

Abstract
Genomic variation includes single-nucleotide variants, small insertions or deletions (indels), and copy number variants (CNVs). CNVs affect gene expression by altering the genome structure and transposable elements within a region. CNVs are greater than 1 kb in size; hence, CNVs can produce more variation than can individual single-nucleotide variations that are detected by next-generation sequencing. Multiple system atrophy (MSA) is an α-synucleinopathy adult-onset disorder. Pathologically, it is characterized by insoluble aggregation of filamentous α-synuclein in brain oligodendrocytes. Generally, MSA is sporadic, although there are rare cases of familial MSA. In addition, the frequencies of the clinical phenotypes differ considerably among countries. Reports indicate that genetic factors play roles in the mechanisms involved in the pathology and onset of MSA. To evaluate the genetic background of this disorder, we attempted to determine whether there are differences in CNVs between patients with MSA and normal control subjects. We found that the number of CNVs on chromosomes 5, 22, and 4 was increased in MSA; 3 CNVs in non-coding regions were considered risk factors for MSA. Our results show that CNVs in non-coding regions influence the expression of genes through transcription-related mechanisms and potentially increase subsequent structural alterations of chromosomes. Therefore, these CNVs likely play roles in the molecular mechanisms underlying MSA.
Electronic supplementary material
The online version of this article (10.1186/s13041-017-0335-6) contains supplementary material, which is available to authorized users.
Keywords: Genomic DNA, Copy number variation, Multiple system atrophy, Array-comparative genome hybridization
Introduction
Multiple system atrophy (MSA) is an adult-onset neurodegenerative disorder that is sporadic and progressive in nature. The estimated incidence is approximately 0.6 per 100,000 individuals per year; this increases to 3 per 100,000 per year in individuals older than 50 years, with a mean age of onset of approximately 60 years [1, 2]. The main clinical features comprise autonomic failure, cerebellar ataxia, levodopa-resistant Parkinsonism, and pyramidal signs with various combinations. According to the predominant motor presentations at examination, the phenotype of MSA is classified as Parkinsonian type (MSA-P) or cerebellar type (MSA-C) [3]. Pathologically, the brains of individuals with MSA exhibit atrophy of the cerebellum, brainstem, and posterolateral putamen, as well as the loss of pigmented neurons in the substantia nigra [1]. The neuropathological hallmark of MSA is the appearance of glial cytoplasmic inclusions, which are mainly composed of insoluble filamentous α-synuclein in oligodendrocytes. Lewy bodies, which also consist of α-synuclein, are found in Parkinson’s disease and in dementia with Lewy bodies, but not in MSA. These three disorders, including MSA, are classified as “synucleinopathies” [4, 5]. α-Synuclein is expressed predominantly in neurons, but it is not normally expressed in mature human oligodendrocytes [6–8]. Recently, it was reported that α-synuclein derived from neurons is released into the extracellular space from exosomes, or through exocytosis, and is incorporated into oligodendroglia [9]. Furthermore, in vitro studies showed that mutant fibrils of α-synuclein promote the fibrilization of α-synuclein and undergo cell-to-cell propagation [10, 11]. Growing evidence suggests that α-synuclein acts in a prion-like manner. However, the origin of α-synuclein in oligodendrocytes and its role in the misfolding or aggregation process in MSA have not been clearly defined.
As described in the 2nd Consensus Criteria, MSA constitutes a sporadic, non-hereditary disorder, although rare cases with familial history of MSA have been reported [3, 12, 13]. The frequencies of phenotypes in MSA vary among different ethnic groups. MSA-P is more common than MSA-C in Europe (62% vs 38%) and North America (60% vs 13%, the remaining 27% presenting with a mixed type) [14, 15]. In contrast, MSA-C is more common than MSA-P in Japan (67% vs 33%) [16, 17]. Thus, genetic predisposition to MSA has been suggested and evaluated. Genetic variations in the α-synuclein, beta-glucosylceramidase, or coenzyme Q2 genes, and individual single-nucleotide polymorphisms (SNPs) have been reported as risk factors for MSA; however, their roles in the etiology of MSA are not fully understood [18–21].
Genomic rearrangements in humans include duplications, deletions, insertions, inversions, and translocations. Structural variations, known as copy-number variations (CNVs), define genomic rearrangements over a range of 1 kb to several Mb. As CNVs cover such a large region of the genome, they may include entire genes and their regulatory regions [22]. In addition, CNVs occur in the presence of specific genomic structures that are highly susceptible to rearrangements, such as region-specific repeat sequences or low copy repeats [23]. Variable structural changes or rearrangements by CNVs directly or indirectly alter gene expression [23, 24].
Genetic disorders lacking heritability may result from low-frequency variants or de novo mutations with low or intermediate penetrance [24]. Through structural changes in genomes, CNVs affect gene dosage, gene expression, and expressed protein structures. Furthermore, the mutation rates of CNVs are at least 100–10,000-fold greater than those of point mutations [25]. Various studies have shown that CNVs are associated with human disease and play an important role in neurodevelopmental, neurodegenerative, and mental health disorders [23, 26, 27]. High-resolution genome-wide association studies, which are powerful tools for detecting disease-related genes, have enabled the identification of numerous single-gene Mendelian disorders. Various SNPs related to MSA have been identified via genome-wide association studies, although these only partially explain the etiology of the disorder [21]. In a study of a monozygotic twins discordant for MSA, i.e., in which one subject was affected by MSA and the other was not, we observed that copy number loss of the SHC2 gene was related to MSA [28]. However, subsequent analysis did not confirm this result [29]. In the present study, we focused on CNVs rather than on SNPs to verify the relationship between MSA and CNVs, and attempted to evaluate the genetic heterogeneity in MSA using array-based comparative genomic hybridization (array-CGH).
Results
Identification of CNVs in 72 individuals
We assayed genomic DNA from 48 Japanese patients with MSA and 24 healthy Japanese controls for genome-wide CNV screening using Agilent 2x400K CNV arrays; the details of the subjects are described as the first set in Table 1. Microarray data were filtered with standard aberration filters and processed. A total of 15,499 autosomal CNVs were detected, 8059 (52%) of which comprised gain CNVs and 7440 (48%) of which were loss CNVs. We compared the numbers of total, gain, and loss CNVs in individuals with MSA-C and MSA-P and in the controls (Fig. 1a–c). The average numbers of CNVs in individuals were 209 in control subjects (n = 24), 215 in those with MSA-C (n = 24), and 222 in those with MSA-P (n = 24). The number of total and gain CNVs was significantly higher in individuals with MSA-P (p = 0.02, p = 0.01; Wilcoxon rank-sum test) compared to that in the control group. In contrast, the number of loss CNVs did not differ between groups. The variance of CNVs in MSA-C was nearly the same as that in the control group, although the number was slightly higher (Additional file 1: Table S1).
Table 1.
Age and gender of Japanese participants
| 1st set | 2nd set | Validation set | ||||||
|---|---|---|---|---|---|---|---|---|
| MSA-C | MSA-P | Control | MSA-C | Control | MSA | Control | ||
| Number of subjects | 24 | 24 | 24 | 40 | 40 | 245 | 212 | |
| Gender (M/F) | 12/12 | 12/12 | 12/12 | 20/20 | 20/20 | 110/135 | 120/92 | |
| Age | Mean ± SE$
p* (vs. control) |
60.3 ± 2.4 0.98 |
64.7 ± 1.3 0.12 |
60.3 ± 2.0 | 62.3 ± 1.4 0.84 |
62.7 ± 1.6 | 63.9 ± 0.6 < 0.01 |
52.8 ± 1.2 |
| Onset | 36–73 | 46–77 | 36–76 | 36 – 83 | ||||
*: student’s t-test p < 0.05: statistically significant $: age at sampling M: male F: female
Fig. 1.
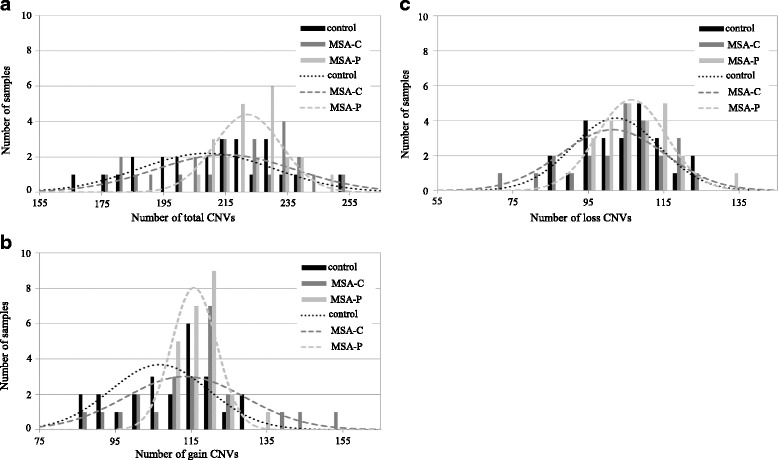
Histogram and distribution curve of each individual CNV in subjects with MSA-C or MSA-P and in controls. a Number of total CNVs (b) Number of gain CNVs (c) Number of loss CNVs
Moreover, we compared the distribution of 15,499 CNVs from chromosome 1 to chromosome 22 in MSA-C, MSA-P, and control subjects (Fig. 2a, b, and Additional file 1: Table S2a). Gain CNV numbers on chromosomes 2, 5, 11, 15, and 22 in MSA-P were significantly higher than those in the control group (chr.2: p = 0.04, chr.5: p < 0.01, chr.11: p = 0.02, chr.15: p = 0.02, chr.22: p < 0.01; Wilcoxon rank-sum test). Loss CNV numbers on chromosome 4 in MSA-P and MSA-C and chromosome 8 in MSA-P were significantly higher than those in the control group (chr.4: p < 0.01, chr.8: p = 0.03; Wilcoxon rank-sum test) (Fig. 2c). The number of gain CNVs was increased in MSA-P and that of loss CNVs on chromosome 4 was increased in all patients with MSA.
Fig. 2.
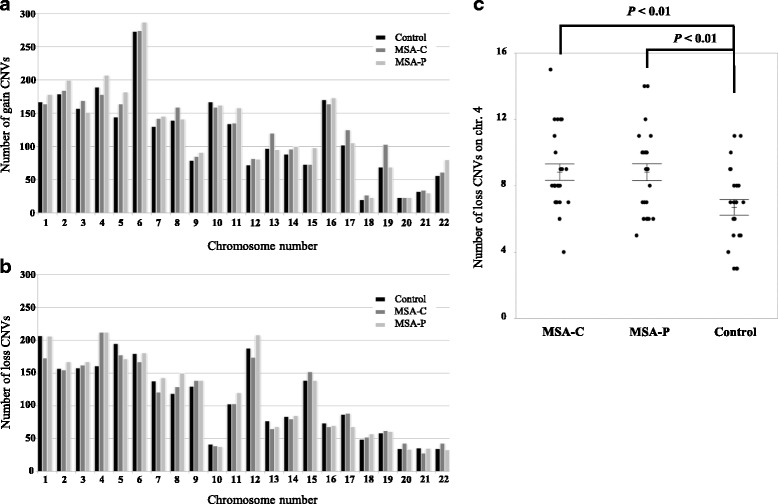
Number of CNVs in autosomal chromosomes in subjects with MSA-C or MSA-P and in controls. a Total number of gain CNVs in each chromosome (b) Total number of loss CNVs in each chromosome (c) Number of loss CNVs in chromosome 4 in subjects with MSA-C or MSA-P and in controls. Statistical significance was analyzed by the Wilcoxon rank-sum test. Error bars indicate ± standard error of the mean (SEM)
CNVs related to MSA
Next, we analyzed CNVs related to MSA by comparing subjects with MSA and control subjects. Subjects in both MSA groups and the control group were matched for age and sex. MSA-C was more common than MSA-P in the Japanese population; therefore, we focused on MSA-C or all patients with MSA. We reanalyzed the microarray data of our 72 samples using the correlation aberration filter (see Materials and Methods for details). Furthermore, to identify CNVs related to MSA, reanalyzed data were filtered as follows in the 1st analysis: the length of the CNV was at least 1 kb; the number of subjects with MSA in which CNV was detected was more than twice that in controls. Based on these criteria, we endeavored to identify conservative CNVs associated with MSA. The aberration ratio on the X chromosome was compared between both males and females. We used CNV data for the X chromosome, but were unable to use CNV data for the Y chromosome. We identified a total of 379 CNVs in this step; of these, 216 were less than 10 kb and 163 were at least 10 kb in length. In the 2nd analysis, to evaluate CNVs of at least 10 kb in length in detail, we prepared an additional 80 Japanese genomic samples (40 unrelated patients with MSA and 40 unrelated adult healthy controls) and designed an Agilent 8x60K custom array with probe spacing of 205 bp in these CNV regions. The additional 80 samples did not overlap with the previous 72 samples. The details are described in the second set of Table 1. The custom arrays were assayed, and 163 CNVs of at least 10 kb in length were re-evaluated and identified. Custom array data were analyzed with the same filters as those used to analyze MSA-related CNVs. These CNVs were selected by increasing the number of samples analyzed, and the length of the regions was changed by short probe spacing during custom analysis. Consequently, they were classified as 30 CNVs less than 10 kb and 65 of at least 10 kb in length.
Overall, we identified a total of 311 CNVs related to MSA, of which 246 (79.1%) were less than 10 kb and 65 (20.9%) were at least 10 kb in length. Only 13 CNVs were greater than 100 kb in length. Furthermore, of the 311 CNVs related to MSA, 142 (45.7%) were in non-coding intergenic regions and 169 (54.3%) were intragenic. Of the 169 CNVs located within gene regions, 72 (42.6%) were in exon-containing regions and 97 (57.4%) were in intronic regions (Fig. 3a). The number of gain and loss CNVs on chromosomes 1–22 was compared between the 311 CNVs related to MSA (Fig. 3b, Additional file 1: Table S3), and their chromosomal locations were determined (Fig. 3c). The number of loss CNVs was 5-fold larger than that of gain CNVs on chromosome 4, whereas only gain CNVs were detected on chromosomes 16 and X. CNVs related to MSA or CNVs of the controls were widely distributed on each chromosome, and were not particularly abundant in the telomere or centromere regions (Additional file 2: Figure S1).
Fig. 3.
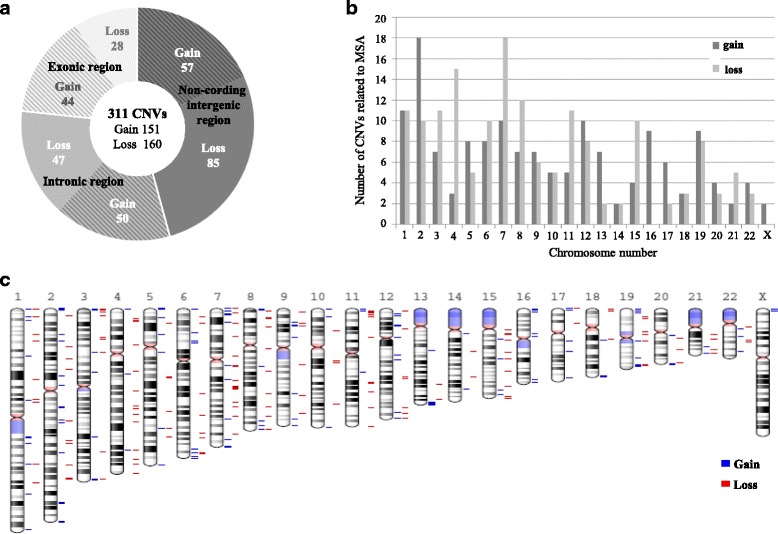
Description of 311 CNVs related to MSA. a Type and genomic region of 311 CNVs (b) Number of CNVs by type on each chromosome (c) Chromosomal location of 311 CNVs
Moreover, we examined the relationships between the 311 CNVs and the samples by cluster analysis, and 29 CNVs were identified in 5 MSA-C samples (Fig. 4, Additional file 1: Table S4a). The age of onset of MSA-C in these patients ranged from 45 to 63 years; the ratio of males to females was 2: 3. We found no genetic similarity in the patients with the 29 CNVs.
Fig. 4.

Cluster analysis of 311 CNVs related to MSA. Cluster heat map of relationships between 311 CNVs and 72 samples by cluster analysis; blue highlighting indicates a cluster of 29 CNVs
Gene set analysis of CNVs related to MSA
Of the 169 CNVs within genes, 72 were in exonic regions of 97 genes, and 97 CNVs were in intronic regions of 89 genes. To determine the relationships, pathways, and larger networks of the extracted CNVs, these 186 genes were evaluated using gene ontology (GO) analysis. A total of 11 GO sets were found by analysis of CNVs contained in exonic regions, with the functional categories significantly related to cell attachment and immune response. In contrast, 294 GO sets were observed via analysis of CNVs contained in intronic regions; these were involved in regulating gamma-aminobutyric acid secretion, D-aspartate transport, and synaptic transmission (Table 2). In addition, approximately 142 CNVs in non-coding intergenic regions were examined using the UCSC Genome Browser. Among these, 130 contained regions corresponding to retrotransposons, simple repeat sequences, CpG islands, large intergenic non-coding RNAs, and DNase I-hypersensitive sites. Only 12 CNVs were located in non-functional regions.
Table 2.
GO process analysis of copy number variants (CNVs) related to MSA
| GO set | Number of set genes | Number of CNV genes | p-value | q-value | |
|---|---|---|---|---|---|
| (a) Results of GO process analysis with CNVs included in exonic regions | |||||
| GO:0007156 | homophilic cell adhesion via plasma-membrane adhesion molecule | 198 | 12 | 8.30e-11 | 1.71e-7 |
| GO:0098742 | cell-cell adhesion via plasma-membrane adhesion molecule | 279 | 12 | 4.19e-9 | 4.31e-6 |
| GO:0007155 | cell adhesion | 1706 | 24 | 3.23e-7 | 1.93e-4 |
| GO:0022610 | biological adhesion | 1720 | 24 | 3.75e-7 | 1.93e-4 |
| GO:0070423 | nucleotide-binding oligomerization domain containing signaling pathway | 41 | 5 | 1.04e-6 | 3.44e-4 |
| GO:0035872 | nucleotide-binding domain, leucine-rich repeat containing receptor signaling pathway | 42 | 5 | 1.17e-6 | 3.44e-4 |
| (b) Results of GO process analysis with CNVs in intronic regions | |||||
| GO:0014054 | positive regulation of gamma-aminobutyric acid secretion | 14 | 5 | 2.48e-9 | 5.48e-6 |
| GO:0070777 | D-aspartate transport | 6 | 4 | 4.65e-9 | 5.48e-6 |
| GO:0070779 | D-aspartate import | 6 | 4 | 4.65e-9 | 5.48e-6 |
| GO:0014052 | regulation of gamma-aminobutyric acid secretion | 19 | 5 | 1.42e-8 | 7.39e-6 |
| GO:0007268 | chemical synaptic transmission | 655 | 16 | 1.72e-8 | 7.39e-6 |
| GO:0099537 | trans-synaptic signaling | 655 | 16 | 1.72e-8 | 7.39e-6 |
Validation of CNVs related to MSA
To determine the frequency and position of extracted CNVs, we assayed the 457 Japanese genomic DNA samples by PCR. The details of the 457 individuals, including 152 whose samples were used for array analysis, are described as the validation set in Table 1. From among the 311 MSA-related CNVs mentioned above, we selected those with lengths of less than 10 kb and prepared a set of primers covering each candidate CNV. Among the selected regions, 42 CNVs were assayed and 12 loss CNVs were successfully verified. The successfully verified CNV data showed the same results by array-CGH and PCR. For these CNVs, an odds ratio of homozygous deletion, or both homozygous and heterozygous deletion, was calculated (Additional file 1: Table S5a). We identified high odds ratios for three CNVs of at least 2-fold higher in MSA than in controls. The three CNVs were located at 3p22.2, 4q34.1, and 15q11.2, and contained the CTD small phosphatase-like (CTDSPL), polypeptide N-acetylgalactosaminyltransferase-like 6 (GALNTL6), and small nuclear ribonucleoprotein polypeptide N (SNRPN) genes, respectively (Table 3, Additional file 2: Figure S2). These CNVs were present in the intronic regions of the genes and caused loss of the intronic region (Fig. 5). The positions of the lost regions completely matched between samples. However, no patients carried more than one of the three CNVs.
Table 3.
Position and frequency of the three CNVs
| Chr | Gene region | Position by sequencing | Control | MSA | Odds ratio for homo del (95% CI) (p value) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Start | Deletion (bp) (Insert, nt) |
Stop | No del | Hetero del | Homo del | No del | Hetero del | Homo del | |||
| 3 | CTDSPL | 37,978,418 | 8,510 (agg) |
37,986,927 | 193 | 19 | 0 | 223 | 18 | 4 | (0.025) |
| 4 | GALNTL6 | 172,988,640 | 4,292 (c) |
172,992,931 | 133 | 75 | 4 | 156 | 79 | 10 | 2.21 (0.73–8.16) (0.166) |
| 15 | SNRPN | 25,107,033 | 2,389 (atcatatcct) |
25,109,421 | 194 | 18 | 0 | 219 | 23 | 3 | (0.053) |
No del: no deletion Hetero del: hetero deletion Homo del: homo deletion
Fig. 5.

Genomic location of three CNVs. The genomic locations of three validated CNV, as displayed by the UCSC Genome Browser (NCBI Build 37/hg19), are shown. The indicated tracks contain information regarding transcriptional regulation (H3K4Me1, H3K4Me3, H3K27Ac, Txn Factor ChIP) from ENCODE. a CTDSPL (b) GALNTL6 (c) SNRPN; the red dotted arrow indicates a genome loss region
Discussion
In this study, we detected and verified CNVs using array-CGH and PCR, and we investigated the relationship between CNVs and MSA. We found that the number of gain CNVs was significantly increased on chromosomes 5 and 22 in patients with MSA-P, whereas the number of loss CNVs was significantly increased on chromosome 4 in all patients with MSA. Further, we identified 311 CNVs related to MSA and a characteristic cluster containing 29 CNVs. Finally, we detected 3 CNVs that may represent risk factors for MSA.
The genomic loci showing a significant increase in CNV numbers are 5p15.33, 5q35.3, 22q12.3, and 22q13.31 in MSA-P and 4q13.1, 4q28.3, 4q32.1, 4q34.1, and 4q35.2 in MSA. This increase in CNVs indicates an increase in genomic recombination events, although the overall recombination rate of these chromosomes was not particularly higher than that of other chromosomes. In addition, the loci of selective chromosomes in MSA-P did not completely match the loci of CNVs reported for Parkinson’s disease [30–32]. Thus, the increase in CNVs at the selected chromosome sites may be characteristic of MSA. Additionally, the increased number of CNVs in MSA was slightly greater in males than that in females (Additional file 1: Table S2b); however, previous studies have not observed any gender-related difference in the incidence of MSA [14–17]. Although various factors increase genomic instability, we cannot clearly explain the cause of the higher sensitivity of genomic instability in certain chromosomes in MSA or why the number of CNVs was slightly greater in males than that in females. In order to clarify these points, more detailed experiments on specific chromosomes and a large-scale familial survey will be necessary.
Large variants of several hundred kb in size affecting numerous genes have been reported as typical rare CNVs in several neurological and neurocognitive disorders [23, 26, 27, 33–35]. In contrast, small and mid-sized variants such as those less than 50 kb in size are regarded as common CNVs associated with disease [36, 37]. Most CNVs related to MSA were small variants of less than 10 kb in size, and only 13 CNVs were 100–500 kb in length (Additional file 1: Table S6). We speculate that the small CNVs are important in MSA because of their higher frequency.
The results of GO analyses indicated an association with MSA that differed from the results obtained in studies of other diseases [26, 38–40]. The GO results of CNVs in intronic regions were more closely related to the molecular mechanism of MSA than those of CNVs in exonic regions. Moreover, the GO results for 29 CNVs identified by cluster analysis indicated a relationship with oligodendrocytes, which play an important role in MSA [41, 42] (Additional file 1: Table S4b). Thus, the identified CNVs are likely related to the onset and pathology of MSA. Additional validation using a larger number of samples is necessary to confirm this hypothesis.
We identified three CNVs with allele frequencies that were significantly different between MSA and control samples. These CNVs were located in the gene bodies of CTDSPL, GALNTL6, and SNRPN. CTDSPL encodes a protein related to neuronal gene silencing in non-neuronal cells [43], GALNTL6 encodes a protein related to cell maturation and differentiation in the brain and testis [44], and SNRPN encodes a protein that plays a role in pre-mRNA processing and tissue-specific alternative splicing events (associated with Prader-Willi syndrome) [45]. Although CNVs in exon-containing regions affect the expression of encoded proteins via a deletion or duplication mechanism, the three CNVs are located in intronic regions. However, these CNVs are contained in genomic regions containing elements such as transcription factor (TF)-binding sites, transposable elements (TEs), or a combination of these sites [46–48]. Deletion of a region by CNVs affects the structure of these elements. Recent reports showed that variations in TF-binding sites, TEs, and non-coding RNA lead to changes in gene expression [49–51] and cause disease [51–54]. Our study showed that approximately 77% of the identified CNVs were in non-coding regions; 95.0% of CNVs in non-coding regions contained active elements, such TF-binding sites, TEs, and non-coding RNA. Because the majority of the human genome comprises non-coding regions, these regions may be responsible for various functions that differ from those of protein-coding regions, which occupy only 1.5% of the human genome [55]. Structural changes in active elements of non-coding regions due to CNVs may affect disorder onset through modulation of gene expression or transcription. Thus, CNVs in non-coding regions are also likely involved in the pathogenesis and onset of MSA.
The detected CNVs in this study comprised similar common variants, with odds ratios between 0.86 and 1.47. We found that some patients had a combination of low-frequency CNVs, and that some combinations of CNVs increased the frequency and specificity of disease compared with the effect of each CNV alone. Prior to the clinical onset of MSA, a long preclinical process likely occurs in the molecular pathology of this disorder. The rate of progression of MSA is likely related to a combination of common CNVs each with small or moderate influence. Moreover, combinations of common CNVs with low frequency may cause non-heritable MSA.
Through CNV analysis, we showed that 1) the frequencies of CNVs were increased in selected chromosomes in MSA, 2) three CNVs potentially act as risk factors for MSA, and 3) CNVs in non-exonic, non-coding regions potentially affect the molecular process underlying MSA. As few studies have analyzed CNVs on a genome-wide scale with sufficient numbers of subjects, disease-associated polymorphisms in non-coding regions should be considered risk factors for MSA.
Methods
Participants and samples
Patients with MSA and control subjects were enrolled in the research study with approval by the Institutional Ethics Committee of Hokkaido University Faculty of Medicine and Graduate School of Medicine. All patients with MSA were neurologically evaluated by board-certified neurologists at the Department of Neurology, Hokkaido University Hospital, and at participating research institutes. The diagnosis of MSA was made based on the 2nd Consensus Criteria [3]. Written informed consent was obtained from all control subjects and all participants (or their families, when patients were unable to provide consent themselves). Among the patients, we selected 212 control subjects who were free from neurological disorders and 245 patients with MSA, and both groups donated blood samples for this study. Genomic DNA extracted from the peripheral blood samples was used in all experiments. We studied 48 patients with sporadic MSA (24 with MSA-C, 24 with MSA-P) and 24 healthy adult controls for genome-wide screening in the primary analysis. Additionally, in order to verify selected regions identified in the primary analysis, 40 patients with MSA-C and 40 healthy adult controls were chosen for secondary analysis using custom arrays. These were not matched to participants from the first analysis. For validation analysis, 245 patients with MSA (163 with MSA-C, 82 with MSA-P) and 212 healthy adult controls, including patients from the first and second analyses, were chosen. The age and gender of patients are listed in Table 1.
Array-CGH assays
Genomic DNA was extracted from the buffy coat of peripheral blood using guanidine, and the samples were purified with 13% polyethylene glycol 6000 (Sigma-Aldrich, St. Louis, MO, USA) in 0.8 M NaCl. The concentration and purity of purified genomic DNA was measured using a Nano Drop 2000 (Thermo Fisher Scientific, Waltham, MA, USA). The concentration of double-strand DNA was measured using the Qubit® dsDNA BR assay with a Qubit® 3.0 fluorometer (Thermo Fisher Scientific). From these measurement results, the ratio of double-strand DNA was calculated, and genomic DNA with a value of at least 0.85 was used. Agilent Sure Print G3 Human CNV (2 × 400 K) arrays and Agilent Sure Print G3 Custom CGH Microarray (8 × 60 K) (Agilent Technologies, Santa Clara, CA, USA) were used for genome-wide array analysis. The Custom CGH Microarray was designed for regions identified to be at least 10 kb in length with the Human CNV (2 × 400 k) arrays using eArray (Agilent). All arrays were designed based on NCBI Build 37/UCSC hg19. Microarray analyses were performed following the manufacturer’s instructions, using 500 ng genomic DNA with a SureTag DNA Labeling Kit (Agilent). NA19000 (Coriell Institute, Camden, NJ, USA) was used as a reference for all microarray experiments. The microarrays were scanned with a Sure Scan Microarray Scanner (Agilent), and raw image data obtained by the scanner were processed using Feature Extraction Software (ver. 11.0.1.1, Agilent). The microarray data, quantified and processed, included quality-control data (QC Report) to ensure that the data was of high quality. According to the manufacturer’s recommendations, the derivative log ratio spread of the robust metric value of QC should be less than 0.30; the derivative log ratio spread of all genomic DNA passed QC with 0.11–0.17. The gender confirmed from the microarray data were all matched with the reported gender of patients whose DNA samples were analyzed.
CNVs and GO analysis
The data processed by Feature Extraction Software were evaluated using the Aberration Detection Method 2 algorithm with a threshold of 6.0 with Agilent CytoGenomics software (ver. 2.7.8.0). To analyze individual arrays, the following standard aberration filters were used: minimum of three contiguous supra-threshold probes and minimum of 0.25 average log2 ratios. Moreover, to analyze CNVs related to MSA, the following correlation aberration filters were applied: minimum of two contiguous supra-threshold probes and minimum of 0.5 average log2 ratios. All arrays were designed based on NCBI Build 37/UCSC hg19. We used the UCSC Genome Browser for physical mapping and annotations of CNVs (http://genome.ucsc.edu/). If necessary, we also used data carried over to GRCh38 using the UCSC Genome Browser. For CNV analysis, we applied data for gene, interspersed repeats, or simple tandem repeats as categorized by the RepeatMasker, Simple Repeats, and large intergenic non-coding RNA tracks by the UCSC Genome Browser. In addition, we used integrated regulation data from the ENCODE track of the UCSC Genome Browser to obtain information relevant to transcriptional regulation. For GO process analysis, MetaCore™ (version 6.30, Thomson Reuters, New York, NY, USA) was used, and the significance levels were set at a p value of 0.01 and q (false discovery rate) of 0.01.
PCR and sequencing
PCR primers were designed based on the results of array analysis (Additional file 1: Table S5b), and amplifications were performed using Tks Gflex™ DNA Polymerase (TaKaRa Bio, Inc., Shiga, Japan) and KOD FX Neo (Toyobo, Osaka, Japan). Amplification conditions are described in Additional file 1: Table S5c. PCR products were separated on a 0.8%, 1.0%, or 3% agarose gel and analyzed, and the PCR products were sequenced to detect deleted regions using the BigDye® Terminator v3.1 cycle Sequencing kit (Thermo Fisher Scientific).
Statistical analysis
Statistical analysis was performed using JMP® Pro statistical software, version 12.0.1 (SAS Institute, Inc., Cary, NC, USA), and p values <0.05 were considered statistically significant. Cluster analysis was performed by complete-linkage hierarchical clustering with Hamming distance implemented in SciPy 0.18.1.
Additional files
Number of CNVs in 72 individuals. Table S2. Number of CNVs on autosomal chromosomes in controls and subjects with MSA-C or MSA-P. Table S3. Chromosome numbers at which the 311 CNVs related to MSA were located. Table S4. Details of the 29 CNVs as obtained by cluster analysis. Table S5. Details of the 12 verified CNVs. Table S6. Large CNVs identified in this study. (DOCX 97 kb)
Gel electrophoresis of PCR products for verification analysis. Figure S2. Chromosomal location of CNVs in control subjects. (PPTX 454 kb)
Acknowledgements
We thank all the individuals and families who kindly cooperated with, and participated in, this study. We would also like to thank the physicians who permitted us to assess patients with MSA. In particular, we express appreciation to the following physicians: Drs. Takahiro Kano and Hideki Houzen (Obihiro-Kousei General Hospital), Makoto Hirotani and Asako Takei (Hokuyukai Neurological Hospital), Kazuto Yoshida (Japanese Red Cross Asahikawa Hospital), Haruo Uesugi (Sapporo Yamanoue Hospital), Toshiyuki Fukazawa (CEReS Incorporated Medical Institution), Kazuya Sako (Nakamura Memorial Hospital), Takeshi Matsuoka (Date Red Cross Hospital), Akihiko Ogata (Hokkaido Neurosurgical Memorial Hospital), and Kinya Hisanaga (Department of Neurology and Clinical Research Center, National Hospital Organization, Miyagi National Hospital), as well as others who supported this study by obtaining informed consent from patients, collecting blood samples, and acquiring clinical information from patients with MSA. We thank Dr. Hidenori Sato (Institute for Promotion of Medical Science Research, Yamagata University Faulty of Medicine) for providing technical assistance with the analysis of CNV data.
Funding
This study was supported by a Grant-in-Aid from the Research Committee for Ataxic Disease (HS), a Grant-in-Aid from the Research Committee of CNS Degenerative Disease, the Ministry of Health, Labour and Welfare of Japan (HS), a grant from the Translational Research Network Program and a grant from the Practical Research Project for Rare / Intractable Diseases, Japan Agency for Medical Research and Development (HS), and in part by Mitsubishi Tanabe Pharma Corporation (HS).
Availability of data and materials
The data generated or analyzed in this study are included in this published article and its Additional files.
Abbreviations
- array-CGH
Array based comparative genomic hybridization
- CNVs
Copy number variants
- CTDSPL
CTD small phosphatase-like
- GALNTL6
Polypeptide N-acetylgalactosaminyltransferase-like 6
- GO
Gene ontology
- MSA
Multiple system atrophy
- MSA-C
Cerebellar type of MSA
- MSA-P
Parkinsonian type of MSA
- SNRPN
Small nuclear ribonucleoprotein polypeptide N
- TEs
Transposable elements
- TFs
Transcription factors
Authors’ contributions
YH designed and performed the experiments, analyzed data, and wrote the manuscript. MK performed experiments. IT performed the cluster analysis, prepared Fig. 4, and provided advice on statistical analysis. IY, MM, IT, TK, and HS performed clinical diagnosis, obtained informed consent from patients, and collected blood samples. JU provided advice on gene set analysis. HS designed and supervised this work and performed critical revisions. All authors read and approved the final manuscript.
Ethics approval and consent to participate
The Institutional Ethics Committee of Hokkaido University Faculty of Medicine and Graduate School of Medicine approved this study. Written informed consent was obtained from all participants (or their families, when patients were unable to provide consent themselves).
Consent for publication
Not applicable.
Competing interests
This study was partly supported by a grant from Mitsubishi Tanabe Pharma Corporation (HS). Masataka Katsu is an employee of Mitsubishi Tanabe Pharma Corporation. The other authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (10.1186/s13041-017-0335-6) contains supplementary material, which is available to authorized users.
Contributor Information
Yuka Hama, Email: hama@pop.med.hokudai.ac.jp.
Masataka Katsu, Email: katsu.masataka@me.mt-pharma.co.jp.
Ichigaku Takigawa, Email: takigawa@ist.hokudai.ac.jp.
Ichiro Yabe, Email: yabe@med.hokudai.ac.jp.
Masaaki Matsushima, Email: mmasaaki@huhp.hokudai.ac.jp.
Ikuko Takahashi, Email: ikukotak@med.hokudai.ac.jp.
Takayuki Katayama, Email: katataka@asahikawa-med.ac.jp.
Jun Utsumi, Email: jutsumi@pop.med.hokudai.ac.jp.
Hidenao Sasaki, Phone: +81-11-706-6028, Email: h-isasak@med.hokudai.ac.jp.
References
- 1.Wenning GK, Colosimo C, Geser F, Poewe W. Multiple system atrophy. Lancet Neurol. 2004;3:93–103. doi: 10.1016/S1474-4422(03)00662-8. [DOI] [PubMed] [Google Scholar]
- 2.Ubhi K, Low P, Masliah E. Multiple system atrophy: a clinical and neuropathological perspective. Trends Neurosci. 2011;34:581–590. doi: 10.1016/j.tins.2011.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gilman S, Wenning GK, Low PA, Brooks DJ, Mathias CJ, Trojanowski JQ, et al. Second consensus statement on the diagnosis of multiple system atrophy. Neurology. 2008;71:670–676. doi: 10.1212/01.wnl.0000324625.00404.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wakabayashi K, Yoshimoto M, Tsuji S, Takahashi H. α-Synuclein immunoreactivity in glial cytoplasmic inclusions in multiple system atrophy. Neurosci Lett. 1998;249:180–182. doi: 10.1016/S0304-3940(98)00407-8. [DOI] [PubMed] [Google Scholar]
- 5.Burn DJ, Jaros E. Multiple system atrophy: cellular and molecular pathology. Mol Pathol. 2001;54:419–426. [PMC free article] [PubMed] [Google Scholar]
- 6.Miller DW, Johnson JM, Solano SM, Hollingsworth ZR, Standaert DG, Young AB. Absence of α-synuclein mRNA expression in normal and multiple system atrophy oligodendroglia. J Neural Transm. 2005;112:1613–1624. doi: 10.1007/s00702-005-0378-1. [DOI] [PubMed] [Google Scholar]
- 7.Asi YT, Simpson JE, Heath PR, Wharton SB, Lees AJ, Revesz T, et al. Alpha-synuclein mRNA expression in oligodendrocytes in MSA. Glia. 2014;62:964–970. doi: 10.1002/glia.22653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Djelloul M, Holmqvist S, Boza-Serrano A, Azevedo C, Yeung MS, Goldwurm S, et al. Alpha-synuclein expression in the oligodendrocyte lineage: an in vitro and in vivo study using rodent and human models. Stem Cell Reports. 2015;5:174–184. doi: 10.1016/j.stemcr.2015.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Reyes JF, Rey NL, Bousset L, Melki R, Brundin P, Angot E. Alpha-synuclein transfers from neurons to oligodendrocytes. Glia. 2014;62:387–398. doi: 10.1002/glia.22611. [DOI] [PubMed] [Google Scholar]
- 10.Masuda-Suzukake M, Nonaka T, Hosokawa M, Oikawa T, Arai T, Akiyama H, et al. Prion-like spreading of pathological α-synuclein in brain. Brain. 2013;136:1128–1138. doi: 10.1093/brain/awt037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yonetani M, Nonaka T, Masuda M, Inukai Y, Oikawa T, Hisanaga S, et al. Conversion of wild-type α-synuclein into mutant-type fibrils and its propagation in the presence of A30P mutant. J Biol Chem. 2009;284:7940–7950. doi: 10.1074/jbc.M807482200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hara K, Momose Y, Tokiguchi S, Shimohara M, Terajima K, Onodera O, et al. Multiplex families with multiple system atrophy. Arch Neurol. 2007;64:545–551. doi: 10.1001/archneur.64.4.545. [DOI] [PubMed] [Google Scholar]
- 13.Wüllner U, Schmitt I, Kammal M, Kretzschmar HA, Neumann M. Definite multiple system atrophy in a German family. J Neurol Neurosurg Psychiatry. 2009;80:449–450. doi: 10.1136/jnnp.2008.158949. [DOI] [PubMed] [Google Scholar]
- 14.Wenning GK, Geser F, Krismer F, Seppi K, Duerr S, Boesch S, et al. The natural history of multiple system atrophy: a prospective European cohort study. Lancet Neurol. 2013;12:264–274. doi: 10.1016/S1474-4422(12)70327-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.May S, Gilman S, Sowell BB, Thomas RG, Stern MB, Colcher A, et al. Potential outcome measures and trial design issues for multiple system atrophy. Mov Disord. 2007;22:2371–2377. doi: 10.1002/mds.21734. [DOI] [PubMed] [Google Scholar]
- 16.Watanabe H, Saito Y, Terao S, Ando T, Kachi T, Mukai E, et al. Progression and prognosis in multiple system atrophy: an analysis of 230 Japanese patients. Brain. 2002;125:1070–1083. doi: 10.1093/brain/awf117. [DOI] [PubMed] [Google Scholar]
- 17.Yabe I, Soma H, Takei A, Fujiki N, Yanagihara T, Sasaki H. MSA-C is the predominant clinical phenotype of MSA in Japan: analysis of 142 patients with probable MSA. J Neurol Sci. 2006;249:115–121. doi: 10.1016/j.jns.2006.05.064. [DOI] [PubMed] [Google Scholar]
- 18.Scholz SW, Houlden H, Schulte C, Sharma M, Li A, Berg D, et al. SNCA variants are associated with increased risk for multiple system atrophy. Ann Neurol. 2009;65:610–614. doi: 10.1002/ana.21685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mitsui J, Matsukawa T, Sasaki H, Yabe I, Matsushima M, Dürr A, et al. Variants associated with Gaucher disease in multiple system atrophy. Ann Clin Transl Neurol. 2015;2:417–426. doi: 10.1002/acn3.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Multiple-System Atrophy Research Collaboration Mutations in COQ2 in familial and sporadic multiple-system atrophy. N Engl J Med. 2013;369:233–244. doi: 10.1056/NEJMoa1212115. [DOI] [PubMed] [Google Scholar]
- 21.Sailer A, Scholz SW, Nalls MA, Shulte C, Federoff M, Price TR, et al. A genome-wide association study in multiple system atrophy. Neurology. 2016;87:1591–1598. doi: 10.1212/WNL.0000000000003221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Feuk L, Carson AR, Scherer SW. Structural variation in the human genome. Nature Rev Genet. 2006;7:85–97. doi: 10.1038/nrg1767. [DOI] [PubMed] [Google Scholar]
- 23.Lee JA, Lupski JR. Genomic rearrangements and gene copy-number alterations as a cause of nervous system disorders. Neuron. 2006;52:103–121. doi: 10.1016/j.neuron.2006.09.027. [DOI] [PubMed] [Google Scholar]
- 24.Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, Hunter DJ, et al. Finding the missing heritability of complex diseases. Nature. 2009;461:747–753. doi: 10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lupski JR. Genomic rearrangements and sporadic disease. Nature Genet. 2007;239(suppl 7):43–47. doi: 10.1038/ng2084. [DOI] [PubMed] [Google Scholar]
- 26.Kushima I, Aleksic B, Nakatochi M, Shimamura T, Shiino T, Yoshimi A, et al. High-resolution copy number variation analysis of schizophrenia in Japan. Mol Psychiatry. 2017;22:430–440. doi: 10.1038/mp.2016.88. [DOI] [PubMed] [Google Scholar]
- 27.Morrow EM. Genomic copy number variation in disorders of cognitive development. J Am Acad Child Adolesc Psychiatry. 2010;49:1091–1104. doi: 10.1016/j.jaac.2010.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sasaki H, Emi M, Iijima H, Ito N, Sato H, Yabe I, et al. Copy number loss of (src homology 2 domain containing)-transforming protein 2 (SHC2) gene: discordant loss in monozygotic twins and frequent loss in patients with multiple system atrophy. Mol Brain. 2011;4 doi:10.1186/1756-6606-4-24. [DOI] [PMC free article] [PubMed]
- 29.Furguson MC, Garland EM, Hedges L, Womack-Nunley B, Hamid R, Phillips IIIJA, et al. SHC2 copy number in multiple system atrophy (MSA) Clin Auton Res. 2014;24:25–30. doi: 10.1007/s10286-013-0216-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, Sutton GG, et al. The sequence of the human genome. Science. 2001;291:1304–1351. doi: 10.1126/science.1058040. [DOI] [PubMed] [Google Scholar]
- 31.Farré M, Micheletti D, Ruiz-Herrera A. Recombination rates and genomic shuffling in human and chimpanzee-a new twist in the chromosomal speciation theory. Mol Biol Evol. 2012;30:853–864. doi: 10.1093/molbev/mss272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cognata VL, Morello G, D’Agata V, Cavallaro S. Copy number variability in Parkinson’s disease: assembling the puzzle through a systems biology approach. Hum Genet. 2017;136:13–37. doi: 10.1007/s00439-016-1749-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sharp AJ, Hansen S, Selzer RR, Cheng Z, Regan R, Hurst JA, et al. Discovery of previously unidentified genomic disorders from the duplication architecture of the human genome. Nature Genet. 2006;38:1038–1042. doi: 10.1038/ng1862. [DOI] [PubMed] [Google Scholar]
- 34.Sebat J, Lakshmi B, Malhotra D, Troge J, Lese-Martin C, Walsh T, et al. Strong association of de novo copy number mutations with autism. Science. 2007;316:445–449. doi: 10.1126/science.1138659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.The International Schizophrenia Consortium. Rare chromosomal deletions and duplications increase risk of schizophrenia. Nature 2008;455:237–241; doi:10.1038/nature07239. [DOI] [PMC free article] [PubMed]
- 36.McCarroll SA, Huett A, Kuballa P, Chilewski SD, Landry A, Goyette P, et al. Deletion polymorphism upstream of IRGM associated with altered IRGM expression and Crohn’s disease. Nature Genet. 2008;40:1107–1112. doi: 10.1038/ng.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.de Cid R, Riveira-Munoz E, Zeeuwen PLJM, Robarge J, Liao W, Dannhauser EN, et al. Deletion of the late cornified envelope LCE3B and LCE3C genes as a susceptibility factor for psoriasis. Nature Genet. 2009;41:211–215. doi: 10.1038/ng.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Boon-Peng H, Jusoh JAM, Marshall CR, Majid F, Danuri N, Basir F, et al. Rare copy number variants identified suggest the regulating pathways in hypertension-related left ventricular hypertrophy. PLoS One. 2016;11:e0148755. doi: 10.1371/journal.pone.0148755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yu L, Wynn J, Ma L, Guha S, Mychaliska GB, Crombleholme TM, et al. De novo copy number variants are associated with congenital diaphragmatic hernia. J Med Genet. 2012;49:650–659. doi: 10.1136/jmedgenet-2012-101135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bendjilali N, Kim H, Weinsheimer S, Guo DE, Kwok P-Y, Zaroff JG, et al. A genome-wide investigation of copy number variation in patients with sporadic brain arteriovenous malformation. PLoS One. 2013;8:e71434. doi: 10.1371/journal.pone.0071434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ettle B, Schlachetzki JCM, Winkler J. Oligodendroglia and myelin in neurodegenerative diseases: more than just bystanders? Mol Neurobiol. 2016;53:3046–3062. doi: 10.1007/s12035-015-9205-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Peferoen L, Kipp M, van der Valk P, van Noort JM. Amor S. Oligodendrocyte-microglia cross-talk in the central nervous system. Immunology. 2013;141:302–313. doi: 10.1111/imm.12163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yao M, Lee S-K, Lee B, Ruiz EC, Pfaff SL, Gill GN. Small CTD phosphatases function in silencing neuronal gene expression. Science. 2005;307:596–600. doi: 10.1126/science.1100801. [DOI] [PubMed] [Google Scholar]
- 44.Peng C, Togayachi A, Kwon Y-D, Xie C, Wu G, Zou X, et al. Identification of a novel human UDP-GalNAc transferase with unique catalytic activity and expression profile. Biochem Biophys Res Commun. 2010;402:680–686. doi: 10.1016/j.bbrc.2010.10.084. [DOI] [PubMed] [Google Scholar]
- 45.Kuslich CD, Kobori JA, Mohapatra G, Gregorio-King C, Donlon TA. Prader-Willi syndrome is caused by disruption of the SNRPN gene. Am J Hum Genet. 1999;64:70–76. doi: 10.1086/302177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Guertin MJ, Lis JT. Mechanisms by which transcription factors gain access to target sequence elements in chromatin. Curr Opin Genet Dev. 2013;23:116–123. doi: 10.1016/j.gde.2012.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stavreva DA, Hager GL. Chromatin structure and gene regulation: a dynamic view of enhancer function. Nucleus. 2015;6:442–448. doi: 10.1080/19491034.2015.1107689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cordaux R, Batzer MA. The impact of retrotransposons on human genome evolution. Nature Rev Genet. 2009;10:691–703. doi: 10.1038/nrg2640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dweep H, Kubikova N, Gretz N, Voskarides K, Felekkis K. Homo Sapiens exhibit a distinct pattern of CNV genes regulation: an important role of miRNAs and SNPs in expression plasticity. Sci Rep. 2015;5:12163. doi: 10.1038/srep12163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Batzer MA, Deininger PL. Alu repeats and human genomic diversity. Nature Rev Genet. 2002;3:370–379. doi: 10.1038/nrg798. [DOI] [PubMed] [Google Scholar]
- 51.Rinn JL, Chang HY. Genome regulation by long noncoding RNAs. Annu Rev Biochem. 2012;81:145–166. doi: 10.1146/annurev-biochem-051410-092902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Beck CR, Garcia-Perez JL, Badge RM, Moran JV. LINE-1 elements in structural variation and disease. Annu Rev Genomics Hum Genet. 2011;12:187–215. doi: 10.1146/annurev-genom-082509-141802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Karczewski KJ, Snyder M, Altman RB, Tatonetti NP. Coherent functional modules improve transcription factor target identification, cooperativity prediction, and disease association. PLoS Genet. 2014;10:e1004122. doi: 10.1371/journal.pgen.1004122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Spielmann M, Mundlos S. Looking beyond the genes: the role of non-coding variants in human disease. Hum Mol Genet. 2016;25(R2):R157–R165. doi: 10.1093/hmg/ddw205. [DOI] [PubMed] [Google Scholar]
- 55.International Human Genome Sequencing Consortium Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Number of CNVs in 72 individuals. Table S2. Number of CNVs on autosomal chromosomes in controls and subjects with MSA-C or MSA-P. Table S3. Chromosome numbers at which the 311 CNVs related to MSA were located. Table S4. Details of the 29 CNVs as obtained by cluster analysis. Table S5. Details of the 12 verified CNVs. Table S6. Large CNVs identified in this study. (DOCX 97 kb)
Gel electrophoresis of PCR products for verification analysis. Figure S2. Chromosomal location of CNVs in control subjects. (PPTX 454 kb)
Data Availability Statement
The data generated or analyzed in this study are included in this published article and its Additional files.