

Abstract
Protein glycosylation is one of the most important posttranslational modifications. Numerous biological functions are related to protein glycosylation. However, analytical challenges remain in the glycoprotein analysis. To overcome the challenges associated with glycoprotein analysis, many analytical techniques were developed in recent years. Enrichment methods were used to improve the sensitivity of detection while HPLC and mass spectrometry methods were developed to facilitate the separation of glycopeptides/proteins and enhance detection, respectively. Fragmentation techniques applied in modern mass spectrometers allow the structural interpretation of glycopeptides/proteins while automated software tools started replacing manual processing to improve the reliability and throughout of the analysis. In this chapter, the current methodologies of glycoprotein analysis were discussed. Multiple analytical techniques are compared, and advantages and disadvantages of each technique are highlighted.
Keywords: Proteomics, Glycoproteomics, Glycoprotein, Enrichment, LC-MS/MS
7.1 Proteomics, posttranslational modifications (PTMs)
Proteins perform numerous functions in living organisms, including, but not limited to, (i) catalyzing reactions, (ii) replicating DNA, (iii) responding to stimuli, and (iv) transporting molecules (Y. Zhang, Fonslow, Shan, Baek, & Yates, 2013). The importance of identifying the proteins present in cell and tissue samples, so called proteomics studies, becomes readily apparent with aberrant cells; often times a splice variant or unique post-translational modification (PTM) lends insight into the mutant phenotype.
The term “proteomics” originated from the combination of “protein” and “genomics” in the 1990s (James, 1997; Wilkins, Pasquali, Appel, Ou, Golaz, Sanchez, et al., 1996). Proteomics focuses on the characterization and quantification of all the proteins originating in a biological system, including cellular location, splice-variants, interactions, and PTMs. The functional capabilities of proteins are often expanded by PTMs which add to the chemical repertoire of proteiiinsss by adding small chemical groups (such as phosphate, simple carbohydrates, acetate, and alkyl groups) or larger molecules (such as ubiquitin, small ubiquitin-like modifier, and complex glycans).
The essential biological functions of protein PTMs require a comprehensive characterization for a complete proteomics analysis. This is a rather daunting task as it has been estimated that the 20,000 genes in the human genome encode approximately 100,000 proteoforms (Gstaiger & Aebersold, 2009; L. M. Smith, Kelleher, & Consortium for Top Down, 2013); to further complicate analysis, over 400 protein PTMs have been identified (Gstaiger et al., 2009). The accurate proteome and PTM characterization of these complex samples remains a challenge.
One of the most commonly used proteomics strategies is bottom-up or shotgun proteomics (Wolters, Washburn, & Yates, 2001; Yates, 2004); this approach analyzes peptides to gain insight into proteins. This is an indirect measurement of proteins through peptides derived from proteolytic digestion of intact proteins. Trypsin is the most commonly used protease in shotgun proteomics studies because of its high cleavage specificity to the carboxyl side of arginine and lysine (Vandermarliere, Mueller, & Martens, 2013). Endoproteinase Lys-C, endoproteinase Glu-C, and elastase are other commonly used proteases; they are typically employed when tryptic peptides do not yield the information desired (Keil, 1987; Siepen, Keevil, Knight, & Hubbard, 2007). The combination of both highly selective and non-selective proteases improves protein and PTMs identification (MacCoss, McDonald, Saraf, Sadygov, Clark, Tasto, et al., 2002; Xu, Wong, Kashina, & Yates, 2009).
Peptide mixtures are routinely analyzed with a liquid chromatography sample separation step coupled to a tandem mass spectrometer detector (LC-MS/MS). Briefly, the complex peptide mixture is separated via liquid chromatography immediately followed by mass spectrometry analysis. The isolated peptide ions are fragmented to yield product ions; these product ions are then used to sequence the peptide. A database search with the peptide sequence yields a protein identification. The quantification is generally achieved by relative quantification, such as isotope labeling strategy and label free quantification.
7.2 Challenges associated with glycoprotein analysis
Glycosylation is one of the most prevalent protein PTMs; it has been estimated that approximately 50% of human proteins are glycosylated (Apweiler, Hermjakob, & Sharon, 1999). Glycans are either covalently bound to asparagine (Asp, N) to form an N-linked glycan or serine (Ser, S) and threonine (Thr, T) residues to form an O-linked glycan (Moremen, Tiemeyer, & Nairn, 2012). Numerous biological functions are related to protein glycosylation, including cell-cell signaling, protein stability, protein localization, and immune response (Helenius & Aebi, 2001; O'Connor & Imperiali, 1996; Varki, 1993). The importance of protein glycosylation is readily apparent in human diseases such as inflammatory diseases (Campbell, Yu, & Rhodes, 2001; Dube & Bertozzi, 2005; Song, Zhu, Hammoud, & Mechref, 2014), rheumatoid arthritis (Elliott, Elliott, Gallagher, McGuire, Field, & Smith, 1997; K. D. Smith, Pollacchi, Field, & Watson, 2002), Alzheimer’s disease (Botella-Lopez, Burgaya, Gavin, Garcia-Ayllon, Gomez-Tortosa, Pena-Casanova et al., 2006; J. Z. Wang, Grundke-Iqbal, & Iqbal, 1996), Pompe disease (Dasouki, Jawdat, Almadhoun, Pasnoor, McVey, Abuzinadah et al., 2014), and cancer (Mechref, Hu, Garcia, & Hussein, 2012; T. H. Tsai, Song, Zhu, Di Poto, Wang, Luo et al., 2015).
Glycoproteomics, a subset of proteomics focused on glycoproteins, focuses on determining changes in glycoprotein macroheterogeneity (glycosylation site occupancy) and microheterogeneity (different glycoforms attached to the same glycosylation site). Currently, several technical challenges are associated with the analysis of glycoproteins derived from biological samples. First, the highly hydrophilic nature of glycans reduces their ionization efficiency, which gives one the impression that they are not abundant. To overcome this reduced ionization efficiency issue, an enrichment step is usually employed.
Second, glycoproteins commonly have more than one glycosylation sites and/or other PTMs, which greatly complicates the characterization of site-specific glycosylation. In order to acquire macroheterogeneity and microheterogeneity information on glycoproteins, a bottom-up strategy is commonly employed; proteases reduce a complex glycoprotein mixture into a simpler glycopeptide sample. LC-MS/MS is currently the most common and powerful analytical platform for the analysis of protein glycosylation via glycopeptide analysis.
Third, the MS/MS spectra of glycopeptides contain information for both the peptide backbone and the glycan moieties. Several MS/MS fragmentation techniques, often sequentially, are utilized to identify the peptide backbone, glycosylation site, and diagnostic product ions indicative of a glycopeptide.
Fourth, the interpretation of MS/MS spectra requires a significant amount of data processing time and experienced personnel; the proteomics data processing workflow is relatively robust whereas the glycoprotemics data processing is still in its infancy. Accurate and precise software tools for automated data analysis are highly desired. Figure 7.1 summarizes the general work flow of glycoproteomics analysis involving (i) cell lysis, (ii) protein extraction, (iii) glycoprotein enrichment, (iv) LC-MS/MS analysis, (v) data analysis, and (vi) validation (Song & Mechref, 2015). Current enrichment techniques, separation platforms, fragmentation methods and software tools will be reviewed in this paper.
Figure 7.1. Flowchart highlighting typical glycoproteomics work flow.
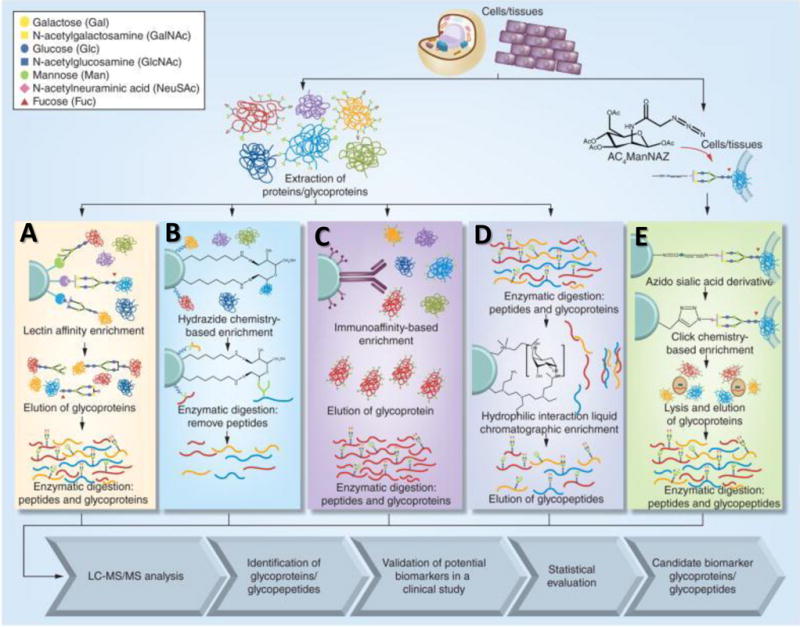
Glycopeptide enrichment techniques: A. lectin affinity, B. hydrazide chemistry, C. immunoaffinity, D. HILIC, and E. click-chemistry. Figure reproduced with permission (Song et al., 2015).
7.3 Enrichment techniques of glycopeptides
The low abundance of glycoproteins in biological samples and the poor ionization efficiency, when analyzed by electrospray ionization, remain challenges in protein glycosylation analysis. In order to overcome these challenges, enrichment techniques are often employed with biological specimens (Ongay, Boichenko, Govorukhina, & Bischoff, 2012); these enrichment techniques are reviewed below.
7.3.1 Lectin affinity chromatographic enrichment
Lectin affinity chromatographic enrichment has been utilized for glycoproteomic analysis of lung (Heo, Lee, Ryoo, Park, & Cho, 2007; Hongsachart, Huang-Liu, Sinchaikul, Pan, Phutrakul, Chuang, et al., 2009), breast (Y. Wang, Ao, Vuong, Konanur, Miller, Goodison, et al., 2008; Z. Yang, Harris, Palmer-Toy, & Hancock, 2006), and liver cancers (Qin, Chen, Sun, Wang, Peng, Xie et al., 2013) Figure 7.1A. Lectins are proteins that specifically bind glycans; the different lectins have different PTMs which differentiate their glycan binding specificity. During enrichment, lectins bind the various glycopeptides through hydrogen bonding and hydrophilic interactions with saccharide units to retain glycopeptides (Madera, Mann, Mechref, & Novotny, 2008; Mechref, Madera, & Novotny, 2008; Ongay et al., 2012). Lectin affinity chromatography selectively enriches for a single type of glycan structure. For example, Concanavalin A (Con A) selectively binds glycans that possess mannose saccharide units, Sambucus Nigra Agglutinin (SNA) will bind N-acetyl neuraminic acid (NeuAc; Sialic Acid) units, and AleuriaAurantia lectin (AAL) will bind fucose units. Table 7.1 lists a few of the lectin materials that are commercially available. The major disadvantage of lectin separation is that for any single lectin, only a single glycopeptide with a specific saccharide unit can be purified. To overcome this, recent studies have used a combination of lectins for broader glycan enrichment (Z. Yang & Hancock, 2004).
Table 1.
Comercially Available Lectin Materials
| Lectin | Source | Glycan Specificity |
|---|---|---|
| Aleuria aurantia lectin (AAL) | Aleuria aurantia mushrooms | Fucα6GlcNAc |
| Concanavalin A (Con A) | Canavalia ensiformis (Jack Bean) seeds | αMan, αGlc |
| Galanthus nivalis agglutinin (GNL, GNA) | Galanthus nivalis (Snowdrop) bulbs | αMan |
| Jacalin lectin (Jacalin, AIL) | Artocarpus integrifolia (Jackfruit) seeds | Galβ3GlcNAc |
| Lens culinaris agglutinin (LCA, LcH) | Lens culinaris (lentil) seeds | αMan, αGlc |
| Maackia amurensis lectin I (MAL I, MAL) | Maackia amurensis seeds | Galβ4GlcNAc |
| Peanut agglutinin (PNA) | Arachis hypogaea peanuts | Galβ3GlcNAc |
| Sambucus nigra agglutinin (SNA, EBL) | Sambucus nigra (Elderberry) bark | Neu5Acα6Gal/GalNAc |
| Wheat Germ agglutinin (WGA) | Triticum vulgaris (wheat germ) | GlcNAc |
Common Vendors: Associates of Cape Cod Inc., Biomeda, Biotinylated Lectins, EMD Biosciences, Europa Bioproducts, EY Laboratories, Glycorex, Glygen Corporation, Invitrogen, Prozyme, Sigma-Aldrich, U.S. Biologicals, Vector Laboratories, Worthington Biochemical Company
Table adapted from [28] with permission.
Ref [29] for a more through review on lectin materials
7.3.2 Covalent hydrazide chemistry enrichment
Hydrazide chemistry enrichment has also been used for glycoprotein purification (Nilsson, Ruetschi, Halim, Hesse, Carlsohn, Brinkmalm, et al., 2009; Ongay et al., 2012; H. Zhang, Li, Martin, & Aebersold, 2003) Figure 7.1B. In this technique, the glycan moieties on glycopeptides are first oxidized to form aldehydes. The glycans are then covalently immobilized on the solid hydrazide support. Following tryptic digestion, the non-glycosylated peptides are washed off while the glycopeptides are retained. The major disadvantage of hydrazide enrichment is that it only provides site specific information and does not allow for determination of the glycan structure due to the oxidative chemical coupling between the glycan and hydrazide solid support after release by PNGase F (Ongay et al., 2012). Hydrazide enrichment has recently been used to identify glycoprotein variations in hepatocellular carcinoma (liver cancer) (R. Chen, Tan, Wang, Wang, Yao, Dong et al., 2011; X. Li, Jiang, Zhao, Wang, Han, Zhao et al., 2013), lung (Q. K. Li, Shah, Li, Aiyetan, Chen, Yung et al., 2013; X. Zeng, Hood, Sun, Conrads, Day, Weissfeld et al., 2010), breast (Whelan, Lu, He, Yan, Saxton, Faull et al., 2009), and prostate cancers(J. Chen, Xi, Tian, Bova, & Zhang, 2013).
7.3.3 Immunoprecipitation enrichment
Immunoaffinity columns can also be used to enrich for specific glycoproteins Figure 7.1C. This type of enrichment allows for characterization of site-specific occupancies or glycan saccharide units. Since this method relies on the capability of available antibodies to recognize target glycoproteins, this approach proves to be more expensive than the previous enrichment techniques. Immunoprecipitation has been used to analyze haptoglobin samples from lung cancer (H. Y. Tsai, Boonyapranai, Sriyam, Yu, Wu, Khoo, et al., 2011; D. Wang, Hincapie, Rejtar, & Karger, 2011).
7.3.4 Hydrophilic interaction liquid chromatography enrichment
Hydrophilic Interaction Liquid Chromatography (HILIC) is an enrichment technique that has gained popularity in the last few years (Buszewski & Noga, 2012) Figure 7.1D. This enrichment technique is based on the hydrophilic interaction between the cellulose of the stationary phase and the hydroxyl groups of the glycoproteins. Peptide washing occurs in an organic solvent followed by glycopeptide elution with an aqueous buffer. HILIC enrichment is beneficial due to the sufficient removal of competing peptides from a digested biological sample. HILIC enrichment is being developed for both off-line and on-line glycoprotein studies.
7.3.5 Click chemistry enrichment
The term “click chemistry” is used to define a reaction process that is quick, easy to employ, results in a product that is easy to purify, and gives high yields (Kolb, Finn, & Sharpless, 2001) Figure 7.1E. Click chemistry was initially developed for pharmaceuticals but has since then expanded into other areas of research (Hein, Liu, & Wang, 2008). One such area is in the field of glycoproteomics. Azide modified glycan residues are inserted into cell membranes during metabolic labeling and then following the pathway of a click chemistry reaction based a copper-catalyzed azide-alkyne cycloaddition; glycoproteins are bound, and the highly abundant cytosolic proteins are removed. Glycoproteins on the cell surface of prostate cancer have been identified using this enrichment technique (L. Yang, Nyalwidhe, Guo, Drake, & Semmes, 2011).
7.3.6 Covalent boronic acid chemistry enrichment
Boronic acid chemistry is another type of a covalent separation technique that is often used in glycopeptide enrichment processes (Ongay et al., 2012) Figure 7.2A. In this method, boronic acid is used to capture 1–2 and 1–3 cis-diol groups of glycopeptides by creating a covalent bond and forming a cyclic bromate ester. The non-covalently bonded species are then washed away, and the glycopeptides can be further analyzed. The benefit of boronic acid chemistry is the ability to reverse the reaction by a change in pH for the release of the glycopeptide. Boronic acid has been functionalized to a variety of surfaces for enrichment procedures including, monoliths, mesoporous silica, magnetic particles, or gold nanoparticles (Ongay et al., 2012). Each functionalized material has shown a high specificity for glycopeptides during the enrichment process.
Figure 7.2. Boronic acid enrichment and electrostatic repulsion hydrophilic interaction chromatography enrichment.
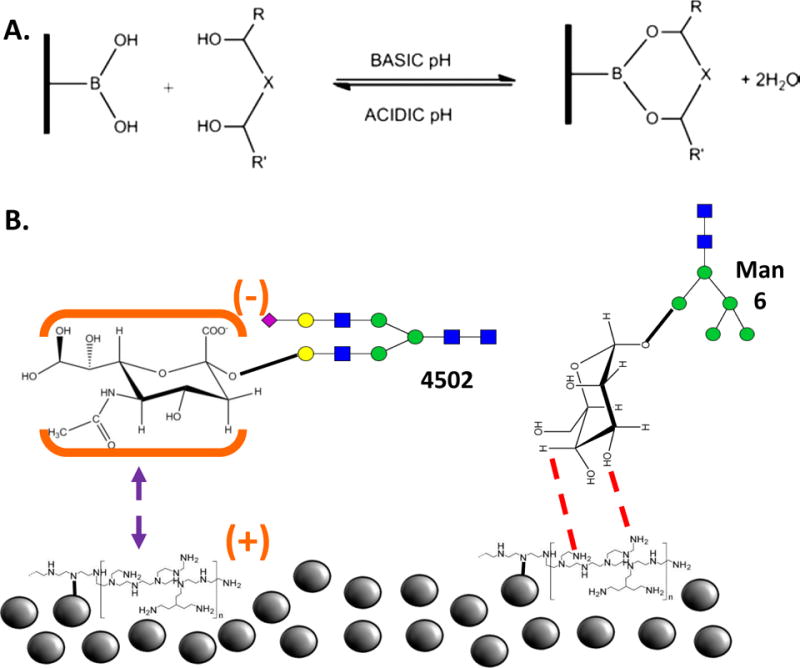
A. A covalent bond between the stationary media and the glycan hydroxyl groups allows for clean separations. Reproduced with permission (Ongay et al., 2012). B. Silica beads functionalized with polyethyleneimine attract uncharged glycans (right) while repelling charged glycans (left). Reproduced with permission (L. H. Zacharias, A. ; Song, E.; Zhao, J.; Zhu, R.; Mirzaei, P.; Mechref, Y., 2016).
7.3.7 Electrostatic repulsion hydrophilic interaction chromatography enrichment
Glycoprotein enrichment by electrostatic repulsion hydrophilic interaction chromatography (ERLIC) is a technique that takes advantage of the electrostatic charge exhibited by the glycan saccharide units (L. G. Zacharias, Hartmann, Song, Zhao, Zhu, Mirzaei et al., 2016) Figure 7.2B. In ERLIC, the stationary phase consists of silica beads with various modified positive, functional groups attached. The negative charge on a sialic acid is attracted to the positive functional groups while peptides with positive charges will be repelled. Additionally, a hydrophilic interaction occurs between the glycan and the functional group. The basic, hydrophobic peptides can, therefore,, be washed out with an organic solvent (such as acetonitrile, ACN) and the captured glycopeptides can then be eluted with a more polar solvent (such as H2O/0.5% formic acid) (Hao, Guo, & Sze, 2011).
7.3.8 Enrichment methods comparison
Many studies have compared the efficiency and specificity of different glycopeptide enrichment methods (Calvano, Zambonin, & Jensen, 2008; Ishihara, Fukuda, Morita, Takinami, Okamoto, Nishimura et al., 2011; Lewandrowski, Moebius, Walter, & Sickmann, 2006; Song et al., 2014; Wohlgemuth, Karas, Eichhorn, Hendriks, & Andrecht, 2009; L. G. Zacharias et al., 2016; C. Zhang, Ye, Xue, Shu, Zhou, Ji et al., 2016; H. Zhang, Guo, Li, Datta, Park, Yang et al., 2010; Y. Zhang, Go, & Desaire, 2008). HILIC based methods have been proved to produce the highest number of glycopeptide identification when compared to other methods (L. G. Zacharias et al., 2016; C. Zhang et al., 2016) Figure 7.3A. However, HILIC and lectin enrichment proved to be less specific than Covalent hydrazide chemistry enrichment (Whelan et al., 2009). On the other hand, different enrichment methods exhibit complimentary to others (Song et al., 2014) Figure 7.3B. Thaysen-Andersen et al. compared the glycopeptide enrichment methods using a broad spectrum of HILIC materials (ZIC-HILIC, PolyHydroxyethyl A, PolySulfethyl A, TSK Amide-80, and LudgerClean S, TiO2), graphitized carbon (Hypersil and LudgerClean EB10), and lectin affinity chromatography (ConA). The comparison showed that a comprehensive glycopeptide profile is achieved by any of the approaches (Thaysen-Andersen, Mysling, & Hojrup, 2009).
Figure 7.3. Comparison of different glycopeptide enrichment methods.
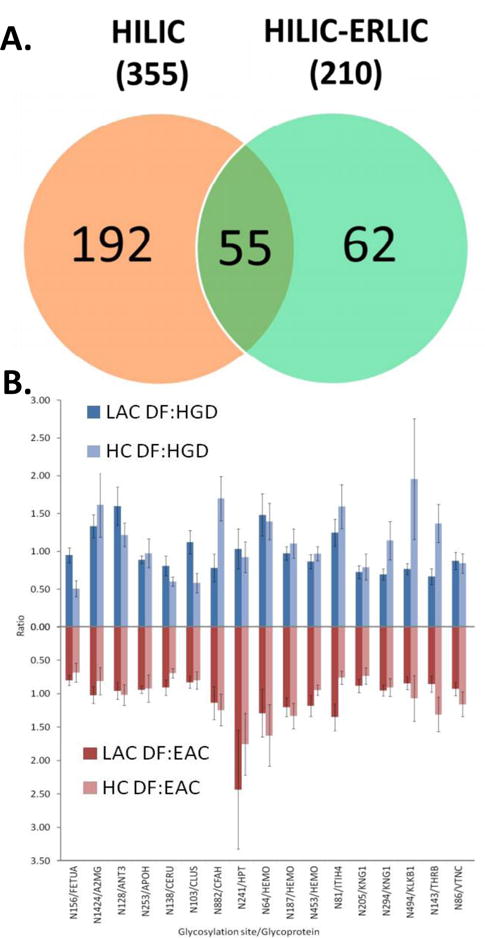
A. The Venn diagram depicts the number of identified unique glycosylation sites for HILIC and HILIC-ERLIC enrichment methods (shown bolded in the parentheses is the total number of glycopeptides identified in each case) and of those, which glycosylation sites were unique to the individual methods. Reproduced with permission (L. G. Zacharias et al., 2016). B. Comparisons of MRM quantitation for 17 common glycosylation site between lectin affinity and hydrazide chemistry enrichments in terms of ratio between different groups of biological samples. (DF: disease-free, HGD: high grade dysplasia, EAC: esophageal adenocarcinoma.) Reproduced with permission (Song et al., 2014).
7.4 High performance liquid chromatography in proteomics and glycoproteomics
High-performance liquid chromatography (HPLC) is the most commonly used separation technique in shotgun proteomic and glycoproteomic analysis because of its high resolving power, high reproducibility, and compatibility with mass spectrometry; HPLC is considered as the most efficient separation technique for large-scale peptide/glycopeptide analysis. Glycopeptide separation remains a challenge because of the intrinsic dichotomy between the hydrophobic component (peptide backbone) and the hydrophilic component (glycan). To address this, a wide range of chromatography approaches have been employed in proteomics and glycoproteomics studies, including reversed phase liquid chromatography (RPLC), hydrophilic interaction liquid chromatography (HILIC) and porous graphitized carbon LC (PGC-LC). Despite all of the improvements in sample separation, the separation of glycopeptides remains a challenge because of the isomeric heterogeneity of the glycan moieties.
7.4.1 Reversed-phase liquid chromatography (RPLC)
Reversed-phase liquid chromatography (RPLC) utilizes a non-polar stationary phase and a polar mobile phase. In the past several decades, RPLC with a C18 stationary phase has been widely used in peptides separation because of the relatively high hydrophobicity of peptides (Y. Zhang et al., 2013). Meanwhile, large scale glycopeptide separations have relied on almost exclusively on C18-RPLC (Anonsen, Vik, Egge-Jacobsen, & Koomey, 2012; Halim, Nilsson, Ruetschi, Hesse, & Larson, 2012; Halim, Ruetschi, Larson, & Nilsson, 2013; Yin, Bern, Xing, Ho, Viner, & Mayr, 2013). Since RPLC is the standard separation system employed by proteomics laboratory, its compatibility with other lines of proteomic research permits the wide use of RPLC in glycopeptide separation (Thaysen-Andersen & Packer, 2014). The retention of glycopeptides in RPLC is directly related to the hydrophobicity of the molecules. In a glycopeptide molecule, the glycan moiety contributes hydrophilicity while the peptide backbone contributes hydrophobicity. Therefore, the hydrophobicity of a glycopeptide is generally lower than its peptide backbone. As a result, those hydrophilic glycopeptides containing relatively larger glycan moieties and smaller peptide moieties might not be retained by C18. Moreover, the hydrophobicity variation of glycan isomers are very limited. Therefore, RPLC offers poor glycan isomer differentiation (Hua, Nwosu, Strum, Seipert, An, Zivkovic, et al., 2012). This limits the use of RPLC in the glycoproteomics analysis.
7.4.2 Hydrophilic interaction liquid chromatography (HILIC)
Hydrophilic interaction liquid chromatography (HILIC) allows analytes to interact with hydrophilic stationary phases such as bare silica particles, amine-, hydroxy-, amide-bonded or zwitterionic (ZIC-HILIC) particles by applying a relative non-polar mobile phase (Alpert, 1990). The capability of separating glycopeptides using HILIC has been shown in several studies (Zauner, Deelder, & Wuhrer, 2011; Zauner, Koeleman, Deelder, & Wuhrer, 2010). Since the mobile phase of HILIC contains high organic solvents, which might lead to solubility problems of glycopeptide mixture, the use of HILIC in large scale glycopeptide separation is limited. On the other hand, HILIC has been proved as the most efficient enrichment method for glycopeptides as abovementioned (Mysling, Palmisano, Hojrup, & Thaysen-Andersen, 2010).
7.4.3 Porous graphitized carbon LC (PGC-LC)
Porous graphitized carbon or PGC was developed as a very insoluble and stable support for HPLC in the 1980s. PGC-LC was first used in the early 1990s to separate glycans isomers (Davies, Smith, Carruthers, Chai, Lawson, & Hounsell, 1993). The combined retention mechanism allows PGC-LC to become one of the major methods to achieved structural isomeric separation of detached glycans (Wuhrer, Deelder, & Hokke, 2005). As an alternative separation technique, PGC-LC has also been applied to glycopeptide analysis (An, Peavy, Hedrick, & Lebrilla, 2003; Hua et al., 2012; Nwosu, Huang, Aldredge, Strum, Hua, Seipert et al., 2013); however, the retention of glycopeptides on PGC might be too strong with vast and hydrophobic peptide moieties especially those glycopeptides with highly sialylated glycan moieties (Alley, Mechref, & Novotny, 2009b; Thaysen-Andersen, Wilkinson, Payne, & Packer, 2011). In order to overcome this issue, non-specific proteases are used to generate glycopeptides with small peptide moieties in several works (An et al., 2003; Froehlich, Barboza, Chu, Lerno, Clowers, Zivkovic et al., 2011; Hua, Hu, Kim, Totten, Oh, Yun et al., 2013; Stavenhagen, Plomp, & Wuhrer, 2015). Nonspecific proteases such as pronase digest glycoproteins into glycopeptides with small peptide backbone (<4~6 amino acids). These glycopeptides have better chance to eluted from PGC-LC and generate more useful MS/MS spectra than those with large peptide backbones. On the other hand, since the non-specific protease will generate multiple peptide backbone of one glycosylation site simultaneously, this quantitative application of this strategy is limited. In addition, compared to the separation of native glycans isomeric selectivity of glycopeptides were also compromised even with a small peptide backbone (Hua et al., 2013). The abovementioned common used HPLC separation techniques are limited to its natural disadvantages in separating glycopeptides. Therefore, the combination of these techniques is applied to glycomic studies (Lam, Lau, Siu, Ng, Kong, Chiu et al., 2011; Liu, Wang, Zhu, Mao, Liu, Cheng et al., 2014; Y. Zhao, Szeto, Kong, Law, Li, Quan et al., 2014). Most of the works have been performed on a PGC and C18-RPLC platform Figure 7.4 (J. Zhao, Song, Zhu, & Mechref, 2016). The C18-RPLC is majorly used for glycopeptide separation which the PGC component is used as purification materials (Liu et al., 2014) or a secondary dimension of separation.(Stavenhagen et al., 2015; Y. Zhao et al., 2014). The combination of different stationary phases with complementary retention mechanisms has been proved to advance glycopeptide analysis.
Figure 7.4. Separation of glycopeptides derived from porcine thyroglobulin (PTG) with C18 analytical column.
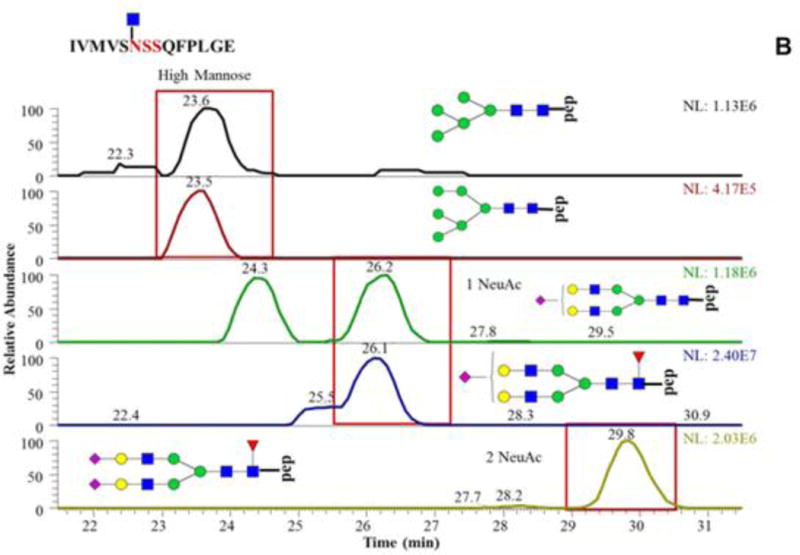
The same peptide backbone is associated with multiple glycan residues highlighting the microheterogeneity at this glycosylation site. Reproduced with permission (J. Zhao et al., 2016).
7.5 Ion Mobility-Mass Spectrometry
Ion mobility-mass spectrometry (IM-MS) has gained the attention of numerous researchers because of its unique ability to separate ions based on size, shape, or charge in addition to be coupled to a high mass accuracy time of flight mass analyzer. There are several different types of ion mobility that have been utilized for glycopeptide analysis. Traveling Wave Ion Mobility Mass Spectrometry (TWIMS-MS) (Giles, Pringle, Worthington, Little, Wildgoose, & Bateman, 2004), the most popular commercially available ion mobility mass spectrometry instrument, has been used to thoroughly characterize an IgG1 mAb’s glycosylation heterogeneity profile with glycopeptides of each glycoform being readily identified (Olivova, Chen, Chakraborty, & Gebler, 2008). Li et al. found that TWIMS could separate peptides from glycopeptides into distinct trend lines that could be used for predicting glycosylation status of other peptides; they also noted that ion mobility reduced chemical noise to allow for the detection of lower abundant ions (H. Li, Bendiak, Siems, Gang, & Hill, 2013). TWIMS-MS has also been used to distinguish epimeric glycopeptides derived from Muc 2 (Both, Green, Gray, Sardzik, Voglmeir, Fontana et al., 2014) Figure 7.5A. Two isomeric glycopeptides, same peptide backbone but differing only in the attachment of either α-GlcNAc or α-GalNAc, were partially separated with TWIMS-MS; interestingly, multiple overlapping conformers were identified for each glycopeptide. The authors confirmed the identity of each glycopeptide by using CID-IMS-MS Figure 7.5B; the diagnostic oxonium ions were separated with TWIMS-MS, highlighting the utility and importance of characterizing product ions.
Figure 7.5. Separation and analysis of glycopeptides by Ion Mobility Mass Spectrometry.
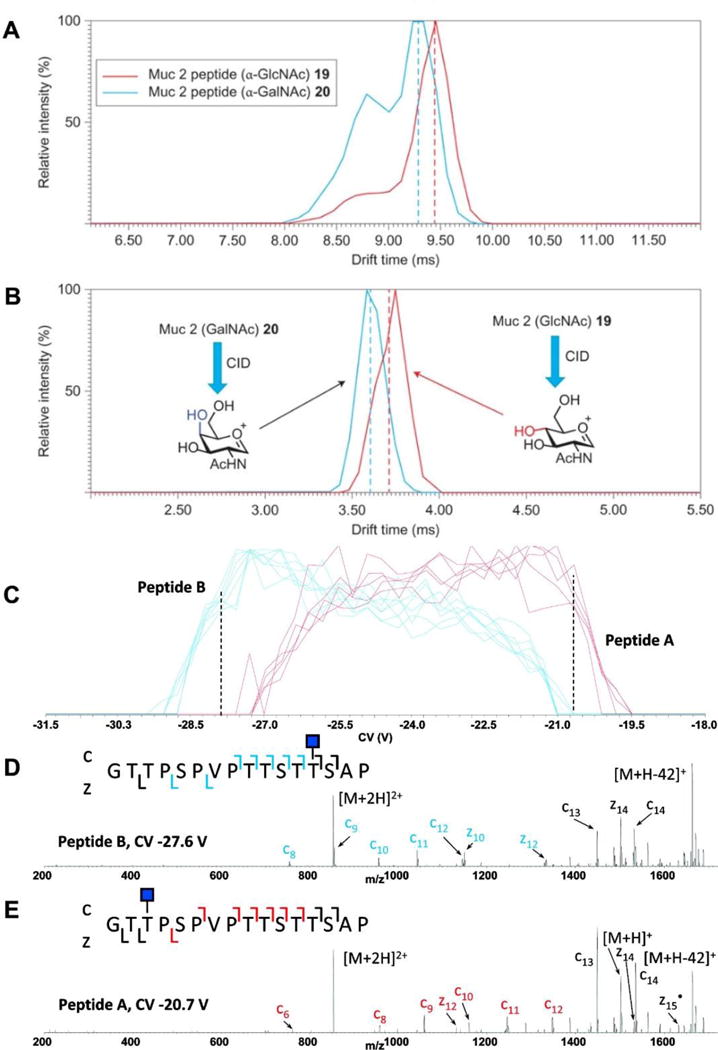
A. Traveling Wave Ion Mobility Mass Spectrometry (TWIMS) identified multiple conformers of the isobaric Muc2 glycopeptides (PTTTPITTTTTVTPTPTPTGTQT with GalNAc 19 and GlcNac 20). TWIMS was not able to differentiate between the two intact glycopeptides; B. however, after CID, TWIMS-MS could distinguish the diagnostic oxonium ions from each Mucin glycopeptide. C. High-field asymmetric wave ion mobility mass spectrometry (FAIMS-MS) separation of two isobaric O-linked glycopeptides, differing only in glycan site attachment. D, E. glycopeptide identity confirmed with ETD MS2; diagnostic c and z ions are indicated in blue and red.
High-field asymmetric wave ion mobility spectrometry (FAIMS) has also been used to rapidly separate co-eluting isomeric O-linked glycopeptides, differing only in glycosylation site Figure 7.5C. The co-eluting peptides were separated by altering the correction voltage (CV) applied. Confirmation of the glycosylation sites was confirmed with ETD Figure 7.5D, E; the two glycosylation sites are readily identified by their diagnostic product ions (Creese & Cooper, 2012). The combination of FAIMS to separate isobaric peptides and ETD to confirm the glycosylation site has proven to be quite useful in analyzing complex glycopeptide mixtures.
7.6 Capillary Electrophoresis
Capillary electrophoresis (CE) has long been employed for the rapid and reproducible separation and analysis of fluorescently labeled glyans and intact glycoproteins. CE has been employed for the rapid separation of glycopeptides before analysis by mass spectrometry. Gimenez et al. (Gimenez, Ramos-Hernan, Benavente, Barbosa, & Sanz-Nebot, 2012) developed a method utilizing CE coupled to ESI-orthogonal accelerating time of flight mass spectrometry (CE-ESI-oaTOF-MS) to rapidly analyze N and O-glycopeptides derived from erythropoietin (EPO), a commonly used glycoprotein hormone abused in athletic competitions. Their method also discovered a novel sulfated sialoform glycopeptide.
Lew et al. developed a rapid method for therapeutic antibody quality control utilizing CE-MS to analyze the tryptic digest of Trastuzumab; the relative abundance of 14-glycoforms on one glycopeptide were readily attained (Lew, Gallegos-Perez, Fonslow, Lies, & Guttman, 2015) Figure 7.6.
Figure 7.6. Capillary electrophoresis mass spectrometry (CE-MS) of therapeutic antibody.
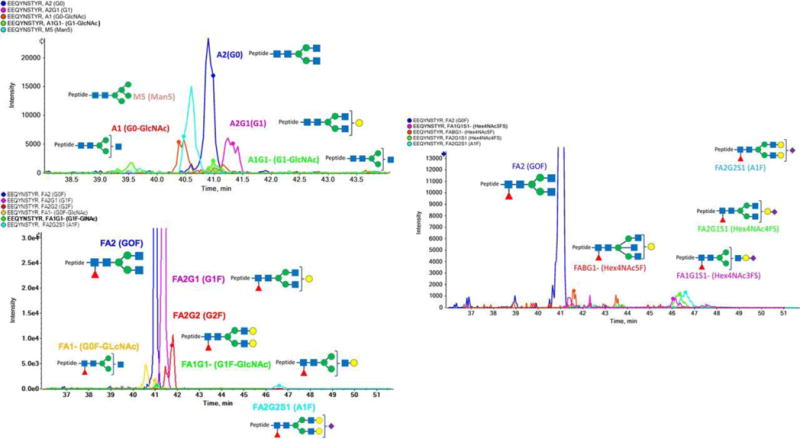
CE-MS quantified 14-glycoforms on one glycopeptide from Trastuzumab. Reproduced with permission (Lew et al., 2015).
Barroso et al. coupled CE-TOF-MS and μLC-TOF-MS for a clinical research application; their method was able to detect the relative glycoform abundance of human Transferrin Tf glycopeptide to predict the alcohol dependence of individuals utilizing a small serum sample (Barroso, Gimenez, Benavente, Barbosa, & Sanz-Nebot, 2013).
7.7 Characterization of intact glycopeptides
In recent years, proteomic and glycoproteomic analysis has benefited from the technological advances of new mass spectrometry techniques. The identification of glycopeptide heavily relies on tandem MS (MS/MS). Informative and complementary fragments created by various fragmentation methods permit the confident interpretation of intact glycopeptides. Three useful tandem MS (MS/MS) techniques are widely used, namely collision induced dissociation (CID), high energy collision dissociation (HCD), and electron transfer dissociation (ETD).
7.7.1 Collision-induced dissociation (CID)
CID MS/MS is the most common and robust fragmentation method used in peptide analysis. It has also been widely used in glycopeptide analysis. During CID process, ions are transmitted through lenses and ion optics from electrospray ion source. In linear ion trap instruments, ions are trapped, isolated, fragmented through resonant-excitation and then scanned by electron multipliers. Since glycosidic linkages of glycoforms are more labile than amide bonds of peptide backbone, a typical CID MS/MS spectrum of glycopeptides is dominated by glycan fragmentation patterns with no or less peptide backbone sequence information (Huddleston, Bean, & Carr, 1993; Mayampurath, Wu, Segu, Mechref, & Tang, 2011). The resulting fragments of glycopeptides are named as B- and Y-type ions (Domon & Costello, 1988).
7.7.2 Higher-energy collision dissociation (HCD)
HCD is a beam-type fragmentation has been demonstrated on LIT instruments and on hybrid instruments with dedicated collision cells (Bereman, Canterbury, Egertson, Horner, Remes, Schwartz et al., 2012; Olsen, Macek, Lange, Makarov, Horning, & Mann, 2007; Ting, Cowley, Hoon, Guilhaus, Raftery, & Cavicchioli, 2009). HCD yields informative low mass fragment of glycopeptides. Specifically, it creates fragmentation pattern of peptides or diagnostic ions of glycan moieties with high intensities at low m/z. The diagnostic ions are also referred as oxonium ions (diagnostic ions), such as m/z 138.0551 (HexNAc-2H2O-CH2O), 204.0875 (HexNAc), 274.0976 (NeuAc-H2O), 366.1395 (Hex+HexNAc), 512.1977 (Hex+HexNAc+dHex) and 657.2353 (NeuAc+Hex+HexNAc) (Mayampurath et al., 2011; Segu, Hammad, & Mechref, 2010). Exists of oxonium ions usually indicate the presence of glycopeptide. The effective characterization of glycopeptides can be performed with the use of oxonium ions in HCD and the glycan fragmentations in CID.
7.7.3 Electron transfer dissociation (ETD)
Alternatively, ETD, as a relatively new invented fragmentation scheme can also be applied to glycopeptide analysis. ETD induces random fragmentation along the peptide backbone of a glycopeptide creating mostly c- and z-type ions (Stephenson & McLuckey, 1998; Syka, Coon, Schroeder, Shabanowitz, & Hunt, 2004). ETD has been recently employed to better identify glycopeptides with mass of glycans. However, the major drawback of ETD is that the efficiency is largely depends on m/z values of glycopeptides, thus limited its use in large scale glycoproteomic analysis (Alley, Mechref, & Novotny, 2009a; S. L. Wu, Huhmer, Hao, & Karger, 2007).
7.8 Characterization of intact glycoproteins
The previous sections all employed a bottom-up proteomics approach involving a protease, usually trypsin, to reduce the large intact protein into smaller peptides before LC-MS/MS analysis. Top-down mass spectrometry eliminates the protease step resulting in the analysis of intact proteins; proteins are fragmented inside the instrument utilizing different dissociation techniques such as ECD, ETD, and UVPD. Top-down mass spectrometry has several advantages over the conventional bottom-up approach in that protein heterogeneity is preserved; that is different PTM combinations are preserved whereas the bottom-up approach loses that connectivity information. Top-down experiments require high mass accuracy and so are performed exclusively on FT based mass analyzer such as FTICR and orbitrap instruments. Middle-down mass spectrometry is similar to the bottom-up approach in that proteins are proteolyzed into smaller peptides but differ in peptide size; middle-down peptides are considerably smaller as they employ a more restrictive enzyme such as IdeS.
Tran et al. combined a top-down and middle-down approach to determine the glycosylation profile of IgG and IgG-fusion protein (Tran, Barton, Feng, Sandjong, Yoon, Awasthi et al., 2016) Figure 7.7. They were able to identify the heterogeneous glycosylation on the Fc portion; glycan heterogeneity matched oligosaccharide profiling. This approach yielded combinatorial information regarding the glycosylation sites that could not have been attained with either a bottom-up approach or an oligosaccharide mapping approach.
Figure 7.7. Combined top-down and middle-down proteomics approach to identifying IgG glycosylation profile.
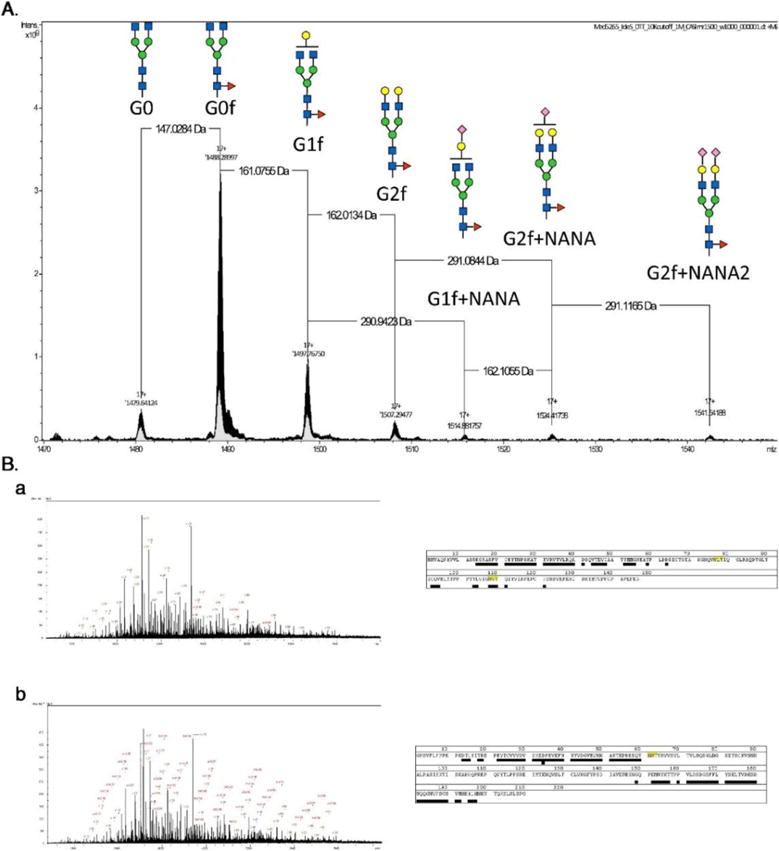
A Top-down native mass spectrometry of intact IgG to readily identify glycoforms. B Middle-down mass spectrometry to identify peptide backbone and any post-translational modifications. Reproduced with permission (Tran et al., 2016).
Nagel et al. employed a top-down approach to rapidly characterize the different sequence polymorphisms (SNPs) as well as assess relative abundances of phosphorylation and glycosylation on fibrinogen in a single experiment; this was done to accurately screen patients for possible cardiovascular risks including stroke (Nagel & Meyer, 2014). They were able to identify primary sequence variants in their clinical samples while also determining the phosphorylation and glycosylation status of each fibrinogen subunit.
Yang et al. employed a native-ms top-down mass spectrometry approach to identify 59 proteoforms of chickecn ovalbumin including multiple phosphorylations and 45 different glycans (Y. Yang, Barendregt, Kamerling, & Heck, 2013). This approach revealed native-ms could rapidly identify the heterogeneity in glycoprotein sample. The authors highlight the advantage of little sample consumption but note that this method cannot yield glycan connectivity information, only intact mass measurements.
Currently, most top-down mass spectrometry approaches aimed at characterizing glycoproteins rely on existing glycan databases to assign a glycan structure to a proteoform. Native-ms and top-down mass spectrometry are well situated to assess the macroheterogeneity of glycoproteins but struggle with identifying the sites of glycosylation on proteins.
7.9 Software tools for MS/MS based glycopeptide characterization
The combination of state of the art mass spectrometers and the accompanying bioinformatics tools have allowed proteomics studies to evolve from single protein identification studies to identifying changes in entire proteomes. The data generated from these entire proteome studies would be impractical to interpret manually but manageable with currently available software. Software for bottom-up proteomics experiments is quite robust in determining peptide sequence and consequently protein identity (Hu, Khatri, & Zaia, 2016); however, accurate quantitation is still an open question being addressed by numerous researchers (Blein-Nicolas & Zivy, 2016).
Glycoproteomics differ from routine proteomics studies in that the glycan cannot be treated as a simple post translational modification, like methylation or phosphorylation, because of the enormous variance of glycan structures. Glycoproteomics studies are generally focused on identifying the different glycans present on glycopeptide, identifying the peptide sequence and consequently the protein’s identity, and determining both the glycan and glycoprotein abundance. To further complicate analysis, more than one glycan can be identified with the same peptide sequence; this is referred to as glycan microheterogeneity. Currenlty, several fragmentation techniques are routinly employed To aid in the identification of glycan and peptide. CID is the most common technique for fragmenting peptides in a bottom-up proteomics approach but it does not work well with glycopeptides; CID fragments the glycan while leaving the peptide predominantly intact. This has been overcome by using a combination of CID and ETD in a data dependent acquisition experiment (Singh, Zampronio, Creese, & Cooper, 2012); CID yields glycan product ions, including the glycopeptide diagnostic oxonium ions, while ETD selectively yields peptide product ions. The data generated from the above mentioned fragmentation techniques can then be analyzed with bioinformatics tools.
The glycoproteomics bioinformatics field is not as well established as the older proteomics field. In addition to the previously mentioned difficulties, glycopeptides with similar glycans tend to co-elute. Numerous tools encompassing different strategies have been developed for various glycoproteomic studies to address these issues. The current software tools annotate glycopeptides utilize precursor ion mass and fragmentation patterns matching with known glycopeptides. For example, Peptoonist (Goldberg, Bern, Parry, Sutton-Smith, Panico, Morris, et al., 2007) combines precursor and fragments information to annotate glycopeptides. On the other hand, GlypID (Y. Wu, Mechref, Klouckova, Mayampurath, Novotny, & Tang, 2010) utilizes the co-elution of N-glycopeptide on RPLC, thus combines co-eluted N-glycopeptide ions from full scans and fragmentation from CID MS/MS scans to identify putative glycoforms associated with the same glycosylation site. In order to improve the confidence of glycopeptide identification, software tools are combined with multiple LC-MS/MS analysis of the same sample (Toghi Eshghi, Shah, Yang, Li, & Zhang, 2015). In this type of experiment, a portion of glycopeptide is deglycosylated first and analyzed to create an experimental peptide back bone database, while another part of the sample is subjected to LC-MS as intact glycopeptide. The experimental database is further utilized by the software tool to identify glycopeptides confidently.
He et al. developed Glycomaster DB to analyze glycopeptides extracted from the human urinary proteome and fragmented with both HCD and ETD (He, Xin, Shan, Lajoie, & Ma, 2014). They were able to identify multiple N-linked glycan forms on the same glysoylation site but were not able to identify any O-linked glycopeptides. Cheng et al. utilized their own program to identify 2200+ unique N-glycopeptides assigned to 1700+ site specific N-glycans on 450+ glycosylation sites in HEK 293T cells; their low false-discovery rate (FDR) indicates a robust identification algorithm for the glycopeptide(Cheng, Chen, Seebun, Ye, Figeys, & Zou, 2014).
Zhu et al. developed GlycoPep Evaluator (GPE) to improve upon existing FDR approaches; GPE generates decoy glycopeptides de novo for every target peptide (Zhu, Su, Go, & Desaire, 2014). Zhang et al. developed c for the specific task of identifying O-linked glycosylations in which Gal or Glc-Gal are attached to hydroxylysine residues commonly found in collagen (Y. Zhang, Yu, Song, Li, Mechref, Tang et al., 2015); this type of glycopeptide is not commonly identified with most software packages. The authors proceeded to identify more glycopeptides in collagen, including two additional glycosylation sites than what had been previously reported.
GlycoSeq was implemented to determine glycopeptides derived from two breast cancer cell lines (Yu, Mayampurath, Zhu, Zacharias, Song, Wang, et al., 2016); the software program utilizes a heuristic iterated glycan sequencing algorithm coupled to biological glycosidic linkage information to readily identify glycans. Jansen et al. created LaCytools to improve glycopeptide identification in therapeutic monoclonal antibodies; a focus on quality control measurements including signal-to-noise, isotope pattern quality, and mass accuracy resulted in a powerful software suite (Jansen, Falck, de Haan, Hipgrave Ederveen, Razdorov, Lauc, et al., 2016). Jansen developed MassyTools to improve glycopeptide data generated from MALDI-MS resulting in the improved calibration and quantitation over existing softwares (Jansen, Reiding, Bondt, Hipgrave Ederveen, Palmblad, Falck, et al., 2015). Park et al. developed a robust software package, Integrated GlycoProteome Analyzer (I-GPA), to analyze and map N-glycoproteomes; their method identified 600+ N-glycopeptides and quantified ~600 N-glycopeptides from human plasma with an astonishing 0% false positive discovery rate manually verified (Park, Kim, Hwang, Lee, Ahn, Lee, et al., 2016). Lih et al. developed MAGIC-web, a web server dedicated to targeted and untargeted N-linked glycan analysis (Lih, Choong, Chen, Cheng, Lin, Chen, et al., 2016). Zeng et al. developed pGlyco to identify N-glycans from HCD-MS/MS and CID-MS/MS data sets; a novel target decoy method was implemented to estimate FDR of glycan identification (W. F. Zeng, Liu, Zhang, Wu, Fang, Peng, et al., 2016).
7.10 Concluding remarks and future directions
In recent years, significant development and improvement of analytical methods have been implemented to glycoproteomics study. Glycopeptides enrichment using lectin affinity, hydrazide chemistry, click chemistry, immunoprecipitation, boronic acid chemistry, HILIC and ERLIC allow for sensitive detection of glycopeptides. LC, CE, and ion mobility mass spectrometry improves the separation of different glycopeptides and increases the identification of micro- and macro heterogeneity of glycoproteins. Multiple mass spectrometry based dissociation mechanisms aid with the structural elucidation of glycopeptides. In addition to advancements in instrumentation, the development of automated annotation software tools for glycopeptide identification and quantitation have significantly improved the analytical throughput.
Despite all of the recent advancements, various hurdles still remain in the analysis of glycopeptides. A comprehensive enrichment technique unique to all glycopeptides has not been developed resulting in an inherent compromise; HILIC based separation is able to enrich and allow for the identification of the highest number of glycopeptides, but it is less specific than hydrazide chemistry enrichment. Accurate glycopeptide structural elucidation results from the use of complementary tandem MS mechanisms to yield accurate information about the peptide backbone (ETD), glycan structure (CID), and oxonium ions (HCD). Although modern mass spectrometers are able to perform CID, HCD and ETD simultaneously, the sensitivity and throughput loss is still considerable. In addition to enrichment and instrumentation short comings, the false positive discovery rate of automated software tools remains a challenge. There is much room for improvement in developing a comprehensive protocol that will yield accurate and quantitative information on all glycopeptides in samples.
Acknowledgments
This work was supported by a grant from the Cancer Prevention Institute of Texas (CPRIT, RP130624) and partially by an NIH grant (1R01GM112490).
References
- Alley WR, Jr, Mechref Y, Novotny MV. Characterization of glycopeptides by combining collision-induced dissociation and electron-transfer dissociation mass spectrometry data. Rapid Commun Mass Spectrom. 2009a;23(1):161–170. doi: 10.1002/rcm.3850. [DOI] [PubMed] [Google Scholar]
- Alley WR, Jr, Mechref Y, Novotny MV. Use of activated graphitized carbon chips for liquid chromatography/mass spectrometric and tandem mass spectrometric analysis of tryptic glycopeptides. Rapid Commun Mass Spectrom. 2009b;23(4):495–505. doi: 10.1002/rcm.3899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alpert AJ. Hydrophilic-interaction chromatography for the separation of peptides, nucleic acids and other polar compounds. J Chromatogr. 1990;499:177–196. doi: 10.1016/s0021-9673(00)96972-3. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/2324207. [DOI] [PubMed] [Google Scholar]
- An HJ, Peavy TR, Hedrick JL, Lebrilla CB. Determination of N-glycosylation sites and site heterogeneity in glycoproteins. Anal Chem. 2003;75(20):5628–5637. doi: 10.1021/ac034414x. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/14710847. [DOI] [PubMed] [Google Scholar]
- Anonsen JH, Vik A, Egge-Jacobsen W, Koomey M. An extended spectrum of target proteins and modification sites in the general O-linked protein glycosylation system in Neisseria gonorrhoeae. J Proteome Res. 2012;11(12):5781–5793. doi: 10.1021/pr300584x. [DOI] [PubMed] [Google Scholar]
- Apweiler R, Hermjakob H, Sharon N. On the frequency of protein glycosylation, as deduced from analysis of the SWISS-PROT database. Biochim Biophys Acta. 1999;1473(1):4–8. doi: 10.1016/s0304-4165(99)00165-8. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/10580125. [DOI] [PubMed] [Google Scholar]
- Barroso A, Gimenez E, Benavente F, Barbosa J, Sanz-Nebot V. Analysis of human transferrin glycopeptides by capillary electrophoresis and capillary liquid chromatography-mass spectrometry. Application to diagnosis of alcohol dependence. Anal Chim Acta. 2013;804:167–175. doi: 10.1016/j.aca.2013.09.044. [DOI] [PubMed] [Google Scholar]
- Bereman MS, Canterbury JD, Egertson JD, Horner J, Remes PM, Schwartz J, MacCoss MJ. Evaluation of front-end higher energy collision-induced dissociation on a benchtop dual-pressure linear ion trap mass spectrometer for shotgun proteomics. Anal Chem. 2012;84(3):1533–1539. doi: 10.1021/ac203210a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blein-Nicolas M, Zivy M. Thousand and one ways to quantify and compare protein abundances in label-free bottom-up proteomics. Biochim Biophys Acta. 2016;1864(8):883–895. doi: 10.1016/j.bbapap.2016.02.019. [DOI] [PubMed] [Google Scholar]
- Botella-Lopez A, Burgaya F, Gavin R, Garcia-Ayllon MS, Gomez-Tortosa E, Pena-Casanova J, Saez-Valero J. Reelin expression and glycosylation patterns are altered in Alzheimer's disease. Proc Natl Acad Sci U S A. 2006;103(14):5573–5578. doi: 10.1073/pnas.0601279103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Both P, Green AP, Gray CJ, Sardzik R, Voglmeir J, Fontana C, Eyers CE. Discrimination of epimeric glycans and glycopeptides using IM-MS and its potential for carbohydrate sequencing. Nat Chem. 2014;6(1):65–74. doi: 10.1038/nchem.1817. [DOI] [PubMed] [Google Scholar]
- Buszewski B, Noga S. Hydrophilic interaction liquid chromatography (HILIC)–a powerful separation technique. Anal Bioanal Chem. 2012;402(1):231–247. doi: 10.1007/s00216-011-5308-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvano CD, Zambonin CG, Jensen ON. Assessment of lectin and HILIC based enrichment protocols for characterization of serum glycoproteins by mass spectrometry. J Proteomics. 2008;71(3):304–317. doi: 10.1016/j.jprot.2008.06.013. [DOI] [PubMed] [Google Scholar]
- Campbell BJ, Yu LG, Rhodes JM. Altered glycosylation in inflammatory bowel disease: a possible role in cancer development. Glycoconj J. 2001;18(11–12):851–858. doi: 10.1023/a:1022240107040. [DOI] [PubMed] [Google Scholar]
- Chen J, Xi J, Tian Y, Bova GS, Zhang H. Identification, prioritization, and evaluation of glycoproteins for aggressive prostate cancer using quantitative glycoproteomics and antibody-based assays on tissue specimens. Proteomics. 2013;13(15):2268–2277. doi: 10.1002/pmic.201200541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen R, Tan Y, Wang M, Wang F, Yao Z, Dong L, Zou H. Development of glycoprotein capture-based label-free method for the high-throughput screening of differential glycoproteins in hepatocellular carcinoma. Mol Cell Proteomics. 2011;10(7):M110.006445. doi: 10.1074/mcp.M110.006445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng K, Chen R, Seebun D, Ye M, Figeys D, Zou H. Large-scale characterization of intact N-glycopeptides using an automated glycoproteomic method. J Proteomics. 2014;110:145–154. doi: 10.1016/j.jprot.2014.08.006. [DOI] [PubMed] [Google Scholar]
- Creese AJ, Cooper HJ. Separation and identification of isomeric glycopeptides by high field asymmetric waveform ion mobility spectrometry. Anal Chem. 2012;84(5):2597–2601. doi: 10.1021/ac203321y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dasouki M, Jawdat O, Almadhoun O, Pasnoor M, McVey AL, Abuzinadah A, Dimachkie MM. Pompe disease: literature review and case series. Neurol Clin. 2014;32(3):751–776. ix. doi: 10.1016/j.ncl.2014.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies MJ, Smith KD, Carruthers RA, Chai W, Lawson AM, Hounsell EF. Use of a porous graphitised carbon column for the high-performance liquid chromatography of oligosaccharides, alditols and glycopeptides with subsequent mass spectrometry analysis. J Chromatogr. 1993;646(2):317–326. doi: 10.1016/0021-9673(93)83344-r. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/8408434. [DOI] [PubMed] [Google Scholar]
- Domon B, Costello C. A systematic nomenclature for carbohydrate fragmentations in FAB-MS/MS spectra of glycoconjugates. Glycoconj J. 1988;5(4):397–409. doi: 10.1007/bf01049915. [DOI] [Google Scholar]
- Dube DH, Bertozzi CR. Glycans in cancer and inflammation–potential for therapeutics and diagnostics. Nat Rev Drug Discov. 2005;4(6):477–488. doi: 10.1038/nrd1751. [DOI] [PubMed] [Google Scholar]
- Elliott MA, Elliott HG, Gallagher K, McGuire J, Field M, Smith KD. Investigation into the concanavalin A reactivity, fucosylation and oligosaccharide microheterogeneity of alpha 1-acid glycoprotein expressed in the sera of patients with rheumatoid arthritis. J Chromatogr B Biomed Sci Appl. 1997;688(2):229–237. doi: 10.1016/s0378-4347(96)00309-x. [DOI] [PubMed] [Google Scholar]
- Froehlich JW, Barboza M, Chu C, Lerno LA, Jr, Clowers BH, Zivkovic AM, Lebrilla CB. Nano-LC-MS/MS of glycopeptides produced by nonspecific proteolysis enables rapid and extensive site-specific glycosylation determination. Anal Chem. 2011;83(14):5541–5547. doi: 10.1021/ac2003888. [DOI] [PubMed] [Google Scholar]
- Giles K, Pringle SD, Worthington KR, Little D, Wildgoose JL, Bateman RH. Applications of a travelling wave-based radio-frequency-only stacked ring ion guide. Rapid Commun Mass Spectrom. 2004;18(20):2401–2414. doi: 10.1002/rcm.1641. [DOI] [PubMed] [Google Scholar]
- Gimenez E, Ramos-Hernan R, Benavente F, Barbosa J, Sanz-Nebot V. Analysis of recombinant human erythropoietin glycopeptides by capillary electrophoresis electrospray-time of flight-mass spectrometry. Anal Chim Acta. 2012;709:81–90. doi: 10.1016/j.aca.2011.10.028. [DOI] [PubMed] [Google Scholar]
- Goldberg D, Bern M, Parry S, Sutton-Smith M, Panico M, Morris HR, Dell A. Automated N-glycopeptide identification using a combination of single- and tandem-MS. J Proteome Res. 2007;6(10):3995–4005. doi: 10.1021/pr070239f. [DOI] [PubMed] [Google Scholar]
- Gstaiger M, Aebersold R. Applying mass spectrometry-based proteomics to genetics, genomics and network biology. Nat Rev Genet. 2009;10(9):617–627. doi: 10.1038/nrg2633. [DOI] [PubMed] [Google Scholar]
- Halim A, Nilsson J, Ruetschi U, Hesse C, Larson G. Human urinary glycoproteomics; attachment site specific analysis of N- and O-linked glycosylations by CID and ECD. Mol Cell Proteomics. 2012;11(4):M111 013649. doi: 10.1074/mcp.M111.013649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halim A, Ruetschi U, Larson G, Nilsson J. LC-MS/MS characterization of O-glycosylation sites and glycan structures of human cerebrospinal fluid glycoproteins. J Proteome Res. 2013;12(2):573–584. doi: 10.1021/pr300963h. [DOI] [PubMed] [Google Scholar]
- Hao P, Guo T, Sze SK. Simultaneous analysis of proteome, phospho- and glycoproteome of rat kidney tissue with electrostatic repulsion hydrophilic interaction chromatography. PLoS One. 2011;6(2):e16884. doi: 10.1371/journal.pone.0016884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He L, Xin L, Shan B, Lajoie GA, Ma B. GlycoMaster DB: software to assist the automated identification of N-linked glycopeptides by tandem mass spectrometry. J Proteome Res. 2014;13(9):3881–3895. doi: 10.1021/pr401115y. [DOI] [PubMed] [Google Scholar]
- Hein CD, Liu XM, Wang D. Click chemistry, a powerful tool for pharmaceutical sciences. Pharm Res. 2008;25(10):2216–2230. doi: 10.1007/s11095-008-9616-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helenius A, Aebi M. Intracellular functions of N-linked glycans. Science. 2001;291(5512):2364–2369. doi: 10.1126/science.291.5512.2364. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/11269317. [DOI] [PubMed] [Google Scholar]
- Heo SH, Lee SJ, Ryoo HM, Park JY, Cho JY. Identification of putative serum glycoprotein biomarkers for human lung adenocarcinoma by multilectin affinity chromatography and LC-MS/MS. Proteomics. 2007;7(23):4292–4302. doi: 10.1002/pmic.200700433. [DOI] [PubMed] [Google Scholar]
- Hongsachart P, Huang-Liu R, Sinchaikul S, Pan FM, Phutrakul S, Chuang YM, Chen ST. Glycoproteomic analysis of WGA-bound glycoprotein biomarkers in sera from patients with lung adenocarcinoma. Electrophoresis. 2009;30(7):1206–1220. doi: 10.1002/elps.200800405. [DOI] [PubMed] [Google Scholar]
- Hu H, Khatri K, Zaia J. Algorithms and design strategies towards automated glycoproteomics analysis. Mass Spectrom Rev. 2016 doi: 10.1002/mas.21487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua S, Hu CY, Kim BJ, Totten SM, Oh MJ, Yun N, An HJ. Glyco-analytical multispecific proteolysis (Glyco-AMP): a simple method for detailed and quantitative Glycoproteomic characterization. J Proteome Res. 2013;12(10):4414–4423. doi: 10.1021/pr400442y. [DOI] [PubMed] [Google Scholar]
- Hua S, Nwosu CC, Strum JS, Seipert RR, An HJ, Zivkovic AM, Lebrilla CB. Site-specific protein glycosylation analysis with glycan isomer differentiation. Anal Bioanal Chem. 2012;403(5):1291–1302. doi: 10.1007/s00216-011-5109-x. [DOI] [PubMed] [Google Scholar]
- Huddleston MJ, Bean MF, Carr SA. Collisional fragmentation of glycopeptides by electrospray ionization LC/MS and LC/MS/MS: methods for selective detection of glycopeptides in protein digests. Anal Chem. 1993;65(7):877–884. doi: 10.1021/ac00055a009. [DOI] [PubMed] [Google Scholar]
- Ishihara T, Fukuda I, Morita A, Takinami Y, Okamoto H, Nishimura S, Numata Y. Development of quantitative plasma N-glycoproteomics using label-free 2-D LC-MALDI MS and its applicability for biomarker discovery in hepatocellular carcinoma. J Proteomics. 2011;74(10):2159–2168. doi: 10.1016/j.jprot.2011.06.010. [DOI] [PubMed] [Google Scholar]
- James P. Protein identification in the post-genome era: the rapid rise of proteomics. Q Rev Biophys. 1997;30(4):279–331. doi: 10.1017/s0033583597003399. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/9634650. [DOI] [PubMed] [Google Scholar]
- Jansen BC, Falck D, de Haan N, Hipgrave Ederveen AL, Razdorov G, Lauc G, Wuhrer M. LaCyTools: A Targeted Liquid Chromatography-Mass Spectrometry Data Processing Package for Relative Quantitation of Glycopeptides. J Proteome Res. 2016;15(7):2198–2210. doi: 10.1021/acs.jproteome.6b00171. [DOI] [PubMed] [Google Scholar]
- Jansen BC, Reiding KR, Bondt A, Hipgrave Ederveen AL, Palmblad M, Falck D, Wuhrer M. MassyTools: A High-Throughput Targeted Data Processing Tool for Relative Quantitation and Quality Control Developed for Glycomic and Glycoproteomic MALDI-MS. J Proteome Res. 2015;14(12):5088–5098. doi: 10.1021/acs.jproteome.5b00658. [DOI] [PubMed] [Google Scholar]
- Keil B. Proteolysis Data Bank: specificity of alpha-chymotrypsin from computation of protein cleavages. Protein Seq Data Anal. 1987;1(1):13–20. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/3447153. [PubMed] [Google Scholar]
- Kolb HC, Finn MG, Sharpless KB. Click Chemistry: Diverse Chemical Function from a Few Good Reactions. Angew Chem Int Ed Engl. 2001;40(11):2004–2021. doi: 10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- Lam MP, Lau E, Siu SO, Ng DC, Kong RP, Chiu PC, Chu IK. Online combination of reversed-phase/reversed-phase and porous graphitic carbon liquid chromatography for multicomponent separation of proteomics and glycoproteomics samples. Electrophoresis. 2011;32(21):2930–2940. doi: 10.1002/elps.201100092. [DOI] [PubMed] [Google Scholar]
- Lew C, Gallegos-Perez JL, Fonslow B, Lies M, Guttman A. Rapid level-3 characterization of therapeutic antibodies by capillary electrophoresis electrospray ionization mass spectrometry. J Chromatogr Sci. 2015;53(3):443–449. doi: 10.1093/chromsci/bmu229. [DOI] [PubMed] [Google Scholar]
- Lewandrowski U, Moebius J, Walter U, Sickmann A. Elucidation of N-glycosylation sites on human platelet proteins: a glycoproteomic approach. Mol Cell Proteomics. 2006;5(2):226–233. doi: 10.1074/mcp.M500324-MCP200. [DOI] [PubMed] [Google Scholar]
- Li H, Bendiak B, Siems WF, Gang DR, Hill HH., Jr Ion Mobility-Mass Correlation Trend Line Separation of Glycoprotein Digests without Deglycosylation. Int J Ion Mobil Spectrom. 2013;16(2):105–115. doi: 10.1007/s12127-013-0127-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li QK, Shah P, Li Y, Aiyetan PO, Chen J, Yung R, Zhang H. Glycoproteomic analysis of bronchoalveolar lavage (BAL) fluid identifies tumor-associated glycoproteins from lung adenocarcinoma. J Proteome Res. 2013;12(8):3689–3696. doi: 10.1021/pr400274w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Jiang J, Zhao X, Wang J, Han H, Zhao Y, Qian X. N-glycoproteome analysis of the secretome of human metastatic hepatocellular carcinoma cell lines combining hydrazide chemistry, HILIC enrichment and mass spectrometry. PLoS One. 2013;8(12):e81921. doi: 10.1371/journal.pone.0081921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lih TM, Choong WK, Chen CC, Cheng CW, Lin HN, Chen CT, Sung TY. MAGIC-web: a platform for untargeted and targeted N-linked glycoprotein identification. Nucleic Acids Res. 2016;44(W1):W575–580. doi: 10.1093/nar/gkw254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Wang F, Zhu J, Mao J, Liu Z, Cheng K, Zou H. Highly efficient N-glycoproteomic sample preparation by combining C(18) and graphitized carbon adsorbents. Anal Bioanal Chem. 2014;406(13):3103–3109. doi: 10.1007/s00216-014-7716-9. [DOI] [PubMed] [Google Scholar]
- MacCoss MJ, McDonald WH, Saraf A, Sadygov R, Clark JM, Tasto JJ, Yates JR., 3rd Shotgun identification of protein modifications from protein complexes and lens tissue. Proc Natl Acad Sci U S A. 2002;99(12):7900–7905. doi: 10.1073/pnas.122231399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madera M, Mann B, Mechref Y, Novotny MV. Efficacy of glycoprotein enrichment by microscale lectin affinity chromatography. J Sep Sci. 2008;31(14):2722–2732. doi: 10.1002/jssc.200800094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayampurath AM, Wu Y, Segu ZM, Mechref Y, Tang H. Improving confidence in detection and characterization of protein N-glycosylation sites and microheterogeneity. Rapid Commun Mass Spectrom. 2011;25(14):2007–2019. doi: 10.1002/rcm.5059. [DOI] [PubMed] [Google Scholar]
- Mechref Y, Hu Y, Garcia A, Hussein A. Identifying cancer biomarkers by mass spectrometry-based glycomics. Electrophoresis. 2012;33(12):1755–1767. doi: 10.1002/elps.201100715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mechref Y, Madera M, Novotny MV. Glycoprotein enrichment through lectin affinity techniques. Methods Mol Biol. 2008;424:373–396. doi: 10.1007/978-1-60327-064-9_29. [DOI] [PubMed] [Google Scholar]
- Moremen KW, Tiemeyer M, Nairn AV. Vertebrate protein glycosylation: diversity, synthesis and function. Nat Rev Mol Cell Biol. 2012;13(7):448–462. doi: 10.1038/nrm3383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mysling S, Palmisano G, Hojrup P, Thaysen-Andersen M. Utilizing ion-pairing hydrophilic interaction chromatography solid phase extraction for efficient glycopeptide enrichment in glycoproteomics. Anal Chem. 2010;82(13):5598–5609. doi: 10.1021/ac100530w. [DOI] [PubMed] [Google Scholar]
- Nagel T, Meyer B. Simultaneous characterization of sequence polymorphisms, glycosylation and phosphorylation of fibrinogen in a direct analysis by LC-MS. Biochim Biophys Acta. 2014;1844(12):2284–2289. doi: 10.1016/j.bbapap.2014.09.021. [DOI] [PubMed] [Google Scholar]
- Nilsson J, Ruetschi U, Halim A, Hesse C, Carlsohn E, Brinkmalm G, Larson G. Enrichment of glycopeptides for glycan structure and attachment site identification. Nat Methods. 2009;6(11):809–811. doi: 10.1038/nmeth.1392. [DOI] [PubMed] [Google Scholar]
- Nwosu CC, Huang J, Aldredge DL, Strum JS, Hua S, Seipert RR, Lebrilla CB. In-gel nonspecific proteolysis for elucidating glycoproteins: a method for targeted protein-specific glycosylation analysis in complex protein mixtures. Anal Chem. 2013;85(2):956–963. doi: 10.1021/ac302574f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connor SE, Imperiali B. Modulation of protein structure and function by asparagine-linked glycosylation. Chem Biol. 1996;3(10):803–812. doi: 10.1016/s1074-5521(96)90064-2. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/8939697. [DOI] [PubMed] [Google Scholar]
- Olivova P, Chen W, Chakraborty AB, Gebler JC. Determination of N-glycosylation sites and site heterogeneity in a monoclonal antibody by electrospray quadrupole ion-mobility time-of-flight mass spectrometry. Rapid Commun Mass Spectrom. 2008;22(1):29–40. doi: 10.1002/rcm.3330. [DOI] [PubMed] [Google Scholar]
- Olsen JV, Macek B, Lange O, Makarov A, Horning S, Mann M. Higher-energy C-trap dissociation for peptide modification analysis. Nat Methods. 2007;4(9):709–712. doi: 10.1038/nmeth1060. [DOI] [PubMed] [Google Scholar]
- Ongay S, Boichenko A, Govorukhina N, Bischoff R. Glycopeptide enrichment and separation for protein glycosylation analysis. J Sep Sci. 2012;35(18):2341–2372. doi: 10.1002/jssc.201200434. [DOI] [PubMed] [Google Scholar]
- Park GW, Kim JY, Hwang H, Lee JY, Ahn YH, Lee HK, Yoo JS. Integrated GlycoProteome Analyzer (I-GPA) for Automated Identification and Quantitation of Site-Specific N-Glycosylation. Sci Rep. 2016;6:21175. doi: 10.1038/srep21175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin X, Chen Q, Sun C, Wang C, Peng Q, Xie L, Li S. High-throughput screening of tumor metastatic-related differential glycoprotein in hepatocellular carcinoma by iTRAQ combines lectin-related techniques. Med Oncol. 2013;30(1):420. doi: 10.1007/s12032-012-0420-8. [DOI] [PubMed] [Google Scholar]
- Segu ZM, Hammad LA, Mechref Y. Rapid and efficient glycoprotein identification through microwave-assisted enzymatic digestion. Rapid Commun Mass Spectrom. 2010;24(23):3461–3468. doi: 10.1002/rcm.4774. [DOI] [PubMed] [Google Scholar]
- Siepen JA, Keevil EJ, Knight D, Hubbard SJ. Prediction of missed cleavage sites in tryptic peptides aids protein identification in proteomics. J Proteome Res. 2007;6(1):399–408. doi: 10.1021/pr060507u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh C, Zampronio CG, Creese AJ, Cooper HJ. Higher energy collision dissociation (HCD) product ion-triggered electron transfer dissociation (ETD) mass spectrometry for the analysis of N-linked glycoproteins. J Proteome Res. 2012;11(9):4517–4525. doi: 10.1021/pr300257c. [DOI] [PubMed] [Google Scholar]
- Smith KD, Pollacchi A, Field M, Watson J. The heterogeneity of the glycosylation of alpha-1-acid glycoprotein between the sera and synovial fluid in rheumatoid arthritis. Biomed Chromatogr. 2002;16(4):261–266. doi: 10.1002/bmc.158. [DOI] [PubMed] [Google Scholar]
- Smith LM, Kelleher NL, Consortium for Top Down, P Proteoform: a single term describing protein complexity. Nat Methods. 2013;10(3):186–187. doi: 10.1038/nmeth.2369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song E, Mechref Y. Defining glycoprotein cancer biomarkers by MS in conjunction with glycoprotein enrichment. Biomark Med. 2015;9(9):835–844. doi: 10.2217/bmm.15.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song E, Zhu R, Hammoud ZT, Mechref Y. LC-MS/MS quantitation of esophagus disease blood serum glycoproteins by enrichment with hydrazide chemistry and lectin affinity chromatography. J Proteome Res. 2014;13(11):4808–4820. doi: 10.1021/pr500570m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stavenhagen K, Plomp R, Wuhrer M. Site-Specific Protein N- and O-Glycosylation Analysis by a C18-Porous Graphitized Carbon-Liquid Chromatography-Electrospray Ionization Mass Spectrometry Approach Using Pronase Treated Glycopeptides. Anal Chem. 2015;87(23):11691–11699. doi: 10.1021/acs.analchem.5b02366. [DOI] [PubMed] [Google Scholar]
- Stephenson JL, Jr, McLuckey SA. Simplification of product ion spectra derived from multiply charged parent ions via ion/ion chemistry. Anal Chem. 1998;70(17):3533–3544. doi: 10.1021/ac9802832. [DOI] [PubMed] [Google Scholar]
- Syka JE, Coon JJ, Schroeder MJ, Shabanowitz J, Hunt DF. Peptide and protein sequence analysis by electron transfer dissociation mass spectrometry. Proc Natl Acad Sci U S A. 2004;101(26):9528–9533. doi: 10.1073/pnas.0402700101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thaysen-Andersen M, Mysling S, Hojrup P. Site-specific glycoprofiling of N-linked glycopeptides using MALDI-TOF MS: strong correlation between signal strength and glycoform quantities. Anal Chem. 2009;81(10):3933–3943. doi: 10.1021/ac900231w. [DOI] [PubMed] [Google Scholar]
- Thaysen-Andersen M, Packer NH. Advances in LC-MS/MS-based glycoproteomics: getting closer to system-wide site-specific mapping of the N- and O-glycoproteome. Biochim Biophys Acta. 2014;1844(9):1437–1452. doi: 10.1016/j.bbapap.2014.05.002. [DOI] [PubMed] [Google Scholar]
- Thaysen-Andersen M, Wilkinson BL, Payne RJ, Packer NH. Site-specific characterisation of densely O-glycosylated mucin-type peptides using electron transfer dissociation ESI-MS/MS. Electrophoresis. 2011;32(24):3536–3545. doi: 10.1002/elps.201100294. [DOI] [PubMed] [Google Scholar]
- Ting L, Cowley MJ, Hoon SL, Guilhaus M, Raftery MJ, Cavicchioli R. Normalization and statistical analysis of quantitative proteomics data generated by metabolic labeling. Mol Cell Proteomics. 2009;8(10):2227–2242. doi: 10.1074/mcp.M800462-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toghi Eshghi S, Shah P, Yang W, Li X, Zhang H. GPQuest: A Spectral Library Matching Algorithm for Site-Specific Assignment of Tandem Mass Spectra to Intact N-glycopeptides. Anal Chem. 2015;87(10):5181–5188. doi: 10.1021/acs.analchem.5b00024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran BQ, Barton C, Feng J, Sandjong A, Yoon SH, Awasthi S, Goo YA. Comprehensive glycosylation profiling of IgG and IgG-fusion proteins by top-down MS with multiple fragmentation techniques. J Proteomics. 2016;134:93–101. doi: 10.1016/j.jprot.2015.10.021. [DOI] [PubMed] [Google Scholar]
- Tsai HY, Boonyapranai K, Sriyam S, Yu CJ, Wu SW, Khoo KH, Chen ST. Glycoproteomics analysis to identify a glycoform on haptoglobin associated with lung cancer. Proteomics. 2011;11(11):2162–2170. doi: 10.1002/pmic.201000319. [DOI] [PubMed] [Google Scholar]
- Tsai TH, Song E, Zhu R, Di Poto C, Wang M, Luo Y, Ressom HW. LC-MS/MS-based serum proteomics for identification of candidate biomarkers for hepatocellular carcinoma. Proteomics. 2015;15(13):2369–2381. doi: 10.1002/pmic.201400364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandermarliere E, Mueller M, Martens L. Getting intimate with trypsin, the leading protease in proteomics. Mass Spectrom Rev. 2013;32(6):453–465. doi: 10.1002/mas.21376. [DOI] [PubMed] [Google Scholar]
- Varki A. Biological roles of oligosaccharides: all of the theories are correct. Glycobiology. 1993;3(2):97–130. doi: 10.1093/glycob/3.2.97. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/8490246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, Hincapie M, Rejtar T, Karger BL. Ultrasensitive characterization of site-specific glycosylation of affinity-purified haptoglobin from lung cancer patient plasma using 10 mum i.d. porous layer open tubular liquid chromatography-linear ion trap collision-induced dissociation/electron transfer dissociation mass spectrometry. Anal Chem. 2011;83(6):2029–2037. doi: 10.1021/ac102825g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JZ, Grundke-Iqbal I, Iqbal K. Glycosylation of microtubule-associated protein tau: an abnormal posttranslational modification in Alzheimer's disease. Nat Med. 1996;2(8):871–875. doi: 10.1038/nm0896-871. [DOI] [PubMed] [Google Scholar]
- Wang Y, Ao X, Vuong H, Konanur M, Miller FR, Goodison S, Lubman DM. Membrane glycoproteins associated with breast tumor cell progression identified by a lectin affinity approach. J Proteome Res. 2008;7(10):4313–4325. doi: 10.1021/pr8002547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whelan SA, Lu M, He J, Yan W, Saxton RE, Faull KF, Chang HR. Mass spectrometry (LC-MS/MS) site-mapping of N-glycosylated membrane proteins for breast cancer biomarkers. J Proteome Res. 2009;8(8):4151–4160. doi: 10.1021/pr900322g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkins MR, Pasquali C, Appel RD, Ou K, Golaz O, Sanchez JC, Hochstrasser DF. From proteins to proteomes: large scale protein identification by two-dimensional electrophoresis and amino acid analysis. Biotechnology (N Y) 1996;14(1):61–65. doi: 10.1038/nbt0196-61. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/9636313. [DOI] [PubMed] [Google Scholar]
- Wohlgemuth J, Karas M, Eichhorn T, Hendriks R, Andrecht S. Quantitative site-specific analysis of protein glycosylation by LC-MS using different glycopeptide-enrichment strategies. Anal Biochem. 2009;395(2):178–188. doi: 10.1016/j.ab.2009.08.023. [DOI] [PubMed] [Google Scholar]
- Wolters DA, Washburn MP, Yates JR., 3rd An automated multidimensional protein identification technology for shotgun proteomics. Anal Chem. 2001;73(23):5683–5690. doi: 10.1021/ac010617e. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/11774908. [DOI] [PubMed] [Google Scholar]
- Wu SL, Huhmer AF, Hao Z, Karger BL. On-line LC-MS approach combining collision-induced dissociation (CID), electron-transfer dissociation (ETD), and CID of an isolated charge-reduced species for the trace-level characterization of proteins with post-translational modifications. J Proteome Res. 2007;6(11):4230–4244. doi: 10.1021/pr070313u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Mechref Y, Klouckova I, Mayampurath A, Novotny MV, Tang H. Mapping site-specific protein N-glycosylations through liquid chromatography/mass spectrometry and targeted tandem mass spectrometry. Rapid Commun Mass Spectrom. 2010;24(7):965–972. doi: 10.1002/rcm.4474. [DOI] [PubMed] [Google Scholar]
- Wuhrer M, Deelder AM, Hokke CH. Protein glycosylation analysis by liquid chromatography-mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 2005;825(2):124–133. doi: 10.1016/j.jchromb.2005.01.030. [DOI] [PubMed] [Google Scholar]
- Xu T, Wong CC, Kashina A, Yates JR., 3rd Identification of N-terminally arginylated proteins and peptides by mass spectrometry. Nat Protoc. 2009;4(3):325–332. doi: 10.1038/nprot.2008.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Nyalwidhe JO, Guo S, Drake RR, Semmes OJ. Targeted identification of metastasis-associated cell-surface sialoglycoproteins in prostate cancer. Mol Cell Proteomics. 2011;10(6):M110.007294. doi: 10.1074/mcp.M110.007294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Barendregt A, Kamerling JP, Heck AJ. Analyzing protein micro-heterogeneity in chicken ovalbumin by high-resolution native mass spectrometry exposes qualitatively and semi-quantitatively 59 proteoforms. Anal Chem. 2013;85(24):12037–12045. doi: 10.1021/ac403057y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Hancock WS. Approach to the comprehensive analysis of glycoproteins isolated from human serum using a multi-lectin affinity column. J Chromatogr A. 2004;1053(1–2):79–88. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/15543974. [PubMed] [Google Scholar]
- Yang Z, Harris LE, Palmer-Toy DE, Hancock WS. Multilectin affinity chromatography for characterization of multiple glycoprotein biomarker candidates in serum from breast cancer patients. Clin Chem. 2006;52(10):1897–1905. doi: 10.1373/clinchem.2005.065862. [DOI] [PubMed] [Google Scholar]
- Yates JR., 3rd Mass spectral analysis in proteomics. Annu Rev Biophys Biomol Struct. 2004;33:297–316. doi: 10.1146/annurev.biophys.33.111502.082538. [DOI] [PubMed] [Google Scholar]
- Yin X, Bern M, Xing Q, Ho J, Viner R, Mayr M. Glycoproteomic analysis of the secretome of human endothelial cells. Mol Cell Proteomics. 2013;12(4):956–978. doi: 10.1074/mcp.M112.024018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu CY, Mayampurath A, Zhu R, Zacharias L, Song E, Wang L, Tang H. Automated Glycan Sequencing from Tandem Mass Spectra of N-Linked Glycopeptides. Anal Chem. 2016;88(11):5725–5732. doi: 10.1021/acs.analchem.5b04858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zacharias LG, Hartmann AK, Song E, Zhao J, Zhu R, Mirzaei P, Mechref Y. HILIC and ERLIC Enrichment of Glycopeptides Derived from Breast and Brain Cancer Cells. J Proteome Res. 2016 doi: 10.1021/acs.jproteome.6b00429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zacharias LHA, Song E, Zhao J, Zhu R, Mirzaei P, Mechref Y. HILIC and ERLIC Enrichment of Glycopeptides Derived from Breast and Brain Cancer Cells. J Proteome Res. 2016 doi: 10.1021/acs.jproteome.6b00429. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zauner G, Deelder AM, Wuhrer M. Recent advances in hydrophilic interaction liquid chromatography (HILIC) for structural glycomics. Electrophoresis. 2011;32(24):3456–3466. doi: 10.1002/elps.201100247. [DOI] [PubMed] [Google Scholar]
- Zauner G, Koeleman CA, Deelder AM, Wuhrer M. Protein glycosylation analysis by HILIC-LC-MS of Proteinase K-generated N- and O-glycopeptides. J Sep Sci. 2010;33(6–7):903–910. doi: 10.1002/jssc.200900850. [DOI] [PubMed] [Google Scholar]
- Zeng WF, Liu MQ, Zhang Y, Wu JQ, Fang P, Peng C, Yang P. pGlyco: a pipeline for the identification of intact N-glycopeptides by using HCD- and CID-MS/MS and MS3. Sci Rep. 2016;6:25102. doi: 10.1038/srep25102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng X, Hood BL, Sun M, Conrads TP, Day RS, Weissfeld JL, Bigbee WL. Lung cancer serum biomarker discovery using glycoprotein capture and liquid chromatography mass spectrometry. J Proteome Res. 2010;9(12):6440–6449. doi: 10.1021/pr100696n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Ye Z, Xue P, Shu Q, Zhou Y, Ji Y, Yang F. Evaluation of different N-glycopeptide enrichment methods for N-glycosylation sites mapping in mouse brain. J Proteome Res. 2016 doi: 10.1021/acs.jproteome.6b00098. [DOI] [PubMed] [Google Scholar]
- Zhang H, Guo T, Li X, Datta A, Park JE, Yang J, Sze SK. Simultaneous characterization of glyco- and phosphoproteomes of mouse brain membrane proteome with electrostatic repulsion hydrophilic interaction chromatography. Mol Cell Proteomics. 2010;9(4):635–647. doi: 10.1074/mcp.M900314-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Li XJ, Martin DB, Aebersold R. Identification and quantification of N-linked glycoproteins using hydrazide chemistry, stable isotope labeling and mass spectrometry. Nat Biotechnol. 2003;21(6):660–666. doi: 10.1038/nbt827. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Fonslow BR, Shan B, Baek MC, Yates JR., 3rd Protein analysis by shotgun/bottom-up proteomics. Chem Rev. 2013;113(4):2343–2394. doi: 10.1021/cr3003533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Go EP, Desaire H. Maximizing coverage of glycosylation heterogeneity in MALDI-MS analysis of glycoproteins with up to 27 glycosylation sites. Anal Chem. 2008;80(9):3144–3158. doi: 10.1021/ac702081a. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Yu CY, Song E, Li SC, Mechref Y, Tang H, Liu X. Identification of Glycopeptides with Multiple Hydroxylysine O-Glycosylation Sites by Tandem Mass Spectrometry. J Proteome Res. 2015;14(12):5099–5108. doi: 10.1021/acs.jproteome.5b00299. [DOI] [PubMed] [Google Scholar]
- Zhao J, Song E, Zhu R, Mechref Y. Parallel data acquisition of in-source fragmented glycopeptides to sequence the glycosylation sites of proteins. Electrophoresis. 2016;37(11):1420–1430. doi: 10.1002/elps.201500562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Szeto SS, Kong RP, Law CH, Li G, Quan Q, Chu IK. Online two-dimensional porous graphitic carbon/reversed phase liquid chromatography platform applied to shotgun proteomics and glycoproteomics. Anal Chem. 2014;86(24):12172–12179. doi: 10.1021/ac503254t. [DOI] [PubMed] [Google Scholar]
- Zhu Z, Su X, Go EP, Desaire H. New glycoproteomics software, GlycoPep Evaluator, generates decoy glycopeptides de novo and enables accurate false discovery rate analysis for small data sets. Anal Chem. 2014;86(18):9212–9219. doi: 10.1021/ac502176n. [DOI] [PMC free article] [PubMed] [Google Scholar]