

Abstract
Background
Array CGH is the criterion standard for identifying copy number variations (CNV), but the restrictive requirement of DNA quality and relatively high cost prevent the use of this method as a general assay in hospitals in developing countries. Our principal objective was to determine whether the semiconductor sequencing platform (SSP) could be an alternative method in CNV detection for spontaneous miscarriage.
Material/Methods
A total of 443 spontaneous miscarriage samples were collected and subjected to low-coverage (0.1X) whole-genome analysis by SSP. These samples were verified by array CGH and 8 low-quality DNA samples were analyzed by SSP and validated by MLPA.
Results
SSP detected 195 chromosomal numerical abnormalities, 74 CNVs, and 9 mosaicisms among the 435 samples. Among 74 CNV abnormalities, SSP detected an equal number (56) of CNVs 56 >1 Mb with array CGH. However, SSP missed more 6 cases CNVs <1 Mb than array CGH (12 vs. 18). SSP detected more mosaicisms than array CGH (9 vs. 7, p=0.5). Interestingly, SSP detected the mosaicism which had only 8% X monosomy, which was much lower than the minimal percentage of monosomy that was detected by array CGH.
Conclusions
SSP is of equivalent efficacy as array CGH in detecting CNVs >1 Mb, and performs better in identifying mosaicism. With the merits of low cost and less demand of input DNA, SSP is a good alternative for use in genetic diagnosis.
MeSH Keywords: Abortion, Spontaneous; Comparative Genomic Hybridization; DNA Copy Number Variations; Mosaicism
Background
Spontaneous miscarriage before 20 weeks of gestation is common (15% of clinically identified pregnancies). Tremendous efforts have been devoted in understanding the mechanisms underlying this phenomenon in the past 50 years [1,2]. Although the etiology of spontaneous miscarriage is still obscure, studies show that chromosomal abnormalities are the principal cause of early pregnancy loss. Among these abnormalities, DNA copy number variation explains 1–13%, while mosaicism accounts for 1–2% of miscarriages [3–5].
Traditionally, the criterion standard G-banding karyotyping has been used to study pregnancy loss; however, it is limited in clinical utility by the need for long-term culture, problems with maternal contamination, limited resolution, and low whole-genome coverage. The American College of Medical Genetics and Genomics (ACMG) Practice Guidelines suggest that array CGH should be the first-line postnatal test when seeking to diagnose intellectual disability or developmental delay [6,7]. However, array CGH requires a large amount of high-quality DNA, and remains expensive in clinical laboratories. The current price of an array CGH test is $550–$700 in China and the price of SSP is predicted to be about $300 once it is commercialized.
Next-generation sequencing (NGS) is a low-cost technique with short turnaround time, unprecedented resolution, and reliable high-throughput, requiring only small amounts of DNA. Both whole-genome and whole-exome sequencing strategies were used to investigate the etiology of miscarriage. Semiconductor sequencing platform (SSP), a type of NGS, has been used clinically for noninvasive prenatal testing (NIPT) and was expanded to identifying CNVs in chorionic villus (CVS) of spontaneous miscarriage in a few studies [8,9].
In the present study, we compared the efficacy of SSP with array CGH in identification of CNV in miscarriage samples, and sought to determine whether SSP could be used to detect mosaicism, and, if so, to determine the lowest level that could be detected.
Material and Methods
Sample collection and DNA extraction
The study was approved by the Clinical Research Ethics Committee of the Guangzhou DAAN Clinical Laboratory Center. All participants gave written informed consent before participation in the study. A total of 443 CVS samples of spontaneous miscarriages occurring within 20 weeks of gestation were collected by the Guangzhou DAAN Clinical Laboratory Center. The maternal age range was 18–46 years with a mean age of 30 years. The gestational weeks were 5–24 weeks, with a mean of 9 weeks 4 days. Samples were rinsed 3 times in PBS following collection, and DNA was extracted using a Qiagen DNeasy Blood and Tissue Kit (Qiagen; Valencia, CA, USA). DNA quality was evaluated with use of the NanoDrop kit (Thermo Scientific; Waltham, MA, USA). All samples were tested for maternal cell contamination using quantitative fluorescent polymerase chain reaction (QF-PCR) based on short tandem repeat (STR) markers for chromosomes 13, 18, 21 and X, Y [10,11].
Construction of an SSP data analysis pipeline
We obtained 5 million raw reads from each sample; 75% (3.5 MB) were unique. For the QC step, multi-mapping reads, low-quality reads (reads less than 50bp and the percentage of Q15 less than 80%), and duplicate reads were removed. The reads were aligned to the human genome assembly of the human genome hg19 (downloaded from UCSC) by TMAP software. For data correction, reads mapped to the area of microsatellites, tandem repeats, and simple repeats were masked.
Each chromosome was divided into 50-kb non-overlapping bins, and the numbers of reads mapping to each bin were calculated. The bin was removed if the amount of the N base on the reference genome was more than 10% of all bases. Otherwise, bins were normalized by median factor. We normalized the GC percentages in each bin by LOWESS regression. Circular binary segmentation (CBS), a reliable algorithm that is widely used in the analysis of comparative genomic hybridization arrays, was optimized to fit SSP. This allowed us to precisely define change points by partitioning chromosomes into regions of equal copy numbers.
Simulation mixture
DNAs from aneuploid and normal samples that had been examined by array CGH and SSP were artificially mixed at the ratios of 12.5%, 25%, 37.5%, 50%, 62.5%, 75%, and 87.5%. We chose trisomy 21 and trisomy 18 as chromosomal gain standards, and X monosomy as the standard for chromosomal loss. After library preparation, the libraries were pooled and sequenced 3 times as described above. The percentage of mosaicism was calculated by scoring the mimic mosaic mixtures. If mosaicism was present, the percentages of mosaicism in miscarriage samples were deduced using normalized read frequencies.
Copy number analysis by SSP
DNA libraries were prepared and sequenced in a blinded manner. End repair, adapter ligation, amplification, and purification all followed the protocol of the Ion Xpress™ Plus gDNA Fragment Library Preparation User Guide (Life Technologies, Carlsbad, CA, USA); we used the Ion Plus Fragment Library V3 and Ion Plus Fragment Library Adapters Kits (Life Technologies). Quality control of size distributions was performed on a 2100 Bioanalyzer (Agilent Technologies) and quantity of DNA libraries was tested by Qubit®2.0 (Invitrogen). A multiplex of 16 libraries (100 pM DNA per sample) was amplified by emulsion PCR on Ion PI™ Ion Sphere™ particles (ISPs) using an Ion OneTouch™ 2 instrument (Life Technologies). Template-positive ISPs were enriched and loaded onto an Ion PI™ Chip v2 (Ion PI™ Sequencing 200 Kit v3; Life Technologies). Reads of up to 200 base pairs were obtained using an SSP. About 3.5×106 reads at a sequencing depth of ~0.1 × were obtained from each sample.
Array CGH validation
DNA (typically 0.5 μg per sample) was labeled and hybridized to a SurePrint G3 Human CGH Microarray, 8×60 K (Agilent Technologies, Palo Alto, CA, USA) consisting of 60 000 oligonucleotides and evaluating the whole genome with an effective backbone resolution of 200 Kb. After washing, slides were scanned using an Agilent SureScan Microarray scanner. Scanned images were analyzed with Agilent Genomic Workbench software.
QF-PCR validation
Probes were designed according to STR markers in chromosomes 13, 18, 21, X, and Y. Validation data were collected by 3500 Dx (ABI, Life Technologies) and analyzed by GeneMapper software (Thermo Fisher Scientific) to decide whether samples were contaminated by maternal cells.
Multiplex ligation-dependent probe amplification validation
Eight samples that failed DNA quality control in the array CGH because of low input or quality analyzed by SSP were validated by multiplex ligation-dependent probe amplification (MRC-Holland, Amsterdam, the Netherlands). Validation data were collected by a use of 3500 (ABI, Life Technologies) and analyzed by Coffalyser software (MRC-Holland, Amsterdam, the Netherlands) after a process of hybridization, ligation, amplification, and electrophoresis according to the protocol.
Statistical analysis
McNemar-Bowker test and Kappa test were used to compare categorical data. SPSS software version 19 was used for analysis.
Results
Simulation of DNA mixtures
A strong positive correlation was evident between the normalized read frequency and the fraction of mosaicism in terms of chromosome gain (r=0.991, p=1.1681e-08), and a strong negative correlation was apparent between the normalized read frequency and the fraction of mosaicism in terms of chromosome loss (r=0.990, p=2.673e-08) (Figures 1, 2).
Figure 1.
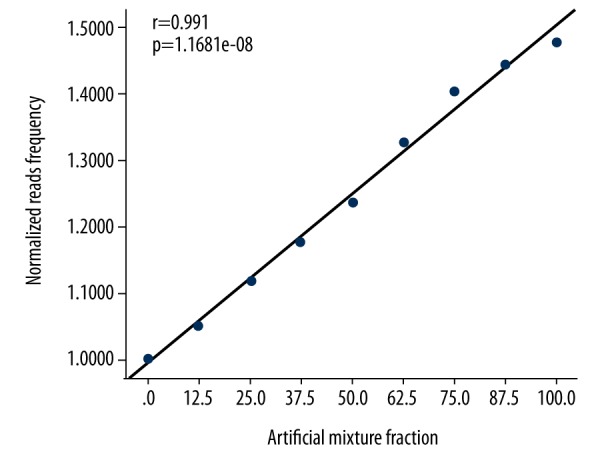
Relationships between the normalized read frequencies and the artificial mixture proportions in terms of chromosome gain.
Figure 2.
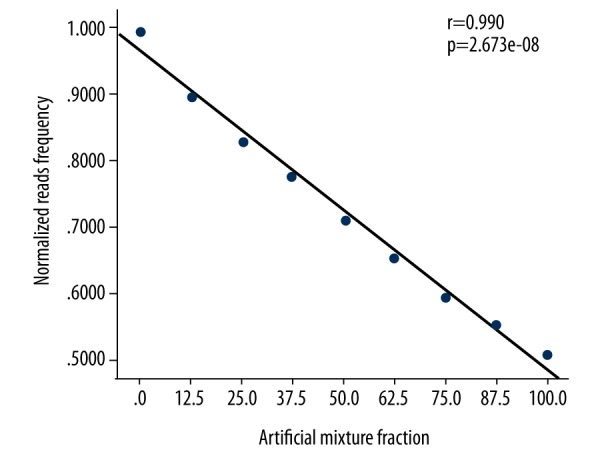
Relationships between the normalized read frequencies and the artificial mixture proportions in terms of X chromosome loss.
Diagnostic yield of miscarriage by SSP
All of the 443 samples were subjected to both Agilent array CGH and SSP. Among them, 8 samples that failed to pass DNA quality control for array CGH protocol were sequenced by SSP. In the remaining 435 samples, 278 (62%) were shown to harbor abnormal chromosomes, including 195 aneuploidies (44%), among which 161 were trisomies and 34 were monosomies. Seventy-four (17%) samples were euploidies with deletions or duplications (from 204 Kb to 147 Mb), among which 56 CNVs were >1 Mb and 12 CNVs <1 Mb. Nine (2%) samples were mosaicism. We detected 157 euploidies (36%) with normal chromosomes, of which 86 were females and 67 were males.
Aneuploidy is the most common abnormality among miscarriage samples; trisomy is much more prevalent than is monosomy. Of the 195 aneuploid samples, trisomy of chromosome 16 was the most common abnormality, followed by trisomies of chromosomes 22 and 13. Chromosomes 1, 5, 6, and 19 were rarely aneuploid (we encountered only 1 example of each); we found no aneuploidy of chromosome 17. Four trisomy samples – D1507090 (trisomy 12 and 15), D1601106 (trisomy 9 and 22), D1601082 (trisomy 3 and 5), and D1604005 (trisomy 8 and 10) – involved 2 chromosomes, and the D1601035 sample (trisomy 13, 14, 21) involved 3 chromosomes. Monosomy was principally confined to the X sex chromosome; we detected only 1 monosomy of chromosome 21 (Figure 3).
Figure 3.
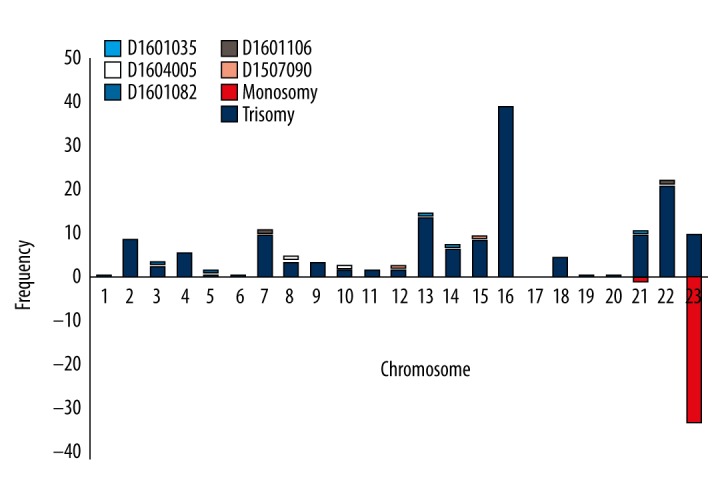
Frequencies of various chromosomal aneuploidies. D1604005 (T8, T10); D1601082 (T3, T5); D1601106 (T7, T22); and D1507090 (T12, T15) were instances of two-chromosome trisomies. D1601035 was a multiple trisomy (3 chromosomes: T13, T14, and T21).
We compared the diagnostic effectiveness and feasibility of SSP and array CGH in identification of chromosomal abnormalities. SSP and array CGH are consistent in identification of both aneuploidy and euploidy, indicating that SSP is reliable for detection of chromosomal numerical abnormalities. Array CGH detected a total of 74 CNVs, including 56 >1 Mb and 18 <1 Mb. SSP identified equal numbers (56) of CNVs >1 Mb and 12 CNVs <1 Mb. The performance of SSP was inferior to array CGH in detecting CNVs at 0.2–1 Mb (p<0.5, Kappa=0.950) (Table 1, Figure 4). For comparing those 2 methods, the area under the receiver operating characteristic curve (AUC) was 0.958, indicating that SSP was performed as well as array CGH when used to identify CNVs (Figure 5). We sought to annotate these 18 CNVs within 0.2–1Mb in the databases. All of them were interpreted as variants of uncertain significance (VOUS) by the databases (Decipher, DGV, ISCA).
Table 1.
Summary of chromosomal abnormalities analysis of CVSs.
| Method | Chromosomal abnormality | Euploidy | Low input/quality DNA | ||
|---|---|---|---|---|---|
| Aneuploidy | CNVs | Mosaicism | |||
| Array CGH | 195 | 74 | 7 | 157 | 0 |
| SSP | 195 | 68 | 9 | 157 | 8 |
Figure 4.

Chromosomal copy number variations analysis by SSP and array CGH. The blue arrow indicates a Del (2q37.1–2q37.3) (10 Mb); a was the result of SSP, and b was the result of array CGH.
Figure 5.
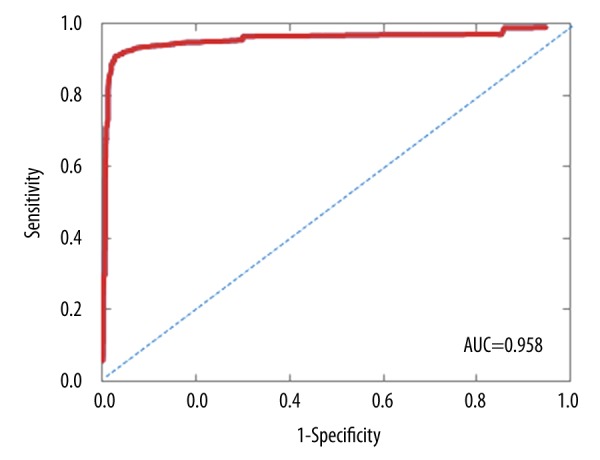
In receiver operating characteristic (ROC) analysis, the area under the curve (AUC) was 0.958.
Comparison of SSP and array CGH in diagnosis of mosaicisms
Nine mosaics in clinical samples, including a 50-Mb microdeletion mosaic in chromosome 7, were detected by SSP (Table 2). Array CGH tested 7 and missed 2 mosaics, which were validated by G-banding karyotyping according to the International System for Human Cytogenetic Nomenclature 2013 (ISCN 2013). However, no statistically significant difference was found between the 2 methods (p=0.5, Kappa=0.873). Then, to facilitate the comparison of the between SSP and karyotyping and between array CGH and karyotyping, we converted the results to normalized read frequency. SSP has a smaller difference than array CGH compared to the criterion standard karyotyping in identification of mosaicism. For SSP, the lowest percentage of mosaicism detected was an 8% X monosomy; the difference was 3% between the results that of karyotyping. When used to identify autosomal and X chromosomal defects, the results produced by SSP exhibited a tiny difference of 0–9% compared to those of karyotyping. For mosaicism in the Y sex chromosome, the difference in results was 14–21% compared to that of karyotyping.
Table 2.
Blind-test results of mosaic and non-mosaic samples.
| Sample # | Abnormality | Aneuploidy % | Converted normalized reads frequency | SSP normalized reads frequency | Variation* | Array CGH normalized reads frequency | Variation** |
|---|---|---|---|---|---|---|---|
| 1 | Mosaic 46 XX/45,X | 60 | 0.7 | 0.776 | 9.84% | 0.562 | 24% |
| 2 | Mosaic 45,X/46,XX | 8 | 0.96 | 0.993 | 3.30% | Undetected | / |
| 3 | Mosaic 45,X | 37 | 0.315 | 0.275 | 14.60% | 0.686 | 54% |
| 4 | Mosaic trisomy 21 | 86 | 1.43 | 1.316 | 8.63% | 1.362 | 5% |
| 5 | Mosaic 7 segment loss (50 Mb) | 86 | 0.57 | 0.657 | 13.18% | 0.699 | 18% |
| 6 | Mosaic 47,XYY/46,XY | 26 | 0.63 | 0.791 | 20.40% | 1.521 | 58% |
| 7 | Mosaic 45,X/46XY | 13 | 0.435 | 0.359 | 21.23% | Undetected | / |
| 8 | Mosaic 45,X/46,XY | 56 | 0.22 | 0.275 | 20.12% | 0.709 | 68% |
| 9 | Mosaic trisomy 8 | 60 | 1.3 | 1.314 | 1.07% | 1.383 | 6% |
The variation between the results of SSP and G-banding karyotyping for mosaicism;
The variation between the results of Array CGH and G-banding karyotyping for mosaicism.
Low-amount DNA samples and validation
SSP accomplished the test with lower DNA input and quality requirements. Eight samples that failed to pass DNA quality control of array CGH were analyzed by SSP, yielding results of 5 trisomies and 3 normal (Table 3). Afterwards, the results were validated by multiplex ligation-dependent probe amplification.
Table 3.
Validation of the results of SSP by MLPA.
| NO | Abnormality by SSP | MLPA |
|---|---|---|
| 1412001 | Trisomy 21 | Yes |
| 1503012 | Normal | Yes |
| 1605031 | Normal | Yes |
| 1510029 | Normal | Yes |
| 1510027 | Trisomy 22 | Yes |
| 1504016 | Trisomy 14 t22 | Yes |
| 1506005 | Trisomy 13 | Yes |
| 1508019 | Trisomy 16 | Yes |
Discussion
In this study, we optimized a method to detect CNVs based on SSP, which relies on GC-bias correction, binary segmentation, and normalized bins for signal filtering to reduce sequence variability and improve accuracy. Information about CNVs and mosaicism is of use in understanding spontaneous miscarriage or in genetic consulting. SSP must be improved in this regard. In the simulated mixture experiment, we determined individual log2 ratios and average log2 segmental variation ratios in samples containing various proportions of a mosaic mixture of trisomy 18 and trisomy 21, and also fractions with various proportions of an X monosomy.
We analyzed 443 miscarriage samples and compared SSP and array CGH to evaluate the performance of our low-coverage whole-genome sequencing approach in identification CNVs and mosaicism in spontaneous miscarriage samples. For identification of chromosomal numerical abnormalities, the low-coverage whole-genome sequencing approach using SSP is equivalent to array CGH.
Among the 443 samples analyzed, aneuploidy was the main cause of miscarriages (195, 44%). As in other studies, the most frequent chromosomal error was single trisomy 16, followed by trisomy 22 [5,12]. Errors also occurred in other chromosomes, but were less frequent; however, we found no error in chromosome 17. Trisomy 17 is known to be rare. Almost all monosomy involved only the sex chromosomes (we encountered 1 case of monosomy 21). Segmental deletions and duplications, combined with normal chromosomes, were found in 74 (17%) samples and 9 (2%) samples were mosaicism.
At 0.1 × depth, the performance SSP is of equivalent sensitivity and resolution in identification of CNVs >1 Mb, while SSP was inferior to array CGH when CNVs were at 0.2–1 Mb. The resolution of SSP depends on the sequencing depth [13]. When a CNV is small, the statistical differences of the reads contained in the bin are too small to be significant from normal chromosomes. A deeper sequencing is needed to detect small CNVs, which entails cost increases. However, in addition to the increased cost with the sequencing depth, the clinical interpretation of these small CNVs will be a concern for VOUS CNVs and might added complexities to counseling, case management, and parental anxiety [14]. In the present study, 18 CNVs identified by array CGH were 0.2–1 Mb, and all of them were interpreted by databases (Decipher, DGV, ISCA) as being uncertain or benign [15]. Taking this into account, this current low-coverage sequencing is the balance point between cost and the information necessary for most genetic diagnosis. However, CNVs smaller than 1 Mb can be pernicious, and will go undetected; thus, the cause of miscarriage will remain unexplained. Under this circumstance, we need to increase the depth of sequencing or choose a finer resolution array CGH chip to make sure a CNV is missed. With the rapid reduction of NGS costs, we will get more information in the near future.
Mosaicism is found in 1–2% of prenatal specimens and in an even higher proportion of IVF-created embryos [16,17]. Our study showed SSP is slightly more sensitive than array CGH (p=0.5). The lowest percentage detected by SSP was 8%, while 26% were detected by array CGH. SSP was more reliable than array CGH compared to karyotyping in detection of mosaicism [18,19]. SSP reliably identified Y chromosomal mosaicism with a difference of karyotyping of 14–21%. However, the interpretation of low-level mosaicisms is still controversial and challenging. In addition, SSP is less demanding in terms of DNA purity and input amount. The results indicate that SSP could be a remedial measure for degraded or very rare samples.
However, neither CMA nor SSP reliably detects polyploidy, affording only hints of sex chromosomal abnormalities such as 69XXY or 69XYY [02,21]. Also, neither technique can identify translocations (such as Robertsonian translocations) at the short arms of chromosomes 13, 14, 15, 21, and 22. Under these circumstances, follow-up testing can determine whether a CNV has arisen de novo or is parental in origin, which facilitates genetic counseling. In addition, over 30% of miscarriages with normal chromosomes were found in our study, and the etiology remains unclear. Early studies showed that if the cytogenetic material was normal, there was increased risk of subsequent pregnancy failure [22]. Maternal endocrine system, immunologic conditions, and maternal illness maybe contribute to the problem [23]. It would be valuable to analyze these cases in a subsequent study. In the present study, CNVs were less common than aneuploidy, and only a limited number of CNVs were analyzed, which might be the underlying reason why no false-positive results were found in the study. Thus, studies with larger sample sizes are needed for clinical validation of this method.
Conclusions
This study adds to the emerging data on validation and utility of SSP, showing the high level of consistency and robust performance of the NGS platform. SSP is therefore likely to move into this diagnostic arena, especially at centers where SSP is already used and where the cost of array CGH is high.
Footnotes
Conflict of Interest
None.
Source of support: This study was supported by the Science and Technology Program of Guangzhou (Grant No. 201604020104), the Science and Technology Program of Guangdong (Grant No. 2015A030401040), and the Key Program for Health Care Collaborative Innovation of Guangzhou (Grant No. 201500000004-4)
References
- 1.Warburton D, Fraser FC. Spontaneous abortion risks in man: Data from reproductive histories collected in a medical genetics unit. Am J Hum Genet. 1964;16:1–25. [PMC free article] [PubMed] [Google Scholar]
- 2.Hardy K, Hardy PJ. 1(st) trimester miscarriage: Four decades of study. Transl Pediatr. 2015;4(2):189–200. doi: 10.3978/j.issn.2224-4336.2015.03.05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zahir FR, Marra MA. Use of affymetrix arrays in the diagnosis of gene copy-number variation. Curr Protoc Hum Genet. 2015;85:8–13. doi: 10.1002/0471142905.hg0813s85. [DOI] [PubMed] [Google Scholar]
- 4.Liu K, Case A, Cheung AP, et al. Advanced reproductive age and fertility. Int J Gynecol Obstet. 2012;117(1):95–102. doi: 10.1016/j.ijgo.2011.11.002. [DOI] [PubMed] [Google Scholar]
- 5.Schaeffer AJ, Chung J, Heretis K, et al. Comparative genomic hybridization – array analysis enhances the detection of aneuploidies and submicroscopic imbalances in spontaneous miscarriages. Am J Hum Genet. 2004;74(6):1168–74. doi: 10.1086/421250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cooley LD, Lebo M, Li MM, et al. American College of Medical Genetics and Genomics technical standards and guidelines: Microarray analysis for chromosome abnormalities in neoplastic disorders. Genet Med. 2013;15(6):484–94. doi: 10.1038/gim.2013.49. [DOI] [PubMed] [Google Scholar]
- 7.Miller DT, Adam MP, Aradhya S, et al. Consensus statement: Chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am J Hum Genet. 2010;86(5):749–64. doi: 10.1016/j.ajhg.2010.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wen J, Hanna CW, Martell S, et al. Functional consequences of copy number variants in miscarriage. Mol Cytogenet. 2015;8:6. doi: 10.1186/s13039-015-0109-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Qiao Y, Wen J, Tang F, et al. Whole exome sequencing in recurrent early pregnancy loss. Mol Hum Reprod. 2016;22(5):364–72. doi: 10.1093/molehr/gaw008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Diego-Alvarez D. Application of quantitative fluorescent PCR with short tandem repeat markers to the study of aneuploidies in spontaneous miscarriages. Hum Reprod. 2005;20(5):1235–43. doi: 10.1093/humrep/deh781. [DOI] [PubMed] [Google Scholar]
- 11.Nagan N, Faulkner NE, Curtis C, Schrijver I. Laboratory guidelines for detection, interpretation, and reporting of maternal cell contamination in prenatal analyses. J Mol Diagn. 2011;13(1):7–11. doi: 10.1016/j.jmoldx.2010.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wapner RJ, Martin CL, Levy B, et al. Chromosomal Microarray Versus Karyotyping For Prenatal Diagnosis. New Engl J Med. 2012;367(23):2175–84. doi: 10.1056/NEJMoa1203382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sims D, Sudbery I, Ilott NE, et al. Sequencing depth and coverage: Key considerations in genomic analyses. Nat Rev Genet. 2014;15(2):121–32. doi: 10.1038/nrg3642. [DOI] [PubMed] [Google Scholar]
- 14.Rajcan-Separovic E. Chromosome microarrays in human reproduction. Hum Reprod Update. 2012;18(5):555–67. doi: 10.1093/humupd/dms023. [DOI] [PubMed] [Google Scholar]
- 15.Riggs ER, Jackson L, Miller DT, Van Vooren S. Phenotypic information in genomic variant databases enhances clinical care and research: The international standards for cytogenomic arrays consortium experience. Hum Mutat. 2012;33(5):787–96. doi: 10.1002/humu.22052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grati FR, Grimi B, Frascoli G, et al. Confirmation of mosaicism and uniparental disomy in amniocytes, after detection of mosaic chromosome abnormalities in chorionic villi. Eur J Hum Genet. 2006;14(3):282–88. doi: 10.1038/sj.ejhg.5201564. [DOI] [PubMed] [Google Scholar]
- 17.Malvestiti F, Agrati C, Grimi B, et al. Interpreting mosaicism in chorionic villi: results of a monocentric series of 1001 mosaics in chorionic villi with follow-up amniocentesis. Prenat Diagn. 2015;35(11):1117–27. doi: 10.1002/pd.4656. [DOI] [PubMed] [Google Scholar]
- 18.Novik V, Moulton EB, Sisson ME, et al. The accuracy of chromosomal microarray testing for identification of embryonic mosaicism in human blastocysts. Mol Cytogenet. 2014;7(1):18. doi: 10.1186/1755-8166-7-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.van Echten-Arends J, Mastenbroek S, Sikkema-Raddatz B, et al. Chromosomal mosaicism in human preimplantation embryos: A systematic review. Hum Reprod Update. 2011;17(5):620–27. doi: 10.1093/humupd/dmr014. [DOI] [PubMed] [Google Scholar]
- 20.Larsen EC, Christiansen OB, Kolte AM, Macklon N. New insights into mechanisms behind miscarriage. BMC Med. 2013;11(1):154. doi: 10.1186/1741-7015-11-154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Krieg S, Westphal L. Immune function and recurrent pregnancy loss. Semin Reprod Med. 2015;33(04):305–12. doi: 10.1055/s-0035-1554917. [DOI] [PubMed] [Google Scholar]
- 22.Ogasawara M, Aoki K, Okada S, Suzumori K. Embryonic karyotype of abortuses in relation to the number of previous miscarriages. Fertil Steril. 2000;73(2):300–4. doi: 10.1016/s0015-0282(99)00495-1. [DOI] [PubMed] [Google Scholar]
- 23.Wang CY, Wang SG, Wang JL, et al. Effect of miRNA-27a and leptin polymorphisms on risk of recurrent spontaneous abortion. Med Sci Monit. 2016;22:3514–22. doi: 10.12659/MSM.897147. [DOI] [PMC free article] [PubMed] [Google Scholar]