

Abstract
Antimalarial compounds with dual therapeutic and transmission-blocking activity are desired as high-value partners for combination therapies. Here, we report the identification and characterization of hexahydroquinolines (HHQs) that show low nanomolar potency against both pathogenic and transmissible intra-erythrocytic forms of the malaria parasite Plasmodium falciparum. This activity translates into potent transmission-blocking potential, as shown by in vitro male gamete formation assays and reduced oocyst infection and prevalence in Anopheles mosquitoes. In vivo studies illustrated the ability of lead HHQs to suppress P. berghei blood-stage parasite proliferation. Resistance selection studies, confirmed by CRISPR/Cas9-based gene editing, identified the digestive vacuole membrane-spanning transporter PfMDR1 as a determinant of parasite resistance to HHQs. Hemoglobin and heme fractionation assays suggest a mode of action that results in reduced hemozoin levels and might involve inhibition of host hemoglobin uptake into intra-erythrocytic parasites. Furthermore, parasites resistant to HHQs displayed increased susceptibility to several first-line antimalarial drugs including lumefantrine, confirming that HHQs have a different mode of action than other antimalarials drugs for which PfMDR1 is known to confer resistance. This work evokes therapeutic strategies that combine opposing selective pressures on this parasite transporter as an approach to countering the emergence and transmission of multidrug-resistant P. falciparum malaria.
Keywords: Plasmodium falciparum, malaria, PfMDR1, high-throughput screens, gametocytes, transmission-blocking, CRISPR/Cas9, drug resistance, combination therapies
Introduction
Malaria remains a global health calamity, with an estimated 212 million cases and 429,000 deaths in 20151 that occur mostly in sub-Saharan Africa and result from infection with the Apicomplexan parasite Plasmodium falciparum (Pf). Improved malaria treatment and control has reduced morbidity and mortality worldwide by 40–50% since 20001, due largely to highly effective artemisinin-based combination therapies and mosquito vector control measures1,2. An important strategy to further advance the malaria elimination agenda is to include transmission-blocking agents in combination therapies3. Such agents inhibit the development of parasite gametocytes (GAMs) that represent an alternative form of intra-erythrocytic development to the pathogenic asexual blood stage (ABS) parasites, and that mature over five stages4. Only stage V GAMS, which may survive days to weeks in circulation5, can continue the Pf lifecycle in Anopheles mosquitoes following ingestion of a parasitized blood meal. In the mosquito midgut, male GAMs exflagellate and produce microgametes that can fuse with female macrogametes to produce zygotes. Following subsequent development as ookinetes and oocysts, sporozoites are formed. Once inoculated in a new host, they will develop in the liver and initiate a new ABS cycle6.
Primaquine is currently the only available drug with potent activity against stage V GAMs7,8, but can be toxic for glucose-6-phosphate dehydrogenase-deficient patients8. Hence, there is a compelling need to develop alternative transmission-blocking drugs. Recent advances in GAM-specific high-throughput screen technology9–19, combined with functional validation assays and drug action/resistance studies19–21, provide a powerful foundation to identify leads with Pf transmission-blocking activity.
In this study, we report a screen of the Novartis-GNF Malaria Box of ABS-active compounds22 on Pf GAM stages and describe the identification and antimalarial properties of hexahydroquinolines (HHQs), a chemical class with considerable transmission-blocking potential.
Results
Identifying HHQs as potent gametocytocidal inhibitors
Using a luciferase (LUC)-based assay, we screened 3,825 compounds from the Novartis-GNF Malaria Box for activity against NF54Pfs16 early-stage GAMs. This screen yielded 797 compounds exhibiting ≥50% inhibition at 2.6 μM. Re-screening confirmed activity for 683 compounds, of which 176 showed ≥50% inhibition at 0.65 μM (Figure 1a). Screening the Novartis-GNF Malaria Box against late-stage GAMs using high-content imaging identified 572 hits with ≥50% inhibition at 2 μM in at least two of the three replicates (Figure 1a; Supplementary Figure 1a).
Figure 1. Identifying HHQs as potent Pf ABS and GAM inhibitors.

(a) Outcomes of the primary screening of the GNF-Novartis Malaria Box library against early- and late-stage Pf NF54Pfs16 GAMs, using luciferase-based and high-content imaging assays, respectively. (b) Scatterplot of early- and late-stage gametocytocidal potency of 63 compounds with dual activity (expressed as pIC50s which equal the −log(IC50); a value of 9 corresponds to 10−9 M, i.e. 1 nM). The red stars correspond to the four compounds selected for the study. Data are based on two independent experiments, each probing both early- and late-stage GAMs. (c) Intra-erythrocytic stage-specific activity of the four selected compounds was obtained from two independent experiments for each parasite stage (Supplementary Table 3). (d) Structure of the 3 HHQs confirmed to have transmission-blocking activity. (e) Impact of PfMDR1 mutations and copy number on HHQ potency. The X-axis depicts the individual mutant lines and their wild-type parental lines, and indicates the GNF-Pf compound number, the clone number and in parentheses the PfMDR1 mutation and its allele balance. Edited implies that the Dd2-B2 line was edited using a CRISPR/Cas9 system to introduce the designated PfMDR1 mutation. Mean ± SEM IC50 values were calculated from three or more independent experiments (detailed in Supplementary Table 5) and significance was assessed by a Welch’s corrected t-test with Bonferroni-Dunn adjusted P-values for multiple comparisons when applicable. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001. Pf: Plasmodium falciparum; ABS: asexual blood stage; GAM: gametocyte.
Late-stage gametocytocidal hits were then prioritized for IC50 determination using both early- and late-stage GAM assays. Limited availability restricted the number of compounds to 149 (Supplementary Figure 1b), yielding 71 and 60 compounds with IC50 values below 1 μM for early- and late-stage GAMs respectively (Supplementary Table 1). Among these, 20 early-GAM and 16 late-GAM active compounds had IC50 values ≤200 nM. In total, 43 compounds showed sub-micromolar activity against both stages (Figure 1b). Reference drug controls showed expected values (Supplementary Table 2).
After deprioritizing chemical classes related to current antimalarial drugs or having liabilities likely to impede potential development, we identified four compounds with potent early- and late-stage gametocytocidal activity: GNF-Pf-5640, GNF-Pf-5660, GNF-Pf-5668 and GNF-Pf-5310 (Figure 1c; Supplementary Table 3). These compounds also showed potent ABS activity (IC50 values <25 nM) against drug-sensitive 3D7 and multidrug-resistant Dd2 parasites, including a PfATP4 mutant line (Supplementary Figure 2, Supplementary Table 3). GNF-Pf-5640, GNF-Pf-5660 and GNF-Pf-5668 share a common scaffold belonging to the HHQ class of compounds (Figure 1d), whereas GNF-Pf-5310 is a muscarinic-like compound (Supplementary Figure 3). Cytotoxicity assays showed no inhibition at 40 μM with GNF-Pf-5660, while the other 3 compounds caused <50% inhibition at that concentration (Supplementary Figure 2), indicating minimal toxicity against mammalian cells.
HHQs demonstrate potent in vitro transmission-blocking activity
Using an in vitro dual gamete formation assay (DGFA), we investigated whether these four potent GAM-active compounds would also inhibit the development of cultured female and male gametes. Results indicated a potent inhibitory effect of the 3 HHQs on exflagellation when GAMs were exposed for 24 h and the compound carried over during the subsequent period of male gamete induction (“carry-over”; Table 1). This effect persisted when compounds were washed out after 24 h, i.e. prior to gamete induction (“wash-out”; Table 1). HHQs were thus effective against both mature male GAMs and developing male gametes, but showed no activity on female gamete formation. The muscarinic-like compound GNF-Pf-5310 showed no activity against either male or female gametes, contrasting with our transmission-blocking control methylene blue 23,24 (Table 1).
Table 1.
Gamete formation inhibitory effect of the 4 selected compounds.
| Mean IC50 (lower 95% CI - upper 95% CI) (nM) | ||||
|---|---|---|---|---|
| Male carry-over | Female carry-over | Male wash-out | Female wash-out | |
| GNF-Pf-5640 | 86.0 (22.0 – 335.1) | > 20 μM | 521.3 (447.3 – 607.6) | > 20 μM |
| GNF-Pf-5660 | 67.7 (40.9 – 112.2) | > 20 μM | 264.2 (142.8 – 488.7) | > 20 μM |
| GNF-Pf-5668 | 88.8 (53.0 – 148.8) | > 20 μM | 138.8 (67.0 – 287.5) | > 20 μM |
| GNF-Pf-5310 | > 20 μM | > 20 μM | > 20 μM | > 20 μM |
| Methylene Blue | 61.5 (38.8 – 97.3) | 161.8 (111.4 – 234.9) | 146.7 (116.3 – 185.1) | 846.2 (490.6 – 1460.0) |
Compounds were carried over from GAM exposure to induction of gamete formation in the carry-over format of the assay, but were washed-out before gamete formation in the wash-out format of the assay. CI: 95% confidence interval based on 4 biological replicates.
GNF-Pf-5660, GNF-Pf-5668 and GNF-Pf-5310 were also tested in a membrane-feeding assay using A. stephensi mosquitoes. This assay more closely mimics the natural parasite lifecycle than the DGFA, since compound-exposed Pf GAMs initiate the entire mosquito vector cycle from gametes to midgut oocysts. Pre-treatment of GAMs with GNF-Pf-5660 or GNF-Pf-5668 at 3x their late-stage IC50 caused a 97% and 57% reduction respectively in the infection prevalence (i.e. the percentage of infected mosquitoes, Figure 2a), whereas the oocyst density per infected mosquito midgut was reduced by 95–99% (Figure 2b). GNF-Pf-5660- and GNF-Pf-5668-exposed GAM cultures used for feeding to A. stephensi showed a 98% and 70% reduction respectively in their male GAM exflagellation rates (Figure 2c), confirming earlier DGFA results. GNF-Pf-5310 was less active at similar exposure levels, causing no reduction in infection prevalence, a 73% reduction in oocyst density, and no effect on male GAM exflagellation rates (Figure 2). As a result, GNF-Pf-5310 was deprioritized for further studies. The positive control methylene blue displayed maximal transmission-blocking activity (Figure 2). Pre-treatment of GAMs with compounds at 1x their late-stage IC50 caused a lower to no effect on the parameters assessed (Figure 2, Supplementary Table 4a). The vehicle control DMSO showed no reductions compared to untreated controls.
Figure 2. HHQs demonstrate potent transmission-blocking activity and inhibit male gamete exflagellation.

For in vitro treatment studies, mosquitoes were fed with compound or DMSO-treated GAMs. Compounds were added at 1′ or 3′ their late-stage GAM IC50, and DMSO vehicle was used as a negative control. Parasites were treated for 3 days and drug then removed for 4 days prior to the blood meal feeds and transmission-blocking studies. For in vivo studies, mosquitoes were fed on P. berghei (Pb)-infected mice that were treated with two doses of 100 mg/kg of the GNF compounds, or solvent as a negative control, or 30 mg/kg DHA as a positive control. Results show (a) Percent reduction in number of infected mosquitoes; (b) Percent reduction in number of oocysts per midgut; and (c) Compound effect on male GAM exflagellation, expressed as the average number of exflagellation centers per field in GAM cultures. Results in panels a–c are shown as mean ± SEM, calculated from two independent experiments (see Methods). (d) Mean percent reduction of blood stage parasites in Pb-infected mice treated with HHQs, or DHA as a positive control. Data are shown as mean ± SEM based on two independent repeats, except for the 100 mg/kg HHQ-treated mice in the 4-day Peters test that were tested once and served as a pilot experiment for the 2-dose test (in order to match the two-dose design used in the transmission-blocking studies shown in panels a–c). Statistical significance was assessed using a Welch’s corrected t-test with Bonferroni-Dunn adjusted P-values for multiple comparisons when applicable: *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001. All data are available in Supplementary Tables 4a–c.
In vivo activity of HHQs
HHQ antimalarial activity was assayed in P. berghei-infected mice using the 4-day Peters test25. Intraperitoneal injection of 70 mg/kg of GNF-Pf-5660 or GNF-Pf-5668 reduced parasitemias by 22% or 43% respectively compared to vehicle-treated mice (Figure 2d). At 100 mg/kg, these agents reduced parasitemia by 65% and 91% respectively (Figure 2d, Supplementary Table 4b). To assess the in vivo transmission-blocking activity of HHQs, infected mice were administered only two 100 mg/kg doses of GNF-Pf-5660 or GNF-Pf-5668. Following blood meals on GNF-Pf-5660-dosed mice, we observed a 25% and 21% reduction in the number of infected mosquitoes and their oocyst burdens respectively (Figure 2a, b). No effect was observed in the GNF-Pf-5668 mosquito cohort, although it displayed a 31% reduction in the numbers of male exflagellation centers at the time of mosquito feeding (Figure 2c, Supplementary Table 4c). Limited in vivo transmission-blocking activity, as compared with our in vitro results, was also observed upon treating mice with two doses of dihydroartemisinin (DHA). Those assays showed 65% and 75% reductions in the number of infected mosquitoes and the oocyst burden respectively, and found no reduction in the numbers of male exflagellation centers (Figure 2a–c). These data are consistent with this two-dosing regimen having less impact on ABS parasitemia than the four days of dosing used in the Peters test (Figure 2d).
Resistance to HHQs is mediated by mutations in PfMDR1 and results in sensitization to clinical antimalarials
To gain insight into the mode of action of HHQs, we selected for Pf ABS resistance in vitro in the Dd2-B2 parasite line, an approach that has proven effective for multiple antimalarial agents26. Whole-genome sequence analysis of the resulting resistant clones identified the PfMDR1 S1075I mutation in the GNF-Pf-5640-selected clone 2C4C3, and PfMDR1 Y290F in the GNF-Pf-5640-selected clone 3H9C7. These resistant clones showed a 77- and 79-fold increase in IC50 values for GNF-Pf-5640 compared to the parental Dd2-B2 line respectively (Figure 1e). The allele balance (AB) of the mutations in these clones was 50–59%, consistent with their acquisition in one of the two pfmdr1 copies present in Dd2-B2. The GNF-Pf-5660-selected clone 2B6H1 and the GNF-Pf-5668-selected clone 3C4D2 showed IC50 increases of 248- and 37-fold respectively, and had acquired the PfMDR1 Y290F (AB 100%) or F1072L (AB 67%) mutations respectively (Figure 1e). Shifts in IC90 values were similar to IC50 increases (Supplementary Tables 5–6).
Whole-genome sequencing data indicated that the GNF-Pf-5668-selected 3C4D2 clone also carried 4 copies of pfmdr1, consistent with further gene amplification compared to the Dd2-B2 parent (that has two pfmdr1 copies in tandem). We were unable to determine whether the F1072L mutation (AB 67%) was mainly located in the additional amplicons. Susceptibility testing of resistant clones indicated that each of the three observed PfMDR1 mutations conferred cross-resistance to all HHQs (Figure 1e).
Because HHQ resistance was the highest in lines harboring the Y290F mutation, we employed a previously derived NF54 line27 that harbored this mutation (NF54selY290F) to verify whether Y290F also conferred resistance at the GAM stage. NF54selY290F early-stage GAMs showed a 40–74-fold IC50 shift for all HHQs compared to wild-type NF54 (Supplementary Table 7). High levels of resistance were also observed at the late GAM stage, but since only very low numbers of NF54selY290F late GAMs could be obtained, accurate late-stage IC50 values could not be calculated.
Structural modeling of PfMDR1 localized amino acid residue 290 to transmembrane domain (TMD) 5 and residues 1072 and 1075 to TMD 12. All three residues resided in the predicted drug-binding pocket cavity of PfMDR128, where they might mediate contact with substrates (Figure 3a–b). Docking studies suggested that each mutation could significantly alter transporter interactions with HHQs. Interestingly, F1072L and Y290F seemed to decrease the minimum estimated energy required for docking (Supplementary Tables 8–12, Supplementary Figure 4). This effect was less pronounced for S1075I, which could be due to this residue not being fully exposed to the drug-binding pocket.
Figure 3. PfMDR1 mutations associated with HHQ resistance locate in its drug binding pocket and can sensitize parasites to first-line antimalarial drugs and the preclinical candidate ACT-451840.

(a) Structural model of PfMDR1 transmembrane domains (TMDs) based on the 4Q4H structure; and (b) visualization of the drug-binding pocket cavity. TMD 7 and TMD 8 were omitted to allow visualization of the mutations. (c) Susceptibility of PfMDR1 mutants, obtained through HHQ selection or genetic editing, to other antimalarials. Mean ± SEM IC50 values were calculated from 3 to 12 independent experiments (Supplementary Table 13). IC90 data followed similar trends as the IC50 data described herein (Supplementary Table 14). Statistical significance was assessed using a Welch’s corrected t-test with Bonferroni-Dunn adjusted P-values for multiple comparisons when applicable: *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001.
To test whether these point mutations in PfMDR1 were causal for resistance, we implemented a pfmdr1-specific CRISPR/Cas9 strategy27. Transfection of parental Dd2-B2 parasites yielded the PfMDR1edF1072L and PfMDR1edS1075I clones that expressed the F1072L and S1075I mutations respectively, observed in both pfmdr1 copies. These clones showed an essentially equivalent gain of resistance to HHQs as the original drug-pressured lines, confirming the causal role of the PfMDR1 mutations in mediating resistance (Figure 1e). Despite repeated efforts, we were unable to introduce the Y290F mutation into the parental line.
Testing KD1mdr1, a pfmdr1 knock-down line previously generated in FCB parasites, showed that a decreased pfmdr1 copy number did not affect HHQ susceptibility (Figure 1e). Similarly, increased pfmdr1 copy number also did not noticeably affect HHQ susceptibility (Figure 1e), as shown by the similar IC50 values of GNF-Pf-5668-3C4D2 (harboring an estimated 4 copies of pfmdr1 in addition to the F1072L mutation) and PfMDR1edF1072L (with the F1072L mutation engineered into the wild-type Dd2-B2 line with 2 pfmdr1 copies).
The impact of the three PfMDR1 mutations on Pf ABS susceptibility to known antimalarials was determined using PfMDR1edF1072L, PfMDR1edS1075I, and GNF-Pf-5660-2B6H1 (also referred to as PfMDR1selY290F) (Figure 3c). For all three lines, no changes in susceptibility were observed for piperaquine, CQ, the active metabolite monodesethyl-CQ (md-CQ), md-amodiaquine (md-ADQ), or ferroquine (Figure 3; Supplementary Tables 13–14). In contrast, PfMDR1edF1072L and PfMDR1selY290F showed a higher susceptibility to both quinine (QN) (3.6- and 3.7-fold, respectively) and mefloquine (MQ) (3.6- and 2.0-fold, respectively), whereas PfMDR1edS1075I showed a 5.1-fold increased QN susceptibility but only a 1.4-fold increased susceptibility to MQ. Lumefantrine (LMF) showed a similar trend, with PfMDR1edF1072L and PfMDR1selY290F showing a higher LMF susceptibility (3.1- and 1.9-fold, respectively), whereas PfMDR1edS1075I remained unchanged. All three mutations significantly increased Pf susceptibility to DHA by ~1.6-fold. Parasite susceptibility to the experimental ACT-451840 compound, a known inhibitor of PfMDR127, was decreased by 16-, 5.5- and 45-fold for PfMDR1edF1072L, PfMDR1edS1075I and PfMDR1selY290F respectively (Figure 3c).
To assess the effects of pfmdr1 copy number variation in combination with the F1072L mutation, we compared the antimalarial susceptibilities of GNF-Pf-5668-3C4D2 (harboring four pfmdr1 copies + F1072L (AB 69%)) to that of PfMDR1edF1072L (having 2 pfmdr1 copies + F1072L (AB 100%)). Results indicated a decreased susceptibility to QN, MQ and LMF (i.e. higher IC50 values) in GNF-Pf-5668-3C4D2 compared to PfMDR1edF1072L (Figure 3c). Importantly, these pfmdr1 copy number-induced changes in GNF-Pf-5668-3C4D2 did not fully compensate for the effects introduced by the F1072L mutation, as this line still showed a increased susceptibility to QN (1.7-fold) and MQ (1.4-fold) compared to the wild-type Dd2-B2 control (Figure 3c). These data provide evidence that the F1072L mutation had a major effect on increasing parasite susceptibility to QN, MQ and LMF that was only partially reversed by increases in pfmdr1 copy number.
HHQs inhibit hemoglobin (Hb) endocytosis
Prior studies have shown that mutant PfMDR1 isoforms can modulate the degree of Pf ABS resistance to several antimalarial drugs, including CQ and ADQ, in lines expressing mutant pfcrt29–31. These 4-aminoquinolines are known to inhibit hemozoin (Hz) formation and cause a buildup in free heme32,33. In light of the finding that PfMDR1 confers Pf resistance to HHQs, we performed subcellular heme fractionation assays to assess whether these agents might also affect Hz formation in ABS parasites (Figure 4, Supplementary Table 15).
Figure 4. GNF-Pf-5660 causes a decrease in Hz and total heme iron levels in wild-type ABS Dd2-B2 and mutant HHQ-resistant PfMDR1selY290F parasites, the latter at significantly higher compound concentrations.

Synchronized parasites were exposed to GNF-Pf-5660 or the reference antimalarials MQ and LMF and fractionated in order to measure Hb, free heme, or Hz iron. Total heme iron represents the sum of these three values. Results show Hb, free heme (which could be loosely associated with lipids or other biomolecules), Hz and total heme iron levels (as fg/cell) in parasites exposed to a range of IC50 values (0.1, 0.25, 0.5, 1.0 and 2.0′). Mean ± SEM amounts were calculated from two independent experiments and statistical significance was assessed by a two-tailed t-test. *P<0.05, **P<0.01, ***P<0.001. All data are available in Supplementary Table 15.
In parental Dd2-B2 parasites, we observed that Hb and free heme concentrations per parasite remained unchanged across all GNF-Pf-5660 exposure levels (up to 2′ IC50) (Figure 4). Hz levels, however, showed a 1.5-fold decrease when parasites were exposed to as little as 0.1′ the GNF-Pf-5660 IC50 value, as compared with untreated controls. A 2.3-fold reduction in Hz levels was observed at higher exposure levels (0.25′ to 2′ IC50, Figure 4). Total heme iron levels also showed similar reductions with increasing concentrations of GNF-Pf-5660 (Figure 4). A similar profile of reduced Hz and total heme iron content per parasite was also observed in PfMDR1selY290F, albeit at >160-fold higher GNF-Pf-5660 exposure levels than in Dd2-B2, consistent with the fold increase in IC50 in the mutant line (Figure 4).
For comparison, we also assessed the heme fractionation profiles of MQ and LMF. In the Dd2-B2 line, MQ and LMF showed little or no effect compared to untreated controls, with the exception of a 1.6-fold decrease in Hz levels in parasites exposed to 2′ the MQ IC50. Intriguingly, PfMDR1selY290F showed MQ profiles similar to those observed with GNF-Pf-5660, i.e. reductions in Hz and total heme iron levels with increasing MQ concentrations. In PfMDR1selY290F, LMF also caused a reduction in Hz and total heme iron amounts at 1–2′ IC50 concentrations (Figure 4).
We also measured the amount of Hb endocytosed by late rings exposed to GNF-Pf-5660, WR99210, MQ or LMF, using Western Blot analysis of wild-type Dd2-B2 or HHQ-resistant PfMDR1selY290F parasites (Figure 5, Supplementary Table 16). Results showed that Hb levels within parasites were reduced by 32% in GNF-Pf-5660-exposed Dd2-B2, but remained unchanged in PfMDR1selY290F. Dd2-B2 parasites showed no significant changes in Hb levels when exposed to WR99210, MQ, or LMF.
Figure 5. HHQs inhibit Hb endocytosis.
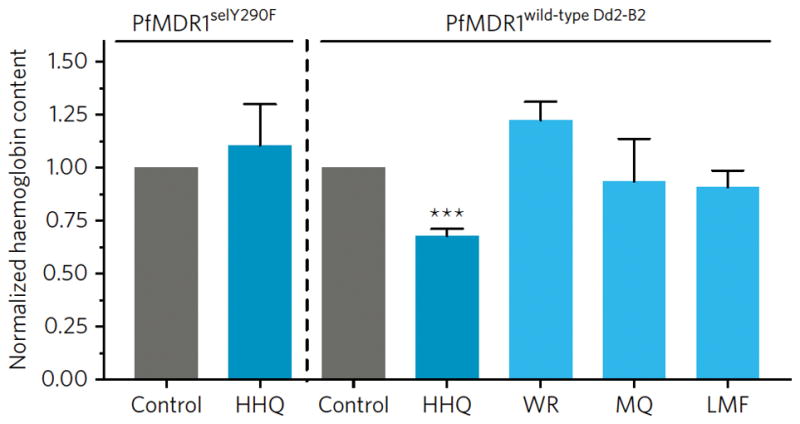
Parasites were exposed to 5′ the IC50 of the HHQ GNF-Pf-5660 or reference antimalarials and Hb content was measured by Western blot analysis, with amounts normalized to mock DMSO-treated control parasites. Mean ± SEM values were calculated from 3 to 4 independent experiments. Statistical significance was assessed by a Welch’s corrected t-test adjusted for multiple comparisons with the Bonferroni-Dunn method. ***P<0.001. HHQ: GNF-Pf-5660; WR: WR99210; MQ: mefloquine; LMF: lumefantrine; CQ: chloroquine. All data are available in Supplementary Table 16 and Supplementary Figures 5a–b.
Discussion
Here we describe the antimalarial properties of HHQs, a class of potent gametocytocidal antimalarials with ABS activity that were identified by screening the Novartis-GNF Malaria Box against early- and late-stage Pf GAMs. Functional assays with Pf GAMs and gametes confirmed potent transmission-blocking activity. Moderate activity with our initial hits was also observed in vivo in P. berghei mouse models. Mode of action and resistance studies implicated a key role for the digestive vacuole (DV) transporter PfMDR1. We also provide evidence that HHQs reduce levels of Hz and total heme iron in treated parasites and inhibit the initial uptake of Hb from host red blood cells (RBCs). Cross-resistance assays also revealed that HHQ-resistant parasites became sensitized to several antimalarials in current clinical use, emphasizing the potential added value of HHQs for combination treatments.
The confirmed hit rates of 18% and 15% obtained from our compound screens of early- and late-stage GAMs respectively (Fig. 1a) highlight the value of screening ABS-active compound libraries as a source for identifying gametocytocidal compounds. The limited overlap (20%) between early- and late-stage GAM hits (Supplementary Figure 1b) suggests that GAMs at different developmental stages express distinct sets of druggable targets, provided that technology-related differences such as incubation times, sensitivity, or specific bias for selecting certain chemical classes were minimal in both screens.
Our HHQ hits GNF-Pf-5640, GNF-Pf-5660 and GNF-Pf-5668 showed potent activity against ABS parasites as well as early- and late-stage GAMs (Figure 1c), with only minimal toxicity against mammalian cells. These compounds were inactive against cultured P. berghei liver-stage parasites16, possibly because Hb import and metabolism (discussed below) does not occur in parasitized hepatocytes.
Results provided herein also document potent in vitro HHQ activity on male GAMs and gametes, which translated into marked transmission-blocking activity in mosquitoes (Table 1 and Figure 2). In P. berghei-infected mice, HHQs showed moderate efficacy: 4-day Peters tests indicated a potent inhibition of ABS growth at 100 mg/kg, whereas the two-dose assay showed only a limited impact on ABS parasitemia and parasite transmission (Figure 2). The high doses required in vivo, compared to the lower doses at which activity was observed in vitro, suggest that HHQs might be rapidly metabolized or otherwise inactivated or eliminated by the host. The ester group present in HHQs, for instance, is likely sensitive to host esterases34. These HHQs are the first hits from this class, and structure-activity relationship studies and chemical optimization will be required. Enhancing HHQ transmission-blocking activity in vivo is an important goal, in light of the limited efficacy observed to date (Figure 2). We note that earlier studies of P. berghei reported that a 30% reduction in oocyst numbers per infected mosquito sufficed to eliminate the parasite from a murine population after several transmission cycles35. In light of these data, HHQs constitute a promising class with transmission-blocking potential that merit pharmacokinetic and pharmacodynamic optimization.
In vitro HHQ resistance selection studies identified three mutations in PfMDR1 (Y290F, F1072L and S1075I), which were confirmed by CRISRP/Cas9 editing to be causal for ABS resistance (Figure 1e). PfMDR1-associated resistance also extended to the GAM stage (Supplementary Table 7). We note that the Pf3k collection of over 2,500 Pf genomes collected worldwide (www.malariagen.net/apps/pf3k) contained no mutations with an AB >0.2 at these PfMDR1 positions, suggesting that they are very rare, if not non-existent, in the field.
Structural modeling studies indicated that all three amino acids co-localize in the predicted drug-binding pocket of PfMDR1 (Figure 3) and generally caused a closer interaction between HHQs and mutant PfMDR1 (Supplementary Tables 8–12). Further studies are required to determine the nature of the interactions between HHQs and wild-type versus mutant PfMDR1 and how this relates to HHQ potency.
PfMDR1, an ATP-binding cassette (ABC) transporter, can mediate the transport of several antimalarial drugs across the DV membrane30,36,37. In the highly acidic DV compartment, Pf proteases degrade Hb from the host RBC, providing a source of amino acids required for protein synthesis and parasite growth38. This catabolic process triggers the formation of highly reactive heme moieties, which Plasmodium normally detoxifies through biomineralization into chemically inert Hz crystals in the DV. Several antimalarials act either by binding toxic heme species and inhibiting Hz formation (CQ or ADQ), or in the case of artemisinins appear to be activated through interaction with heme iron39. Mutations in PfMDR1, or the chloroquine resistance transporter PfCRT that also resides on the DV membrane, are known to affect in vitro parasite susceptibility to these drugs, presumably by affecting drug transport, and are associated with an increased risk of parasite recrudescence or treatment failure in patients30,40–42.
Heme fractionation studies showed a different profile in parasites treated with GNF-Pf-5660 as compared to CQ32, ADQ32, MQ or LMF (Figure 4), indicating a distinct mode of action. HHQs did not directly inhibit heme conversion into Hz, but were found to reduce total amounts of intracellular heme iron. These data suggest that HHQs act upstream of the Hb digestion pathway by interfering with parasite-mediated uptake of host Hb, which delivers the substrate for Hz formation. Western blot data showed a reduction of total Hb levels in HHQ-treated wild-type Dd2-B2 but not HHQ-resistant PfMDR1selY290F parasites (Figure 5), supporting an effect of HHQs on parasite-mediated Hb endocytosis.
Parasite lines resistant to CQ, pyrimethamine and PfATP4 inhibitors remained sensitive to HHQs (Supplementary Table 3), whereas HHQ-resistant lines showed an increased Pf ABS susceptibility to QN, MQ, DHA and LMF (except for PfMDR1edS1075I that showed no difference for LMF). Susceptibility to CQ, md-CQ, md-ADQ, ferroquine and piperaquine remained unaltered in HHQ-resistant parasites (Figure 3, Supplementary Table 13). QN, MQ, DHA and LMF are thought to interact at some level with heme but also act outside of the DV, raising the possibility that they might engage with the Hb import and catabolism process that begins with endocytosis33,43–45. We speculate that the PfMDR1 mutations causing HHQ resistance might sensitize parasites to MQ and LMF by preventing their sequestration by wild-type PfMDR1 in the DV, away from their primary site of action46.
HHQ-resistant ABS parasites showed no substantial change in their susceptibility to the heme-binding drugs CQ or md-ADQ that act primarily in the DV41. Interestingly, PfMDR1 Y290F was also recently identified in selections using ACT-451840, a piperazine-based pre-clinical candidate with low nanomolar activity against Pf ABS parasites and GAMs27. Studies with the ACT-213615 analog earlier showed direct binding to PfMDR1, identifying this as a potential target47. The unusually high levels of resistance to both HHQs and ACT-451840 conferred by each of the three mutations (Figure 1e, Figure 3c) suggest that both compound series likely share a similar mode of action. These data also evoke the possibility that PfMDR1 might itself be a target of HHQs, a hypothesis that can be investigated using HHQ analogs to study protein interactions with PfMDR147 in cultured parasites or heterologous expression systems48.
Stage-specific profiling showed that pfmdr1 is maximally transcribed around the time of merozoite egress (see www.PlasmoDB.org), suggesting that PfMDR1 is already present in early- to mid-stage rings, well before ABS parasites have formed their mature DV. This timing, supported by Western blot data49, suggests that PfMDR1 might be involved in vesicle-mediated endocytosis of host Hb from the RBC en route to the developing DV. Such a role for PfMDR1 would be consistent with our observations that HHQ inhibition reduces intra-parasitic total heme iron and Hz levels in trophozoites and reduces Hb levels in late-stage rings (Figures 4–5).
Further studies into the stage-dependency and subcellular site of action of HHQs, and of drugs that are impacted by HHQ-selected mutations in PfMDR1 such as the first-line agent LMF, should yield interesting insights into how these agents act and how to leverage this information into identifying promising drug combinations. Ideally, such combinations would exert opposing selective pressures on mutant and wild-type PfMDR1, in an effort to stall the emergence and spread of multidrug resistance. This approach is currently being used in clinical trials where artemether + LMF is being combined with ADQ (see Clinical Trials NCT02453308), based on earlier reports that LMF and ADQ exert opposing selective pressures on wild-type and mutant forms of both pfmdr1 and pfcrt40,50. In addition, the potent activity of HHQs against both ABS parasites and GAMs highlights the promise of developing this chemical class into a partner for future antimalarial combination therapies with transmission-blocking activity.
Methods
Parasite cultures and strains
Pf ABS parasites were cultured at 3–4% hematocrit (Hct) in human O+ RBCs in RPMI-1640 media, supplemented with 25 mM HEPES, 50 μM hypoxanthine, 2 mM L-glutamine, 2 g/l sodium bicarbonate, 0.5% (w/v) AlbuMAXII (Invitrogen) and 10 μg/ml gentamycin, in 5% O2, 5% CO2 and 90% N2 at 37°C. For GAM cultures, Albumax was replaced with 10% human serum. 3D7 and Dd2 parasites were originally obtained from Dr. Thomas Wellems (NIAID, NIH). Dd2-B2 is a genetically homogenous parasite line that was cloned from Dd2 by limiting dilution. The NITD609-resistant parasites (generated in the Dd2 background) and the NF54Pfs16 line have been previously reported23,51. The NF54selY290F and the FCB pfmdr1 knock-down line (KD1mdr1) and their corresponding wild-type lines were obtained in two earlier studies27,52. A list of all parasite lines generated in this study is available in Supplementary Table 17. Parasite lines were screened by PCR for Mycoplasma every 2–3 months.
Compound screening against gametocytes
The Novartis-GNF Malaria Box consisted of 3,825 known inhibitors of ABS Pf parasites with a median IC50 of 0.53 μM (25th percentile: 0.23 μM, 75th percentile: 1.09 μM), as determined by a SYBR Green-based 72-h growth inhibition assay53, and was obtained as 10 μl of 1 mM stock solutions in 100% DMSO. These compounds were independently screened against early- and late-stage GAMs (stages I and IV respectively), as described below. Screening was performed with the NF54Pfs16 line that expresses a green fluorescent protein (GFP)–LUC fusion under the pfs16 promoter that is active throughout gametocytogenesis23. Earlier studies have reported the optimization of high-throughput early- and late-stage GAM screens using this line in LUC- or GFP-based assays54–56. For the LUC-based screens with early-stage GAMs, compounds were diluted 1.5-fold in 100% DMSO, and further diluted in water to a final assay concentration of 2.6 μM in 0.4% DMSO. Hits, defined as compounds that showed ≥ 50% inhibition at 2.6 μM, were retested for confirmation at 2.6, 1.3 and 0.65 μM. For the GFP high-content imaging-based screens with late-stage GAMs, compounds were diluted to a final assay concentration of 2 μM in 0.4% DMSO and screened in triplicate. Due to limitations in the availability of the screening hits, not all hits could be retested for confirmation. Fresh batches of 125 hits from the late-stage screens were obtained as 2 mM 100% DMSO stock solutions from GNF-Novartis, and 24 additional hits were purchased as solids from various commercial sources (Supplementary Table 1).
Luciferase-based early-stage gametocyte inhibition assay
LUC-based screening with stage I GAMs was conducted as previously described13. Briefly, NF54Pfs16 parasites were synchronized by two cycles of 5% sorbitol treatment, and gametocytogenesis was induced by applying multiple known stimulant stress factors (a drop in Hct, higher parasitemia, and spent media). After one replication cycle under these conditions, the resulting stage I GAMs and asexual rings were separated from residual Hz-containing and later-stage GAMs by paramagnetic purification over a MACS column (Miltenyi Biotech). Medium containing 50 mM N-acetyl-D-glucosamine (to eliminate ABS parasites) was provided and refreshed daily. On day 1 of gametocytogenesis, which corresponds to day 0 of the early-stage GAM assay, the parasitemia was adjusted to 10% and the Hct was reduced to 0.5%. Parasites were seeded under sterile conditions to a volume of 45 μl per well in 384-well white luminescence plates (CulturPlate, PerkinElmer) using a Multidrop dispenser (Thermo Scientific). Experimental compounds were added to the plates at the desired concentrations in a volume of 5 μl with a final DMSO concentration of 0.4% per assay well. Two columns of each plate were used as in-plate controls, containing 0.4% DMSO (negative control, 0% inhibition) or 5 μM puromycin (positive control, 100% inhibition), respectively. Plates were sealed with gas-exchange membranes (Breathe-easy, Sigma) and incubated for 72 h at 37°C in 5% O2, 5% CO2 and 90% N2. On day 4 of gametocytogenesis, 25 μl of supernatant was removed from each well and replaced with 15 μl Steadylite solution (luciferase assay kit, PerkinElmer), using a Biomek FX (Beckman Coulter) high-throughput liquid handling system. Plates were sealed and incubated at room temperature for 60 minutes to allow for complete cell lysis, after which the plates were read using a MicroBeta Trilux luminometer (PerkinElmer).
Image-based late-stage gametocyte inhibition assay
The effect of compounds on stage IV GAMs was determined using an image-based assay54. Briefly, day 8 post-induction stage IV NF54Pfs16 GAMs were purified over a MACS column and adjusted to 10% parasitemia and 0.1% Hct. Parasites were dispensed in 45 μl volumes into 384-well imaging plates containing 5 μl of diluted compounds per well, with a final DMSO concentration of 0.4%. After 72 h incubation at 37°C in 5% O2, 5% CO2 and 90% N2, 5 μl of 0.07 μg/ml MitoTracker Red CM-H2XRos (Invitrogen) in PBS was added and incubated overnight. Parasites were imaged on an Opera QEHS confocal imaging system (PerkinElmer). Image analysis using an Acapella-based script determined the number of viable mature GAMs per imaged area of each well, as described previously54. Controls included 5 μM puromycin. Experiments were performed with triplicate technical replicates on three independent occasions.
Gametocytocidal potency
Stage-specific potency data were generated using LUC and imaging-based assays for early- and late-stage GAMs, respectively, as described above. Relevant parasite stages were exposed to serial dilutions of compounds, typically with a maximal concentration of 40 μM, in 14 to 21 serial dilution steps with three concentrations per log dose. Assays were repeated on two independent occasions. Reference compounds (puromycin, DHA, CQ, pyrimethamine and primaquine) were used as positive and negative controls and yielded IC50 values (Supplementary Table 2) that were consistent with earlier reports 13,55.
Imaging-based asexual blood stage high-throughput assay
Hit compounds with confirmed gametocytocidal activity were also evaluated for ABS activity using our imaging-based high-throughput assay57. Briefly, sorbitol-synchronized Pf parasite lines were seeded at 2% parasitemia and 0.3% Hct in 384-well clear-bottom imaging plates (CellCarrier, PerkinElmer). For this work we used 3D7 (drug-sensitive), Dd2 (resistant to CQ, MQ and pyrimethamine) and NITD609-RDd2 clone #2 (resistant to the PfATP4 inhibitor now termed KAE60951). Parasites were incubated with compounds for 72 h at 37°C in 5% O2, 5% CO2 and 90% N2. Parasites were then permeabilized using 0.01% Triton-X100 and 0.001% saponin plus 5 mM EDTA, and stained using 0.5 μg/mL 4′,6-diamidino-2-phenylindole (DAPI). Plates were kept at room temperature in the dark for a minimum of 6 h and were then imaged on an Opera confocal microplate system (PerkinElmer) using a 20′ water objective with a 405-nm excitation and 420–490-nm band pass emission filter and an exposure time of 80 msec. The experiment was performed with two independent cultures. Parasite numbers, calculated based on spot size, intensity and fluorescence over background, were determined using Acapella software57.
HEK293 cytotoxicity
Cytotoxicity of the selected gametocytocidal hits was assessed against the HEK293 human embryonic kidney cell line (ATCC #CRL-1573), maintained in DMEM medium (Life Technologies) supplemented with 10% fetal calf serum (Gibco). Cells were trypsinized, seeded into sterile 384-well plates (Falcon) at 2,000 cells per well in 45 μl media, and incubated overnight at 37°C in 5% CO2 and 95% humidity to allow adhesion. A resazurin-based metabolic assay was utilized to determine cell viability58. Briefly, test compounds were serially diluted and added to the plates to generate a dose-response with a maximal concentration of 40 μM, in wells containing a final DMSO concentration of 0.4%. Plates were incubated for 72 h under standard conditions, after which the medium was removed and replaced with 35 μl of serum-free DMEM containing 44 μM resazurin. The plates were then incubated for a further 4–6 h under standard conditions before being read on an EnVision® plate reader (PerkinElmer) using 530 nm/595 nm fluorescence excitation/emission settings. Two biological replicates were performed. The cells were certified by ATCC as being Mycoplasma-free at source.
Dual gamete formation assay
The effects of GNF-Pf-5310, GNF-Pf-5640, GNF-Pf-5660 and GNF-Pf-5668 on the formation of male and female gametes were determined using the DGFA56. Compounds were tested across a range of 12 concentrations (0.84 nM – 20 μM) and IC50 values were calculated using non-linear regression (Graphpad Prism 6). Briefly, stage V GAMs were exposed to compounds for 24 h, after which gamete formation was triggered in the presence (carry-over format) or absence (wash-out format, three washes) of compound. Male gamete formation was assessed by automated microscopic detection of exflagellation, and female gamete formation was evaluated by live immunostaining for Pfs25 surface expression59. Data were calculated from four biological replicates, with methylene blue included as a control.
Membrane-feeding assay
Late-stage GAM (12 days post GAM induction) were treated for 3 days with GNF-Pf-5660, GNF-Pf-5668 or GNF-Pf-5310 at 1′ or 3′ their late-stage GAM IC50 values (with daily medium changes including compound refreshment), and then maintained without compound for another 4 days. Methylene blue was used as a positive control at a concentration of 3′ the GAM IC50. GAMs treated with 0.25% DMSO were used as the negative control23. Stage V GAM cultures at 50% Hct and 0.01% to 0.16% parasitemia were exposed to Anopheles stephensi mosquitoes (4–6 days post-emergence) for 30 minutes using an artificial membrane feeding apparatus. For each compound and controls, 2 containers holding 25–35 parasite-exposed mosquitoes each were maintained at 26°C, 78% relative humidity with a 12 h day/night cycle. Seven or 8 days post feeding (for experiments 1 and 2, respectively), the midguts from blood-fed female mosquitoes (n=17–21 per container) were dissected and stained with 0.2% Mercurochrome to count oocyst numbers. Infection prevalence was defined as the percentage of mosquitoes harboring one or more midgut oocysts, and infection intensity was defined as the mean number of oocysts per mosquito. Midguts of mosquitoes fed with DMSO vehicle-treated GAMs showed an average of 19 oocysts. All samples were blinded for both dissection and oocyst counting.
In vivo potency of HHQs on ABS parasites
For our 4-day Peters test, four female CD-1 mice (Taconic; 5 weeks old, weighing 22–25g) per group were infected intraperitoneally with 1x106 P. berghei ANKA MRA868 ABS parasites. Treatments with 70 or 100 mg/kg of GNF-Pf-5660 or GNF-Pf-5668, 30 mg/kg DHA or solvent-only (20% DMSO in water) were administered intraperitoneally 2 h, 24 h, 48 h and 72 h after infection. Blood stage parasitemias were assessed at 96 h post infection by microscopy. Plasma compound concentrations were not measured. All experiments involving mice were reviewed and approved by the Columbia University Institutional Animal Care and Use Committee. Sample size was determined in accordance with published literature60,61 and to minimize the number of animals under experiment. All research animals were randomly allocated into treated and control groups and groups were blinded for collection and analysis of data. This research is compliant with federally regulations detailed in the Animal Welfare Act (AWA) (7 U.S.C. 2131, seq.) and the Health Research Extension Act of 1985 (Public Law 99–158).
In vivo transmission-blocking effect of HHQs
To assess the in vivo transmission-blocking effect of HHQs, four female CD-1 mice were treated with two doses of 100 mg/kg GNF-Pf-5660 or GNF-Pf-5668, 30 mg/kg DHA, or solvent only. Doses were spaced 12 h apart, starting when blood stage parasitemia after infection with 5×106 P. berghei ANKA MRA868 parasites reached 1%, in order to measure drug impact only on GAMs. We note that in P. berghei GAMs develop and become transmissible within a 24 h period. Twelve hours after the last dose, both blood stage parasitemia and the level of inhibition of male GAM exflagellation were assessed. For the exflagellation test, a tail blood sample was collected in a heparinized tube, mixed with ookinete medium (RMPI medium, 25 mM HEPES, 50 mg/l hypoxanthine, 2 g/l sodium bicarbonate and 0.5% Albumax) containing 100 μM xanthurenic acid, and evaluated by microscopy after a 10 min incubation at 20°C. Shortly after, 25–30 Anopheles stephensi mosquitoes were fed on individual mice that were anesthetized by intraperitoneal injection of 200 μl of a mixture of ketamine (17.5 mg/ml) and xylazine (2.5 mg/ml). Unfed mosquitoes were removed and engorged mosquitoes were fed daily with a 10% sugar solution and maintained at 20°C at 80% humidity for 10 days. Midguts of 10–20 mosquitoes fed per mouse were then dissected and stained with 0.5% mercurochrome for oocyst counting by microscopy. Data were analyzed in a similar manner as for the membrane-feeding assay (see above).
Selections for HHQ resistance
To select for Pf resistance to GNF-Pf5640, GNF-Pf-5660 and GNF-Pf-5668, triplicate flasks of 109 Dd2-B2 parasites were exposed to compounds at 5′ their ABS IC50 values. Briefly, medium containing the compound of interest was refreshed daily during the first 7 days, while cultures were carefully monitored to avoid overgrowth and ensure that sensitive parasites were effectively cleared. RBCs (0.5% Hct) were added on day 7 and the total culture volume was reduced by 25% each subsequent week until a 5 ml volume was obtained. Fresh RBCs (1% Hct) were added on a weekly basis from day 14 onwards, always maintaining a total Hct level of 3–4%. Once cultures appeared negative, the medium containing the compound of interest was refreshed every other day until at least 1% parasitemia was reached, after which medium was replenished daily. Resistant clones were obtained by limiting dilution62. DNA was extracted with the DNAeasy Blood & Tissue kit (Qiagen) and sequenced using paired-end reads on a Illumina HiSeq 2500 platform27. Whole-genome sequence analysis was performed at UC San Diego and Columbia University, using previously-described customized pipelines and the GATK toolkit26,63,64.
Gene editing
pfmdr1 point mutations were introduced into Dd2-B2 parasites using a two-plasmid CRISPR/Cas9 targeted gene-editing strategy27. The Cas9 plasmid encoded the Cas9 nuclease, a single guide RNA (5′-GAAAATTAATGTCCTTAAA for F1072L and S1075I, and either 5′-ATTTTAGTTTCTTATGCATT or 5′-TTACATATAGGTTTAATAAA for Y290F) that complexes the Cas9 nuclease with the target sequence, and a human dhfr cassette that mediates resistance to the WR99210 selection agent27. The donor plasmid contained the homologous pfmdr1 sequence (nucleotides 1871–3421 of the Pf3D7_0523000 coding sequence for the mutations at amino acid position 1072 and 1075, and nucleotides 383–1034 for the mutation at amino acid position 290) harboring the mutation of interest as well as 8 synonymous mutations to prevent the Cas9 nuclease from cleaving the edited gene sequence, and the blasticidin S deaminase gene cassette that mediates resistance to blasticidin. Ring-stage cultures were transfected with the Cas9 and donor plasmids as described27. Twenty-four h after electroporation, blasticidin (2 μg/ml) and WR99210 (2.5 nM) selection was initiated. Medium containing these drugs was refreshed daily for 12 days. From day 13 onwards, cultures were maintained under blasticidin pressure until parasites were microscopically observed. pfmdr1-modified clones were obtained by limiting dilution and gene editing was verified by PCR and Sanger sequencing. Transfections were performed in duplicate on 1–3 occasions until editing was achieved.
Drug susceptibility profiling of HHQ-resistant lines in ABS parasites
To assess antimalarial susceptibilities, we exposed ring-stage cultures (at an initial parasitemia of 0.2% and 1% Hct) for 72 h to a range of 10 concentrations (plated in duplicate), in a 96-well format. Dd2-B2 parasites were included as a parental control. Parasite survival was assessed by flow cytometry on an Accuri C6 (BD Biosciences) using SYBR Green and MitoTracker Deep Red FM (Life technologies) as nuclear and vital dyes65. Assays were performed on 3–10 separate occasions (Supplementary Table 5). Growth inhibition data were plotted using non-linear regression to calculate IC50 values (using Graphpad Prism 6). IC50 values were compared between the selected or edited lines and their parental controls (Dd2-B2, FCB or NF54Pfs16) using an unequal variance t-test, also referred to as the Welch’s t-test, to correct for differences in number of repeats per group. A Bonferroni-Dunn correction for multiple comparisons was performed when applicable.
Drug susceptibility of HHQ-resistant lines at GAM stages
The NF54selY290F line was tested together with a wild-type NF54 parental control for GAM susceptibility to the 3 HHQs using a previously described parasite lactate dehydrogenase-based assay66.
PfMDR1 modeling
The Pf 3D7 PfMDR1 amino acid sequence (accession number XP_001351787) was used to develop a structural model. Membrane protein topology was predicted using TOPCONS67. Residues 51–350 and 781–1090, based on the topology prediction, were used for homology detection and structure prediction with the HHpred tool68. The best ranked hits (Prob=100) were considered and the ABC transporter structure of Thermotoga maritima (PDB 4Q4H) was used to model PfMDR1, using MODELLER because of its apo state69. PfMDR1 TMDs were refined using WinCoot70 and assembled by superimposition on the 4Q4H template using the visualization software Yasara71. Docking simulations of compounds GNF-Pf-5640, GNF-Pf-5660 and GNF-Pf-5668 were performed using DockingServer72, with residues 290, 1072 and 1075 seeded in a 15 ångström box at default conditions.
Heme fractionation assay
Heme fractionation analysis was performed as described32. Briefly, early ring-stage sorbitol-synchronized Dd2-B2 and PfMDR1selY290F parasites were exposed to GNF-Pf-5660, MQ or LMF at concentrations corresponding to 0.1, 0.25, 0.5, 1 and 2′ the ABS IC5032. After a 32 h incubation, cells were saponin-lysed and trophozoites were subjected to a series of lysis, solubilization, and differential centrifugation steps to obtain fractions that contain Hb, free heme or Hz. The UV-visible spectra of heme as Fe(III)heme-pyridine was analyzed in each of these fractions and compared to a heme standard curve for accurate quantification. These quantities were subsequently normalized using the number of analyzed cells, as determined by flow cytometry of an aliquot of exposed cells. Two-tailed t-tests were used to assess significance32.
Western blotting
Sorbitol-synchronized parasites were exposed to 5′ the IC50 of GNF-Pf-5660, WR99210, MQ, or LMF for 5 h starting at the late ring stage, i.e. before parasites entered the mid-stage trophozoites that have maximal rates of Hb degradation and Hz formation. RBCs were then saponin-lysed and parasites washed five times in 1′PBS. The parasite pellet was solubilized in SDS-PAGE sample buffer and sonicated for 10 cycles of 10 s. Samples were then boiled for 10 min at 90°C and 3.5′106 parasites were loaded on a 4–20% Criterion TGX precast midi protein gel (Bio-Rad). After transferring the resolved proteins to a nitrocellulose membrane, membranes were blocked for 1.5 h in TBS-Tween 0.1% (TBS-T) + 5% blocking grade blocker (Bio-Rad). Membranes were cut in two at the 37 kDa marker position and the lower and upper part were incubated overnight at 4°C in TBS-T + 1% blocker and a 1:5,000 dilution of rabbit anti-human Hb antibody (Sigma #H4890, lot 124M4788V) or a 1:2,000 dilution of β-actin monoclonal antibody (Fisher #AM4302), respectively. After washing in TBS-T, membranes were incubated in TBS-T + 1% blocker and a 1:10,000 dilution of goat anti-rabbit IgG conjugated to horseradish peroxidase (GE Healthcare #RPN4301) to detect Hb, or a 1:10,000 dilution of sheep anti-mouse Ig coupled to horseradish peroxidase (GE Healthcare #NXA931) to detect β-actin. Proteins bands were detected using the Pierce ECL Western Blotting substrate (ThermoScientific), blue basic autoradiography film (Genemate) and the Nikon X-OMAT 2000. Hb band intensities were calculated in ImageJ, normalized for loading with the β-actin signal and compared to the untreated controls. Experiments were repeated on 3–4 independent occasions.
Data and materials availability
All data generated or analyzed during this study are included in this published article and its supplementary information files. Requests for materials should be addressed to the corresponding author.
Supplementary Material
Acknowledgments
Funding information
We gratefully acknowledge the Bill and Melinda Gates Foundation (grant OPP1040399 to DAF and VMA and grant OPP1054480 to EAW and DAF), the National Institutes of Health (grant R01 AI103058 to EAW and DAF and R01 AI110329 to TJE), the Australian Research Council (LP120200557 to VMA), and the Medicines for Malaria Venture for their continued support. PEF and MIV are supported by the Northern Portugal Regional Operational Programme (NORTE 2020), under the Portugal 2020 Partnership Agreement, through the European Regional Development Fund (FEDER).
We gratefully thank Thierry T. Diagana (Novartis Institute for Tropical Diseases, Singapore) for provision of the compounds and the Red Cross (Australia and US) for the provision of human blood for cell cultures. We would also like to thank Dr. Graeme Stevenson for assistance with the triaging of compounds following screening.
Footnotes
Contributions
L.L. and S.D. screened the Novartis-GNF Malaria Box against early- and late-stage GAMs using LUC or GFP-imaging technologies, assayed ABS parasites and mammalian cells, and confirmed compound potency. L.L., S.D. and V.M.A. analyzed screening data and selected the HHQs for further evaluation. T.L., K.L.S. and S.L.H. contributed in vitro membrane feeding and male gametocyte exflagellation assay data. A.R., R.E.S. and M.D. contributed the DGFA data. M.V. and T.R.S.K. performed in vivo efficacy studies. K.R., M.V. and T.R.S.K. performed in vivo transmission-blocking studies. S.G. performed selections for HHQ-resistant lines, which were cloned by O.L. and M.V. Whole-genome sequence analysis was performed by V.C., P.P.H. and E.A.W. Gene editing of pfmdr1 was performed by M.V. with help from C.N. and D.A.F. Susceptibility assays on resistant lines with clinical and experimental antimalarials was performed by M.V. with help from J.M.M. Early- and late-stage GAM susceptibility testing was performed by G.S. and P.A. Heme fractionation data were provided by J.M.C. and T.J.E. PfMDR1 modeling studies were performed by P.E.F and M.I.V. Data were compiled by M.V., L.L., and D.A.F., who wrote the manuscript with input from V.M.A. All authors approved of the final version.
Competing interests
The authors declare no competing financial interests.
References
- 1.World Health Organisation. World Malaria Report 2016. 2016 http://www.who.int/malaria/publications/world-malaria-report-2016.
- 2.Wells TNC, van Huijsduijnen RH, Van Voorhis WC. Malaria medicines: a glass half full? Nat Rev Drug Discov. 2015;14:424–442. doi: 10.1038/nrd4573. [DOI] [PubMed] [Google Scholar]
- 3.Sinden RE. Targeting the parasite to suppress malaria transmission. Adv Parasitol. 2017;97:147–185. doi: 10.1016/bs.apar.2016.09.004. [DOI] [PubMed] [Google Scholar]
- 4.Josling GA, Llinás M. Sexual development in Plasmodium parasites: knowing when it’s time to commit. Nat Rev Microbiol. 2015;13:573–587. doi: 10.1038/nrmicro3519. [DOI] [PubMed] [Google Scholar]
- 5.Bousema T, Drakeley C. Epidemiology and infectivity of Plasmodium falciparum and Plasmodium vivax gametocytes in relation to malaria control and elimination. Clin Microbiol Rev. 2011;24:377–410. doi: 10.1128/CMR.00051-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cowman AF, Healer J, Marapana D, Marsh K. Malaria: Biology and disease. Cell. 2016;167:610–624. doi: 10.1016/j.cell.2016.07.055. [DOI] [PubMed] [Google Scholar]
- 7.Dicko A, et al. Primaquine to reduce transmission of Plasmodium falciparum malaria in Mali: a single-blind, dose-ranging, adaptive randomised phase 2 trial. Lancet Infect Dis. 2016;16:674–684. doi: 10.1016/S1473-3099(15)00479-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.White NJ. Primaquine to prevent transmission of falciparum malaria. Lancet Infect Dis. 2013;13:175–81. doi: 10.1016/S1473-3099(12)70198-6. [DOI] [PubMed] [Google Scholar]
- 9.Tanaka TQ, et al. A quantitative high throughput assay for identifying gametocytocidal compounds. Mol Biochem Parasitol. 2013;188:20–25. doi: 10.1016/j.molbiopara.2013.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cevenini L, et al. Multicolor bioluminescence boosts malaria research: Quantitative dual-color assay and single-cell imaging in Plasmodium falciparum parasites. Anal Chem. 2014;86:8814–8821. doi: 10.1021/ac502098w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sanders NG, Sullivan DJ, Mlambo G, Dimopoulos G, Tripathi AK. Gametocytocidal screen identifies novel chemical classes with Plasmodium falciparum transmission blocking activity. PLoS One. 2014;9:1–13. doi: 10.1371/journal.pone.0105817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bolscher JM, et al. A combination of new screening assays for prioritization of transmission-blocking antimalarials reveals distinct dynamics of marketed and experimental drugs. J Antimicrob Chemother. 2014;70:1357–1366. doi: 10.1093/jac/dkv003. [DOI] [PubMed] [Google Scholar]
- 13.Lucantoni L, Duffy S, Adjalley SH, Fidock DA, Avery VM. Identification of MMV malaria box inhibitors of Plasmodium falciparum early-stage gametocytes using a luciferase-based high-throughput assay. Antimicrob Agents Chemother. 2013;57:6050–6062. doi: 10.1128/AAC.00870-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sun W, et al. Chemical signatures and new drug targets for gametocytocidal drug development. Sci Rep. 2014;4:3743. doi: 10.1038/srep03743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bowman JD, et al. Antiapicoplast and gametocytocidal screening to identify the mechanisms of action of compounds within the malaria box. Antimicrob Agents Chemother. 2014;58:811–819. doi: 10.1128/AAC.01500-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Plouffe DM, et al. High-throughput assay and discovery of small molecules that interrupt malaria transmission. Cell Host Microbe. 2016;19:114–126. doi: 10.1016/j.chom.2015.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Miguel-Blanco C, et al. Imaging-based high-throughput screening assay to identify new molecules with transmission-blocking potential against Plasmodium falciparum female gamete formation. Antimicrob Agents Chemother. 2015;59:3298–3305. doi: 10.1128/AAC.04684-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lucantoni L, Loganathan S, Avery VM. The need to compare: assessing the level of agreement of three high-throughput assays against Plasmodium falciparum mature gametocytes. Sci Rep. 2017;7:45992. doi: 10.1038/srep45992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Delves MJ, et al. Routine in vitro culture of P falciparum gametocytes to evaluate novel transmission-blocking interventions. Nat Protoc. 2016;11:1668–80. doi: 10.1038/nprot.2016.096. [DOI] [PubMed] [Google Scholar]
- 20.de Koning-Ward TF, Gilson PR, Crabb BS. Advances in molecular genetic systems in malaria. Nat Rev Microbiol. 2015;13:373–387. doi: 10.1038/nrmicro3450. [DOI] [PubMed] [Google Scholar]
- 21.Flannery EL, Fidock DA, Winzeler EA. Using genetic methods to define the targets of compounds with antimalarial activity. J Med Chem. 2013;56:7761–7771. doi: 10.1021/jm400325j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Plouffe D, et al. In silico activity profiling reveals the mechanism of action of antimalarials discovered in a high-throughput screen. Proc Natl Acad Sci USA. 2008;105:9059–64. doi: 10.1073/pnas.0802982105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Adjalley SH, et al. Quantitative assessment of Plasmodium falciparum sexual development reveals potent transmission-blocking activity by methylene blue. Proc Natl Acad Sci USA. 2011;108:E1214–E1223. doi: 10.1073/pnas.1112037108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Coulibaly B, et al. Efficacy and safety of triple combination therapy with artesunate-amodiaquine-methylene blue for falciparum malaria in children: a randomized controlled trial in Burkina Faso. J Infect Dis. 2015;211:689–97. doi: 10.1093/infdis/jiu540. [DOI] [PubMed] [Google Scholar]
- 25.Fidock DA, Rosenthal PJ, Croft SL, Brun R, Nwaka S. Antimalarial drug discovery: efficacy models for compound screening. Nat Rev Drug Discov. 2004;3:509–20. doi: 10.1038/nrd1416. [DOI] [PubMed] [Google Scholar]
- 26.Corey VC, et al. A broad analysis of resistance development in the malaria parasite. Nat Commun. 2016;7:11901. doi: 10.1038/ncomms11901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ng CL, et al. CRISPR-Cas9-modified pfmdr1 protects Plasmodium falciparum asexual blood stages and gametocytes against a class of piperazine-containing compounds but potentiates artemisinin-based combination therapy partner drugs. Mol Microbiol. 2016;101:381–93. doi: 10.1111/mmi.13397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ferreira PE, et al. PfMDR1: Mechanisms of transport modulation by functional polymorphisms. PLoS One. 2011;6:3–10. doi: 10.1371/journal.pone.0023875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sidhu ABS, Valderramos SG, Fidock D. A pfmdr1 mutations contribute to quinine resistance and enhance mefloquine and artemisinin sensitivity in Plasmodium falciparum. Mol Microbiol. 2005;57:913–26. doi: 10.1111/j.1365-2958.2005.04729.x. [DOI] [PubMed] [Google Scholar]
- 30.Veiga MI, et al. Globally prevalent PfMDR1 mutations modulate Plasmodium falciparum susceptibility to artemisinin-based combination therapies. Nat Commun. 2016;7:11553. doi: 10.1038/ncomms11553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sá JM, et al. Geographic patterns of Plasmodium falciparum drug resistance distinguished by differential responses to amodiaquine and chloroquine. Proc Natl Acad Sci USA. 2009;106:18883–9. doi: 10.1073/pnas.0911317106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Combrinck JM, et al. Optimization of a multi-well colorimetric assay to determine haem species in Plasmodium falciparum in the presence of anti-malarials. Malar J. 2015;14:253. doi: 10.1186/s12936-015-0729-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Combrinck JM, et al. Insights into the role of heme in the mechanism of action of antimalarials. ACS Chem Biol. 2013;8:133–137. doi: 10.1021/cb300454t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fukami T, Yokoi T. The emerging role of human esterases. Drug Metab Pharmacokinet. 2012;27:466–477. doi: 10.2133/dmpk.dmpk-12-rv-042. [DOI] [PubMed] [Google Scholar]
- 35.Blagborough AM, et al. Transmission-blocking interventions eliminate malaria from laboratory populations. Nat Commun. 2013;4:1812–1817. doi: 10.1038/ncomms2840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.de Mello CX, et al. Comparison of the sensitivity of imprint and scraping techniques in the diagnosis of American tegumentary leishmaniasis in a referral centre in Rio de Janeiro, Brazil. Parasitol Res. 2011;109:927–33. doi: 10.1007/s00436-011-2335-2. [DOI] [PubMed] [Google Scholar]
- 37.Gil JP, Krishna S. pfmdr1 (Plasmodium falciparum multidrug drug resistance gene 1): a pivotal factor in malaria resistance to artemisinin combination therapies. Expert Rev Anti Infect Ther. 2017;15:527–543. doi: 10.1080/14787210.2017.1313703. [DOI] [PubMed] [Google Scholar]
- 38.Sigala PA, Goldberg DE. The peculiarities and paradoxes of Plasmodium heme metabolism. Annu Rev Microbiol. 2014;68:259–278. doi: 10.1146/annurev-micro-091313-103537. [DOI] [PubMed] [Google Scholar]
- 39.Tilley L, Straimer J, Gnädig NF, Ralph SA, Fidock DA. Artemisinin action and resistance in Plasmodium falciparum. Trends Parasitol. 2016;32:682–696. doi: 10.1016/j.pt.2016.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Venkatesan M, et al. Polymorphisms in Plasmodium falciparum chloroquine resistance transporter and multidrug resistance 1 genes: parasite risk factors that affect treatment outcomes for P falciparum malaria after artemether-lumefantrine and artesunate-amodiaquine. Am J Trop Med Hyg. 2014;91:833–43. doi: 10.4269/ajtmh.14-0031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Petersen I, Eastman R, Lanzer M. Drug-resistant malaria: molecular mechanisms and implications for public health. FEBS Lett. 2011;585:1551–62. doi: 10.1016/j.febslet.2011.04.042. [DOI] [PubMed] [Google Scholar]
- 42.Petersen I, et al. Balancing drug resistance and growth rates via compensatory mutations in the Plasmodium falciparum chloroquine resistance transporter. Mol Microbiol. 2015;97:381–95. doi: 10.1111/mmi.13035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Roberts L, Egan TJ, Joiner KA, Hoppe HC. Differential effects of quinoline antimalarials on endocytosis in Plasmodium falciparum. Antimicrob Agents Chemother. 2008;52:1840–1842. doi: 10.1128/AAC.01478-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wunderlich J, Rohrbach P, Dalton JP. The malaria digestive vacuole. Front Biosci (Schol Ed) 2012;1:1424. doi: 10.2741/s344. [DOI] [PubMed] [Google Scholar]
- 45.Rohrbach P, et al. Genetic linkage of pfmdr1 with food vacuolar solute import in Plasmodium falciparum. EMBO J. 2006;25:3000–3011. doi: 10.1038/sj.emboj.7601203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Reiling SJ, Rohrbach P. Monitoring PfMDR1 transport in Plasmodium falciparum. Malar J. 2015;14:270. doi: 10.1186/s12936-015-0791-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Brunner R, et al. UV-triggered affinity capture identifies interactions between the Plasmodium falciparum multidrug resistance protein 1 (PfMDR1) and antimalarial agents in live parasitized cells. J Biol Chem. 2013;288:22576–22583. doi: 10.1074/jbc.M113.453159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pleeter P, Lekostaj JK, Roepe PD. Purified Plasmodium falciparum multi-drug resistance protein (PfMDR 1) binds a high affinity chloroquine analogue. Mol Biochem Parasitol. 2010;173:158–61. doi: 10.1016/j.molbiopara.2010.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cowman AF, Karcz S, Galatis D, Culvenor JG. A P-glycoprotein homologue of Plasmodium falciparum is localized on the digestive vacuole. J Cell Biol. 1991;113:1033–1042. doi: 10.1083/jcb.113.5.1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tumwebaze P, et al. Changing antimalarial drug resistance patterns identified by surveillance at three sites in Uganda. J Infect Dis. 2017;215:631–635. doi: 10.1093/infdis/jiw614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rottmann M, et al. Spiroindolones, a potent compound class for the treatment of malaria. Science. 2010;329:1175–1180. doi: 10.1126/science.1193225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sidhu SBA, Uhlemann A, Valderramos SG, Krishna S, Fidock DA. Decreasing pfmdr1 copy number in Plasmodium falciparum malaria heightens susceptibility to mefloquine, lumefantrine, halofantrine, quinine and artemisinin. J Infect Dis. 2006;194:528–535. doi: 10.1086/507115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Novartis GNF Malariabox. https://www.ebi.ac.uk/chemblntd.
- 54.Duffy S, Avery VM. Identification of inhibitors of Plasmodium falciparum gametocyte development. Malar J. 2013;12:408. doi: 10.1186/1475-2875-12-408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lucantoni L, Fidock DA, Avery VM. A luciferase-based, high-throughput assay for screening and profiling transmission-blocking compounds against Plasmodium falciparum gametocytes. Antimicrob Agents Chemother. 2016;60:2097–2107. doi: 10.1128/AAC.01949-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ruecker A, et al. A male and female gametocyte functional viability assay to identify biologically relevant malaria transmission-blocking drugs. Antimicrob Agents Chemother. 2014;58:7292–7304. doi: 10.1128/AAC.03666-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Duffy S, Avery VM. Development and optimization of a novel 384-well anti-malarial imaging assay validated for high-throughput screening. Am J Trop Med Hyg. 2012;86:84–92. doi: 10.4269/ajtmh.2012.11-0302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fletcher S, Avery VM. A novel approach for the discovery of chemically diverse anti-malarial compounds targeting the Plasmodium falciparum coenzyme A synthesis pathway. Malar J. 2014;13:343. doi: 10.1186/1475-2875-13-343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Delves MJ, et al. Male and female Plasmodium falciparum mature gametocytes show different responses to antimalarial drugs. Antimicrob Agents Chemother. 2013;57:3268–3274. doi: 10.1128/AAC.00325-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.McNamara CW, et al. Targeting Plasmodium PI(4)K to eliminate malaria. Nature. 2013;504:248–53. doi: 10.1038/nature12782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pereira MR, et al. In vivo and in vitro antimalarial properties of azithromycin-chloroquine combinations that include the resistance reversal agent amlodipine. Antimicrob Agents Chemother. 2011;55:3115–24. doi: 10.1128/AAC.01566-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Goodyer ID, Taraschi TF. Plasmodium falciparum: a simple, rapid method for detecting parasite clones in microtiter plates. Exp Parasitol. 1997;86:158–60. doi: 10.1006/expr.1997.4156. [DOI] [PubMed] [Google Scholar]
- 63.Manary MJ, et al. Identification of pathogen genomic variants through an integrated pipeline. BMC Bioinformatics. 2014;15:63–76. doi: 10.1186/1471-2105-15-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hameed PS, et al. Triaminopyrimidine is a fast-killing and long-acting antimalarial clinical candidate. Nat Commun. 2015;6:6715. doi: 10.1038/ncomms7715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ekland EH, Schneider J, Fidock DA. Identifying apicoplast-targeting antimalarials using high-throughput compatible approaches. FASEB J. 2011;25:3583–93. doi: 10.1096/fj.11-187401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.D’Alessandro S, et al. A Plasmodium falciparum screening assay for anti-gametocyte drugs based on parasite lactate dehydrogenase detection. J Antimicrob Chemother. 2013;68:2048–2058. doi: 10.1093/jac/dkt165. [DOI] [PubMed] [Google Scholar]
- 67.Tsirigos KD, Peters C, Shu N, Lukas K, Elofsson A. The TOPCONS web server for consensus prediction of membrane protein topology and signal peptides. Nucleic Acids Res. 2015;43:W401–407. doi: 10.1093/nar/gkv485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Alva V, et al. The MPI bioinformatics Toolkit as an integrative platform for advanced protein sequence and structure. Nucleic Acids Res. 2016;44:W410–415. doi: 10.1093/nar/gkw348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sali A, Potterton L, Yuan F, Van Vlijmen H, Karplus M. Evaluation of Comparative Protein Modeling by MODELLER. Proteins. 1995;23:318–326. doi: 10.1002/prot.340230306. [DOI] [PubMed] [Google Scholar]
- 70.Emsley P, Lohkamp B. Features and development of Coot. Acta Crystallogr Sect D. 2010;D66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Krieger E, Vriend G. Models @ Home: distributed computing in bioinformatics using a screensaver based approach. Bioinformatics. 2002;18:315–318. doi: 10.1093/bioinformatics/18.2.315. [DOI] [PubMed] [Google Scholar]
- 72.Bikadi Z, Hazai E. Application of the PM6 semi-empirical method to modeling proteins enhances docking accuracy of AutoDock. J Cheminform. 2009;1:15. doi: 10.1186/1758-2946-1-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.