

Abstract
Enhancing the thermal stability of proteins is an important task for protein engineering. There are several ways to increase the thermal stability of proteins in biology, such as greater hydrophobic interactions, increased helical content, decreased occurrence of thermolabile residues, or stable hydrogen bonds. Here, we describe a well-defined polymer based on β-helical polyisocyanotripeptides (TriPIC) that uses biological approaches, including hydrogen bonding and hydrophobic interactions for its exceptional thermal stability in aqueous solutions. The multiple hydrogen bonding arrays along the polymer backbone shield the hydrophobic core from water. Variable temperature CD and FTIR studies indicate that, on heating, a better packed polymer conformation further stiffens the backbone. Driven by hydrophobic interactions, TriPIC solutions give fully reversible hydrogels that can withstand high temperatures (80 °C) for extended times. Cryo-scanning electron microscopy (cryo-SEM), small-angle X-ray scattering (SAXS), and thorough rheological analysis show that the hydrogel has a bundled architecture, which gives rise to strain stiffening effects on deformation of the gel, analogous to many biological hydrogels.
Introduction
To build up its architectures, nature uses well-defined structures as building blocks, such as α-helices and β-sheets, which are the principal structural components of proteins and of the double helix of DNA. These stable, robust architectural motifs are responsible for the tertiary structure and mechanical strength and thus play a crucial role in the function of all biological systems. The stability of such structural elements is particularly important for hyperthermophilic organisms that need to survive demanding thermal conditions.1−3 Understanding and enhancing the thermal stability of proteins is not only academically interesting but also important for industrial applications.4,5 Great example are Taq DNA polymerase, derived from Thermus aquaticus, that is the commonly used enzyme in polymerase chain reaction (PCR) and DNA sequencing by virtue of its high thermal stability6 and the thermally stable cytoskeleton in thermophilic archaea.7 As a consequence, many studies have focused on the adaptation of thermophilic proteins to high temperatures. Both experimental and computational works have demonstrated that the proteins in a thermophilic cell take advantage of greater hydrophobic interactions, increased helical content, decreased occurrence of thermolabile residues, stable hydrogen bonds, salt bridge formation, better atom packing, and optimizing of the surface charges to deal with the extra heat in their environments.8−11
The architecture of biopolymers is attractive to mimic with synthetic polymers due to their high degree of function and their potential applications in the fields of material and supramolecular sciences.12,13 Among many artificial polymers, polyisocyanopeptides (PICs) are particularly interesting because of their stable helical backbones with a high helix inversion barrier.14−18 They adopt an ∼41 (four repeats per turn) β-helical conformation stabilized by a β-sheet-like peptidic hydrogen bond network along the polymer backbone between monomers n and n + 4.19,20 Moreover, this helical structure plays a vital role in the high stiffness of the polymer.21 In earlier work, we showed that gels made of polyisocyanodipeptides grafted with ethylene glycol side chains mimic biological gels22,23 and more complex cytoskeletal composites24 in nearly all aspects. The ultrasensitive mechanical responses that these gels demonstrate originate from the network architecture of semiflexible bundles that is formed when a polymer solution is heated into the mesophilic region (37 °C).23,25 Furthermore, these gels decorated with GRGDS peptides can serve as a 3D matrix to manipulate the differentiation of stem cells.26 To the best of our knowledge, however, very few examples of synthetic biomimetic polymers with high thermal stability have been reported. In this work, we use a truly synthetic polymer based on the helical PIC backbone and mimic thermophilic proteins using biological approaches. This model system will aid in understanding and developing the underlying design principles of thermally stable aqueous biomimetic materials or even thermophilic proteins.
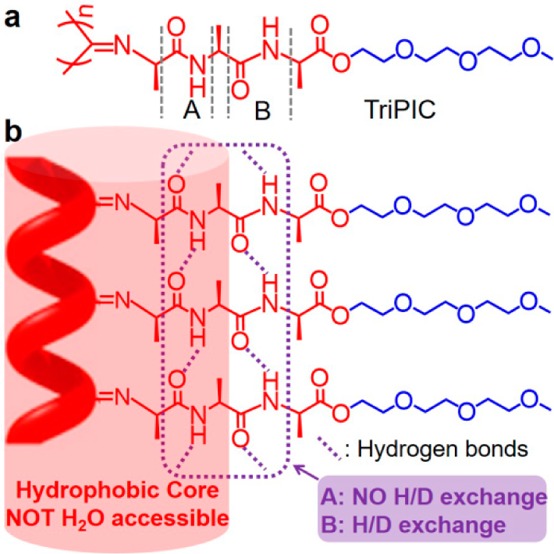
The exceptional stability of oligo(ethylene glycol)-decorated polyisocyanotripeptides (TriPIC, Figure 1) is based on three adjacent alanine groups. The rigid, helical backbone of TriPIC is stabilized by two parallel well-defined hydrogen-bonding arrays between the two amide groups, which run parallel to the polyisocyanide backbone. The amide regions are partially inaccessible to water, and consequently, a hydrophobic core is constructed around the polymer backbone. On heating, the first hydrogen bond (A, NOT H2O accessible) keeps stable which mainly holds the helical architecture, whereas the second one (B) is stretched, endowing the polymer with a better defined helix and stiffer backbone. Beyond a gelation temperature, single polymers assemble and bundle into a network, driven by the hydrophobic interactions of the glycol tails, which induces an unusual thermal stability of the gels, which are stable for many hours at 80 °C.
Figure 1.
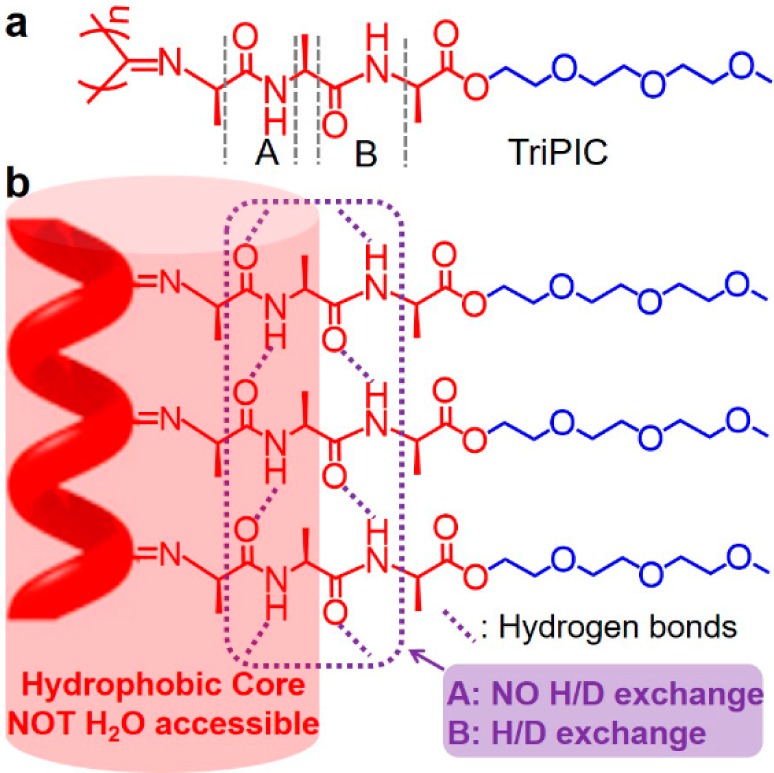
(a) Molecular structure of the tri(ethylene glycol)-functionalized polyisocyanotripeptides (TriPIC). (b) Schematic representation of the key groups that stabilize the structure against thermal agitation.
Results and Discussion
Polymer Synthesis and Characterization
The synthesis of the oligo(ethylene glycol)-functionalized polyisocyanotripeptide (TriPIC) pursues a standard peptide coupling strategy, followed by subsequent dehydration and polymerization reactions27 (Scheme 1). A detailed description of the synthesis and characterization (1H and 13C NMR, MS, HRMS) of the intermediate and final products is given in the Experimental Section and the Supporting Information (Figures S1–S6). The isocyanide monomers were polymerized through nickel(II)-catalyzed polymerization using ethanol as the initiator in freshly distilled chloroform. In the formation of the helix, both hydrogen bonding and steric interactions (from the methyl of the alanine group) play crucial roles.28 In our hands, polymers with different amino acids such as glycine or valine either did not polymerize or were unable to from self-supporting hydrogels. Here, we use the TriPIC with alanines in the l,d,l-configuration. In fact, we were unable to even polymerize the equivalent diastereomeric d,d,l-isocyanotripeptide monomer, most likely due to unfavorable steric interactions. The polymer length was regulated by adjusting the molar ratio between the catalyst and the monomer. In this way, a series of polymers with different lengths (TriPIC-a-g) were obtained. The molecular weights (Mv) were estimated by viscometry based on the Mark–Houwink equation,29 and the apparent polymer contour length (L) was calculated from the measured Mv.30 In addition, polymers prepared at higher monomer-to-catalyst ratio were much longer than the ones at lower ratios, as was clearly observed by atomic force microscopy (AFM, Figure S7). The results are consistent with the molecular weights (Mv) and the used starting conditions.
Scheme 1. (a) Synthetic Route to TriPIC Polymers.

Reagents and conditions: (i) HCl, EtOAc, room temperature, 2 h; (ii) l-HO-Ala-Boc, EDC·HCl, HOBt, DiPEA, room temperature, 16 h, yield = 65% for two steps; (iii) HCl, EtOAc, room temperature, 2 h; (iv) HCO2Na/HCO2Et, reflux, 16 h, yield = 68% for two steps; (v) diphosgene, N-methylmorpholine, CH2Cl2, −40 °C, 1 h, yield = 80%; (vi) Ni(ClO4)2·6H2O, EtOH/CHCl3, room temperature, 2–7 days, yield = 85%. [M]/[cat.] = monomer-to-catalyst ratio, Mv = viscosity-derived molecular weight; degree of polymerization n and the polymer contour length L are calculated based on Mv.
Single Polymer Chain Stability
The helical backbone conformation is stabilized by hydrogen-bonding arrays, and as such, water will have a large impact on the stability and the rigidity of the helix. We used H/D exchange experiments in conjunction with FTIR and ATR spectroscopy to study the solvent accessibility of the polymer core. Figure 2a shows the FTIR spectrum of TriPIC-b, together with that of its monomer, both recorded in D2O solution. The absorption band at 1735 cm–1 (light blue) is due to the ester C=O stretching vibration, and the band at 1645 cm–1 (purple) corresponds to the amide I modes of the two amide groups. This band shows a strong narrowing upon polymerization. The area under the band stays constant. In the monomer, the amide groups form hydrogen bonds with water with a large variation in hydrogen-bond strength, leading to a broad absorption band. Upon polymerization, the amide A groups form intrachain hydrogen bonds, which have a much smaller distribution in hydrogen-bond strength, thus leading to a much narrower absorption band. The shoulder at 1620 cm–1 (which is observed for the polymer only) is attributed to the C=N stretch vibration. Interestingly, we also observe a strong amide II (mainly NH-bending character) absorption band at 1523 cm–1 for the polymer, and this band is completely absent in the monomer spectrum. This band points to the presence of protonated NH groups in the polymer. These NH groups are apparently extremely well shielded from the solvent because they show no sign of H/D exchange even after storing the polymer in D2O for several weeks. In order to test whether all NH groups in the polymer are inaccessible to the solvent, we exchanged the D2O solvent for a 50/50 mixture of H2O/D2O and recorded ATR spectra (Figure 2b). The amide II band (1523 cm–1) increases in intensity when H2O is added to the solvent while a second band at 1440 cm–1 (due to the amide II′ absorption of deuterated amide groups) decreases in intensity. This indicates that the polymer contains not only solvent-inaccessible NH groups but also solvent-accessible NH groups. Interestingly, the ATR spectra also show two well-resolved bands in the amide I region (at 1645 and 1671 cm–1). The band at 1671 cm–1 blue-shifts a few cm–1 when H2O is added to the solvent whereas the band at 1645 cm–1 is unaffected. This leads us to conclude that the two absorption bands correspond to the two types of amide groups (A and B): apparently, the amide groups closest to the helical backbone (A) absorb at 1645 cm–1 and are solvent inaccessible, whereas the amide groups further away from the central helix (B) and close to the poly(ethylene glycol) chain, absorb at 1671 cm–1 (in D2O), and are accessible to the solvent.
Figure 2.
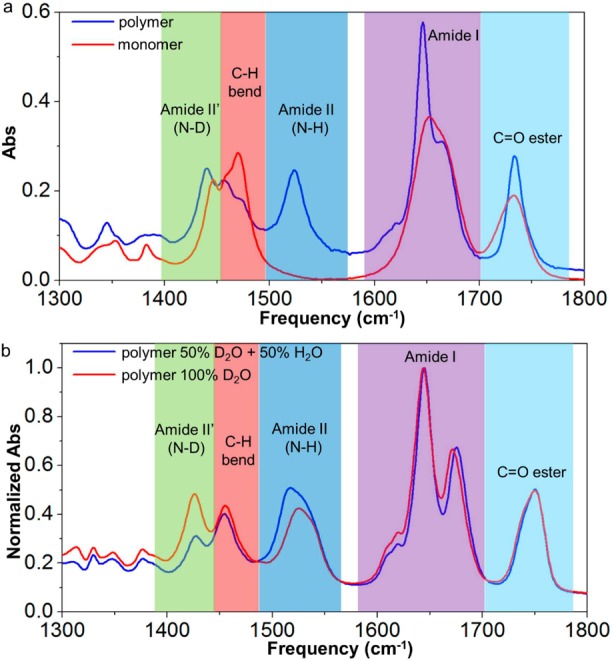
(a) FTIR spectra of polymer TriPIC-b (blue line) and the corresponding monomer (red line) between 1300 and 1800 cm–1 in D2O at room temperature; c = 17.0 g L–1. (b) ATR spectra of polymer TriPIC-b in D2O (red line) and in 50/50 D2O/H2O (blue line) at room temperature. The different color coded absorptions are discussed in the main text.
Hydrogel Formation
Equivalent to the corresponding dipeptide polyisocyanides,22 the TriPIC polymers display a lower critical solution temperature (LCST). Heating beyond the LCST causes the oligo(ethylene glycol) tails to dehydrate, which induce bundle formation of the polymers and the formation of hydrogels. The gelation temperature Tgel coincides with the LCST of the aqueous polymer solution.
We studied the thermal responsiveness and stability of TriPIC hydrogels using rheological analysis (Figure 3). When an aqueous TriPIC-f solution is heated in a rheometer, the storage modulus G′, defined as the ratio between the applied stress σ and the measured strain γ, increases as a network is formed (Figure 3a), even at concentration as low as c = 0.1 g L–1 (about 0.01 wt %). The gel stiffness is retained when the hydrogel is kept for more than 10 h at elevated temperatures T = 80 °C, which demonstrates a remarkable thermal stability. At elevated temperatures, both the storage G′ and the loss modulus G″ are independent of frequency (Figure S8). We find that gelation is fully reversible (Figure S9a); cooling the sample results in a low viscous solution with minimal thermal hysteresis. Repeated heating and cooling cycles have virtually no effect on the mechanical properties of the TriPIC hydrogels (Figure S9b).
Figure 3.

Linear and nonlinear rheological analyses of TriPIC hydrogels: (a) Time sweep of TriPIC-f (c = 2.0 g L–1), first heating to 80 °C, then conditioning at 80 °C for 10 h. (b) Storage modulus G′ as a function of temperature for TriPIC-f at different concentrations between c = 0.1–4 g L–1. (c) Differential modulus K′ = ∂σ/∂γ against stress σ for TriPIC-f gels at 60 °C for different polymer concentrations. (d) Storage modulus G′ as a function of temperature ramps of TriPIC-f hydrogels (2 g L–1) for different NaCl concentrations.
The gelation temperature, Tgel, as defined as the onset point of the increase in the G′ as a function of the temperature, was found to be 50 °C for TriPIC-f, which is identical to those of the gels made of polymers with different molecular weights TriPICa-g (Figure S10a) and also those of the gels prepared at different polymer concentrations (Figure 3b). The shear modulus in the gel phase, however, increases sharply with both the concentration c and the polymer contour length L. Over the measured concentration regime c = 0.1–4.0 g L–1, the storage modulus scales as G′ ∝ c2, which is in line with experimental and theoretical results of the semiflexible networks.31,32 For such networks it is expected that at high stresses, i.e., beyond a critical stress level σc, the hydrogel becomes stiffer. Many biological networks, such as actin, collagen, and fibrin, display this so-called strain-stiffening behavior, which plays a key role in tissue protection and cell–cell communication and may influence other cellular functions. The nonlinear regime is readily probed by a prestress protocol,33 where the rheometer applies a constant stress to the sample and on top of that a small oscillatory stress, yielding the differential modulus defined as K′ = ∂σ/∂γ.22,34 At small deformations, K′ = G′, but beyond a critical stress σc, the differential modulus scales to the applied stress K′∝ σm, where m is the stiffening index that indicates the mechanical responsiveness of the hydrogel.
Indeed, TriPIC hydrogels of different polymer length (Figure S10b) and at different concentrations (Figure 3c) clearly show strain-stiffening. At the highest concentrations (or contour length), the plateau in K′ is followed by a nonlinear regime with m ≈ 1, a value similar to what is experimentally found for collagen but lower than the theoretical limit m = 3/2, which was found for the dipeptide PIC gels.23 At lower concentrations, the critical stress is so low that even below σ < 1 Pa the network already responds nonlinearly and the gel becomes progressively stiffer. In these samples, the critical stress, i.e., the threshold for the nonlinear regime, is comparable to that of biological hydrogels and are of the order to what cells can apply to their extracellular environments.
A third approach to manipulate the mechanical properties of TriPIC hydrogels is the use of salts. Commonly, the addition of salts has a significant influence on the LCST of thermoresponsive polymers and proteins, an effect known as the Hofmeister effect.35,36 In the case of TriPIC gels (Figure 3d), Tgel decreases (linearly) with increasing the NaCl concentration, and as a result, the mechanical parameters (K′ and σc) in the linear and the nonlinear regime can be readily tuned by adding the salts (Figure S11). The nature of the salt determines the amplitude of the Hofmeister effect and of the effect on the gel mechanics.37
Architectural Analysis
The biomimetic mechanical properties of TriPIC hydrogels are the direct result of their fibrillar or bundle architecture. Bundle formation is a design criterion in both of mesophilic and thermophilic biological hydrogel networks, which intrinsically gives rise to gels at low concentrations, with large pore sizes and with strong mechanical repsonses.38,39 We studied the gel architecture in situ by using cryo-scanning electron microscopy (cryo-SEM) and small-angle X-ray scattering (SAXS). Cryo-SEM images (Figure 4a,b) show a homogeneous and branched network of polymer bundles with pore sizes of the order of 100 nm at c = 4.0 g L–1. Quantification of the bundle diameter is not easy from the SEM images but can be derived from the SAXS patterns. We recorded scattering of the TriPIC-f (4 g L–1) as a function of temperature (Figure 4c): at low T molecularly dissolved and beyond the LCST (50 °C) in the gel phase, where the scattering curves start to deviate. Fitting the scattering data with appropriate models allows us to extract the architectural information on the network. In solution (at T < Tgel), we fitted the data to the semiflexible polymer model of Kholodenko (Figure 4d, red solid line), yielding a polymer diameter R = 1.0 nm and a persistent length lp = 48 nm. The contour length fixed at 160 nm is outside the experimental window. Upon heating the sample to 70 °C, however, the curve of the scattering profile changes because of bundle formation and is fitted excellently by a composite Kholodenko/Ornstein–Zernike (KOZ) model (pink solid line),25 with a bundle radius RB = 2.96 nm. The persistence length of the polymer bundles Lp,B is now outside the experimental window and is fixed on 800 nm. The Ornstein–Zernike term yields a correlation length ξOZ = 110 nm that corresponds to the average pore size of the network. The results agree markedly well to the cryo-SEM images.
Figure 4.

Lower (a) and higher magnification (b) cryo-SEM images of TriPIC (TriPIC-f, c = 4.0 g L–1) hydrogels. The accelerating voltage was 3.0 kV. (c) SAXS curves measured at different temperature for a TriPIC-f hydrogel with c = 4.0 g L–1. (d) SAXS curves for a 4.0 g L–1 TriPIC-f sample in the liquid state at low temperature (5 °C) and in the gel state at high temperature (70 °C). The solid lines represent the best fit to the Kholodenko model for the low-temperature data and the best fit to the composite model with a Kholodenko term and an Ornstein–Zernike term for the high-temperature data.
Bundling is crucial in the formation of semiflexible networks and their unique properties but is still poorly understood experimentally. To obtain deep insights into the mechanism of the bundling of TriPIC polymers, we carried out temperature-dependent FTIR experiments (Figure 5a) and specifically followed the vibrations of the two hydrogen-bond-forming amide groups. Note that the hydrogen bonds formed by the amide closest to the isocyanide backbone (amide A) is thought to maintain the helical architecture of the polymer,28 while the hydrogen bonds of the second amide group (amide B) further stabilizes and stiffens the TriPIC polymer backbone. This is consistent with the present results regarding the different solvent accessibilities of these two amide groups. The intensities of the C=O stretch vibrations of both amides and the ester (Figure 5a) change with temperatures, which indicates that the local chemical environment of these groups changes as a function of temperature.
Figure 5.

(a) FTIR spectra of the TriPIC-b in D2O (c = 17.0 g L–1) as a function of temperature. The amide I and ester regions of the spectra were fitted with six Gaussian-curves centered at 1622, 1646, 1661, 1674, 1734, and 1739 cm–1. Inset: the amplitude changes of amide and ester groups with temperature at 1646, 1661, 1674, 1734, and 1739 cm–1. At Tgel (dashed line), most of the intensities show an infliction point. (b) CD spectra of TriPIC-b as a function of temperature. (c) Evaluation of the CD signal at λ = 315 nm of TriPIC-b with temperature. The CD measurements were performed in H2O (c = 0.5 g L–1).
To analyze the data more quantitatively, the spectra were deconvoluted by globally fitting six Gaussian peaks with peak centers at 1622, 1646, 1661, 1674, 1734, and 1739 cm–1 (Figure S12 shows additional information). We fitted the amide I,A band with one Gaussian and the amide I,B band and the ester vibrational band each with two Gaussians as this allows us to model the blue-shift and broadening of the bands with increasing temperature. The areas of the Gaussians are shown in the inset. We observe that there is an inflection point at Tgel = 50 °C, above which the intensity variation of the bands becomes stronger. Interestingly, the ester C=O and the amide I,B vibrational bands show a blue-shift with increasing temperature while the center position of the amide I,A band is independent of temperature. The ester C=O groups and part of the amide I,B groups are hydrogen-bonded to their aqueous solvation shell, and these hydrogen-bonds become weaker with increasing temperature, leading to the observed blue-shift and peak broadening of the amide I,B and ester bands. The amide A hydrogen-bond strength does not change: these groups are already so tightly bound and shielded from the solvent that polymer bundling does not alter their already highly ordered conformation. We note that the spectral changes with temperature of the aqueous TriPIC polymer solution or gel are fully reversible: on cooling the original spectrum is obtained again (Figure S13). As a control, we do not observe any spectral changes when heating a solution of the monomer (Figure S14).
The transition to the gel phase is also readily identified using UV–vis spectroscopy (Figure S15) and circular dichroism (CD) spectroscopy (Figure 5b,c). Heating the solution shows a clear step in the ellipticity Δε at λ = 315 nm, right at the transition temperature Tgel = 50 °C, which implies an increased helical content and a better-defined helical architecture are formed as a result of the stretching of the hydrogen bonds and increasingly stiffer backbones on heating. This is also supported by the observed changes in the UV–vis spectra that show a large jump in the absorbance at λ = 315 nm. Again, these transitions are completely reversible upon cooling with minimum hysteresis effects. It is interesting to note that the large effects in UV–vis and CD were not observed in the corresponding dipeptide polyisocyanide.27 Apparently, the introduction of the second hydrogen bond in the polymer has a large effect on its molecular structure and rigidity and, as such, plays a very important role in gelation and thermal stability.
Conclusion
In summary, we presented a thermal stable and well-defined helical polymer based on the β-helix polyisocyanotripeptide that mimics biological features of thermophilic proteins. This exceptional helical architecture is stabilized by a double hydrogen-bonding array between the amide groups, which plays a key role in the bundle formation, gelation, and thermal stability. Consequently, the TriPIC polymers have several significant and unusual characteristics. First, the hydrophobic core, which is constructed by the multiple hydrogen bonds, is partly shielded from water. The increased helical definition on heating the polymer in solution in an important design feature toward its thermal stability. Second, on heating aqueous TriPIC solution, the polymers assemble into bundled networks driven by hydrophobic interactions between the ethylene glycol tails. The formed gels show excellent thermal stability at temperatures as high as 80 °C. Third, the mechanical properties of TriPIC gels in both the linear and nonlinear regimes, including the storage modulus G′ and critical stress σc, are readily tailored by the polymer concentration, their length, and by salts present in solution. In short, we have presented a truly synthetic polymer that is able to deal with excessive thermal energy using biological approaches such as hydrogen bonding and hydrophobic interactions, even without the changing of surface electrostatics which is a common natural strategy. This approach can be extended to design principles for future thermophilic proteins and synthetic polymers.
Experimental Section
Materials and Instrumentation
All chemicals were purchased from Sigma-Aldrich, Acros, or Alfa Aesar and used as received unless specially stated. Dichloromethane and chloroform were distilled over CaCl2. Tetrahydrofuran and toluene were distilled over sodium in the presence of benzophenone. 1H and 13C NMR spectra were recorded on a Varian Inova 400 or a Bruker Avance III 500 MHz. Rheology measurements were measured with stress-controlled and parallel-plate rheometer (Discovery HR-2, TA Instruments). AFM images were taken by a Nanoscope IV in tapping mode. The absorption spectra were taken on a JASCO V-630 UV–vis spectrophotometer, and the CD spectra were carried out on a JASCO J-810. Cryo-scanning electron microscopy (cryo-SEM) was performed on a JEOL 6330 cryo-scanning electron microscope. IR spectra were performed on a Bruker VERTEX 80v vacuum FT-IR spectrometer.
Synthesis of 3EG-l-Ala-d-Ala-l-Ala-Boc
3EG-l-Ala-d-Ala-Boc (4.0 g, 9.84 mmol), as synthesized according to the literature,27 was dissolved in 20 mL of HCl solution in ethyl acetate (2.3 mol/L) and stirred at rt for 2 h. After the Boc-protecting group was totally removed as monitored by TLC (3% of methanol in DCM as eluent), the solvent and the excess HCl were removed by rotary evaporator invacuo. The crude product was dissolved in 250 mL of distilled DCM, to which l-HO-Ala-Boc (1.9 g, 10 mmol) and hydroxybenzotriazole (1.4 g, 10 mmol) were added. N-Ethyldiisopropylamine (1.3 g, 10 mmol) was added slowly until the reaction mixture was basic and clear in appearance, and 1-[3-(dimethylamino)propyl]-3-ethylcarbodiimide hydrochloride (EDC.HCl) (1.9 g, 10 mmol) was added. The reaction mixture was stirred at rt overnight. The reaction mixture was washed successively with citric acid solution (6%, 100 mL), water (100 mL), saturated solution of sodium bicarbonate (100 mL), and water (100 mL × 2) and dried over sodium sulfate. The solvent was removed by rotary evaporation, and the crude product was purified by a silica column chromatography (2% of methanol in DCM as an eluent). The product was collected as a colorless oil, with a yield of 65%. Analysis: 1H NMR (500 MHz, CDCl3) δ 7.36 (d, J = 6.6 Hz, 1H), 7.22 (d, J = 5.5 Hz, 1H), 5.57 (d, J = 5.3 Hz, 1H), 4.53–4.40 (m, 2H), 4.22–4.03 (m, 3H), 3.60–3.56 (m, 2H), 3.55–3.50 (m, 6H), 3.46–3.42 (m, 2H), 3.26 (s, 3H), 1.32 (s, 9H), 1.31–1.23 (m, 9H). 13C NMR (125 MHz, CDCl3) δ 173.01, 172.57, 171.96, 155.46, 79.59, 71.77, 70.45, 70.42, 70.36, 68.77, 64.22, 58.81, 50.30, 48.48, 48.05, 28.23, 18.23, 17.89, 17.69. MS (ES+): m/z (%) = 500.1 [M + Na]+. HRMS (ES+): calcd for C21H39N3O9Na 500.2579, found 500.2585.
Synthesis of 3EG-l-Ala-d-Ala-l-Ala-For
3EG-l-Ala-d-Ala-l-Ala-Boc (2 g, 4.94 mmol) was dissolved in 10 mL of HCl solution in ethyl acetate (2.3 mol/L) and stirred at rt for 2 h. After the Boc-protecting group was totally removed as monitored by TLC (3% of methanol in DCM as eluent), the solvent and the excess HCl were removed by rotary evaporator in vacuo. The crude product was mixed with 30 mL of ethyl formate, to which mixture sodium formate (1.34 g, 19.76 mmol) was added. The reaction was stirred under reflux overnight, after which it was cooled down to rt, and the solid was filtered off and washed thoroughly with CHCl3. The solvent was removed from the filtrate, and the crude product was purified with a silica column chromatography (3% of methanol in DCM as an eluent) to yield 3EG-l-Ala-d-Ala-l-Ala-For as a colorless solid, with a yield of 68%. Analysis: 1H NMR (δ ppm, CDCl3, 500 MHz): 8.18 (s, 1H), 7.04 (d, J = 7.1 Hz, 1H), 6.94 (d, J = 6.6 Hz, 1H), 6.65 (d, J = 4.4 Hz, 1H), 4.51–4.60 (m, 3H), 4.33–4.21 (m, 2H), 3.71–3.67 (m, 2H), 3.67–3.63 (m, 6H), 3.57–3.54 (m, 2H), 3.38 (s, 3H), 1.42 (d, J = 7.1 Hz, 6H), 1.38 (d, J = 7.0 Hz, 3H). 13C NMR (δ ppm, CDCl3, 125 MHz): 172.85, 172.38, 172.04, 161.59, 71.75, 70.43, 70.41, 70.35, 68.75, 64.29, 58.83, 48.75, 48.09, 47.85, 18.15, 18.01, 17.68. MS (ES+): m/z (%) = 428.3 [M + Na]+. HRMS (ES+): calcd for C17H31N3O8Na 428.2003, found 428.2009.
Synthesis of Monomer
3EG-l-Ala-d-Ala-l-Ala-For (1,215.6 mg, 3.0 mmol) and N-methylmorpholine (NMM; 831 μL, 7.5 mmol) were dissolved in 300 mL of freshly distilled CH2Cl2 and cooled down to −40 °C (dry ice and acetone bath) under an argon atmosphere. A solution of diphosgene (185 μL, 1.5 mmol) in 40 mL of freshly distilled CH2Cl2 was added dropwise under argon over 1 h with the temperature kept at −40 °C. The reaction was quenched by adding sodium bicarbonate (5 g). The quenched mixture was stirred for 5 min at −40 °C. The reaction mixture was purified by the silica column (CH2Cl2/methanol, 2%), to yield monomer as a pale solid, with a yield of 80%. Analysis: 1H NMR (δ ppm, CDCl3, 500 MHz): 7.20 (d, 1H), 6.89 (d, 1H), 4.61 (m, 1H), 4.53 (m, 1H), 4.32 (m, 3H), 3.72 (m, 2H), 3.66 (s, 6H), 3.57 (m, 2H), 3.39 (s, 3H), 1.67 (d, J = 7.1 Hz, 3H), 1.44 (m, 6H). 13C NMR (δ ppm, CDCl3, 125 MHz): 172.39, 170.84, 166.30, 161.05, 71.88, 70.60, 70.54, 70.45, 68.93, 64.45, 58.96, 48.88, 48.35, 19.79, 18.13, 17.87. MS (ES+): m/z (%) = 410.2 [M + Na]+. HRMS (ES+): calcd for C17H29N3O7Na 410.1898, found 410.1901.
Synthesis of Polymer
A fresh 1.0 mM catalyst stock solution was prepared by dissolving 18.3 mg of Ni(ClO4)2·6H2O (Sigma-Aldrich) in 5.0 mL of absolute ethanol, followed by a dilution with freshly distilled chloroform to a total volume of 50 mL. 300 mg of the monomer was dissolved in 6 mL of freshly distilled chloroform. The length of the polymers were controlled by different molar ratio of catalyst over monomer (500:1, 1,000:1, 2,000:1, 5000:1, 8000:1, 12000:1, 15000:1). The reaction mixture was stirred for 2–7 days at room temperature, and the isocyanide peak was monitored by ATR-FTIR. When the reaction was complete, the reaction mixture was precipitated in a large volume of diisopropyl ether for three times.
Characterizations of Polymer
The molecular weight (Mv) of the polymer was measured by viscometry in acetonitrile at 25 °C, according to the Mark–Houwink equation, and the apparent polymer contour length (L) was calculated from the measured Mv.30 The polymer was observed by the atomic force microscopy (AFM) in the tapping mode. The solutions of polymers (CHCl3) were spin-coated onto the freshly cleaved Mica substrate.
Rheology
To prepare the TriPIC hydrogels, 4.0 mg of polymers was dissolved in 1.0 mL of precooled Milli-Q water by stirring for 12 h at 4 °C, resulting in a 4.0 g L–1 transparent gel solution. Rheological measurements were performed on a stress-controlled rheometer (Discovery HR-2, TA Instruments), which was equipped with of parallel-plate geometry (40 mm diameter; aluminum) and temperature controller. In order to minimize sample evaporation at high temperature, an oil sealing was used. For the linear viscoelastic regime, all experiments were performed at a fixed frequency ω = 1 Hz and an oscillatory deformation of amplitude strain (4%). The gelation temperature values were determined by the onset of the increase of storage modulus G′. The nonlinear regime was studied by a prestress protocol.33 In this case, the gels were subjected to a constant prestress (σ0) with a small oscillatory stress (σ < 0.1 σ0) superposed upon it, at ω = 10–0.1 Hz and a desired temperature. The differential modulus K′ was defined as K′ = ∂σ/∂γ and plotted as a function of applied prestress at ω = 1 Hz.
Cryo-SEM
All the apparatus used in this experiment were supposed to preheat at 70 °C, such as tweezers and cryo-SEM sample holder. The gel solutions were incubated at 70 °C for 3 min, and then the gelation samples were mounted onto a preheated holder and suddenly plunged into a freezing liquid nitrogen bath. In order to study the inside of the gel, the samples were fractured. SEM images were examined on a JEOL 6330 cryo-scanning electron microscope at an accelerating voltage of 3.0 kV.
Small-Angle X-ray Scattering (SAXS)
SAXS measurements and fitting of the SAXS curves were performed according to the literature procedure.24 In order to prevent water evaporation at high temperature, the capillaries were melted.
Fourier Transform Infrared (FTIR)
17.0 mg of totally dry TriPIC polymer was dissolved in 1.0 mL of fresh D2O by stirring for 24 h in a cold room at 4 °C under N2. IR transmission measurements were performed on a Bruker VERTEX 80v vacuum FT-IR spectrometer as a function of temperature at a resolution of 2 cm–1. The cell temperature was stabilized for at least 3 min before the spectra were taken, and a minimum of 50 scans were averaged. As a background, the spectrum of pure D2O was performed.
Acknowledgments
This work was supported by the National Nature Science Foundation of China (NSFC, 51503143, 21574037, and 21761132007), Tianjin Natural Science Foundation (16JCQNJC05000 and 15JCYBJC17500), Innovation Foundation of Tianjin University (2016XRX0017), China International Science and Technology Project (2016YFE0114900), Natural Science Foundation of Hebei Province (B2015202330 and B2017202051), Tianjin 1000 Youth Talents Plan, and Netherlands Organization for Scientific Research (NWO) with a Veni Grant (680-47-437) and a Gravitation Grant (024.001.035). Y. Duan is acknowledged for preparing some of the intermediate compounds.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.macromol.7b01832.
Chemical characterizations (1H and 13C NMR spectra of the monomer and intermediates), AFM images and analysis of TriPIC-a-g, additional FTIR and rheology experimental results (PDF)
Author Present Address
R.H.: Department of Tumor Immunology, Radboud Institute for Molecular Life Sciences, Radboud University Medical Center, Geert Grooteplein 26, 6525 GA Nijmegen, The Netherlands.
The authors declare no competing financial interest.
Supplementary Material
References
- Rudnicki M. S.; Cirka H. A.; Aghvami M.; Sander E. A.; Wen Q.; Billiar K. L. Nonlinear Strain Stiffening Is Not Sufficient to Explain How Far Cells Can Feel on Fibrous Protein Gels. Biophys. J. 2013, 105, 11–20. 10.1016/j.bpj.2013.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Searcy D. G.; Hixon W. G. Cytoskeletal Origins in Sulfur-Metabolizing Archaebacteria. BioSystems 1991, 25, 1–11. 10.1016/0303-2647(91)90008-9. [DOI] [PubMed] [Google Scholar]
- Pack S. P.; Yoo Y. J. Protein Thermostability: Structure-Based Difference of Amino Acid between Thermophilic and Mesophilic Proteins. J. Biotechnol. 2004, 111, 269–277. 10.1016/j.jbiotec.2004.01.018. [DOI] [PubMed] [Google Scholar]
- Urbieta M. S.; Donati E. R.; Chan K.-G.; Shahar S.; Sin L. L.; Goh K. M. Thermophiles in the Genomic Era: Biodiversity, Science, and Applications. Biotechnol. Adv. 2015, 33, 633–647. 10.1016/j.biotechadv.2015.04.007. [DOI] [PubMed] [Google Scholar]
- Singh B.; Poças-Fonseca M. J.; Johri B.; Satyanarayana T. Thermophilic Molds: Biology and Applications. Crit. Rev. Microbiol. 2016, 42, 985–1006. 10.3109/1040841X.2015.1122572. [DOI] [PubMed] [Google Scholar]
- Innis M. A.; Myambo K. B.; Gelfand D. H.; Brow M. DNA Sequencing with Thermus Aquaticus DNA Polymerase and Direct Sequencing of Polymerase Chain Reaction-Amplified DNA. Proc. Natl. Acad. Sci. U. S. A. 1988, 85, 9436–9440. 10.1073/pnas.85.24.9436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ettema T. J.; Lindås A. C.; Bernander R. An Actin-Based Cytoskeleton in Archaea. Mol. Microbiol. 2011, 80, 1052–1061. 10.1111/j.1365-2958.2011.07635.x. [DOI] [PubMed] [Google Scholar]
- Kumar S.; Nussinov R. How Do Thermophilic Proteins Deal with Heat?. Cell. Mol. Life Sci. 2001, 58, 1216–1233. 10.1007/PL00000935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gromiha M. M.; Pathak M. C.; Saraboji K.; Ortlund E. A.; Gaucher E. A. Hydrophobic Environment Is a Key Factor for the Stability of Thermophilic Proteins. Proteins: Struct., Funct., Genet. 2013, 81, 715–721. 10.1002/prot.24232. [DOI] [PubMed] [Google Scholar]
- Szilágyi A.; Závodszky P. Structural Differences between Mesophilic, Moderately Thermophilic and Extremely Thermophilic Protein Subunits: Results of a Comprehensive Survey. Structure 2000, 8, 493–504. 10.1016/S0969-2126(00)00133-7. [DOI] [PubMed] [Google Scholar]
- Tompa D. R.; Gromiha M. M.; Saraboji K. Contribution of Main Chain and Side Chain Atoms and Their Locations to the Stability of Thermophilic Proteins. J. Mol. Graphics Modell. 2016, 64, 85–93. 10.1016/j.jmgm.2016.01.001. [DOI] [PubMed] [Google Scholar]
- Li Q.; Barrett D. G.; Messersmith P. B.; Holten-Andersen N. Controlling Hydrogel Mechanics Via Bio-Inspired Polymer–Nanoparticle Bond Dynamics. ACS Nano 2016, 10, 1317–1324. 10.1021/acsnano.5b06692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C.; Mcadams D. A.; Grunlan J. C. Nano/Micro-Manufacturing of Bioinspired Materials: A Review of Methods to Mimic Natural Structures. Adv. Mater. 2016, 28, 6292–6321. 10.1002/adma.201505555. [DOI] [PubMed] [Google Scholar]
- Schwartz E.; Koepf M.; Kitto H. J.; Nolte R. J.; Rowan A. E. Helical Poly(Isocyanides): Past, Present and Future. Polym. Chem. 2011, 2, 33–47. 10.1039/C0PY00246A. [DOI] [Google Scholar]
- Yashima E.; Ousaka N.; Taura D.; Shimomura K.; Ikai T.; Maeda K. Supramolecular Helical Systems: Helical Assemblies of Small Molecules, Foldamers, and Polymers with Chiral Amplification and Their Functions. Chem. Rev. 2016, 116, 13752–13990. 10.1021/acs.chemrev.6b00354. [DOI] [PubMed] [Google Scholar]
- Maeda K.; Wakasone S.; Shimomura K.; Ikai T.; Kanoh S. Chiral Amplification in Polymer Brushes Consisting of Dynamic Helical Polymer Chains through the Long-Range Communication of Stereochemical Information. Macromolecules 2014, 47, 6540–6546. 10.1021/ma501612e. [DOI] [Google Scholar]
- Ihara E.Polyisocyanides, Polyisocyanates. In Encyclopedia of Polymeric Nanomaterials; Kobayashi S., Müllen K., Eds.; Springer: Berlin, 2015; pp 1851–1859. [Google Scholar]
- Lifson S.; Andreola C.; Peterson N.; Green M. Macromolecular Stereochemistry: Helical Sense Preference in Optically Active Polyisocyanates. Amplification of Conformational Equilibrium Deuterium Isotope Effect. J. Am. Chem. Soc. 1989, 111, 8850–8858. 10.1021/ja00206a013. [DOI] [Google Scholar]
- Millich F.; Baker G. K. Polyisonitriles. Iii. Synthesis and Racemization of Optically Active Poly (Α-Phenylethylisonitrile). Macromolecules 1969, 2, 122–128. 10.1021/ma60008a003. [DOI] [Google Scholar]
- Cornelissen J. J.; Donners J. J.; de Gelder R.; Graswinckel W. S.; Metselaar G. A.; Rowan A. E.; Sommerdijk N. A.; Nolte R. J. Β-Helical Polymers from Isocyanopeptides. Science 2001, 293, 676–680. 10.1126/science.1062224. [DOI] [PubMed] [Google Scholar]
- van Buul A. M.; Schwartz E.; Brocorens P.; Koepf M.; Beljonne D.; Maan J. C.; Christianen P. C.; Kouwer P. H.; Nolte R. J.; Engelkamp H.; Blank K.; Rowan A. E. Stiffness Versus Architecture of Single Helical Polyisocyanopeptides. Chem. Sci. 2013, 4, 2357–2363. 10.1039/c3sc50552a. [DOI] [Google Scholar]
- Kouwer P. H.; Koepf M.; Le Sage V. A.; Jaspers M.; van Buul A. M.; Eksteen-Akeroyd Z. H.; Woltinge T.; Schwartz E.; Kitto H. J.; Hoogenboom R.; et al. Responsive Biomimetic Networks from Polyisocyanopeptide Hydrogels. Nature 2013, 493, 651–655. 10.1038/nature11839. [DOI] [PubMed] [Google Scholar]
- Jaspers M.; Dennison M.; Mabesoone M. F.; MacKintosh F. C.; Rowan A. E.; Kouwer P. H. Ultra-Responsive Soft Matter from Strain-Stiffening Hydrogels. Nat. Commun. 2014, 5, 5808. 10.1038/ncomms6808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaspers M.; Vaessen S. L.; van Schayik P.; Voerman D.; Rowan A. E.; Kouwer P. H. J. Nonlinear mechanics of hybrid polymer networks that mimic the complex mechanical environment of cells. Nat. Commun. 2017, 8, 15478. 10.1038/ncomms15478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaspers M.; Pape A.; Voets I. K.; Rowan A. E.; Portale G.; Kouwer P. H. Bundle Formation in Biomimetic Hydrogels. Biomacromolecules 2016, 17, 2642–2649. 10.1021/acs.biomac.6b00703. [DOI] [PubMed] [Google Scholar]
- Das R. K.; Gocheva V.; Hammink R.; Zouani O. F.; Rowan A. E. Stress-Stiffening-Mediated Stem-Cell Commitment Switch in Soft Responsive Hydrogels. Nat. Mater. 2015, 15, 318–325. 10.1038/nmat4483. [DOI] [PubMed] [Google Scholar]
- Koepf M.; Kitto H. J.; Schwartz E.; Kouwer P. H.; Nolte R. J.; Rowan A. E. Preparation and Characterization of Non-Linear Poly (Ethylene Glycol) Analogs from Oligo (Ethylene Glycol) Functionalized Polyisocyanopeptides. Eur. Polym. J. 2013, 49, 1510–1522. 10.1016/j.eurpolymj.2013.01.009. [DOI] [Google Scholar]
- Metselaar G. A.; Adams P.; Nolte R. J.; Cornelissen J. J.; Rowan A. E. Polyisocyanides Derived from Tripeptides of Alanine. Chem. - Eur. J. 2007, 13, 950–960. 10.1002/chem.200600928. [DOI] [PubMed] [Google Scholar]
- Chuah H.; Lin-Vien D.; Soni U. Poly (Trimethylene Terephthalate) Molecular Weight and Mark–Houwink Equation. Polymer 2001, 42, 7137–7139. 10.1016/S0032-3861(01)00043-X. [DOI] [Google Scholar]
- Mandal S.; Eksteen-Akeroyd Z. H.; Jacobs M. J.; Hammink R.; Koepf M.; Lambeck A. J.; van Hest J. C.; Wilson C. J.; Blank K.; Figdor C. G.; Rowan A. E. Therapeutic Nanoworms: Towards Novel Synthetic Dendritic Cells for Immunotherapy. Chem. Sci. 2013, 4, 4168–4174. 10.1039/c3sc51399h. [DOI] [Google Scholar]
- MacKintosh F.; Käs J.; Janmey P. Elasticity of Semiflexible Biopolymer Networks. Phys. Rev. Lett. 1995, 75, 4425. 10.1103/PhysRevLett.75.4425. [DOI] [PubMed] [Google Scholar]
- Gardel M.; Shin J.; MacKintosh F.; Mahadevan L.; Matsudaira P.; Weitz D. Elastic Behavior of Cross-Linked and Bundled Actin Networks. Science 2004, 304, 1301–1305. 10.1126/science.1095087. [DOI] [PubMed] [Google Scholar]
- Broedersz C.; MacKintosh F. Molecular Motors Stiffen Non-Affine Semiflexible Polymer Networks. Soft Matter 2011, 7, 3186–3191. 10.1039/c0sm01004a. [DOI] [Google Scholar]
- Lin Y.-C.; Yao N. Y.; Broedersz C. P.; Herrmann H.; MacKintosh F. C.; Weitz D. A. Origins of Elasticity in Intermediate Filament Networks. Phys. Rev. Lett. 2010, 104, 058101. 10.1103/PhysRevLett.104.058101. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Furyk S.; Bergbreiter D. E.; Cremer P. S. Specific Ion Effects on the Water Solubility of Macromolecules: Pnipam and the Hofmeister Series. J. Am. Chem. Soc. 2005, 127, 14505–14510. 10.1021/ja0546424. [DOI] [PubMed] [Google Scholar]
- Deyerle B. A.; Zhang Y. Effects of Hofmeister Anions on the Aggregation Behavior of Peo–Ppo–Peo Triblock Copolymers. Langmuir 2011, 27, 9203–9210. 10.1021/la201463g. [DOI] [PubMed] [Google Scholar]
- Jaspers M.; Rowan A. E.; Kouwer P. H. J. Tuning Hydrogel Mechanics Using the Hofmeister Effect. Adv. Funct. Mater. 2015, 25, 6503–6510. 10.1002/adfm.201502241. [DOI] [Google Scholar]
- Claessens M.; Semmrich C.; Ramos L.; Bausch A. Helical Twist Controls the Thickness of F-Actin Bundles. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 8819–8822. 10.1073/pnas.0711149105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker B. M.; Chen C. S. Deconstructing the Third Dimension–How 3d Culture Microenvironments Alter Cellular Cues. J. Cell Sci. 2012, 125, 3015–3024. 10.1242/jcs.079509. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.