statistical tests were performed with Tukey’s correction for pairwise analysis. Survival of tumor-bearing mice was analyzed by log-rank test. P < 0.05 was considered statistically significant.
Study approval.
This study was performed in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals (National Academies Press. 2011.). The study protocol was approved by the Institutional Animal Care and Use Committee at Vanderbilt University (protocol no. M/07/061).
Author contributions
SPS, JK, WGSF, HT, MRD, AEP, and MKW conducted experiments, acquired data, analyzed data. SPS, JK, WGSF, HT, RHC, NAJ, NGC, RJC, and ABR designed experiments. SPS, JK, MT, DMP, RJC, and ABR prepared the manuscript.
Supplementary Material
Acknowledgments
This work was funded by the following grants: NIH R01-CA55724, NIH R01-CA111947, F31 CA165667-01, and Vanderbilt GI SPORE 50 CA95103. The authors wish to acknowledge the outstanding support of the Vanderbilt Antibody and Protein Resource and Biostatistics Shared Resource for generation of Kaiso pAbs and assistance with TCGA mRNA analysis, respectively.
Version 1. 11/13/2017
Electronic publication
Version 2. 11/14/2017
Corrected formatting error in references.
Version 3. 12/01/2017
Print issue publication
Footnotes
JK’s present address is: Department of Biochemistry, Osaka International Cancer Institute, Osaka, Japan.
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information:J Clin Invest. 2017;127(12):4462–4476.https://doi.org/10.1172/JCI77217.
Contributor Information
Jumpei Kondo, Email: jumpeko@gmail.com.
Haruna Takeda, Email: htakeda@kanazawa-med.ac.jp.
Manish Tripathi, Email: mtripath@uthsc.edu.
D. Michael Payne, Email: paynem2006@yahoo.com.
Robert J. Coffey, Email: robert.coffey@vanderbilt.edu.
References
- 1.Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61(5):759–767. doi: 10.1016/0092-8674(90)90186-I. [DOI] [PubMed] [Google Scholar]
- 2.Vogelstein B, et al. Genetic alterations during colorectal-tumor development. N Engl J Med. 1988;319(9):525–532. doi: 10.1056/NEJM198809013190901. [DOI] [PubMed] [Google Scholar]
- 3.Clevers H, Nusse R. Wnt/β-catenin signaling and disease. Cell. 2012;149(6):1192–1205. doi: 10.1016/j.cell.2012.05.012. [DOI] [PubMed] [Google Scholar]
- 4.Schepers A, Clevers H. Wnt signaling, stem cells, and cancer of the gastrointestinal tract. Cold Spring Harb Perspect Biol. 2012;4(4):a007989. doi: 10.1101/cshperspect.a007989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Perl AK, Wilgenbus P, Dahl U, Semb H, Christofori G. A causal role for E-cadherin in the transition from adenoma to carcinoma. Nature. 1998;392(6672):190–193. doi: 10.1038/32433. [DOI] [PubMed] [Google Scholar]
- 6.Birchmeier W, Behrens J. Cadherin expression in carcinomas: role in the formation of cell junctions and the prevention of invasiveness. Biochim Biophys Acta. 1994;1198(1):11–26. doi: 10.1016/0304-419x(94)90003-5. [DOI] [PubMed] [Google Scholar]
- 7.Jeanes A, Gottardi CJ, Yap AS. Cadherins and cancer: how does cadherin dysfunction promote tumor progression? Oncogene. 2008;27(55):6920–6929. doi: 10.1038/onc.2008.343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yap AS. The morphogenetic role of cadherin cell adhesion molecules in human cancer: a thematic review. Cancer Invest. 1998;16(4):252–261. doi: 10.3109/07357909809039774. [DOI] [PubMed] [Google Scholar]
- 9.Takeichi M. Morphogenetic roles of classic cadherins. Curr Opin Cell Biol. 1995;7(5):619–627. doi: 10.1016/0955-0674(95)80102-2. [DOI] [PubMed] [Google Scholar]
- 10.Kourtidis A, Lu R, Pence LJ, Anastasiadis PZ. A central role for cadherin signaling in cancer. Exp Cell Res. 2017;358(1):78–85. doi: 10.1016/j.yexcr.2017.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Corso G, Marrelli D, Roviello F. Familial gastric cancer and germline mutations of E-cadherin. Ann Ital Chir. 2012;83(3):177–182. [PubMed] [Google Scholar]
- 12.Berx G, et al. E-cadherin is inactivated in a majority of invasive human lobular breast cancers by truncation mutations throughout its extracellular domain. Oncogene. 1996;13(9):1919–1925. [PubMed] [Google Scholar]
- 13.Derksen PW, et al. Somatic inactivation of E-cadherin and p53 in mice leads to metastatic lobular mammary carcinoma through induction of anoikis resistance and angiogenesis. Cancer Cell. 2006;10(5):437–449. doi: 10.1016/j.ccr.2006.09.013. [DOI] [PubMed] [Google Scholar]
- 14.Nollet F, Berx G, van Roy F. The role of the E-cadherin/catenin adhesion complex in the development and progression of cancer. Mol Cell Biol Res Commun. 1999;2(2):77–85. doi: 10.1006/mcbr.1999.0155. [DOI] [PubMed] [Google Scholar]
- 15.Behrens J, et al. Loss of epithelial differentiation and gain of invasiveness correlates with tyrosine phosphorylation of the E-cadherin/beta-catenin complex in cells transformed with a temperature-sensitive v-SRC gene. J Cell Biol. 1993;120(3):757–766. doi: 10.1083/jcb.120.3.757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hülsken J, Behrens J, Birchmeier W. Tumor-suppressor gene products in cell contacts: the cadherin-APC-armadillo connection. Curr Opin Cell Biol. 1994;6(5):711–716. doi: 10.1016/0955-0674(94)90098-1. [DOI] [PubMed] [Google Scholar]
- 17.Chaffer CL, Weinberg RA. A perspective on cancer cell metastasis. Science. 2011;331(6024):1559–1564. doi: 10.1126/science.1203543. [DOI] [PubMed] [Google Scholar]
- 18.Thoreson MA, et al. Selective uncoupling of p120(ctn) from E-cadherin disrupts strong adhesion. J Cell Biol. 2000;148(1):189–202. doi: 10.1083/jcb.148.1.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Davis MA, Ireton RC, Reynolds AB. A core function for p120-catenin in cadherin turnover. J Cell Biol. 2003;163(3):525–534. doi: 10.1083/jcb.200307111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gottardi CJ, Gumbiner BM. Adhesion signaling: how beta-catenin interacts with its partners. Curr Biol. 2001;11(19):R792–R794. doi: 10.1016/S0960-9822(01)00473-0. [DOI] [PubMed] [Google Scholar]
- 21.Rimm DL, Koslov ER, Kebriaei P, Cianci CD, Morrow JS. Alpha 1(E)-catenin is an actin-binding and -bundling protein mediating the attachment of F-actin to the membrane adhesion complex. Proc Natl Acad Sci U S A. 1995;92(19):8813–8817. doi: 10.1073/pnas.92.19.8813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Desai R, Sarpal R, Ishiyama N, Pellikka M, Ikura M, Tepass U. Monomeric α-catenin links cadherin to the actin cytoskeleton. Nat Cell Biol. 2013;15(3):261–273. doi: 10.1038/ncb2685. [DOI] [PubMed] [Google Scholar]
- 23.Sarpal R, Pellikka M, Patel RR, Hui FY, Godt D, Tepass U. Mutational analysis supports a core role for Drosophila α-catenin in adherens junction function. J Cell Sci. 2012;125(Pt 1):233–245. doi: 10.1242/jcs.096644. [DOI] [PubMed] [Google Scholar]
- 24.Ireton RC, et al. A novel role for p120 catenin in E-cadherin function. J Cell Biol. 2002;159(3):465–476. doi: 10.1083/jcb.200205115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xiao K, et al. Cellular levels of p120 catenin function as a set point for cadherin expression levels in microvascular endothelial cells. J Cell Biol. 2003;163(3):535–545. doi: 10.1083/jcb.200306001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nanes BA, et al. p120-catenin binding masks an endocytic signal conserved in classical cadherins. J Cell Biol. 2012;199(2):365–380. doi: 10.1083/jcb.201205029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wildenberg GA, et al. p120-catenin and p190RhoGAP regulate cell-cell adhesion by coordinating antagonism between Rac and Rho. Cell. 2006;127(5):1027–1039. doi: 10.1016/j.cell.2006.09.046. [DOI] [PubMed] [Google Scholar]
- 28.Smith AL, Dohn MR, Brown MV, Reynolds AB. Association of Rho-associated protein kinase 1 with E-cadherin complexes is mediated by p120-catenin. Mol Biol Cell. 2012;23(1):99–110. doi: 10.1091/mbc.E11-06-0497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Anastasiadis PZ. p120-ctn: A nexus for contextual signaling via Rho GTPases. Biochim Biophys Acta. 2007;1773(1):34–46. doi: 10.1016/j.bbamcr.2006.08.040. [DOI] [PubMed] [Google Scholar]
- 30.Wang T, Pei X, Zhan J, Hu J, Yu Y, Zhang H. FERM-containing protein FRMD5 is a p120-catenin interacting protein that regulates tumor progression. FEBS Lett. 2012;586(19):3044–3050. doi: 10.1016/j.febslet.2012.07.019. [DOI] [PubMed] [Google Scholar]
- 31.Fagotto F, Funayama N, Gluck U, Gumbiner BM. Binding to cadherins antagonizes the signaling activity of beta-catenin during axis formation in Xenopus. J Cell Biol. 1996;132(6):1105–1114. doi: 10.1083/jcb.132.6.1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kim NG, Koh E, Chen X, Gumbiner BM. E-cadherin mediates contact inhibition of proliferation through Hippo signaling-pathway components. Proc Natl Acad Sci U S A. 2011;108(29):11930–11935. doi: 10.1073/pnas.1103345108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li P, Silvis MR, Honaker Y, Lien WH, Arron ST, Vasioukhin V. αE-catenin inhibits a Src-YAP1 oncogenic module that couples tyrosine kinases and the effector of Hippo signaling pathway. Genes Dev. 2016;30(7):798–811. doi: 10.1101/gad.274951.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Takahashi K, Suzuki K. Density-dependent inhibition of growth involves prevention of EGF receptor activation by E-cadherin-mediated cell-cell adhesion. Exp Cell Res. 1996;226(1):214–222. doi: 10.1006/excr.1996.0221. [DOI] [PubMed] [Google Scholar]
- 35.Qian X, Karpova T, Sheppard AM, McNally J, Lowy DR. E-cadherin-mediated adhesion inhibits ligand-dependent activation of diverse receptor tyrosine kinases. EMBO J. 2004;23(8):1739–1748. doi: 10.1038/sj.emboj.7600136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Morris ZS, McClatchey AI. Aberrant epithelial morphology and persistent epidermal growth factor receptor signaling in a mouse model of renal carcinoma. Proc Natl Acad Sci U S A. 2009;106(24):9767–9772. doi: 10.1073/pnas.0902031106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Padua D, Massagué J. Roles of TGFbeta in metastasis. Cell Res. 2009;19(1):89–102. doi: 10.1038/cr.2008.316. [DOI] [PubMed] [Google Scholar]
- 38.Perez-Moreno M, Davis MA, Wong E, Pasolli HA, Reynolds AB, Fuchs E. p120-catenin mediates inflammatory responses in the skin. Cell. 2006;124(3):631–644. doi: 10.1016/j.cell.2005.11.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stairs DB, et al. Deletion of p120-catenin results in a tumor microenvironment with inflammation and cancer that establishes it as a tumor suppressor gene. Cancer Cell. 2011;19(4):470–483. doi: 10.1016/j.ccr.2011.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Thoreson MA, Reynolds AB. Altered expression of the catenin p120 in human cancer: implications for tumor progression. Differentiation. 2002;70(9-10):583–589. doi: 10.1046/j.1432-0436.2002.700911.x. [DOI] [PubMed] [Google Scholar]
- 41.Davis MA, Reynolds AB. Blocked acinar development, E-cadherin reduction, and intraepithelial neoplasia upon ablation of p120-catenin in the mouse salivary gland. Dev Cell. 2006;10(1):21–31. doi: 10.1016/j.devcel.2005.12.004. [DOI] [PubMed] [Google Scholar]
- 42.Smalley-Freed WG, et al. p120-catenin is essential for maintenance of barrier function and intestinal homeostasis in mice. J Clin Invest. 2010;120(6):1824–1835. doi: 10.1172/JCI41414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bartlett JD, et al. Targeted p120-catenin ablation disrupts dental enamel development. PLoS One. 2010;5(9):e12703. doi: 10.1371/journal.pone.0012703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Oas RG, et al. p120-Catenin is required for mouse vascular development. Circ Res. 2010;106(5):941–951. doi: 10.1161/CIRCRESAHA.109.207753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Smalley-Freed WG, et al. Adenoma formation following limited ablation of p120-catenin in the mouse intestine. PLoS One. 2011;6(5):e19880. doi: 10.1371/journal.pone.0019880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kurley SJ, et al. p120-catenin is essential for terminal end bud function and mammary morphogenesis. Development. 2012;139(10):1754–1764. doi: 10.1242/dev.072769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fodde R, et al. A targeted chain-termination mutation in the mouse Apc gene results in multiple intestinal tumors. Proc Natl Acad Sci U S A. 1994;91(19):8969–8973. doi: 10.1073/pnas.91.19.8969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Moser AR, Pitot HC, Dove WF. A dominant mutation that predisposes to multiple intestinal neoplasia in the mouse. Science. 1990;247(4940):322–324. doi: 10.1126/science.2296722. [DOI] [PubMed] [Google Scholar]
- 49.Barker N, et al. Crypt stem cells as the cells-of-origin of intestinal cancer. Nature. 2009;457(7229):608–611. doi: 10.1038/nature07602. [DOI] [PubMed] [Google Scholar]
- 50.van de Wetering M, et al. The beta-catenin/TCF-4 complex imposes a crypt progenitor phenotype on colorectal cancer cells. Cell. 2002;111(2):241–250. doi: 10.1016/S0092-8674(02)01014-0. [DOI] [PubMed] [Google Scholar]
- 51.Sellin JH, Wang Y, Singh P, Umar S. beta-Catenin stabilization imparts crypt progenitor phenotype to hyperproliferating colonic epithelia. Exp Cell Res. 2009;315(1):97–109. doi: 10.1016/j.yexcr.2008.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dohn MR, Brown MV, Reynolds AB. An essential role for p120-catenin in Src- and Rac1-mediated anchorage-independent cell growth. J Cell Biol. 2009;184(3):437–450. doi: 10.1083/jcb.200807096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.March HN, et al. Insertional mutagenesis identifies multiple networks of cooperating genes driving intestinal tumorigenesis. Nat Genet. 2011;43(12):1202–1209. doi: 10.1038/ng.990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mann MB, Jenkins NA, Copeland NG, Mann KM. Sleeping Beauty mutagenesis: exploiting forward genetic screens for cancer gene discovery. Curr Opin Genet Dev. 2014;24:16–22. doi: 10.1016/j.gde.2013.11.004. [DOI] [PubMed] [Google Scholar]
- 55.Takeda H, Rust AG, Ward JM, Yew CC, Jenkins NA, Copeland NG. Sleeping Beauty transposon mutagenesis identifies genes that cooperate with mutant Smad4 in gastric cancer development. Proc Natl Acad Sci U S A. 2016;113(14):E2057–E2065. doi: 10.1073/pnas.1603223113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Alimonti A, et al. A novel type of cellular senescence that can be enhanced in mouse models and human tumor xenografts to suppress prostate tumorigenesis. J Clin Invest. 2010;120(3):681–693. doi: 10.1172/JCI40535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Veitia RA. Exploring the etiology of haploinsufficiency. Bioessays. 2002;24(2):175–184. doi: 10.1002/bies.10023. [DOI] [PubMed] [Google Scholar]
- 58.Janssen KP, et al. APC and oncogenic KRAS are synergistic in enhancing Wnt signaling in intestinal tumor formation and progression. Gastroenterology. 2006;131(4):1096–1109. doi: 10.1053/j.gastro.2006.08.011. [DOI] [PubMed] [Google Scholar]
- 59.Shibata H, et al. Alpha-catenin is essential in intestinal adenoma formation. Proc Natl Acad Sci U S A. 2007;104(46):18199–18204. doi: 10.1073/pnas.0705730104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gonzalez-Billault C, Muñoz-Llancao P, Henriquez DR, Wojnacki J, Conde C, Caceres A. The role of small GTPases in neuronal morphogenesis and polarity. Cytoskeleton (Hoboken) 2012;69(7):464–485. doi: 10.1002/cm.21034. [DOI] [PubMed] [Google Scholar]
- 61.Noren NK, Arthur WT, Burridge K. Cadherin engagement inhibits RhoA via p190RhoGAP. J Biol Chem. 2003;278(16):13615–13618. doi: 10.1074/jbc.C200657200. [DOI] [PubMed] [Google Scholar]
- 62.Yu W, et al. Involvement of RhoA, ROCK I and myosin II in inverted orientation of epithelial polarity. EMBO Rep. 2008;9(9):923–929. doi: 10.1038/embor.2008.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Myant KB, et al. ROS production and NF-κB activation triggered by RAC1 facilitate WNT-driven intestinal stem cell proliferation and colorectal cancer initiation. Cell Stem Cell. 2013;12(6):761–773. doi: 10.1016/j.stem.2013.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.el Marjou F, et al. Tissue-specific and inducible Cre-mediated recombination in the gut epithelium. Genesis. 2004;39(3):186–193. doi: 10.1002/gene.20042. [DOI] [PubMed] [Google Scholar]
- 65.Sato T, et al. Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett’s epithelium. Gastroenterology. 2011;141(5):1762–1772. doi: 10.1053/j.gastro.2011.07.050. [DOI] [PubMed] [Google Scholar]
- 66.Wu J, Mariner DJ, Thoreson MA, Reynolds AB. Production and characterization of monoclonal antibodies to the catenin p120ctn. Hybridoma. 1998;17(2):175–183. doi: 10.1089/hyb.1998.17.175. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.