

Abstract
Gas–liquid mass transfer of gaseous reactants is a major limitation for high space–time yields, especially for O2‐dependent (bio)catalytic reactions in aqueous solutions. Herein, oxygenic photosynthesis was used for homogeneous O2 supply via in situ generation in the liquid phase to overcome this limitation. The phototrophic cyanobacterium Synechocystis sp. PCC6803 was engineered to synthesize the alkane monooxygenase AlkBGT from Pseudomonas putida GPo1. With light, but without external addition of O2, the chemo‐ and regioselective hydroxylation of nonanoic acid methyl ester to ω‐hydroxynonanoic acid methyl ester was driven by O2 generated through photosynthetic water oxidation. Photosynthesis also delivered the necessary reduction equivalents to regenerate the Fe2+ center in AlkB for oxygen transfer to the terminal methyl group. The in situ coupling of oxygenic photosynthesis to O2‐transferring enzymes now enables the design of fast hydrocarbon oxyfunctionalization reactions.
Keywords: biocatalysis, oxidoreductases, oxyfunctionalization, oxygen mass transfer, photosynthesis
Gas–liquid mass transfer defines the performance and efficiency of reactions in liquids with gaseous reactants. This is especially true for (bio)catalysts operating in aqueous solutions.1 O2 is one of the most prominent gaseous reactants. As an oxidant for oxidative catalysis, O2 is of great importance for the production of value‐added chemicals and pharmaceuticals.2 For the efficient use of O2 as a reactant, harsh reaction conditions with high temperatures and/or pressures are typically necessary. Such conditions may lead to severe safety and selectivity issues, often resulting in low reaction yields. They typically also necessitate highly regulated, elaborate, and thus expensive process control regimes.2a, 3 Mild reaction conditions, high selectivities, and high yields are generally desirable for oxidative production processes and achieved most efficiently by enzyme catalysis.4 However, low gas–liquid mass transfer rates unfortunately constitute major limitations under such mild conditions.1d Furthermore, the application of enzymes in whole cells, which is advantageous for oxygenases, suffers from a competition for O2 between the target reaction and respiration.5 A technical solution for increasing the O2 gas–liquid mass transfer rate under ambient conditions is the utilization of O2‐enriched air.6 Yet, O2 mass transfer is basically limiting the space–time yields of processes with high oxidation rates, especially in the production of bulk chemicals.1d, 5a, 7 To improve O2 mass transfer, various reactor concepts with different modes of gaseous reactant supply have been proposed.2a Examples include the utilization of bubble columns, gas‐permeable membranes, segmented flow microreactors, or falling film microreactors.7a, 8
Herein, we report a novel concept based on oxygenic photosynthesis for the homogeneous supply of O2 to an oxidation reaction. To date, several studies have investigated the coupling of light‐driven electron activation to (enzymatic) reactions, both chemically and biotechnologically.9 However, light‐driven water oxidation has not been considered for the homogeneous supply of O2. Photosynthesis generates O2 in situ within an aqueous liquid phase from water. This has the potential to basically overcome gas–liquid mass transfer limitations. Light‐driven photosynthetic water oxidation is the core of our concept, delivering O2 homogeneously within cells to the catalytically active oxygenase enzyme, thus driving the oxyfunctionalization reaction (Figure 1).
Figure 1.
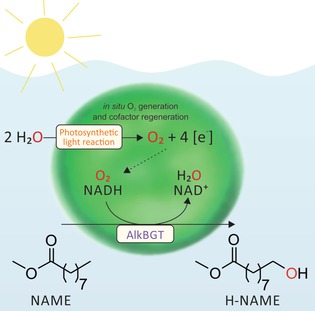
Homogenous O2 evolution coupled to an oxygenase‐catalyzed oxyfunctionalization reaction. Water is oxidized by the photosynthetic cyanobacterium Synechocystis sp. PCC6803, yielding O2 and activated reduction equivalents. The heterologously introduced alkane monooxygenase system AlkBGT captures both O2 and the reduction equivalents, and catalyzes the regiospecific oxyfunctionalization of nonanoic acid methyl ester (NAME) to ω‐hydroxynonanoic acid methyl ester (H‐NAME).
The well‐studied phototrophic cyanobacterium Synechocystis sp. PCC 6803 was chosen as the source for delivering O2. It was engineered for the synthesis of alkane monooxygenase AlkBGT originating from Pseudomonas putida GPo1 (hereinafter referred to as Syn6803 pAH042; see the Supporting Information for experimental procedures).10 The highly regioselective terminal oxyfunctionalization of nonanoic acid methyl ester served as the model oxidation reaction. It constitutes an industrially relevant example for the production of polymer building blocks from renewables (Figure 1).11
Syn6803 pAH042 produced ca. 65 μm ω‐hydroxynonanoic acid methyl ester (H‐NAME) from 10 mm nonanoic acid methyl ester (NAME) within 20 min under constant illumination. This translates into a specific oxidation rate of 1.5±0.2 μmol min−1 gCDW −1 (Table 1) and demonstrates the functionality of the biocatalyst. However, a specific oxidation rate of 1.3±0.1 μmol min−1 gCDW −1 was still measured in the dark, showing that reduction equivalents were supplied at almost the same rate with and without light (Table 1). Obviously, the catabolism of storage compounds enabled substantial NAD(P)H regeneration in the dark.
Table 1.
Specific rates for the hydroxylation of nonanoic acid methyl ester to ω‐hydroxynonanoic acid methyl ester and O2 evolution of Syn6803 pAH042.
| Conditions | Specific production rate [μmol min−1 gCDW −1] |
|---|---|
| Aerobic, irradiated[a] | 1.5±0.2 |
| Aerobic, in the dark[a] | 1.3±0.1 |
| Anaerobic, irradiated[b] | 0.9±0.1 |
| Anaerobic, in the dark[b] | 0.0 |
| Anaerobic, irradiated, OER[c] | 3.7±0.5 |
Specific product formation rates are given with respect to the product formed after [a] 20 or [b] 30 min. [c] The specific O2 evolution rate (OER) was determined within the aqueous phase in a sealed, gas‐free glass chamber in the absence of substrate. Average values and standard deviations of at least two independent biological replicates are given.
Upon successful construction of the functional phototrophic whole‐cell biocatalyst, we evaluated the oxidation reaction for exclusive utilization of photosynthetically generated O2. The terminal hydroxylation of NAME by Syn6803 pAH042 was studied under anaerobic, but otherwise identical conditions. H‐NAME formation depended directly upon illumination and thus water oxidation. Product formation was not observed in the absence of light (Figure 2). The specific oxidation rate obtained under anaerobic conditions and illumination was 0.9±0.1 μmol min−1 gCDW −1 (Table 1), de facto driven by O2 generated in the photosynthetic light reaction.
Figure 2.
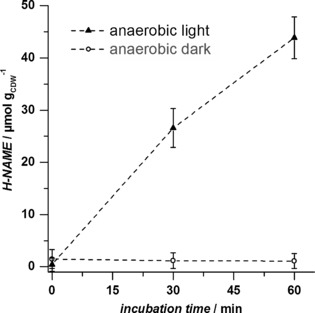
In situ supply of photosynthetically generated O2 to the oxidizing enzyme AlkBGT in Syn6803 pAH042. The biotransformation experiment was performed under anaerobic conditions under irradiation (‐ ‐ ‐ ‐▴‐ ‐ ‐ ‐) or in the dark (‐ ‐ ‐ ‐○‐ ‐ ‐ ‐). Average values and standard deviations of two independent biological replicates are given. CDW=cell dry weight.
The specific O2 evolution rate of Syn6803 pAH042 was determined separately in the absence of the substrate NAME, for assessing the fraction of photosynthetically generated O2 captured by the monooxygenase (Table 1). With an O2 evolution rate of 3.7±0.5 μmol min−1 gCDW −1, corresponding to 100 % of O2 available in the system (assuming no photorespiration), nearly 25 % of the photosynthetically generated O2 was captured for terminal hydroxylation of NAME.
Diffusion of photosynthetically generated O2 may affect the reaction efficiency of the terminal hydroxylation and theoretically results in gas–liquid mass transfer processes within the assay system. The specific O2 accumulation rate in the aqueous phase was calculated to be 0.01 μmol min−1 gCDW −1 assuming immediate O2 diffusion from the aqueous to the gaseous phase (aqueous/gaseous ratio 1:10, Henry volatility for O2 in water: H cc=c aq/c gas=0.0297 at 25 °C).12 Thus the effective O2 concentration does not exceed 0.6 μm within 30 min of reaction time (applied biomass concentration: 2 gCDW L−1). In contrast, Michaelis constants (K M) of oxygenases with respect to O2 are typically in the range of 10–60 μm.5a This, together with the high fraction of O2 captured by the monooxygenase (25 %), suggests that the photosynthetically generated O2 is concentrated within the microbial cell and captured in situ by the monooxygenase before diffusing out of the cell. Although O2 can in principle diffuse across cellular membranes, the lipid bilayer system seems to pose a physical barrier that is beneficial for the intracellular oxidation process. These results are proof of concept for the in situ coupling of photosynthetic O2 evolution to O2‐dependent oxidation reactions. The photosynthetic light reaction was used for the intracellular supply of both activated reduction equivalents and O2.
These results might be the starting point for the development of various efficient photosynthesis‐driven oxyfunctionalization reactions. In the present case, future optimizations include an increase in the AlkBGT level in the cyanobacterial whole‐cell biocatalyst.13 This is obvious from comparing the transformation rates of NAME into H‐NAME catalyzed by E. coli W3110 carrying the very plasmid pAH042 (10.0±0.1 μmol min−1 gCDW −1; see S4 in the Supporting Information) with those of E. coli that strongly express alkBGT (104–128 μmol min−1 gCDW −1).14 Other targets are electron channeling and improved cultivation and bioreactor concepts. The cyanobacterial photosynthetic metabolism supports the supply of activated reduction equivalents at high rates (123 μmol min−1 gCDW −1).9b Yet, the O2 evolution rate determined in this study implies a photosynthetic activity of only 3.7 μmol min−1 gCDW −1. This corresponds to a specific NAD(P)H regeneration rate of 7.4 μmol min−1 gCDW −1. The theoretical maximum of this rate was estimated to be 850 μmol min−1 gCDW −1 (assumptions for PSII: k cat=1000 s−1, 10 mg gCDW −1, M W=350 kDa).9b, 15 With high biomass concentrations (40 gCDW L−1), a theoretical maximum of 2040 mmol L−1 h−1 would be possible for the oxygen supply rate. This translates into a volumetric mass transfer coefficient k L A of 4533 h−1 for a bioreactor operated at 2.5 atm, 30 °C, and a residual O2 concentration of 100 μm (typical conditions for large‐scale bioreactor operation).5a In contrast, the k L A values of large‐scale bioreactors are on the order of 200 h−1.5a In addition, the use of photoautotrophic instead of chemoheterotrophic organisms largely relieves the competition for O2 between oxygenation and respiration.
The development of photobioreactors enabling the generation of high biomass concentrations with high oxygen evolution activity is key for the future applicability of the presented concept.16 Biofilm cultivation in capillary microreactors constitutes one possible solution to increase the cyanobacterial biomass concentration.17 Stable cyanobacterial biofilm cultivation has recently been achieved over several weeks with retention of the photosynthetic activity throughout the biofilm. Reaction optimization addressing the key issue of photobioreactor development has the potential to facilitate currently oxygen‐transfer‐limited selective hydroxylation processes for the biocatalytic functionalization of hydrocarbons.5 In summary, the in situ coupling of oxygenic photosynthesis to oxidizing enzymes provides a novel and safe access to O2 as a reactant for designing new reactions for oxidation catalysis.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank Birke Brumme, Lisa‐Marie Bangen (DBFZ, Leipzig, Germany), and Dr. Sabine Kleinsteuber (UMB, UFZ, Leipzig, Germany) for assistance and laboratory infrastructure. The group of Victor de Lorenzo (Madrid, Spain) and Prof. Peter Lindblad (Uppsala University, Sweden) kindly provided the plasmids pSEVA251 and pPMQAK1 and pSB1AC3_PrnpB:lacI and pSB1AC3_Ptrc1O:GFP, respectively. We acknowledge the use of the facilities of the Centre for Biocatalysis (MiKat) at the Helmholtz Centre for Environmental Research, which is supported by European Regional Development Funds (EFRE, Europe funds Saxony) and the Helmholtz Association.
A. Hoschek, B. Bühler, A. Schmid, Angew. Chem. Int. Ed. 2017, 56, 15146.
References
- 1.
- 1a. Chaudhari R., Bhattacharya A., Bhanage B., Catal. Today 1995, 24, 123–133; [Google Scholar]
- 1b. Wachsen O., Himmler K., Cornils B., Catal. Today 1998, 42, 373–379; [Google Scholar]
- 1c. Cornils B., Herrmann W. A., Aqueous-phase organometallic catalysis: concepts and applications, Wiley, Chichester, 2004, pp. 207; [Google Scholar]
- 1d. Law H., Baldwin C., Chen B., Woodley J., Chem. Eng. Sci. 2006, 61, 6646–6652; [Google Scholar]
- 1e. Park J. B., J. Microbiol. Biotechnol. 2007, 17, 379–392. [PubMed] [Google Scholar]
- 2.
- 2a. Gavriilidis A., Constantinou A., Hellgardt K., Hii K. K. M., Hutchings G. J., Brett G. L., Kuhn S., Marsden S. P., React. Chem. Eng. 2016, 1, 595–612; [Google Scholar]
- 2b. Shi Z., Zhang C., Tang C., Jiao N., Chem. Soc. Rev. 2012, 41, 3381–3430; [DOI] [PubMed] [Google Scholar]
- 2c. Piera J., Bäckvall J. E., Angew. Chem. Int. Ed. 2008, 47, 3506–3523; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 3558–3576; [Google Scholar]
- 2d. Bühler B., Bollhalder I., Hauer B., Witholt B., Schmid A., Biotechnol. Bioeng. 2003, 82, 833–842. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Schuchardt U., Cardoso D., Sercheli R., Pereira R., Da Cruz R. S., Guerreiro M. C., Mandelli D., Spinacé E. V., Pires E. L., Appl. Catal. 2001, 211, 1–17; [Google Scholar]
- 3b. Osterberg P. M., Niemeier J. K., Welch C. J., Hawkins J. M., Martinelli J. R., Johnson T. E., Root T. W., Stahl S. S., Org. Process Res. Dev. 2015, 19, 1537–1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.
- 4a. Bordeaux M., Galarneau A., Drone J., Angew. Chem. Int. Ed. 2012, 51, 10712–10723; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 10870–10881; [Google Scholar]
- 4b. Schmid A., Dordick J. S., Hauer B., Kiener A., Wubbolts M., Witholt B., Nature 2001, 409, 258–268. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a. Duetz W. A., van Beilen J. B., Witholt B., Curr. Opin. Biotechnol. 2001, 12, 419–425; [DOI] [PubMed] [Google Scholar]
- 5b. Schrewe M., Julsing M. K., Bühler B., Schmid A., Chem. Soc. Rev. 2013, 42, 6346–6377. [DOI] [PubMed] [Google Scholar]
- 6. Hilker I., Baldwin C., Alphand V., Furstoss R., Woodley J., Wohlgemuth R., Biotechnol. Bioeng. 2006, 93, 1138–1144. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a. Gemoets H. P., Su Y., Shang M., Hessel V., Luque R., Noël T., Chem. Soc. Rev. 2016, 45, 83–117; [DOI] [PubMed] [Google Scholar]
- 7b. Garcia-Ochoa F., Gomez E., Biotechnol. Adv. 2009, 27, 153–176. [DOI] [PubMed] [Google Scholar]
- 8.
- 8a. Karande R., Schmid A., Buehler K., Adv. Synth. Catal. 2011, 353, 2511–2521; [Google Scholar]
- 8b. Greene J. F., Preger Y., Stahl S. S., Root T. W., Org. Process Res. Dev. 2015, 19, 858–864; [Google Scholar]
- 8c. Tomaszewski B., Lloyd R. C., Warr A. J., Buehler K., Schmid A., ChemCatChem 2014, 6, 2567–2576; [Google Scholar]
- 8d. Kantarci N., Borak F., Ulgen K. O., Process Biochem. 2005, 40, 2263–2283; [Google Scholar]
- 8e. Bolivar J. M., Krämer C. E., Ungerböck B., Mayr T., Nidetzky B., Biotechnol. Bioeng. 2016, 113, 1862–1872. [DOI] [PubMed] [Google Scholar]
- 9.
- 9a. Hisatomi T., Kubota J., Domen K., Chem. Soc. Rev. 2014, 43, 7520–7535; [DOI] [PubMed] [Google Scholar]
- 9b. Köninger K., Gomez Baraibar A., Mügge C., Paul C. E., Hollmann F., Nowaczyk M. M., Kourist R., Angew. Chem. Int. Ed. 2016, 55, 5582–5585; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 5672–5675; [Google Scholar]
- 9c. Lassen L. M., Nielsen A. Z., Ziersen B., Gnanasekaran T., Moller B. L., Jensen P. E., ACS Synth. Biol. 2014, 3, 1–12; [DOI] [PubMed] [Google Scholar]
- 9d. Okeefe D. P., Tepperman J. M., Dean C., Leto K. J., Erbes D. L., Odell J. T., Plant Physiol. 1994, 105, 473–482; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9e. Mifsud M., Gargiulo S., Iborra S., Arends I. W., Hollmann F., Corma A., Nat. Commun. 2014, 5, 3145; [DOI] [PubMed] [Google Scholar]
- 9f. Hollmann F., Taglieber A., Schulz F., Reetz M. T., Angew. Chem. Int. Ed. 2007, 46, 2903–2906; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2007, 119, 2961–2964; [Google Scholar]
- 9g. Balcerzak L., Lipok J., Strub D., Lochyński S., J. Appl. Microbiol. 2014, 117, 1523–1536; [DOI] [PubMed] [Google Scholar]
- 9h. Yu Y., You L., Liu D., Hollinshead W., Tang Y. J., Zhang F., Mar. Drugs 2013, 11, 2894–2916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Peterson J. A., Basu D., Coon M. J., J. Biol. Chem. 1966, 241, 5162–5164. [PubMed] [Google Scholar]
- 11.
- 11a. Schrewe M., Julsing M. K., Lange K., Czarnotta E., Schmid A., Bühler B., Biotechnol. Bioeng. 2014, 111, 1820–1830; [DOI] [PubMed] [Google Scholar]
- 11b. Ladkau N., Assmann M., Schrewe M., Julsing M. K., Schmid A., Bühler B., Metab. Eng. 2016, 36, 1–9; [DOI] [PubMed] [Google Scholar]
- 11c. Schaffer S., Haas T., Org. Process Res. Dev. 2014, 18, 752–766; [Google Scholar]
- 11d.Evonik Industries AG, Elements 45—Quarterly Science Newsletter 2013, 45, 13.
- 12. Sander R., Atmos. Chem. Phys. 2015, 15, 4399–4981. [Google Scholar]
- 13.
- 13a. Oliver J. W. K., Atsumi S., Photosynth. Res. 2014, 120, 249–261; [DOI] [PubMed] [Google Scholar]
- 13b. Ruffing A. M., Bioeng. Bugs 2011, 2, 136–149. [DOI] [PubMed] [Google Scholar]
- 14.
- 14a. Julsing M. K., Schrewe M., Cornelissen S., Hermann I., Schmid A., Bühler B., Appl. Environ. Microbiol. 2012, 78, 5724–5733; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14b. Schrewe M., Magnusson A. O., Willrodt C., Bühler B., Schmid A., Adv. Synth. Catal. 2011, 353, 3485–3495. [Google Scholar]
- 15.
- 15a. Dismukes G. C., Brimblecombe R., Felton G. A., Pryadun R. S., Sheats J. E., Spiccia L., Swiegers G. F., Acc. Chem. Res. 2009, 42, 1935–1943; [DOI] [PubMed] [Google Scholar]
- 15b. Shen J. R., Annu. Rev. Plant. Biol. 2015, 66, 23–48. [DOI] [PubMed] [Google Scholar]
- 16. Kumar K., Dasgupta C. N., Nayak B., Lindblad P., Das D., Bioresour. Technol. 2011, 102, 4945–4953. [DOI] [PubMed] [Google Scholar]
- 17. David C., Bühler K., Schmid A., J. Ind. Microbiol. Biotechnol. 2015, 42, 1083–1089. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary