

Abstract
Drought stress results in significant crop yield losses. Comparative transcriptome analysis between tolerant and sensitive species can provide insights into drought tolerance mechanisms in jute. We present a comprehensive study on drought tolerance in two jute species—a drought tolerant species (Corchorus olitorius L., GF) and a drought sensitive species (Corchorus capsularis L., YY). In total, 45,831 non-redundant unigenes with average sequence length of 1421 bp were identified. Higher numbers of differentially expressed genes (DEGs) were discovered in YY (794) than in GF (39), implying that YY was relatively more vulnerable or hyper-responsive to drought stress at the molecular level; the two main pathways, phenylpropanoid biosynthesis and peroxisome pathway, significantly involved in scavenging of reactive oxygen species (ROS) and 14 unigenes in the two pathways presented a significant differential expression in response to increase of superoxide. Our classification analysis showed that 1769 transcription factors can be grouped into 81 families and 948 protein kinases (PKs) into 122 families. In YY, we identified 34 TF DEGs from and 23 PK DEGs, including 19 receptor-like kinases (RLKs). Most of these RLKs were downregulated during drought stress, implying their role as negative regulators of the drought tolerance mechanism in jute.
Introduction
Crop production is greatly affected by drought, one of the major abiotic stresses that cause significant yield loss1,2. Drought is further aggravated by population growth, global scarcity of water resources, and climate abnormalities. Therefore, it is of crucial importance to clarify the mechanisms of drought stress tolerance in crops in order to solve or alleviate the problems caused by this stress2.
Plant response to drought stress is a complex biological process involving physiological, biochemical, and molecular changes. During this process, the expression profiles of a large number of genes are altered; these genes relate mainly to two classes of genes. The first class includes genes encoding ‘effector proteins’, which directly protect plants from abiotic stress including membrane protein genes; genes involved in biosynthesis of various osmoprotectants; photosynthesis-related genes; genes involved in growth and development; chaperones-encoding genes; and genes encoding detoxification enzymes3,4 etc. The second class of genes encode ‘regulatory proteins’ that regulate the expression of downstream target genes in the stress response3 and include protein kinases (PKs), such as mitogen-activated PKs, ribosomal PKs, and receptor PKs1, and transcription factors (TFs), such as AP2/ERF, AREB/ABF, bZIP, DREB, MYC/MYB, and WRKY3,5,6.
As well, many plant metabolism pathways participate in response to abiotic stress, such as photosynthetic, phenylalanine metabolism, peroxidase, MAPK signalling pathways, etc. A delicate balance between the multiple pathways is necessary for plant growth and development7. However, the balance might be be broken when the plant is underlying abiotic stress. For example, ROS, such as O− 2, H2O2, etc. can be used as signaling molecule when keeping at low levels under normal conditions. Production and balance of ROS is related to photosynthetic, phenylalanine metabolism, peroxidase pathways8,9.Underlying stress, the electrons of photosynthetic pathway leak to O2 and result in generation of a mass of ROS. In order to cope with the crisis, numerous effector genes involved in phenylalanine metabolism and peroxidase pathways present differential expression, such as ferulic acid10, sinapic acid11, isocitrate dehydrogenase (ICDH), superoxide dismutase (SOD), catalase (CAT)7, etc. If these pathways are able to reconstruct the new balance and preserve ROS at a relativly low level, the tissues and cells may be prevented from damage and death because of oxidation.
Advancements in high-throughput sequencing technology resulted in cost reductions, rendering transcriptome sequencing the most direct and effective way to explore stress resistance mechanism in plants. Recent studies12,13 have shown that comparative analysis of transcriptomes between a tolerant and a sensitive genotype can effectively elucidate the molecular mechanism of abiotic stress. After cotton, jute (Corchorus spp.) is the second most important global biodegradable natural fibre crop. It is a diploid annual crop (2n = 14) distributed in the tropics, subtropics, and warm temperate regions of the world (mainly in Asia and Africa)14. It includes two cultivated species, C. capsularis L., a drought sensitive species, and C. olitorius L., a drought tolerant species. Recently, the demand for jute has increased worldwide because of its broad-spectrum application and eco-friendly characteristics15,16.
Several studies on drought tolerance in jute plants focused mainly on drought-resistance evaluation of germplasm17,18 and morphological19, physiological20, and biochemical21 changes during the response period. However, to our knowledge, to date only Sawarkar et al.22 and Das et al.23 have studied the genetics of jute under drought conditions, and a comprehensive and high-throughput study on drought tolerance is yet to be conducted. Here, we carried out transcriptome sequencing in C. capsularis and C. olitorius exposed to drought stress to explore the drought resistance mechanism in these cultivated species.
Results
Transcriptome sequencing and assembly
Twelve GF and YY samples were used for transcriptome sequencing and analysis, generating a total of 608,395,184 raw reads (Table 1). After quality control of these reads, 587,249, 098 (96.52%) clean reads were obtained from the raw data with an average GC content of 43.37% and Q20 average bases quality score of 97.14%, accounting for 88.1 Gb of sequencing data, which were used for de novo assembly of transcriptome. A total of 123,327 transcripts and 45,831 non-redundant unigenes were assembled using all clean reads. The length of the transcripts and unigenes varied from 201 to 15,890 bp, with averages of 1716 and 1421 bp, respectively (Table 2). The sequence raw reads and obtained in the present study are available in the NCBI Sequence Read Archive (SRA) under BioProject number PRJNA378897.
Table 1.
Details of the raw data and clean data of twelve transcriptomes from drought-tolerant Corchorus olitorius (GF) and drought-sensitive C. capsularis (YY).
| Species | Raw reads | Clean reads | Clean bases (Gb) | Mapped reads avg (%) | Q20 avg (%) | GC avg (%) | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| Control | Drought | Control | Drought | Control | Drought | Control | Drought | |||
| GF | 147,199,898 | 161,498,660 | 142,004,554 | 156,115,186 | 21.3 | 23.42 | 74.08 | 74.12 | 97.14 | 43.37 |
| YY | 157,609,856 | 142,086,770 | 152,431,084 | 136,698,274 | 22.88 | 20.5 | 76.87 | 74.29 | ||
| total | 608,395,184 | 587,249,098 (96.52%) | 88.1 | — | — | |||||
Table 2.
Assembly output statistics of all clean data using Trinity assembler.
| Min Length | Mean Length | Max Length | N50 | Number | Total Nucleotides | |
|---|---|---|---|---|---|---|
| Transcripts | 201 | 1716 | 15,573 | 2327 | 123,327 | 211,649,647 |
| Unigenes | 201 | 1421 | 15,573 | 2131 | 45,831 | 65,148,415 |
Functional annotation of unigenes
Sequence alignments of all 45,831 unigenes to the NCBI non-redundant (Nr), NCBI nucleotide (Nt), Kyoto Encyclopedia of Genes and Genomes (KEGG) Orthology (KO), gene ontology (GO), eukaryotic ortholog group (KOG), protein family (Pfam), and SwissProt databases revealed that 30,799 (75.79%) unigenes were successfully annotated in at least one databases and 6,520 (14.22%) unigenes were annotated in all seven databases. The largest number of unigenes (30, 799; 67.2%) was aligned to the Nr database, whereas more than 50% of unigenes were aligned to each the SwissProt, Pfam, GO, and Nt databases. The lowest number of unigenes (12,305; 26.84%) was annotated in the KO database (Supplemental Fig. 1).
Meanwhile, unigenes within GO annotation were classified into 46 terms involved in cellular components, biological processes, and molecular function (Supplemental Fig. 2). Comparison to the KEGG database revealed that the successfully annotated unigenes were assigned to 280 KEGG pathways, which were grouped into 32 classifications based on pathway hierarchy 2 (Supplemental Fig. 3). Unigenes successfully annotated in KOG were aligned to 26 KOG classifications (Supplemental Fig. 4).
Differential gene expression in response to polyethylene glycol treatment
The analysis of GF drought-stressed (GFD) vs. GF control (GFC) and YY drought-stressed (YYD) vs. YY control (YYC) revealed 39 (Fig. 1) and 794 (Fig. 2) significantly differentially expressed unigenes (DEGs) in GF and YY, respectively (Supplementary file 1). Of those, 7 and 567 DEGs were upregulated and 32 and 227 were down-regulated in GFD vs. GFC and YYD vs. YYC, respectively. Between GF and YY, there were only 7 common DEGs, all of which were down-regulated.
Figure 1.
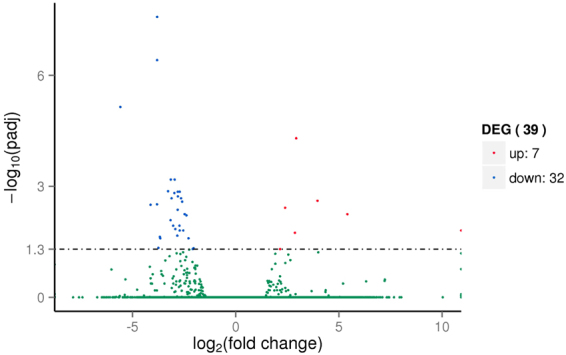
Analysis of differentially expressed unigenes in GFD (GF drought-stressed) vs. GFC (GF control).
Figure 2.
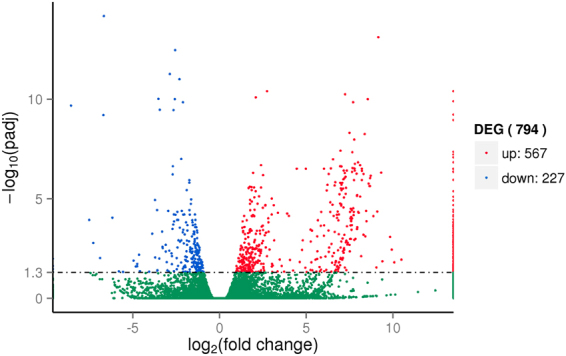
Analysis of differentially expressed unigenes in YYD (YY drought-stressed) vs. YYC (YY control).
GO and KEGG analysis of differentially expressed unigenes
We carried out GO annotation of 39 and 794 DEGs identified in GF and YY, respectively. In the drought tolerant species, only 28 genes were significantly enriched in catalytic activity under the molecular function (Fig. 3A). In the drought sensitive species, under the biological process (BP) category, metabolic process, translation, oxidation-reduction process, cell wall organisation or biogenesis, cell morphogenesis, and ribosome biogenesis etc. were prominently represented (Fig. 3B). Under the cellular component (CC) category, a large number of unigenes were enriched in cell wall and ribosome etc. (Fig. 3B), whereas for the molecular function (MF) category, oxidoreductase activity, peroxidase activity, structural constituent of ribosome, and coenzyme binding etc. were significantly enriched (Fig. 3B).
Figure 3.

Gene ontology analysis showed that the differentially expressed unigenes (DEGs) enrichment categories in GFD (GF drought-stressed) vs. GFC (GF control) (A) and in YYD(YY drought-stressed) vs. YYC (YY control) for biological process (BP) (B), cellular component (CC) (B), and molecular function (MF) (B).
The KEGG pathway analysis showed that DEGs in YY were mainly located in ribosome, carbon metabolism, protein processing in endoplasmic reticulum, oxidative phosphorylation, biosynthesis of amino acids, phenylpropanoid biosynthesis, glycolysis/gluconeogenesis, glyoxylate and dicarboxylate metabolism, carbon fixation in photosynthetic organisms, MAPK signalling pathway, lysosome, citrate cycle (TCA cycle), plant–pathogen interaction, phenylalanine metabolism, and arginine and proline metabolism (Table 3). In contrast, in GF, only the fatty acid metabolism pathway was enriched for three unigenes.
Table 3.
Main KEGG enriched pathway of differentially expressed genes (DEGs) in drought-sensitive Corchorus capsularis.
| Pathway | KEGG pathway ID | DEGs number |
|---|---|---|
| Ribosome | ko03010 | 74 |
| Carbon metabolism | ko01200 | 24 |
| Protein processing in endoplasmic reticulum | ko04141 | 24 |
| Oxidative phosphorylation | ko00190 | 20 |
| Biosynthesis of amino acids | ko01230 | 16 |
| Phenylpropanoid biosynthesis | ko00940 | 14 |
| Glycolysis/Gluconeogenesis | ko00010 | 14 |
| Glyoxylate and dicarboxylate metabolism | ko00630 | 13 |
| Carbon fixation in photosynthetic organisms | ko00710 | 13 |
| MAPK signalling pathway | ko04010 | 13 |
| Lysosome | ko04142 | 13 |
| Citrate cycle (TCA cycle) | ko00020 | 12 |
| Plant-pathogen interaction | ko04626 | 12 |
| Phenylalanine metabolism | ko00360 | 11 |
| Arginine and proline metabolism | ko00330 | 11 |
| Pyruvate metabolism | ko00620 | 9 |
| Endocytosis | ko04144 | 9 |
| Valine, leucine and isoleucine degradation | ko00280 | 8 |
| Starch and sucrose metabolism | ko00500 | 8 |
| RNA transport | ko03013 | 8 |
| Pentose and glucuronate interconversions | ko00040 | 7 |
| Peroxisome | ko04146 | 7 |
| Cysteine and methionine metabolism | ko00270 | 7 |
Differential expression of TFs
A total of 1769 unigenes encoding TFs were classified into 81 TF families (Supplementary file 2). The highest number of unigenes was in the MYB family, with 143 unigenes, followed by AP2-EREBP, Orphans, bHLH, and NAC families, each including more than 80 unigenes. In YY under drought stress, 34 DEGs encoding TFs belonged to 20 TF families: AP2-EREBP, MYB, C2C2-Dof, HB, bHLH, TCP, CCAAT, Orphans, ARID, HSF, C2H2, bZIP, C2C2-CO-like, MBF1, GRAS, C3H, BBR/BPC, NAC, HMG, and TRAF (Table 4). Of these, AP2-EREBP included the highest number of DEGs (5), followed by MYB, C2C2-Dof, and HB.
Table 4.
Expression and function annotation of 34 differentially expressed genes (DEGs) encoding transcription factors (TFs) in drought-sensitive Corchorus capsularis.
| Unigenes | TF family | log2(FC) | Function annotation |
|---|---|---|---|
| c81341_g1 | AP2-EREBP | −1.1847 | regulation of transcription, DNA-dependent |
| c88904_g1 | AP2-EREBP | 1.3936 | regulation of transcription, DNA-dependent//viral infectious cycle//signal transduction |
| c83083_g1 | AP2-EREBP | 1.7811 | regulation of transcription, DNA-dependent |
| c87442_g1 | AP2-EREBP | 2.3182 | sensory perception of chemical stimulus//regulation of transcription, DNA-dependent |
| c80754_g1 | AP2-EREBP | 2.8227 | pathogenesis//regulation of transcription, DNA-dependent |
| c84857_g2 | ARID | −1.4303 | tRNA aminoacylation for protein translation |
| c88699_g1 | BBR/BPC | −1.2835 | metabolic process//photosynthesis |
| c83837_g1 | bHLH | −1.7142 | — |
| c82269_g1 | bHLH | −1.4277 | — |
| c81311_g1 | bZIP | 1.7312 | DNA replication//autophagy//regulation of transcription, DNA-dependent//cell cycle |
| c81026_g2 | C2C2-CO-like | −2.2776 | — |
| c81214_g4 | C2C2-Dof | −1.3781 | pathogenesis//oxidation-reduction process//ion transport//regulation of transcription |
| c89585_g4 | C2C2-Dof | −1.08 | regulation of transcription, DNA-dependent |
| c79713_g4 | C2C2-Dof | 1.7456 | regulation of transcription, DNA-dependent |
| c82698_g1 | C2H2 | 3.0106 | — |
| c89112_g2 | C3H | −1.3664 | tRNA processing//oxidation-reduction process//carotenoid biosynthetic process |
| c78769_g3 | CCAAT | 1.0078 | DNA-dependent transcription, initiation |
| c77173_g1 | CCAAT | 7.0214 | — |
| c85510_g1 | GRAS | −1.2846 | regulation of transcription, DNA-dependent |
| c80964_g1 | HB | −1.8847 | regulation of transcription, DNA-dependent |
| c88433_g2 | HB | −1.7419 | regulation of transcription, DNA-dependent |
| c87626_g1 | HB | −1.2914 | glycolipid transport//barrier septum assembly//regulation of transcription |
| c76701_g2 | HMG | 10.065 | regulation of transcription, DNA-dependent//transport |
| c86122_g1 | HSF | 1.5553 | regulation of transcription, DNA-dependent//spindle assembly |
| c85671_g1 | MBF1 | 2.1036 | oxidation-reduction process//metabolic process |
| c80089_g1 | MYB | −2.1779 | — |
| c89680_g1 | MYB | 1.3701 | — |
| c80040_g2 | MYB | 1.7617 | transport//drug transmembrane transport//transmembrane transport//drug transport |
| c76176_g1 | NAC | 2.3436 | oxidation-reduction process//regulation of transcription, DNA-dependent |
| c89867_g2 | Orphans | −1.4272 | — |
| c83081_g1 | Orphans | 1.6443 | phosphorelay signal transduction system |
| c80837_g1 | TCP | −1.3654 | pantothenate biosynthetic process//malate transport//oxidation-reduction process |
| c79759_g2 | TCP | −1.1567 | sucrose metabolic process//starch metabolic process//galactose metabolic process |
| c68683_g1 | TRAF | 5.6375 | modulation by virus of host morphology or physiology |
FC: fold change.
Protein kinases in response to drought stress
Eukaryotic genomes have a large number of PK genes and play an important role in phosphorylation events that activate and inactivate the downstream signalling pathway. A total of 948 PK genes, belonging to 122 PK families, were identified (Supplementary file 3). The most abundant family was RLK-Pelle_DLSV with 93 members, followed by RLK-Pelle_LRR-XI-1 with 53 members. The families RLK-Pelle_DLSV, RLK-Pelle_LRR-XI-1, RLK-Pelle_L-LEC, RLK-Pelle_CrRLK1L-1, CAMK_CDPK, RLK-Pelle_SD-2b, RLK-Pelle_LRR-III, RLK-Pelle_LRR-XII-1, RLK-Pelle_RLCK-VIIa-2, CAMK_CAMKL-CHK1, and TKL-Pl-4 each included more than 20 members. 15 families included 23 DEGs in YY, and the highest number of DEGs (19) was assigned to the receptor-like kinases (RLK) class. Most of the DEGs were downregulated by drought stress (Table 5).
Table 5.
Analysis of protein kinase differentially expressed genes (DEGs) in drought-sensitive Corchorus capsularis (YY) under drought stress (YYD) and normal (YYC) conditions.
| PK families | DEGs | LogFC (YYD vs. YYC) | Annotation |
|---|---|---|---|
| AGC-Pl | c77309_g1 | 5.214 | AGC protein kinase |
| AGC_RSK-2 | c88811_g2 | −1.268 | Phototropin 1 isoform 1 |
| RLK-Pelle_CR4L | c89074_g1 | −1.7142 | Crinkly 4 |
| RLK-Pelle_CrRLK1L-1 | c84515_g1 | −1.0488 | Malectin/receptor-like protein kinase family protein |
| RLK-Pelle_CrRLK1L-1 | c84566_g1 | −2.2044 | Malectin/receptor-like protein kinase family protein |
| RLK-Pelle_CrRLK1L-1 | c86137_g3 | −1.4011 | Kinase superfamily protein, putative isoform 1 |
| RLK-Pelle_DLSV | c83696_g1 | −3.7212 | S-locus lectin protein kinase family protein |
| RLK-Pelle_DLSV | c84899_g1 | −1.682 | Serine/threonine kinases, protein kinases, ATP binding, sugar |
| RLK-Pelle_DLSV | c89207_g1 | −2.2017 | Leucine-rich repeat transmembrane protein kinase |
| RLK-Pelle_DLSV | c89313_g1 | −1.5947 | Cysteine-rich RLK 10 |
| RLK-Pelle_DLSV | c90903_g5 | −2.4626 | S-locus lectin protein kinase family protein |
| RLK-Pelle_L-LEC | c84451_g1 | −2.6991 | Kinase, putative |
| RLK-Pelle_LRR-III | c89009_g1 | −1.2063 | Leucine-rich repeat protein kinase family protein isoform 1 |
| RLK-Pelle_LRR-VI-1 | c89569_g1 | −1.4033 | Leucine-rich repeat protein kinase family protein isoform 1 |
| RLK-Pelle_LRR-VII-1 | c89862_g1 | −1.9284 | Leucine-rich repeat protein kinase family protein isoform 1 |
| RLK-Pelle_LRR-VIII-1 | c88539_g1 | 1.6051 | LRR receptor-like serine/threonine-protein kinase, putative |
| RLK-Pelle_LRR-XI-1 | c88948_g1 | −1.1307 | HAESA-like 1 isoform 1 |
| RLK-Pelle_LRR-XII-1 | c90920_g2 | −1.619 | Leucine-rich repeat protein kinase family protein, putative |
| RLK-Pelle_LysM | c88139_g2 | 2.2853 | PREDICTED: chitin elicitor receptor kinase 1-like |
| RLK-Pelle_SD-2b | c85931_g1 | 1.7428 | Receptor protein kinase 1 |
| RLK-Pelle_SD-2b | c86648_g4 | −3.273 | Receptor protein kinase 1 |
| STE_STE11 | c77570_g1 | 1.8479 | Mitogen-activated protein kinase kinase kinase 15 |
| STE_STE11 | c85006_g6 | −2.6717 | PREDICTED: mitogen-activated protein kinase kinase kinase 3-like |
FC: fold change.
A comprehensive scavenging pathway of ROS
The accumulation of ROS can damage to DNA, RNA, proteins and lipids and then result in disrupting normal metabolism and even tissues and cell death. In order to cope with the toxicity of ROS, plants have evolved an array of efficient cooperative system included enzymatic and nonenzymatic antioxidants and involved in multiple metabolic pathway. In YY, under drought stress, the two main pathways, phenylpropanoid biosynthesis and peroxisome pathway, significantly involved in scavenging of ROS, and 14 unigenes in the two pathways presented a significant differential expression in response to increase of superoxide (Fig. 4): one SOD (upregulated, 104.42 fold change), one ascorbate (upregulated, 3.26 fold change), one CAT unigene (upregulated, 28.89 fold change), one ICDH (upregulated, 147.54 fold change), eight NADP+ (five upregulated, 8.37~83.87 fold change; three dowregulated, −2.35~−2.84), one aldehyde dehydrogenase (upregulated, 3.61 fold change) and one alcohol dehydrogenase (downregulated, −2.32 fold change).
Figure 4.

Fourteen differentially expressed unigenes (DEGs) involved in phenylpropanoid biosynthesis and peroxisome pathway showed a comprehensive pathway in response to increase of superoxide. PSI: photosystem I, SOD: superoxide dismutase, ICDH: isocitrate dehydrogenase, CAT: catalase, ROS: reactive oxygen species.
SNP marker identification
In the study, a total of 31 1906 SNP sites with mutated codon in 22 873 unigenes were discovered in both jute species (12 plants). Of which, 18 964 unigenes had SNP sites with mutated amino acid and the number of those for each unigene varied from 1 to 92 (Fig. 5A). Unigenes with such a SNP were the most common (3756) followed by those with two (2659), while there were only 7.5% unigenes with more than 15 SNP sites with mutated amino acid. In total, 546 DEGs with SNP sites resulted in amino acid change were discovered. The the most common DEGs (107) were those with a SNP with mutated amino acid (Fig. 5B).
Figure 5.

Satistics of SNP sites with mutated amino acid and unigenes included those SNP sites (A); Satistics of differentially expressed unigenes (DEGs) included SNP sites with mutated amino acid and those SNP sites (B).
Validation of the differential gene expression
To validate the differential expression results of the transcriptome sequencing data analysis, real-time quantitative PCR (qRT-PCR) was carried out in YY and GF under drought stress and control conditions. A total of eight DEGs were selected randomly for the qRT-PCR analysis. All the selected DEGs were significantly differentially expressed, and the expression profiles were consistent with the results of transcriptome sequencing. Fold change values of differential expression for each DEG obtained by qRT-PCR analysis in YY or GF are displayed in Fig. 6.
Figure 6.

qRT-PCR analysis of eight differentially expressed unigenes. Data represent the fold change of relative quantification of each DEG in GFD (GF drought-stressed) vs. GFC (GF control) or YYD (YY drought-stressed) vs. YYC (YY control); the error bar represents the standard deviation.
Discussion
Drought is one of the major environmental factors affecting the geographical distribution of plants in nature. It causes significant yield loss in crop plants and threatens food security in many tropical and subtropical countries24. Therefore, it is essential to understand the molecular mechanisms of plant tolerance to drought to lay the foundation for crop molecular breeding. Jute (Corchorus spp.) is one of the most important bast fibre crops in the world. Although the jute genome25 have been published in this year, Yet, to our knowledge, to date, only two studies implementing transcriptome sequencing were conducted on jute (C. capsularis)26,27. The lack of gene sequences and functional annotation hindered the analysis of the genetic basis of molecular mechanisms triggered by abiotic stress in jute. In recent years, the accelerated development of sequencing platforms has made it possible to narrow the gap in our knowledge of the molecular mechanisms involved in abiotic stress in jute compared to those in other plants.
Overall, the number of detected DEGs was greater in YY than in GF, suggesting that the drought-sensitive jute species was relatively more vulnerable or hyper-responsive to drought stress at the molecular level. Similar results have been reported in studies on drought stress response in other plants12,28,29 and may be explained by the fact that compared to tolerant species, sensitive species undergo greater changes in phenotype, physiological and biochemical properties when mitigating the effects of stress conditions.
The GO annotation in GF showed DEG enrichment only in catalytic activity, which may contribute to the plant’s adaptation to drought stress through modifying the physiological and biochemical processes. Various environmental stresses such as drought and salt stresses can cause accumulation of reactive oxygen species (ROS), causing damages to cell wall and membrane tissues24. For example, ROS accumulation can cause crosslinking of phenolics and cell-wall glycoproteins, leading to cell-wall stiffening30. Antioxidant defence system plays a crucial role in scavenging ROS and protecting plants from oxidative damage. In YY, numerous DEGs were enriched in the antioxidant defence system terms: oxidation reduction process, oxidoreductase activity, peroxidase activity etc.; in addition, many pathways have participated in the complex regulatory and interaction network of ROS. The two pathways that are worth mentioning are phenylalanine metabolism and peroxidase pathways. Under drought stress, the leakage of photosynthetic electrons to the O2 is increased31; it is reported that the rate can reach to 50% as compared to unstressed in wheat32; which results in a mass of superoxide (O2 −). And then ascorbate can function as an antioxidant directly to scavenge singlet oxygen and superoxide etc.7,31; while SOD (upregulated 104.42 fold change) can firstly dismutate superoxide to H2O2 which can promptly attacks thiol proteins33. Furtherly. CAT can scavenge H2O2 without a reductant7. And an increase of the activity of the main antioxidative systems (such as enzymes of the ascorbate-glutathione cycle) is generally accompanied by significant rise of activity and protein expression of NADP-ICDH34. The NADP-ICDH upregulated in YY can involve in thioredoxin reductase system and glutathione peroxidase system etc. to scavenge H2O2 10. Phenolic acids are powerful antioxidants presented widely in plants. Of which, Ferulic acid (FA) and sinapic acid arising from the metabolism of phenylalanine have ability to quench ROS11. In particular, FA can enhance the activities of SOD and CAT to decrease content of ROS and involved in proline biosynthesis to regulate osmosis pressure under environment stress. And aldehyde dehydrogenase can synthesize FA and sinapic acid with coniferylaldehycle and sinapapaldehyde, respectively; while alcohol dehydrogenase can convert coniferylaldehycle and sinapapaldehyde into coniferylalcohol and sinapyl alcohol. However, in YY, c85671_g2 encoding aldehyde dehydrogenase was upregulated and c84589_g3 encoding alcohol dehydrogenase was downregulated, which might be conducive to deal with drought stress.
The primary plant cell wall consists of cellulose fibrils interconnected by hemicellulose tethers, such as arabinoxylan and xyloglucan, and embedded in pectin24,30. The plant cell wall undergoes extensive remodelling for survival under stress conditions35 to repair cell wall damage caused by ROS and maintain the osmotic pressure within the cell. The present study revealed the enrichment of 20 genes in the cell wall term: 17 were upregulated and 3 downregulated. Of these unigenes, 5 encoding for pectinesterase, which can remodel the cell wall, were significantly altered by drought stress in other plants36,37. In addition, the KEGG analysis revealed that many unigenes were also located in the biosynthesis pathways of various osmoprotectants, such as biosynthesis of amino acids, arginine and proline metabolism, and pentose and glucuronate interconversions, which support plant survival under different osmotic conditions, stabilise the membranes and proteins, and reduce the osmotic potential of membranes to prevent dehydration inside the cell38,39. Further, a large number of DEGs were located in the phenylpropanoid biosynthesis, citrate cycle, carbon fixation in photosynthetic organisms, and MAPK signalling pathways, which were enriched under various stresses and play crucial roles in plant survival under stress conditions40–42.
In the present study, 1769 unigenes were classified into 81 TF families. Of those, 34 DEGs from 20 TF families were found only in YY exposed to drought stress. A previous study12 on banana plants showed that the number of DEGs encoding TFs was higher in the sensitive genotype than in the tolerant genotype. The TF families, including MYB, AP2, bHLH, NAC, bZIP, C2C2-Dof, and HSFs, were differentially expressed under drought stress in the present study; these results were consistent with the results reported for other plants3. In addition, we found the largest number of DEGs (5) in the AP2-EREBP family, most of which were induced by drought stress; this family plays an important role in the regulation of transcription and signal transduction, such as phenylalanine metabolism and peroxidase pathways.
A large number of PK genes play an important role in phosphorylation events, which activate and deactivate the downstream signalling cascades under stresses43. For example, a study on Arabidopsis showed that the gene expression of some of the RLK members changes under water stress44. In the present study, 23 PK DEGs, including 19 RLK members, were identified in YY. Most of these DEGs were downregulated by drought stress, suggesting that the RLK members function as a negative regulator of drought tolerance in jute; our results were consistent with those reported by45 for rice.
With development of high-throughput sequencing, the molecular research of jute obtains a golden opportunity to grow up. However, compared with other crops, development of molecular markers and genes or QTLs mapping are still lacking because of a lag in molecular research on jute. To date, the densest genetic map includes only 913 polymorphic markers46, which greatly limits molecular marker-assisted selection and gene clones. Gene and QTL mapping involved in jute drought stress is not reported. In the study, The markers of SNP will be applicable in QTL and gene mapping in jute, particularly, the SNP sites located in DEGs can be used as developing markers which can be used directly as candidate drought-tolerance genes mapping or association analysis to improve efficiency of gene mapping.
In summary, we present the first comprehensive research on drought tolerance in two jute species, a drought-tolerant and a drought-sensitive species, exposed to drought stress conditions and elucidate the molecular basis of the drought tolerance mechanism. We identified 45,831 non-redundant unigenes and inferred the relationship between jute plants and other plant species based on unigene annotation by using molecular databases. The study analysed the DEGs in both jute species under drought stress condition and explored the GO terms and KEGG pathways related to drought tolerance. The results of the DEGs analysis showed that compared to the drought sensitive species, the drought tolerant species is less affected by drought stress. Further, 1769 TFs and 948 PKs were identified in the present study. Of these, 23 PK DEGs, including 19 RLK members, and 34 DEGs, encoding TFs from 20 TF families were detected in YY. Overall, we believe that the data presented herein will be useful to study the drought tolerance mechanism in plants, clone drought tolerance genes, and breed drought-tolerant jute cultivars.
Materials and Methods
Plant materials, drought stress treatment, and RNA isolation
Two jute species, drought-tolerant C. olitorius (Gangfengchangguo, GF) and drought-sensitive C. capsularis (Yueyuan5hao, YY), were used in this study. Sixty plants of each species were cultivated in a greenhouse at 25–28 °C in a hydroponic culture with Yoshida nutrient solution. Six disease-free seedlings at the 9-leaf stage, with strong and uniform growth, were selected from each species and equally divided into two groups: one was transferred to a control pot containing only Yoshida nutrient solution and the other was transferred to a pot containing a solution of equal parts of 10% polyethylene glycol and Yoshida nutrient solution (for drought treatment). After 24 h, the leaves and roots of every plant in the control and treatment pots were collected and used for RNA extraction.
Total RNA was isolated from the control and drought-stressed leaf and root samples using a Trizol reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. The total RNA isolated from the leaves and from the root of a single plant was pooled in equal volume and concentration to prepare sequencing libraries. In total, twelve RNA sequencing libraries were prepared (three each GFD, GFC, YYD, and YYC; three independent biological replicates were prepared for each treatment of each species in this study). RNA degradation and contamination was monitored on 1% agarose gels, and RNA purity was checked using a NanoPhotometer® spectrophotometer (Implen, Inc., Westlake Village, CA, USA). RNA concentration was measured using a Qubit® RNA Assay Kit in a Qubit® 2.0 Flurometer (Life Technologies, Carlsbad, CA, USA). RNA integrity was assessed using an RNA Nano 6000 Assay Kit and the Agilent Bioanalyzer 2100 system (Agilent Technologies, Santa Clara, CA, USA).
Transcriptome sequencing
Sequencing libraries were generated using a NEBNext® Ultra™ RNA Library Prep Kit for Illumina® (New England Biolabs Inc., Ipswich, MA, USA) following the manufacturer’s recommendations, and index codes were incorporated to assign sequences to each sample. The quality of all libraries was assessed in the Agilent Bioanalyzer 2100 system. The clustering of the index-coded samples was performed on a cBot Cluster Generation System using a TruSeq PE Cluster Kit v3-cBot-HS (Illumina, USA) for RNA libraries according to the manufacturer’s instructions. After cluster generation, the library preparations were sequenced on an Illumina HiSeq X Ten platform and paired-end reads were generated for transcriptome sequencing.
Transcriptome data analysis and annotation
Quality control of raw data was carried out using in-house perl scripts to remove reads containing adapter and poly-N sequences and low quality reads. All downstream analyses were based on clean, high-quality data. Transcriptome assembly for all clean data was accomplished using Trinity47 with min_kmer_cov set to 2 and all other parameters set to default. In addition, after obtaining the transcripts, all clean reads were mapped to the transcripts and the transcripts with less than 5X coverage were removed. Gene function was annotated based on the following public databases using the e-value cut-off listed parenthetically: Nr (e-value = 1e-5), Nt (e-value = 1e-5), Pfam (e-value = 0.01), KOG (e-value = 1e-5), Swiss-Prot (e-value = 1e-5), KO (e-value = 1e-10), and GO (e-value = 1e-6).
Differential gene expression analysis and biological analysis of DEGs
Gene expression levels were estimated by RSEM48 for each sample. Clean data were mapped back onto the assembled transcriptome, and the read-count for each gene was obtained from the mapping results. Differential expression analysis of the two treatments groups of each species was performed using the DESeq R package (1.10.1)49. Genes with an adjusted P-value < 0.05 found by DESeq were assigned as differentially expressed. GO enrichment analysis of the DEGs was implemented by the GOseq R packages based on Wallenius non-central hyper-geometric distribution50. KOBAS51 software was used to test the statistical enrichment of DEGs in KEGG pathways.
SNP calling
The assembled unigenes were used as reference transcriptome, and all of clean reads were mapped to the reference transcriptome. Identification of SNP refered to the method reported by Zhang et al.27. Raw vcf files were filtered with GATK52 standard filter method and parameters, and only SNPs with distance >5 were retained. The unigenes with SNP and differential expression were found out using in-house perl scripts.
qRT-PCR analysis
To validate the results of the high-throughput sequencing, qRT-PCR of the same samples used for transcriptome sequencing was performed in an AB GeneAmp PCR System 9700 (Applied Biosystems, Foster City, CA, USA). The qRT-PCR was carried out in a two-step procedure according to the method by reported by Yangs53. The thermal cycle consisted of an initial denaturation at 95 °C for 10 min followed by 40 cycles at 95 °C for 10 s and 58 °C for 30 s. The relative expression levels were analysed according to a protocol described by Livak and Schmittgen54. Eight DEGs randomly selected from the RNA-seq results were used for validation; the jute ELF gene was selected as the endogenous control55. Each PCR reaction was conducted in triplicates. The primer sequence of DEGs and ELF gene are listed in Supplementary File 4.
Electronic supplementary material
Acknowledgements
This study was financially supported by the National Natural Science Foundation of China (grant number 31601351), the Science and Technology Innovation Project of Chinese Academy of Agricultural Sciences (grant number ASTIP-IBFC01), The National Key Technology Support Program (grant number 2013BAD01B03-13), The Germplasm Resources Protection Project (grant number 2016NWB044).
Author Contributions
Conceived and designed the experiments: Jianguang Su. Performed the experiments: Zemao Yang, Zhigang Dai, Ruike Lu. The acquisition, analysis, and interpretation of data for the work: Zemao Yang, Bibo Wu, Qing Tang, Ying Xu. Drafting the work or revising it: Zemao Yang, Chaohua Cheng.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s41598-017-16812-5.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Bedada G, et al. Transcriptome sequencing of two wild barley (Hordeum spontaneum L.) ecotypes differentially adapted to drought stress reveals ecotype-specific transcripts. BMC Genomics, 15,1(2014-11-19) 2014;15:995. doi: 10.1186/1471-2164-15-995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Müller BSDF, et al. Differentially Expressed Genes during Flowering and Grain Filling in Common Bean (Phaseolus vulgaris) Grown under Drought Stress Conditions. Plant Molecular Biology Reporter. 2014;32:438–451. doi: 10.1007/s11105-013-0651-7. [DOI] [Google Scholar]
- 3.Ramalingam, A., Kudapa, H., Pazhamala, L. T., Garg, V. & Varshney, R. K.Gene Expression and Yeast Two-Hybrid Studies of 1R-MYB Transcription Factor Mediating Drought Stress Response in Chickpea (Cicer arietinum L.). Frontiers in Plant Science 6 (2015). [DOI] [PMC free article] [PubMed]
- 4.Xu Y, et al. Transcriptome sequencing and whole genome expression profiling of chrysanthemum under dehydration stress. Bmc Genomics. 2013;14:1–15. doi: 10.1186/1471-2164-14-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu T, Zhu S, Tang Q, Yu Y, Tang S. Identification of drought stress-responsive transcription factors in ramie (Boehmeria nivea L. Gaud) Bmc Plant Biology. 2013;13:130. doi: 10.1186/1471-2229-13-130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Agarwal G, et al. Genome-wide dissection of AP2/ERF and HSP90 gene families in five legumes and expression profiles in chickpea and pigeonpea. Plant Biotechnology Journal. 2016;14:1563. doi: 10.1111/pbi.12520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Miller G, Nobuhiro, Ciftci-Yilmaz S, Mittler R. Reactive oxygen species homeostasis and signalling during drought and salinity stresses. Plant Cell & Environment. 2010;33:453–467. doi: 10.1111/j.1365-3040.2009.02041.x. [DOI] [PubMed] [Google Scholar]
- 8.Foyer CH, Noctor G. Redox sensing and signalling associated with reactive oxygen in chloroplasts, peroxisomes and mitochondria. Physiologia Plantarum. 2003;119:355–364. doi: 10.1034/j.1399-3054.2003.00223.x. [DOI] [Google Scholar]
- 9.Li DM, et al. Ferulic acid pretreatment enhances dehydration-stress tolerance of cucumber seedlings. Biologia Plantarum. 2013;57:711–717. doi: 10.1007/s10535-013-0326-0. [DOI] [Google Scholar]
- 10.Sofo A, Scopa A, Nuzzaci M, Vitti A. Ascorbate Peroxidase and Catalase Activities and Their Genetic Regulation in Plants Subjected to Drought and Salinity Stresses. International Journal of Molecular Sciences. 2015;16:13561–13578. doi: 10.3390/ijms160613561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Szalai G, Janda T. Effect of Salt Stress on the Salicylic Acid Synthesis in Young Maize (Zea mays L.) Plants. Journal of Agronomy & Crop Science. 2009;195:165–171. doi: 10.1111/j.1439-037X.2008.00352.x. [DOI] [Google Scholar]
- 12.Muthusamy M, Uma S, Backiyarani S, Saraswathi MS, Chandrasekar A. Transcriptomic Changes of Drought-Tolerant and Sensitive Banana Cultivars Exposed to Drought Stress. Frontiers in Plant Science. 2016;7:1609. doi: 10.3389/fpls.2016.01609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shankar R, Bhattacharjee A, Jain M. Transcriptome analysis in different rice cultivars provides novel insights into desiccation and salinity stress responses. Scientific Reports. 2016;6:23719. doi: 10.1038/srep23719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ghosh, S., Meena, K., Sinha, M. K. & Karmakar, P. G. Genetic Diversity in Corchorus olitorius Genotypes Using Jute SSRs. Proceedings of the National Academy of Sciences India, 1-10 (2015).
- 15.M IM. Biochemistry, Medicinal and Food values of Jute (Corchorus capsularis L. and C. olitorius L.) leaf: A Review. Int J Enhanc Res Sci Technol Eng. 2013;2:135–144. [Google Scholar]
- 16.Ogunkunle CO, Ziyath AM, Adewumi FE, Fatoba PO. Bioaccumulation and associated dietary risks of Pb, Cd, and Zn in amaranth (Amaranthus cruentus) and jute mallow (Corchorus olitorius) grown on soil irrigated using polluted water from Asa River, Nigeria. Environmental Monitoring & Assessment. 2015;187:281. doi: 10.1007/s10661-015-4441-6. [DOI] [PubMed] [Google Scholar]
- 17.Yang, L., Zhou, R. Y. & Jin, S. Y. Effect of PEG induced drought stress on the germination indices of different varieties of Corchorus capsularis L. Journal of Southern Agriculture (2011).
- 18.Yao, Y. F., Hong, J. J., Zeng, R. Q. & Yang, Y. X. Drought Resistance of Various Jutes (Corchorus L.)in Seedling Under PEGStress. Fujian Journal of Agricultural Sciences (2013).
- 19.Mitra, G. C. & Basu, N. C. Studies on the size and distribution of stomata in jute (Corchorus olitorius and Corchorus capsularis) and its bearing on resistance to drought. Acta Agron Budap (1974).
- 20.Ayodele VI, Fawusi MOA. Studies on drought susceptibility of Corchorus olitorius L. II. Effects of moisture stress at different physiological stages on vegetative growth and seed yield of C. olitorius cv. ‘Oniyaya’. Biotronics. 1990;19:33–37. [Google Scholar]
- 21.Abass AA. Morphological characters and ascorbic acid content of an elite genotype of: the influence of moisture stress. South African Journal of Plant & Soil. 2013;30:113–117. doi: 10.1080/02571862.2013.811300. [DOI] [Google Scholar]
- 22.Sawarkar AG. Genetic Divergence of Tossa Jute (Corchorus olitorius L.) for Fibre Yield and its Related Component Characters Under Moisture Stress Condition. 2015;28:263–266. [Google Scholar]
- 23.Das A, Ray R, Mandal N, Chakrabarti K. An analysis of transcripts and enzyme profiles in drought stressed jute (Corchorus capsularis) and rice (Oryza sativa) seedlings treated with CaCl 2, hydroxyapatite nano-particle and β-amino butyric acid. Plant Growth Regulation. 2016;79:401–412. doi: 10.1007/s10725-015-0144-9. [DOI] [Google Scholar]
- 24.Zhu J, Jianping NI. Abiotic Stress Signaling and Responses in Plants. Cell. 2016;167:313. doi: 10.1016/j.cell.2016.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Islam MS, et al. Comparative genomics of two jute species and insight into fibre biogenesis. Nature Plants. 2017;3:16223. doi: 10.1038/nplants.2016.223. [DOI] [PubMed] [Google Scholar]
- 26.Chakraborty A, Sarkar D, Satya P, Karmakar PG, Singh NK. Pathways associated with lignin biosynthesis in lignomaniac jute fibres. Molecular Genetics & Genomics. 2015;290:1523. doi: 10.1007/s00438-015-1013-y. [DOI] [PubMed] [Google Scholar]
- 27.Zhang L, et al. De novo transcriptome sequence and identification of major bast-related genes involved in cellulose biosynthesis in jute (Corchorus capsularis L.) Bmc Genomics. 2015;16:1062. doi: 10.1186/s12864-015-2256-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yates SA, et al. De novo assembly of red clover transcriptome based on RNA-Seq data provides insight into drought response, gene discovery and marker identification. BMC Genomics,15,1(2014-06-09) 2014;15:453. doi: 10.1186/1471-2164-15-453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fracasso A, Trindade LM, Amaducci S. Drought stress tolerance strategies revealed by RNA-Seq in two sorghum genotypes with contrasting WUE. Bmc Plant Biology. 2016;16:1–18. doi: 10.1186/s12870-016-0800-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tenhaken R. Cell wall remodeling under abiotic stress. Frontiers in Plant Science. 2015;5:771. doi: 10.3389/fpls.2014.00771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Carvalho MHCD. Drought stress and reactive oxygen species: Production, scavenging and signaling. Plant Signaling & Behavior. 2008;3:156–165. doi: 10.4161/psb.3.3.5536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Biehler K, Fock H. Evidence for the Contribution of the Mehler-Peroxidase Reaction in Dissipating Excess Electrons in Drought-Stressed Wheat. Plant Physiology. 1996;112:265–272. doi: 10.1104/pp.112.1.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Trost, P., Fermani, S., Calvaresi, M. & Zaffagnini, M. Biochemical Basis of Sulphenomics: How Protein Sulphenic Acids may be Stabilized by the Protein Microenvironment. Plant Cell & Environment (2016). [DOI] [PubMed]
- 34.Leterrier M, et al. Peroxisomal NADP-isocitrate dehydrogenase is required for Arabidopsis stomatal movement. Protoplasma. 2016;253:403–415. doi: 10.1007/s00709-015-0819-0. [DOI] [PubMed] [Google Scholar]
- 35.Le GH, et al. Cell Wall Metabolism in Response to AbioticStress. Plants. 2015;4:112. doi: 10.3390/plants4010112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhuang Y, et al. Effects of water-deficit stress on the transcriptomes of developing immature ear and tassel in maize. Plant Cell Reports. 2007;26:2137. doi: 10.1007/s00299-007-0419-3. [DOI] [PubMed] [Google Scholar]
- 37.Iovieno P, et al. Transcriptomic Changes Drive Physiological Responses to Progressive Drought Stress and Rehydration in Tomato. Frontiers in Plant Science. 2016;7:371. doi: 10.3389/fpls.2016.00371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hussain WS, Brajendra SN, Athokpam H, Iqbal MJ. Compatible Solute Engineering in Plants for Abiotic Stress Tolerance - Role of Glycine Betaine. Current Genomics. 2013;14:157–165. doi: 10.2174/1389202911314030001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Singh M, Kumar J, Singh S, Singh VP, Prasad SM. Roles of osmoprotectants in improving salinity and drought tolerance in plants: a review. Reviews in Environmental Science & Bio/technology. 2015;14:407–426. doi: 10.1007/s11157-015-9372-8. [DOI] [Google Scholar]
- 40.Dixon RA, Paiva NL. Stress-Induced Phenylpropanoid Metabolism. Plant Cell. 1995;7:1085. doi: 10.1105/tpc.7.7.1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Haitao S, Tiantian Y, Zhulong C. Comparative proteomic responses of two bermudagrass (Cynodon dactylon (L). Pers.) varieties contrasting in drought stress resistance. Plant Physiology & Biochemistry. 2014;82:218–228. doi: 10.1016/j.plaphy.2014.06.006. [DOI] [PubMed] [Google Scholar]
- 42.Jia X, et al. Integrating transcriptomics and metabolomics to characterise the response of Astragalus membranaceus Bge. var. mongolicus (Bge.) to progressive drought stress. Bmc Genomics. 2016;17:1–16. doi: 10.1186/s12864-015-2294-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ye Y, et al. The role of receptor-like protein kinases (RLKs) in abiotic stress response in plants. Plant Cell Reports. 2017;36:1–8. doi: 10.1007/s00299-016-2084-x. [DOI] [PubMed] [Google Scholar]
- 44.Tanaka H, et al. Abiotic stress-inducible receptor-like kinases negatively control ABA signaling in Arabidopsis. Plant Journal. 2012;70:599. doi: 10.1111/j.1365-313X.2012.04901.x. [DOI] [PubMed] [Google Scholar]
- 45.Wu F, et al. Plasma membrane receptor-like kinase leaf panicle 2 acts downstream of the Drought and Salt Tolerance transcription factor to regulate drought sensitivity in rice. Journal of Experimental Botany. 2015;66:271–281. doi: 10.1093/jxb/eru417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tao A, et al. High-density genetic map construction and QTLs identification for plant height in white jute (Corchorus capsularis L.) using specific locus amplified fragment (SLAF) sequencing. Bmc Genomics. 2017;18:355. doi: 10.1186/s12864-017-3712-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Grabherr MG, et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nature Biotechnology. 2011;29:644. doi: 10.1038/nbt.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. Bmc Bioinformatics. 2011;12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biology. 2010;11:R106. doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Young MD, Wakefield MJ, Smyth GK, Oshlack A. Gene ontology analysis for RNA-seq: accounting for selection bias. Genome Biology. 2010;11:1–12. doi: 10.1186/gb-2010-11-2-r14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mao X, Cai T, Olyarchuk JG, Wei L. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics. 2005;21:3787–3793. doi: 10.1093/bioinformatics/bti430. [DOI] [PubMed] [Google Scholar]
- 52.Mckenna A, et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Research. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yang, Z. et al. Salt-Stress Response Mechanisms Using de Novo Transcriptome Sequencing of Salt-Tolerant and Sensitive Corchorus spp. Genotypes. Genes8, 10.3390/genes8090226 (2017). [DOI] [PMC free article] [PubMed]
- 54.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2012;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 55.Ferdous AS, Islam MT, Alam SS, Khan H. Identification of stable reference genes for quantitative PCR in jute under different experimental conditions: an essential assessment for gene expression analysis. Australian Journal of Crop Science. 2015;ume 9:646–655. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.