

Abstract
The rise of multi- and even totally antibiotic resistant forms of Mycobacterium tuberculosis underlines the need for new antibiotics. The pathogen is resistant to β-lactam compounds due to its native serine β-lactamase, BlaC. This resistance can be circumvented by administration of a β-lactamase inhibitor. We studied the interaction between BlaC and the inhibitor clavulanic acid. Our data show hydrolysis of clavulanic acid and recovery of BlaC activity upon prolonged incubation. The rate of clavulanic acid hydrolysis is much higher in the presence of phosphate ions. A specific binding site for phosphate is identified in the active site pocket, both in the crystalline state and in solution. NMR spectroscopy experiments show that phosphate binds to this site with a dissociation constant of 30 mM in the free enzyme. We conclude that inhibition of BlaC by clavulanic acid is reversible and that phosphate ions can promote the hydrolysis of the inhibitor.
Mycobacterium tuberculosis (Mtb) is one of the most lethal pathogens worldwide with 1.8 million deaths estimated in 2015.1 Despite promising recent developments,2 treatment of the tuberculosis disease (TB) remains associated with long duration and poor patient adherence. These factors have accelerated the rise of multidrug resistant (MDR), extensively drug resistant (XDR), and, recently, even totally drug resistant (TDR) strains of Mtb.3 These strains pose a serious threat to public healthcare, underlining the need for continued research into Mtb resistance mechanisms and potential new medicines. One potential drug strategy that has received attention is the use of β-lactam/β-lactamase inhibitor combinations.4−6
Mtb possesses a native Ambler class A β-lactamase, BlaC, that hydrolyses a broad spectrum of β-lactam antibiotics. The innate resistance that BlaC confers to Mtb, together with the availability of more effective treatment options, resulted in the β-lactam class of antibiotics not being employed in the treatment of Mtb infections. However, with antibiotic effectiveness decreasing more rapidly than drug development can counter, the broad range, proven safety, and ample availability of β-lactam antibiotics may provide new opportunities for treatment of Mbt infection. In fact, resistance to β-lactams can be circumvented by inhibition of BlaC by β-lactam-like suicide substrates. The most common of these inhibitors is clavulanic acid, and indeed, combinations of clavulanic acid with β-lactam antibiotics were found to be bactericidal against even XDR Mtb.7−10
Clavulanic acid inhibits BlaC in a substrate-like fashion, forming a covalent bond with the catalytic serine (Ser-70 by standard Ambler notation11). Generally, in class A β-lactamases, it can then form a variety of covalently bound fragmentation products in the active site, leaving the enzyme either transiently or irreversibly inactivated.12 For BlaC, several of these products have been found.13−15 Formation of these inactive forms was initially reported to be irreversible in BlaC,13 but slow recovery of activity was reported thereafter.16
A crystal structure of a covalent adduct formed between BlaC and clavulanic acid was published by Tremblay et al.14 Interestingly, this structure models a well resolved phosphate ion in the carboxylate binding site, immediately adjacent to the bond between enzyme and adduct. At the same position, a phosphate ion can also be found in several other BlaC crystal structures.5,17−20 In fact, in 26 of the 29 BlaC crystal structures that have been published to date, this position was found to be occupied by either a phosphate ion or a carboxylate group of the ligand that was used for cocrystallization. Additionally, Xu et al. noticed that in their structure of BlaC with avibactam (PDB: 4DF6), the sulfate group of the inhibitor occupied this position.16 The authors of these studies either do not mention the active site phosphate they model or assume that it is an artifact of the high phosphate concentration under crystallization conditions. We investigated the role of the phosphate ion and demonstrate that it affects the rate of recovery from clavulanic acid inhibition. We also show that a phosphate ion binds to the enzyme in solution in the active site.
Materials and Methods
Materials
NMR analysis indicated that the nitrocefin purchased from BioVision Inc. and Oxoid Limited was significantly purer than that from Cayman Chemicals. The BioVision nitrocefin was used in this study. Several values have been reported for the change in extinction coefficient upon hydrolysis of nitrocefin (e.g., refs (15, 21)). To determine this value independently, a stock solution containing 5.0 mg of nitrocefin was diluted to a range of seven concentrations from 10 to 75 μM in 100 mM sodium phosphate buffer, pH 6.4. The A486 values before and after complete hydrolysis by 20 min incubation with 5 nM BlaC were determined. The slope of a linear fit of ΔA486 against nitrocefin concentration yielded Δε486. The procedure was performed in duplicate, yielding a Δε486 of 17 ± 1 mM–1 cm–1. Clavulanic acid powder is hygroscopic, so its concentration was determined by the absorbance at 256 nm in NaOH. The extinction coefficient was determined by quantitative NMR versus a standard of trimethylsilylpropanoic acid. For ChemCruz and Matrix clavulanic acid, which are sold in a cellulose matrix, we found identical UV–vis spectra, yielding an absorbance at 256 nm of 20.0 ± 0.1 mM–1 cm–1. For TRC clavulanic acid, which is a pure powder, the UV–vis spectrum is clearly different, and the NMR spectrum shows impurities. The extinction coefficient at 256 nm is 18.7 mM–1 cm–1.
Production and Purification of BlaC
The blaC gene, lacking codons for the N-terminal 42 amino acids that constitute the signal peptide and with the addition of a C-terminal histidine tag (Uniprot P9WKD3 modified as specified in Figure S1), was expressed using a host optimized sequence (ThermoFisher Scientific), cloned in the pET28a vector in Escherichia coli BL21 (DE3) pLysS cells. The cells were cultured in LB medium at 310 K until the optical density at 600 nm reached 0.6, at which point expression was induced with 1 mM IPTG and incubation continued at 289 K overnight. For the production of isotope labeled proteins for NMR experiments, LB medium was replaced with M9 medium (Table S1) containing 15N ammonium chloride (0.3 g/L) as the sole nitrogen source and, where necessary, 13C D-glucose (4.0 g/L) and 2H2O (99.8%) as the carbon and hydrogen source, respectively. Cells were harvested by centrifugation and lysed with a French press in a buffer of 50 mM Tris-HCl pH 7.5 containing 500 mM NaCl. After centrifugation, the soluble fraction was loaded on a HisTrap Nickel column (GE Healthcare) and eluted with a gradient of 0–250 mM imidazole in the same buffer. A Superose 12 10/300 GL size exclusion chromatography column (GE Healthcare) was used for further purification. Protein concentration and buffer exchange were performed using 10 kDa cutoff Amicon Ultra centrifugal filter units (Merck Millipore Ltd.). Protein purity was determined by SDS-PAGE (Figure S3), and concentrations were determined by the absorption at 280 nm, using the theoretical extinction coefficient ε280 = 29 910 M–1 cm–1.22 BlaC with a TEV-cleavable His-tag (sequence specified in Figure S2) was produced in the same way, with additional cleavage of the purified protein by His-tagged TEV protease. Subsequent repurification using another HisTrap Nickel column (GE Healthcare) yielded pure, His-tag-less BlaC in the flowthrough. Cleavage was confirmed by MS using a Waters Synapt spectrometer yielding a mass of 28 637 ± 1 Da, corresponding with 100% cleavage and 96% efficiency of the applied 15N-labeling. No other protein forms, such as the noncleaved construct (expected mass 31 770 Da at 96% 15N labeling), were detected in the final sample.
Kinetics
All kinetic measurements were performed by measuring hydrolysis of the chromogenic reporter substrate nitrocefin at 486 nm, using a PerkinElmer Lambda 800 UV–vis spectrometer thermostated at 298 K. To determine Michaelis–Menten kinetic constants, initial nitrocefin hydrolysis rates by 5.4 nM BlaC were measured in 100 mM of the specified buffers, in triplicate. OriginPro 9.1 was used to fit standard Michaelis–Menten curves to these data. Reported are, for each condition, the average and standard deviation of the three independent fits.
The apparent first-order rate constants of inhibition (kiso) were obtained by fitting the hydrolysis of 125 μM nitrocefin by 2 nM BlaC in the presence of various concentrations of clavulanic acid against eq 1.13
| 1 |
[P] Is the concentration of product in μM, vs and vi are the final and initial reaction velocities in the presence of inhibitor in μM s–1, respectively, t is time in s, and kiso is the apparent first-order rate constant for the progression from vi to vs in s–1. Subsequently, the rate constants of inhibition were obtained by fitting these data against eq 2,16 in which k3 and k2 are the rate constants for step 3 and 2 in the conversion model (7) (see Results), respectively, while Ki is the ratio k–1/k1 in that model.
| 2 |
The data were also simulated using GNU Octave 3.2.4 and numerical simulations of the differential equations derived from the following model.
| 3 |
| 4 |
where eqs 3 and 4 describe the conversion of nitrocefin (N) and clavulanic acid (C), respectively. E is the enzyme, NE and CE are the noncovalent complexes, and Ii and Pi represent the covalent intermediates and the products, respectively. An example script is provided as Supporting Information. Equation 4 is equivalent to the conversion model, discussed in the Results (7).
Inhibition Recovery
All samples for inhibition recovery experiments were thermostated at 298 K, at the concentrations indicated in Table 3. Activity measurements were performed by dilution in buffer without inhibitor to a final concentration of 2 nM BlaC with 100 μM nitrocefin. The time between initial dilution and measurement was kept <5 min, and the reported time is that of the measurement. Separate incubations were performed to test the stability of BlaC without clavulanic acid, as well as clavulanic acid without BlaC.
Table 3. Rates of BlaC Activity Recovery from Clavulanic Acid Inhibition.
| buffera | [BlaC] (μM) | [clavulanic acid] (μM) | ratio | 50% recovery time (h) | turnover rate (10–4 s–1) |
|---|---|---|---|---|---|
| MESb | 20 | 100 | 1:5 | 14 ± 1 | 1.03 ± 0.07 |
| MESc | 100 | 100 | 1:1 | 1.8 ± 0.8 | 1.5 ± 0.7 |
| MESc | 100 | 300 | 1:3 | 7 ± 0.8 | 1.2 ± 0.1 |
| MESc | 100 | 500 | 1:5 | 13.8 ± 0.8 | 1.01 ± 0.06 |
| MESc | 100 | 1000 | 1:10 | 28 ± 0.8 | 0.99 ± 0.03 |
| MESc | 100 | 1500 | 1:15 | 43.5 ± 1 | 0.96 ± 0.02 |
| MESd | 300 | 1500 | 1:5 | 6.0 ± 0.6 | 2.3 ± 0.2 |
| MES + 100 mM acetic acidc | 100 | 500 | 1:5 | 36 ± 3 | 0.38 ± 0.03 |
| MES + 100 mM Na2SO4c | 100 | 500 | 1:5 | 2.0 ± 0.5 | 7 ± 2 |
| NaPib | 20 | 100 | 1:5 | 0.63 ± 0.06 | 22 ± 2 |
| NaPid | 300 | 1500 | 1:5 | 0.82 ± 0.01 | 16.8 ± 0.3 |
Buffers were all 100 mM, pH 6.4.
Errors are the standard deviations over four replicates.
Errors are the estimated error in half-time determination of single experiments.
Errors are the standard deviations over two replicates.
Mass Spectrometry
Samples for whole-protein mass spectrometry were flash-frozen in liquid nitrogen and stored at 193 K. Upon thawing, they were transferred to 10 mM ammonium acetate buffer pH 6.8 using Micro Bio-Spin chromatography columns (Bio-Rad), loaded on a C4 polymeric reversed phase UPLC column and then analyzed using either an LTQ-Orbitrap mass spectrometer (ThermoScientific) or a Synapt G2-Si mass spectrometer (Waters), 10–25 min after thawing. Data were deconvoluted for charge using Thermo Xcalibur.
Crystallization and Soaking Conditions
Crystallization conditions for BlaC at a concentration of 20 mg mL–1 were screened by sitting-drop vapor-diffusion using the JCSG+ and PACT premier (Molecular Dimensions) screens at 293 K with 500-nL drops. The reservoir solution (75 μL) was pipetted by a Genesis RS200 robot (Tecan), and the drops were made by an Oryx6 robot (Douglas Instruments). Initial hits were obtained in conditions D2 and D3 of the PACT premier screen, which consist of 0.1 M 1:2:2-d/l-malic acid:MES:Tris base pH 5.0 and 6.0, respectively, and 25% polyethylene glycol (PEG) 1500. Buffer optimization led to small needles in 0.1 M sodium acetate buffer, pH 5.0, 25% PEG1500, which were resuspended in 50 μL of the reservoir solution and crushed using a Seed Bead-HR2–320 (Hampton). Crushed needles were diluted 1:10 in the same reservoir solution and used for seeding freshly plated, 2-μL drops in the same conditions. Crystals useful for X-ray diffraction grew within 4 days. The crystals were mounted on cryoloops and cryo-protected by flash-cooling using liquid nitrogen, either directly in mother liquor and 15% glycerol, or after soaking for 50 min in mother liquor, 100 mM NH4H2PO4, and 15% glycerol.
X-ray Data Collection and Structure Solving
X-ray data collection was performed at the ESRF synchrotron-radiation facility (Grenoble, France) on beamline ID30A-3 using a PIXEL, Eiger_4 M (DECTRIS) X-ray detector. For structure 5OYO, a total of 4500 frames were collected, with an oscillation of 0.05°, an exposure time of 0.02 s, total 90 s. For structure 5NJ2, a total of 3000 frames were collected, with an oscillation of 0.1°, an exposure time of 0.01 s, total 30 s. The data sets were autoprocessed by the EDNA23 Autoprocessing package that used XDS(24) to integrate the intensities and AIMLESS(25) to scale and merge the intensities in the mxCuBE26 to a resolution of 2.1 and 1.19 Å for 5OYO and 5NJ2, respectively. The structures were solved by molecular replacement with MOLREP(27) using 2GDN28 as a search model from the CCP4 suite29 and manually refined using REFMAC(30) and Coot.31 Data collection and refinement statistics are presented in Table S2.
Nuclear Magnetic Resonance Spectroscopy Experiments
Samples for backbone assignment contained 0.75 mM [15N,13C,2H] BlaC in 20 mM MES pH 6.0 with 1 mM DTT and 6% D2O (NMR buffer) at 298 K. A set of standard HNCA, HNCACB, HNcoCACB, HNCO, and HNcaCO experiments was recorded on a Bruker AVIII HD 850 MHz spectrometer equipped with a TCI cryoprobe for backbone assignment. All other NMR spectra, unless stated otherwise, were recorded on ca. 0.35 mM [15N] BlaC samples in the same buffer at 298 K, on the same spectrometer. Data were processed with Topspin 3.2 (Bruker Biospin, Leiderdorp) and analyzed using CCPNmr Analysis.32
NMR titrations were performed by addition of an increasing volume of 0.9 M sodium phosphate or sodium chloride stock in NMR buffer to the sample, decreasing protein concentration from 0.35 to 0.25 mM during the titration. Nonlinear regression fitting with a shared association constant (KA) and individual maximal chemical shift perturbations (CSP) values (CSPmax) in Origin 9.1 was used to fit the CSP data of selected residues to eq 5, in which R is the ratio of phosphate concentration over enzyme concentration, Cstock is the concentration of the phosphate stock solution used for titrating and Ei is the initial concentration of enzyme in the sample. The phosphate titration was performed in duplicate. As the interexperimental variation between samples was found to be larger than the intraexperimental variation between reporter peaks, data from the two experiments were fitted separately. Reported are the average and standard deviation of the two fits.
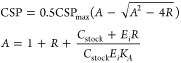 |
5 |
Samples for NMR visualization of BlaC recovery from clavulanic acid inhibition contained 0.3 mM [15N] BlaC and 1.5 mM clavulanic acid in 100 mM MES or sodium phosphate buffer, pH 6.4, with 1 mM DTT and 6% D2O, at 298 K. Activity measurements as described above were performed at various time points to check the relation between spectra and functional states. Separate incubations were performed as controls on the stability of BlaC without clavulanic acid, as well as clavulanic acid without BlaC.
Results
BlaC is normally produced by M. tuberculosis with an N-terminal Tat-type signal peptide that is used to locate BlaC as a lipoprotein on the outside of the cell membrane.33−35 To obtain soluble protein for in vitro experiments, a BlaC gene encoding only the soluble beta-lactamase domain supplemented with a C-terminal 6xHistidine purification tag was expressed in E. coli. The protein was isolated and purified using immobilized metal affinity chromatography and subsequent size exclusion chromatography to yield ca. 30 mg BlaC per liter of culture medium.
The Michaelis–Menten kinetic parameters of nitrocefin hydrolysis by BlaC were determined in buffers with and without phosphate (Table 1, Figure S4). The measured Michaelis constant Km is higher in phosphate buffer than in the other tested buffers. This effect appears to be somewhat compensated by a higher catalytic efficiency at pH 6.
Table 1. Buffer Dependence of Nitrocefin Hydrolysis by BlaCa.
| buffer | kcat (s–1) | Km (μM) | kcat/Km (× 105 M–1 s–1) |
|---|---|---|---|
| NaPi pH 6.0 | 107 ± 6 | 147 ± 14 | 7.3 ± 0.4 |
| NaPi pH 7.0 | 64 ± 6 | 153 ± 23 | 4.2 ± 0.2 |
| MES pH 6.0 | 69 ± 1 | 38 ± 7 | 18 ± 3 |
| BIS-TRIS pH 6.0 | 83 ± 3 | 61 ± 7 | 14 ± 1 |
Buffer concentrations were each 100 mM. Errors represent the standard deviation over triplicate measurements.
Our results are in agreement with previously published kinetic values for BlaC nitrocefin hydrolysis, which have kcat/Km in the range of (4–18) × 105 M–1 s–1.5,13,15,21 Similar to most of these studies, further experiments have been performed at pH 6.4, which is the optimum pH for BlaC. The work in this study was performed on BlaC with a C-terminal His-tag (Figure S1). To ensure that the tag has no effect on the activity, we also prepared BlaC with a cleavable N-terminal His-tag (Figure S2). The kinetic parameters of nitrocefin hydrolysis by BlaC without His-tag were then compared to those of the His-tagged BlaC used in this study and were found to be the same, at kcat/Km of 10 × 105 M–1 s–1 and 11 × 105 M–1 s–1, respectively, in 100 mM MES pH 6.4 (Table S3).
Next, BlaC inhibition by clavulanic acid was studied. For this, Hugonnet and Blanchard proposed a reactivation model, eq 6, including fast binding of the inhibitor I to the enzyme E to form the EI complex, followed by slower conversion to the long-lived EI* complex.13
| 6 |
Using this approach, they determined the affinity constant for BlaC clavulanic acid inhibition (Ki = k–1/k1) to be 12.1 μM, the inactivation rate k2 2.7 s–1 and reactivation rate k–2 indistinguishable from zero. This led to the conclusion that clavulanic acid inhibition of BlaC is irreversible. To study the variation of inhibition kinetics with buffer conditions, we used the reactivation model with the adjustment proposed by Xu et al.16 (7), in which conversion of covalently bound clavulanic acid EI* into a product P is allowed with a rate constant k3, rather than reversal of the covalent linkage to the active site serine residue with a rate constant k–2.
| 7 |
As clavulanic acid acts as a slow-onset inhibitor for BlaC, reaction rates can be estimated from the rate at which enzymatic activity decreases upon administration of the inhibitor. The resulting inhibition curves are plotted in Figure 1 for phosphate buffer and Figure S5 for MES buffer. These data were fitted to eq 1 to obtain the apparent first-order rate constants (kiso) of inhibition for each clavulanic acid concentration, which were then fitted to eq 2 to estimate the rate constants of inhibition (Table 2). The mathematical description of kiso given in eq 2 is the same for the reactivation model of Hugonnet and Blanchard13 and the conversion model of Xu et al.,16 except k–2 is replaced by k3. However, the latter model predicts that in time clavulanic acid will be degraded and BlaC will regain activity, whereas the former model predicts that an equilibrium is reached and BlaC will remain inhibited. The latter model applies, as is discussed below.
Figure 1.

(a) Inhibition curves of BlaC nitrocefin hydrolysis with increasing concentrations of clavulanic acid in 100 mM NaPi, pH 6.4. Green lines represent experimental data, black lines are fits using eq 1. (b) Plot of kiso values obtained from the fit of each inhibition curve against the respective clavulanic acid concentration, for MES (red circles) as well as NaPi (black squares) buffer. The solid lines represent the fits to eq 2.
Table 2. Rate Constants of BlaC Inhibition with Clavulanic Acida.
| approach | Ki (μM) | k2 (10–2 s–1) | k3 (10–4 s–1) | |
|---|---|---|---|---|
| NaPi pH 6.4 | fit to eqs 1 and 2 | 32 ± 2 | 2.9 ± 0.1 | 4 ± 1 |
| simulation | 20 | 4.5 | 18 | |
| MES pH 6.4 | fit to eqs 1 and 2 | 35 ± 4 | 4.9 ± 0.3 | 6 ± 3 |
| simulation | 20 | 4.5 | 0.25 |
Errors represent the standard errors of the fit.
The fits of the inhibition curves follow the data remarkably accurately. However, variation of the parameters of the fit shows that they are correlated and other combinations can be obtained that yield equally accurate fits. To evaluate the quality of the data and the values of the parameters, the data were also simulated by solving the differential equations underlying the kinetic model numerically using GNU Octave software. Good simulations (Figure S6) were obtained with the values listed in Table 2. All parameters are listed in Table S4. These simulations show that curves contain small errors in the offset and in the shape due to measuring artifacts, which are faithfully fitted in the first approach. This suggests that the parameters derived from the kiso curves may not be very accurate, due to overfitting. It is clear, however, that the slopes of the last parts of the curves (i.e., the vs values in eq 1) are close to zero in the presence of high concentrations of clavulanic acid in MES buffer, but not in phosphate buffer. This is reflected in the k3 value obtained in the simulations, which is much larger for the inhibition in phosphate buffer than in MES buffer. The phosphate buffer data cannot be simulated with the k3 values obtained for the MES buffer data, indicating that this difference is clearly significant. This finding implies that hydrolysis of clavulanic acid by BlaC is much faster in phosphate buffer.
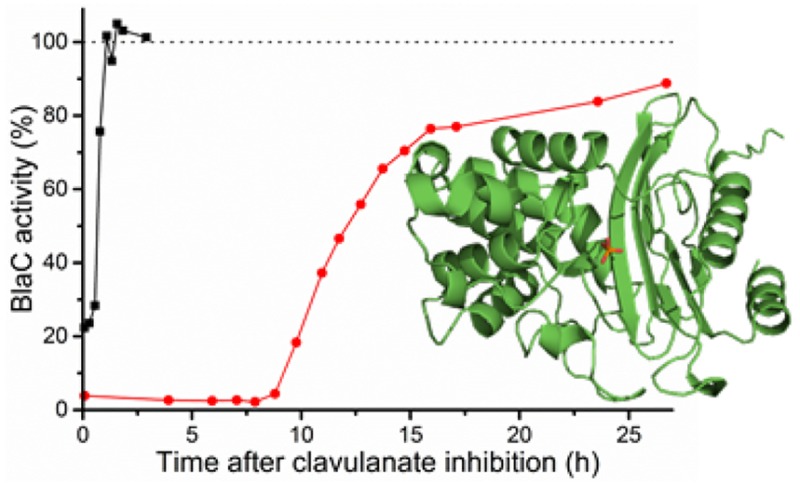
To test the rate of clavulanic acid hydrolysis in a more direct manner, the recovery of activity after inhibition was assayed. BlaC was incubated with a 5-fold excess of clavulanic acid, and samples were taken over time and tested for nitrocefin hydrolase activity. Enzyme activity was observed to return after a characteristic delay time, reproducible over different batches of enzyme and inhibitor but dependent on reaction conditions (Figure 2, Table 3). At 20 μM BlaC with 100 μM clavulanic acid in 100 mM MES pH 6.4, Hugonnet and Blanchard13 observed no return of activity within 12 h. We find that recovery occurs after ca. 14 h. Moreover, recovery was ca. 22 times faster in phosphate buffer than in MES buffer under the same conditions. Addition of sulfate to the MES buffer resulted in ca. 7 times faster recovery, whereas addition of acetate slowed the recovery down ca. 2.6 times. Turnover rates were defined as the number of clavulanic acid molecules inactivated per enzyme molecule per second and were derived from the 50% recovery times. The turnover rates, listed in Table 3, are close to those derived from the simulations of the inhibition data (k3 values in Table 2). Interestingly, at a high concentration of BlaC (300 μM), in 100 mM MES, recovery was found to be about two times faster than that at 100 or 20 μM, with the same clavulanic acid-to-enzyme ratio of 5:1. To investigate whether this was due to an allosteric effect of clavulanic acid at high concentration, the recovery time was measured for solutions with 100 μM BlaC and increasing concentrations of clavulanic acid, from 100 to 1500 μM. The total recovery time increased linearly with the concentration of clavulanic acid (Figures S7 and S8), indicating that the turnover rate of BlaC was constant. Thus, it is concluded that the increase in turnover rate only occurs at very high BlaC concentration (300 μM), perhaps due to a weak protein–protein interaction. However, this effect is unlikely to be relevant under physiological conditions and was not investigated further. No significant dependence of the turnover rate was seen in phosphate buffer.
Figure 2.
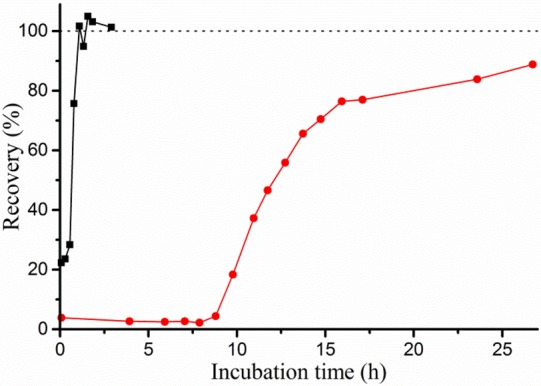
Example curves of BlaC recovery from clavulanic acid inhibition. 20 μM BlaC was incubated with 100 μM clavulanic acid in 100 mM NaPi (black squares) or MES (red circles) buffer, pH 6.4. Samples were taken at various time points, diluted to 2.0 nM BlaC and tested for hydrolase activity using 100 μM nitrocefin. BlaC and clavulanic acid separately were each stable throughout the experiments (data not shown). The activity of inhibited enzyme is nonzero due to recovery taking place in the time between initial dilution and activity measurement of the samples (∼5 min).
To gain insight in the inhibition intermediates that arise, samples were analyzed using whole-protein mass spectrometry (Figure 3). Before inhibition, the enzyme was present in three forms, with the theoretical mass of BlaC as the main species and minor additional species with relative masses of ca. −19 and −35 present. It is unclear how these latter two species differ from the native enzyme or whether this is an artifact of the sample treatment and MS analysis. Upon inhibition with clavulanic acid, these peaks diminish and species of ca. +35, +51, and +70 appear instead. The consistent relative intensities and mass differences of +70 relative to the three unbound peaks (−19 + 70 = +51; – 35 + 70 = +35) suggest that these peaks represent a single inhibition intermediate. This is likely the propionaldehyde ester that was previously reported upon inhibition of BlaC with clavulanic acid.13,14 Several other peaks also appear, including ca. +86, +136, and +154 species. The ca. +86 intermediate has not been observed for inhibition of BlaC with clavulanic acid before, but may correspond to the hydrated propionaldehyde (+88) that was observed upon clavulanic acid inhibition of related β-lactamases.12 The ca. +154 and +136 adducts were previously observed as BlaC clavulanic acid intermediates, and these were proposed to represent a decarboxylated trans-enamine adduct and its dehydrated variant, respectively.13,14 Interestingly, although the observed masses are similar to previous results, their relative intensities are not. The BlaC +154 enamine was previously reported as the major dead-end product. However, we observe mainly the +70 aldehyde adduct that was previously reported as minor and decreasing over time. We observed only minor changes in relative intensities upon prolonged incubation, even upon incubating at higher clavulanic acid/BlaC ratios to achieve higher turnover numbers (Figure 3, right). Instead, concurrent with the return of activity, all species were observed to diminish and the original masses returned. Remaining peaks could indicate either irreversible inhibition products or incomplete recovery, but these peaks were relatively low in intensity. Furthermore, although the time required for enzyme recovery was influenced greatly by the presence or absence of phosphate, no effect on the type of intermediates was observed (Figure S9).
Figure 3.

Charge-deconvoluted mass spectra of BlaC before and during incubation at 100 μM BlaC with 500 (left) or 1500 (right) μM clavulanic acid in 100 mM MES, pH 6.4. Upon inhibition with clavulanic acid, the main species contain covalently bound adducts. After prolonged incubation, the enzyme returns to its free form. The lowest spectra on either side correspond to recovered enzyme activity in the samples. The MS data were obtained using a Waters Synapt mass spectrometer. Each spectrum was normalized to the total signal intensity.
As we found phosphate to promote recovery, we then sought to characterize its interaction with BlaC. To further corroborate the binding location in the active site, a BlaC crystal was soaked in phosphate buffer and diffraction analysis was performed. This yielded a 1.19 Å resolution structure, which is the highest resolution obtained for BlaC to date. It is available in the Protein Data Bank as entry 5NJ2.
The two protein molecules in the asymmetric unit each display a well-resolved phosphate group with a conserved position and orientation in the active site, at hydrogen bonding distance from the Ser-70, Ser-130, Thr-235, and Thr-237 (Figure 4a). In both subunits, extra density is observed on the oxygen moiety of the phosphate that is turned toward Ser-70 and the conserved active site water molecule, which could indicate the presence of a hydrogen atom. However, we have not built this atom, because placement and refinement of hydrogen atoms typically only occurs at resolutions better than 1.0 Å. Only one other phosphate group was modeled in the crystal structure. This group is also well-resolved, but it does not show any extra densities, further suggesting specificity of the phosphate hydrogen bridge for the substrate binding site. For comparison, BlaC was also crystallized in the presence of acetate, without subsequent soaking with phosphate. This yielded a 2.1 Å structure (Protein Data Bank entry 5OYO). It shows that acetate occupies the carboxylate binding site and forms hydrogen bridges with residues Ser-130, Thr-235, and Thr-237 (Figure 4b). Unlike phosphate, it does not form a hydrogen bridge with Ser-70.
Figure 4.

Close-up on the carboxylate binding sites of BlaC crystal structures 5NJ2, chain A (a) and 5OYO chain B (b). Several catalytically important residues and a conserved active site water molecule are indicated. Distances (in Å) of proposed hydrogen bridges (purple dashed lines) involving the phosphate group and acetate are indicated in red. The mesh shows the 2Fo – Fc electron density map contoured at 1.5σ (a) or 1.0σ (b).
To establish whether BlaC also interacts with phosphate in the solution state, we used nuclear magnetic resonance spectroscopy (NMR). As NMR studies of BlaC have not been reported before, we recorded a set of standard three-dimensional (3D) NMR spectra to perform sequential backbone assignment. With these, 98% of the BlaC backbone H–N moieties were assigned to a resonance peak in the corresponding 1H–15N heteronuclear single quantum coherence (HSQC) spectrum (Figure S10). Interestingly, the four residues at hydrogen-bonding distance from the active site phosphate (Figure 4a) were the only nonproline, nonterminal residues whose backbone resonances could not be identified in the spectra, suggesting that their amide nuclei are in intermediate exchange, causing line broadening of the NMR resonances. The assignment data are available at the Biological Magnetic Resonance Bank under ID 27067.
We then acquired two-dimensional (2D) HSQC spectra of BlaC at phosphate concentrations varying from 0–250 mM (Figure S11). Multiple resonances were found to shift position, indicating that the corresponding residues experience fast exchange between different chemical environments. At low phosphate concentrations only a few peaks were affected, showing large chemical shift perturbations (CSPs), whereas CSPs for many resonances were observed at higher phosphate concentrations (>50 mM). When mapped on the protein structure, it becomes apparent that the large CSPs arise for nuclei close to the position in the active site where phosphate is present in the crystal structure (Figure 5, left). The smaller CSPs that appear at high phosphate concentration represent much weaker phosphate binding at other sites. The data allowed us to determine the binding affinity in solution. By analyzing the magnitude of active-site CSPs as a function of the phosphate/BlaC ratio, a binding affinity (KD) of 27 ± 11 mM was found for phosphate binding to BlaC (Figure 5, right). A separate titration with sodium chloride (Figure S12) did not result in any CSPs with the magnitude, colocalization, or affinity of those found for phosphate, confirming that the observed effects are caused by a specific interaction.
Figure 5.

BlaC–phosphate interaction. (a) Crystal structure 5NJ2 is shown with residues that are affected by phosphate binding in solution highlighted. Residues of which the amide backbone experienced CSPs over 0.075, 0.10, and 0.15 ppm are displayed in yellow, orange, and red, respectively, whereas the ones for which no data were available are displayed in gray and those with no or small CSPs are colored green. The phosphate as observed in the crystal structure is indicated in red. (b) Binding curves. The plot shows the CSPs upon phosphate titration for five selected amide resonances plotted against the ratio of the phosphate and BlaC concentrations. Data points are shown with an estimated peak picking error of ±0.02 ppm, error in KD is the standard deviation over duplicate titrations.
NMR spectroscopy was also used to study the BlaC–clavulanic acid interaction. Upon addition of a 5-fold excess of clavulanic acid, several peaks disappeared and new peaks appeared nearby. Unsurprisingly, the corresponding nuclei were located in the active site (Figure 6a). Upon prolonged incubation, the peaks of the unbound state reappeared and the peaks of the bound state disappeared. This observation indicates that the free and bound forms are not in exchange on the chemical shift time scale (exchange rate ≪100 s–1), in line with the expected formation of a covalent intermediate. The recovery times of free BlaC after incubation with 5-fold excess of clavulanic acid based on the NMR peak intensities (Figure 6b) were in agreement with the kinetics and MS data (Table 3), including data taken directly on the NMR samples.
Figure 6.

Effect of clavulanic acid on BlaC as measured by NMR. (a) Crystal structure 3CG5,14 highlighting residues of which NMR resonances are affected by addition of clavulanic acid. Residues of which the amide backbone resonance experienced CSPs over 0.01 ppm and over 0.05 ppm are displayed in yellow and red, respectively, while the ones for which no data were available are displayed in gray and the remaining residues in green. The bound reacted adduct of clavulanic acid as was observed by Tremblay et al.14 is shown in purple sticks. (b) Effect of clavulanic acid on BlaC over time, in 100 mM MES (red circles) and NaPi (black squares) buffer, pH 6.4. Data points show average and standard deviation of relative signal intensities from the native (open symbols) and inhibited (filled symbols) resonances of residues Cys-69, Ala-74, Asp-131, Ala-146, and Tyr-241.
Discussion
In this work, BlaC and its interaction with clavulanic acid were further characterized in vitro. The Michaelis–Menten kinetic values found for nitrocefin hydrolysis are largely consistent with previous observations, although an elevation of the Michaelis constant was observed in the presence of phosphate. This may point toward competition between phosphate and substrate for occupation of the carboxylate binding site. This explanation would lend further credence to the suggestion by Kurz et al.36 that the same competition may explain their observation of an oddly placed carbonyl moiety in cocrystallization of BlaC with a boronic acid transition state inhibitor. To determine the reaction rates with clavulanic acid, the model proposed by Xu et al.16 was used (eq 7). It should be noted that clavulanic acid chemistry may be more complicated than this model suggests, as various inhibition intermediates have been reported13,14 and observed in this study. Presumably, different inhibition intermediates will have different rates of formation and decomposition.
The approach of fitting inhibition curves to initial and final velocities (vi and vs) and an exponential decay constant (kiso) that describes the time to reach a steady state inhibition level has been used frequently. Our simulations of the curves suggest that the method may lead to overfitting of the parameters and should be used with caution. In particular the rate constant for hydrolysis of the covalent intermediate (k3) is poorly defined. This rate is very low and its value depends heavily on the fits of the lowest inhibitor concentrations. The parameters of clavulanic acid inhibition onset that we found are in reasonable agreement with the Ki = 12.1 μM and k2 = 2.7 s–1 found by Hugonnet and Blanchard,13 who used the same approach described here except for their assumption that the covalent intermediate is not hydrolyzed (k3 = 0). We found that BlaC slowly converts clavulanic acid to regain activity, consistent with the observations of Xu et al.16 The return to the native active form of the protein was demonstrated by activity assays, NMR spectroscopy, and mass spectrometry.
The main covalent intermediate of inhibition was observed to harbor a ca. + 70 mass compared to the noninhibited protein, corresponding to the adducts observed by Hugonnet and Blanchard13 upon inhibition with each of the inhibitors clavulanic acid, sulbactam, and tazobactam and proposed by them and others to be a hydrolyzable aldehyde adduct. However, the +136 and +154 clavulanic acid enamine adducts observed as main, dead-end, reaction products by Hugonnet and Blanchard13 as well as Xu et al.14 were observed in only minor quantities in our analysis. We demonstrate that the rate of recovery is highly dependent on reaction conditions. Phosphate ions enhance the rate, yet the composition of the inhibition intermediates is not affected. This indicates that phosphate promotes the release of covalently bound clavulanic acid adducts from the active site and does not change the direction of the initial chemistry. NMR experiments support phosphate ion binding in the active site and show that the dissociation constant is 3 × 10–2 M. This affinity requires that at crystallization conditions of 2 M phosphate, the site should be fully occupied. This is consistent with the observations published so far. We note that structure 2GDN is the only BlaC structure that was modeled with an empty carboxylate binding site, despite a high phosphate concentration in the crystallization buffer, but the data do show density that suggests the presence of a phosphate ion there. Our high resolution structure shows that the phosphate is in hydrogen bond distance to several important active site residues and may be protonated at the phosphate oxygen close to Ser-70.
To formulate a hypothesis about the role of phosphate in promoting hydrolysis, we examined the structure of BlaC covalently bound to a cleavage product of clavulanic acid (PDB entry 3CG5,14Figure 7). It should be noted that the intermediate in structure 3CG5 is not the dominant species observed in our work, of +70 Da, but the ester bond to Ser-70 is likely to be in a similar place in all intermediates. In this structure, a phosphate (PO4 309) is present in the same location as in the structure of substrate free BlaC, although it is slightly displaced by the presence of the adduct. Most likely, the carboxylate binding site is also involved in the initial interaction between BlaC and the carboxy group of clavulanic acid, but it becomes available upon decarboxylation of the acyclic adduct. This reaction occurs rapidly and appears to create enough space to allow for diffusion of phosphate into the carboxylate binding site. In the second step of the reaction, a nucleophilic attack on the carbon (atom CAG in structure 3CG5) of the adduct forming the ester bond with Ser-70 occurs, the bond between Ser-70 and the adduct is broken, and Ser-70 Oγ is reprotonated. The nucleophilic attack can be executed by water 441, located at 3.1 Å underneath the ester bond plane and hydrogen bonded to Glu-166. This is the standard mechanism of β-lactamase hydrolysis. However, O3 of the phosphate is located also at 3.1 Å of the CAG carbon and right above the ester bond plane. So, alternatively, the phosphate could carry out the nucleophilic attack. After release from the enzyme, the phospho-adduct would probably be rapidly hydrolyzed.
Figure 7.

Schematic representation of catalytically important groups and their hydrogen-bonding network, as present in structure 3CG514 of BlaC with a clavulanic acid cleavage product (indicated in blue) covalently bound to Ser-70. Distances (in Å) between heavy atoms involved in potential H-bonds are shown in red. Residues are numbered according to the Ambler consensus,11 and other numbers represent the internal numbering of the published structure.
An alternative role of the phosphate could be in reprotonation of the Ser-70 Oγ. The distance between the phosphate oxygen O3 and the Ser-70 Oγ is larger (3.5 Å) than in the substrate-free structure (2.9 Å, Figure 4a), but it is close to the Oγ of Ser-130 (2.5 Å). If the nucleophilic attack is performed by the water, a proton would be donated to Glu-166. This glutamate is hydrogen bonded to the amine of Lys-73, which also forms an H-bond with Ser-70 Oγ, allowing for proton transfer to Ser. Alternatively, the Ser-70 Oγ could be reprotonated by accepting the proton from Ser-130 (O–O distance 2.7 Å), which in its turn accepts a proton from O3 of the phosphate (Figure 7). The phosphate is in contact with waters on the protein surface (HOH 492, 500, and 516). In this case, Glu-166 could release its proton by a flip of the carboxy group and donation of a proton to the nearby water chain (HOH 586 and HOH 587).
Our data show that sulfate can also accelerate hydrolysis of clavulanic acid, albeit less than phosphate. It can be expected that the sulfate ion is fully deprotonated at pH 6.4, so it could not act as a hydrogen donor to Ser-130. However, it could act as a nucleophile to attack carbon CAG. One BlaC structure, PDB 3ZHH,37 shows sulfate bound in the carboxylate binding site, in a similar location as the phosphate. The limited resolution does not allow for a detailed analysis. We also solved the structure of BlaC with excess acetate. Acetate in MES buffer has a negative effect on the hydrolysis rate as compared to MES buffer only. This observation cannot be readily explained, but the data show acetate binding in the carboxylate binding site. It forms a hydrogen bond with Ser-130, so could act as a hydrogen bond donor. In an overlay of 3CG5 and our structure 5OYO, it can be seen that the closest oxygen of the acetate is at 4.2 Å of the CAG carbon, making a nucleophilic attack unlikely. We cannot exclude that MES buffer (2-(N-morpholino)ethanesulfonic acid) can bind to the carboxylate binding site as well, via its sulfonate group, having a weak positive effect on the rate of hydrolysis. Acetate could be competing with MES, leading to a reduction of the rate. These considerations point to a role of phosphate and sulfate as alternative nucleophiles. We emphasize, however, that the proposed mechanisms are speculative. Further research is required to understand the influence of anions on the hydrolysis rate of clavulanic acid. Clearly, the binding site is promiscuous, and various ions have quite different effects on hydrolysis.
It is worthwhile to consider whether the effect we observe is specific for BlaC, or if it may signify a more general mechanism in class A β-lactamases. Indeed, related proteins such as TEM-1, SHV-1, and CTX-M-1 have very similar carboxylate binding sites. However, most of these enzymes harbor an Ala at position 237, where BlaC has a Thr, which contributes a hydrogen bond to the binding of phosphate (Figure 4a). Indeed, although phosphates and sulfates have been observed to occupy analogous positions in at least TEM-1,38 CTX-M-9,39 and L2,40 many X-ray structures of these enzymes with empty carboxylate binding site have also been reported (e.g., structure 3CMZ41 of TEM-1 and structure 1SHV42 of SHV-1). These observations suggest that the affinities between these proteins and phosphate-like groups may be lower than in the case of BlaC. We therefore expect any effect of phosphate in related enzymes to be less significant. Whether the role of phosphate and other anions in the breakdown of clavulanic acid by BlaC is relevant under physiological conditions is unclear. M. tuberculosis is known to prevent maturation of lysosomes by blocking phagolysosomal fusion and has been shown to live at slightly acidic pH.43 The experiments reported here have been conducted at pH 6.4, which is the optimal pH of BlaC and can thus be expected to be physiologically relevant. The concentration of phosphate ions in M. tuberculosis within macrophages is unknown. Total prokaryotic and eukaryotic intracellular phosphate ion concentrations, however, are in the 1–10 mM range.44 Additionally, sulfate ions were observed to have a similar effect, and it cannot be excluded that other compounds, such as ATP, can also interact with BlaC. Thus, it is reasonable to expect that a substantial fraction of BlaC molecules binds a phosphate-like group specifically in the active site.
Acknowledgments
We would like to thank Qing Miao for her design of Figure 7 and Aleksandra Chikunova for her assistance with experimental preparations. Furthermore, we acknowledge the European Synchrotron Radiation Facility for provision of synchrotron radiation facilities, and we would like to thank Christoph Mueller Dieckmann and David von Stetten for assistance in using beamline ID30A-3. Parts of this work were previously presented on posters at the CHAINS conferences 2015 and 2016 in Veldhoven, The Netherlands.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.biochem.7b00556.
Composition of M9 medium (Table S1). Data collection and refinement statistics of crystal structures (Table S2). Michaelis–Menten constants of BlaC with and without His-tag (Table S3). Parameters of inhibition simulation (Table S4). Sequences of BlaC variants (Figures S1–S2). SDS-PAGE of protein purification (Figure S3). Michaelis–Menten curves in different buffers (Figure S4). Inhibition curves in MES buffer (Figure S5). Simulation of inhibition curves (Figure S6). Dependence of recovery time on the ratio of clavulanic acid to BlaC (Figures S7–S8). MS spectra of recovery experiment in MES and sodium phosphate buffers (Figure S9). Backbone assignment of BlaC HSQC spectrum (Figure S10). Overlay of HSQC spectra of BlaC upon titration with sodium phosphate (Figure S11). Chemical shift perturbation per residue upon titration with sodium phosphate and sodium chloride (Figure S12). Octave script for simulation of inhibition curves.
Author Contributions
† W.E. and R.T. contributed equally to this work.
This study was supported by internal funding.
The authors declare no competing financial interest.
Supplementary Material
References
- World Health Organization . (2016) Global Tuberculosis Report 2016, Geneva. [Google Scholar]
- Cohen J. (2017) Easier cure for resistant TB. Science 355, 677–677. 10.1126/science.355.6326.677. [DOI] [PubMed] [Google Scholar]
- Smith T.; Wolff K. A.; Nguyen L. (2012) Molecular biology of drug resistance in Mycobacterium tuberculosis. Curr. Top. Microbiol. Immunol. 374, 53–80. 10.1007/82_2012_279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers H. F.; Moreau D.; Yajko D.; Miick C.; Wagner C.; Hackbarth C.; Kocagöz S.; Rosenberg E.; Hadley W. K.; Nikaido H. (1995) Can penicillins and other beta-lactam antibiotics be used to treat tuberculosis?. Antimicrob. Agents Chemother. 39, 2620–2624. 10.1128/AAC.39.12.2620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurz S. G.; Wolff K. A.; Hazra S.; Bethel C. R.; Hujer A. M.; Smith K. M.; Xu Y.; Tremblay L. W.; Blanchard J. S.; Nguyen L.; Bonomo R. A. (2013) Can inhibitor-resistant substitutions in the Mycobacterium tuberculosis β-Lactamase BlaC lead to clavulanate resistance?: a biochemical rationale for the use of β-lactam-β-lactamase inhibitor combinations. Antimicrob. Agents Chemother. 57, 6085–6096. 10.1128/AAC.01253-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurz S. G.; Bonomo R. A. (2012) Reappraising the use of β-lactams to treat tuberculosis. Expert Rev. Anti-Infect. Ther. 10, 999–1006. 10.1586/eri.12.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers H. F.; Kocagöz T.; Sipit T.; Turner J.; Hopewell P. C. (1998) Activity of amoxicillin/clavulanate in patients with tuberculosis. Clin. Infect. Dis. 26, 874–877. 10.1086/513945. [DOI] [PubMed] [Google Scholar]
- Hugonnet J.-E.; Tremblay L. W.; Boshoff H. I.; Barry C. E.; Blanchard J. S. (2009) Meropenem-clavulanate is effective against extensively drug-resistant Mycobacterium tuberculosis. Science 323, 1215–1218. 10.1126/science.1167498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- England K.; Boshoff H. I. M.; Arora K.; Weiner D.; Dayao E.; Schimel D.; Via L. E.; Barry C. E. (2012) Meropenem-clavulanic acid shows activity against Mycobacterium tuberculosis in vivo. Antimicrob. Agents Chemother. 56, 3384–3387. 10.1128/AAC.05690-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forsman L. D.; Giske C. G.; Bruchfeld J.; Schön T.; Juréen P.; Ängeby K. (2015) Meropenem-clavulanic acid has high in vitro activity against multidrug-resistant Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 59, 3630–3632. 10.1128/AAC.00171-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambler R. P.; Coulson A. F. W.; et al. (1991) A standard numbering scheme for the Class A beta-lactamases. Biochem. J. 276, 269–272. 10.1042/bj2760269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drawz S. M.; Bonomo R. A. (2010) Three decades of β-lactamase inhibitors. Clin. Microbiol. Rev. 23, 160–201. 10.1128/CMR.00037-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hugonnet J.-E.; Blanchard J. S. (2007) Irreversible inhibition of the Mycobacterium tuberculosis beta-lactamase by clavulanate. Biochemistry 46, 11998–12004. 10.1021/bi701506h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremblay L. W.; Hugonnet J.-E.; Blanchard J. S. (2008) Structure of the covalent adduct formed between Mycobacterium tuberculosis beta-lactamase and clavulanate. Biochemistry 47, 5312–5316. 10.1021/bi8001055. [DOI] [PubMed] [Google Scholar]
- Soroka D.; Li de la Sierra-Gallay I.; Dubée V.; Triboulet S.; Van Tilbeurgh H.; Compain F.; Ballell L.; Barros D.; Mainardi J. L.; Hugonnet J. E.; Arthur M. (2015) Hydrolysis of clavulanate by Mycobacterium tuberculosis β-lactamase BlaC harboring a canonical SDN motif. Antimicrob. Agents Chemother. 59, 5714–5720. 10.1128/AAC.00598-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H.; Hazra S.; Blanchard J. S. (2012) NXL104 irreversibly inhibits the β-lactamase from Mycobacterium tuberculosis. Biochemistry 51, 4551–4557. 10.1021/bi300508r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mire J. A. (2013) BlaC E166A Faropenem Acyl-Intermediate Complex, Protein Data Bank, 10.2210/pdb4ebl/pdb. [DOI] [Google Scholar]
- Tremblay L. W., and Blanchard J. S. (2011) Crystal Structure of BlaC-E166A Covalently Bound with Cefuroxime, Protein Data Bank, 10.2210/pdb3nbl/pdb. [DOI] [Google Scholar]
- Hazra S.; Kurz S. G.; Wolff K.; Nguyen L.; Bonomo R. A.; Blanchard J. S. (2015) Kinetic and structural characterization of the interaction of 6-methylidene penem 2 with the beta-lactamase from Mycobacterium tuberculosis. Biochemistry 54, 5657–5664. 10.1021/acs.biochem.5b00698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie H.; Mire J.; Kong Y.; Chang M.; Hassounah H. A.; Thornton C. N.; Sacchettini J. C.; Cirillo J. D.; Rao J. (2012) Rapid point-of-care detection of the tuberculosis pathogen using a BlaC-specific fluorogenic probe. Nat. Chem. 4, 802–809. 10.1038/nchem.1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow C.; Xu H.; Blanchard J. S. (2013) Kinetic characterization of hydrolysis of nitrocefin, cefoxitin, and Meropenem by β-lactamase from Mycobacterium tuberculosis. Biochemistry 52, 4097–4104. 10.1021/bi400177y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artimo P.; Jonnalagedda M.; Arnold K.; Baratin D.; Csardi G.; de Castro E.; Duvaud S.; Flegel V.; Fortier A.; Gasteiger E.; Grosdidier A.; Hernandez C.; Ioannidis V.; Kuznetsov D.; Liechti R.; Moretti S.; Mostaguir K.; Redaschi N.; Rossier G.; Xenarios I.; Stockinger H. (2012) ExPASy: SIB bioinformatics resource portal. Nucleic Acids Res. 40, W597–W603. 10.1093/nar/gks400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Incardona M.-F.; Bourenkov G. P.; Levik K.; Pieritz R. A.; Popov A. N.; Svensson O. (2009) EDNA : a framework for plugin-based applications applied to X-ray experiment online data analysis. J. Synchrotron Radiat. 16, 872–879. 10.1107/S0909049509036681. [DOI] [PubMed] [Google Scholar]
- Kabsch W. (2010) XDS. Acta Crystallogr., Sect. D: Biol. Crystallogr. 66, 125–132. 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans P. R.; Murshudov G. N. (2013) How good are my data and what is the resolution?. Acta Crystallogr., Sect. D: Biol. Crystallogr. 69, 1204–1214. 10.1107/S0907444913000061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabadinho J.; Beteva A.; Guijarro M.; Rey-Bakaikoa V.; Spruce D.; Bowler M. W.; Brockhauser S.; Flot D.; Gordon E. J.; Hall D. R.; Lavault B.; McCarthy A. A.; McCarthy J.; Mitchell E.; Monaco S.; Mueller-Dieckmann C.; Nurizzo D.; Ravelli R. B. G.; Thibault X.; Walsh M. A.; Leonard G. A.; McSweeney S. M. (2010) MxCuBE: a synchrotron beamline control environment customized for macromolecular crystallography experiments. J. Synchrotron Radiat. 17, 700–707. 10.1107/S0909049510020005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vagin A.; Teplyakov A. (1997) MOLREP: an automated program for molecular replacement. J. Appl. Crystallogr. 30, 1022–1025. 10.1107/S0021889897006766. [DOI] [Google Scholar]
- Wang F.; Cassidy C.; Sacchettini J. C. (2006) Crystal structure and activity studies of the Mycobacterium tuberculosis β-lactamase reveal its critical role in resistance to β-lactam antibiotics. Antimicrob. Agents Chemother. 50, 2762–2771. 10.1128/AAC.00320-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winn M. D.; Ballard C. C.; Cowtan K. D.; Dodson E. J.; Emsley P.; Evans P. R.; Keegan R. M.; Krissinel E. B.; Leslie A. G. W.; McCoy A.; McNicholas S. J.; Murshudov G. N.; Pannu N. S.; Potterton E. A.; Powell H. R.; Read R. J.; Vagin A.; Wilson K. S. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr., Sect. D: Biol. Crystallogr. 67, 235–242. 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov G. N.; Skubák P.; Lebedev A. A.; Pannu N. S.; Steiner R. A.; Nicholls R. A.; Winn M. D.; Long F.; Vagin A. A. (2011) REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr., Sect. D: Biol. Crystallogr. 67, 355–367. 10.1107/S0907444911001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P.; Lohkamp B.; Scott W. G.; Cowtan K. (2010) Features and development of Coot. Acta Crystallogr., Sect. D: Biol. Crystallogr. 66, 486–501. 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vranken W. F.; Boucher W.; Stevens T. J.; Fogh R. H.; Pajon A.; Llinas M.; Ulrich E. L.; Markley J. L.; Ionides J.; Laue E. D. (2005) The CCPN data model for NMR spectroscopy: Development of a software pipeline. Proteins: Struct., Funct., Genet. 59, 687–696. 10.1002/prot.20449. [DOI] [PubMed] [Google Scholar]
- McDonough J. A.; McCann J. R.; Tekippe E. M.; Silverman J. S.; Rigel N. W.; Braunstein M. (2008) Identification of functional Tat signal sequences in Mycobacterium tuberculosis proteins. J. Bacteriol. 190, 6428–6438. 10.1128/JB.00749-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutcliffe I. C.; Harrington D. J. (2004) Lipoproteins of Mycobacterium tuberculosis: An abundant and functionally diverse class of cell envelope components. FEMS Microbiol. Rev. 28, 645–659. 10.1016/j.femsre.2004.06.002. [DOI] [PubMed] [Google Scholar]
- Gu S.; Chen J.; Dobos K. M.; Bradbury E. M.; Belisle J. T.; Chen X. (2003) Comprehensive proteomic profiling of the membrane constituents of a Mycobacterium tuberculosis strain. Mol. Cell. Proteomics 2, 1284–1296. 10.1074/mcp.M300060-MCP200. [DOI] [PubMed] [Google Scholar]
- Kurz S. G.; Hazra S.; Bethel C. R.; Romagnoli C.; Caselli E.; Prati F.; Blanchard J. S.; Bonomo R. A. (2015) Inhibiting the beta-lactamase of Mycobacterium tuberculosis (Mtb) with novel boronic acid transition-state inhibitors (BATSIs). ACS Infect. Dis. 1, 234–242. 10.1021/acsinfecdis.5b00003. [DOI] [PubMed] [Google Scholar]
- Feiler C.; Fisher A. C.; Boock J. T.; Marrichi M. J.; Wright L.; Schmidpeter P. A. M.; Blankenfeldt W.; Pavelka M.; DeLisa M. P. (2013) Directed evolution of Mycobacterium tuberculosis β-lactamase reveals gatekeeper residue that regulates antibiotic resistance and catalytic efficiency. PLoS One 8, e73123 10.1371/journal.pone.0073123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jelsch C.; Mourey L.; Masson J.-M.; Samama J.-P. (1993) Crystal structure of Escherichia coli TEM1 β-lactamase at 1.8 Å resolution. Proteins: Struct., Funct., Genet. 16, 364–383. 10.1002/prot.340160406. [DOI] [PubMed] [Google Scholar]
- Chen Y.; Bonnet R.; Shoichet B. K. (2007) The acylation mechanism of CTX-M β-lactamase at 0.88 Å resolution. J. Am. Chem. Soc. 129, 5378–5380. 10.1021/ja0712064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pernot L., Petrella S., and Sougakoff W. (2004) Crystal Structure of the Class A Beta-Lactamse L2 from Stenotrophomonas maltophilia at 1.51 angstrom. Protein Data Bank, 10.2210/pdb1o7e/pdb. [DOI] [Google Scholar]
- Marciano D. C.; Pennington J. M.; Wang X.; Wang J.; Chen Y.; Thomas V. L.; Shoichet B. K.; Palzkill T. (2008) Genetic and structural characterization of an L201P global suppressor substitution in TEM-1 β-lactamase. J. Mol. Biol. 384, 151–164. 10.1016/j.jmb.2008.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzin A. P.; Nukaga M.; Nukaga Y.; Hujer A. M.; Bonomo R. A.; Knox J. R. (1999) Structure of the SHV-1 β-Lactamase. Biochemistry 38, 5720–5727. 10.1021/bi990136d. [DOI] [PubMed] [Google Scholar]
- Levitte S.; Adams K. N.; Berg R. D.; Cosma C. L.; Urdahl K. B.; Ramakrishnan L. (2016) Mycobacterial acid tolerance enables phagolysosomal survival and establishment of tuberculous infection in vivo. Cell Host Microbe 20, 250–258. 10.1016/j.chom.2016.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson D. L., and Cox M. M. (2005) Lehninger Principles of Biochemistry, 4th ed., W. H. Freeman and Company: New York, p 497. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.