

Abstract
A set of bifunctional oxidase–peroxidases has been prepared by fusing four distinct oxidases to a peroxidase. Although such fusion enzymes have not been observed in nature, they could be expressed and purified in good yields. Characterization revealed that the artificial enzymes retained the capability to bind the two required cofactors and were catalytically active as oxidase and peroxidase. Peroxidase fusions of alditol oxidase and chitooligosaccharide oxidase could be used for the selective detection of xylitol and cellobiose with a detection limit in the low‐micromolar range. The peroxidase fusions of eugenol oxidase and 5‐hydroxymethylfurfural oxidase could be used for dioxygen‐driven, one‐pot, two‐step cascade reactions to convert vanillyl alcohol into divanillin and eugenol into lignin oligomers. The designed oxidase–peroxidase fusions represent attractive biocatalysts that allow efficient biocatalytic cascade oxidations that only require molecular oxygen as an oxidant.
Keywords: biocatalysis, domino reactions, enzymes, protein engineering, sensors
In nature, most enzymes take part in metabolic pathways in which each formed product is a substrate for the next enzymatic reaction. To optimize the efficiency of such intricate biocatalytic cascades, the enzymes are often brought together to form enzyme complexes, for example, the pyruvate dehydrogenase complex, the microbial type I fatty acid synthase complex, and the cellulosome.1, 2, 3 The cellulosome is found in anaerobic microorganisms and consists of a scaffolding protein, which brings together the required hydrolytic enzymes to degrade cellulosic biomass.3 In some cases, this has even led to the fusion of two or more enzymes to create a bi‐/multifunctional protein,4 for example, pyrroline‐5‐carboxylate synthase, which features both glutamate kinase and γ‐glutamyl phosphate reductase activities, or the pentafunctional AROM complex from Aspergillus nidulans, which is involved in aromatic amino acid biosynthesis.5, 6
Inspired by the latter observation, various artificial enzyme fusions have been created in recent years to engineer efficient multifunctional biocatalysts. The first artificial bifunctional fusion enzyme, a histidinol dehydrogenase/aminotransferase, was published in 1970.7 Several fusion enzymes have been made since.4, 8, 9 For example, a fusion between a fatty acid decarboxylase cytochrome P450 (OleTJE) and alditol oxidase (AldO) was made to fuel the reactions of OleTJE with hydrogen peroxide produced by the oxidase.10 To enable efficient cofactor regeneration, we have shown that various nicotinamide adenine dinucleotide (phosphate) (NAD(P)H)‐dependent monooxygenases can be produced fused to phosphite dehydrogenases, which efficiently regenerate NAD(P)H.11, 12
Fusion enzymes have several advantages over separate enzymes. They are cheaper and less labor intensive concerning their production because only one enzyme needs to be expressed and purified. Another advantage is the close proximity of the catalytic sites, which enables substrate channeling.4, 9, 13, 14 Substrate channeling circumvents diffusion of the intermediate product in the solution, and hence, increases the combined reaction rate.
We were particularly inspired by the interplay between oxidases and peroxidases also found in nature. Oxidases and peroxidases are often coexpressed. Oxidases produce hydrogen peroxide, which again is a substrate for peroxidases. Well‐known examples of such interplay between oxidases and peroxidases are found in fungi.15, 16 Many fungi secrete specialized peroxidases (e.g., lignin peroxidase and manganese peroxidase) that aid in biomass degradation.15 Except for secreting these heme‐containing enzymes, these fungi also secrete various oxidases (e.g., pyranose oxidase and aryl alcohol oxidase) to serve as hydrogen peroxide producing enzymes to fuel the peroxidases.15, 16 The consecutive reactions of oxidases and peroxidases are also applied in enzyme activity screening approaches and biosensors. Numerous assays and biosensors are based on the combination of an oxidase and a peroxidase, for instance, for the detection of glucose or uric acid levels in blood serum.17, 18, 19 The activities of various oxidases were studied in coupled assays, in which, typically, horseradish peroxidase (HRP) is employed.20, 21, 22, 23 HRP, however, is still extracted from horseradish because of difficulties in the heterologous expression of this plant peroxidase.24 Therefore, for this study, we selected a recently discovered bacterial peroxidase, SviDyP, which was easily produced by Escherichia coli.25, 26
Fusion enzymes between oxidases and peroxidases have not been made previously, although they form catalytically logical combinations because the oxidase‐formed hydrogen peroxide will drive the fused peroxidase (Figure 1).
Figure 1.
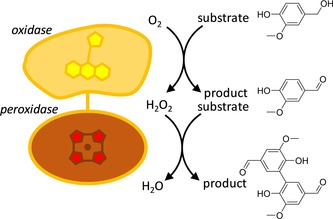
Fused oxidase–peroxidases (P‐oxidases) enable O2‐driven oxidative cascade reactions. The cascade reaction from vanillyl alcohol to divanillin is shown as an example.
Herein, we fused a bacterial peroxidase (SviDyP from Saccharomonospora viridis DSM 43017, EC 1.11.1.19) to four different bacterial oxidases (EC 1.1.3.x).25, 26 SviDyP belongs to the family of DyP‐type peroxidases, which are known for their activity on dyes and phenolic compounds.27, 28, 29, 30 The peroxidase is easily expressed in a bacterial host and is also a very robust enzyme. SviDyP was fused to two flavin adenine dinucleotide (FAD)‐containing oxidases that were active towards sugars: alditol oxidase (HotAldO) from Acidothermus cellulolyticus 11B and chitooligosaccharide oxidase (ChitO) from Fusarium graminearum.20, 23 In this study, a ChitO triple mutant, Q268R/G270E/S410R (ChitO*), was used because of its increased catalytic efficiency towards glucose, lactose, cellobiose, and maltose. In addition to these oxidases, SviDyP was fused to two other flavoprotein oxidases that featured a partially overlapping substrate/product scope to SviDyP: eugenol oxidase (EugO) from Rhodococcus sp. strain RHA1 and 5‐hydroxymethylfurfural oxidase (HMFO) from Methylovorus sp. strain MP688.21, 22 This overlap in substrate/product scope, with both fusion partners active on phenolic compounds, would allow one‐pot cascade reactions. Thus, we were able to produce four novel bifunctional fusion biocatalysts that could either serve a role in biosensing or act as a catalyst for one‐pot, two‐step cascade reactions.
The four DyP‐type peroxidase/oxidase fusion enzymes (which we termed P‐oxidases) were made by cloning the genes of the individual oxidases ChitO*, EugO, HMFO, and HotAldO C terminally to the gene encoding for His‐tagged SviDyP. The resulting fusion enzymes were overexpressed and subsequently purified by affinity chromatography to yield 26–60 mg of enzyme per liter of culture broth medium. The fusion enzymes displayed an intense red–brown color that was indicative of binding of the heme and flavin cofactors. Analysis by UV/Vis absorbance spectroscopy revealed absorbance maxima at λ=280 (protein) and 406 nm (heme) for all enzymes. The Reinheitszahl (R z value) of the fusion enzymes varied between 0.61 and 0.97, and suggested effective incorporation of the heme cofactor. The typical absorbance maxima of FAD, λ≈350–385 and 440–460 nm,20, 21, 22, 31 could not be observed due to the high absorbance of the heme cofactor. To confirm binding of the FAD cofactor, the purified ChitO*, EugO, and Hot‐ AldO fusion enzymes were analyzed for in‐gel fluorescence after SDS‐PAGE. This revealed that all three fusion enzyme contained a covalently bound flavin cofactor. Such analysis was not feasible for the HMFO fusion enzyme because this flavoprotein oxidase contained a dissociable FAD. Nevertheless, activity measurements (see below) confirmed that this fusion enzyme was also functional as an oxidase, thus confirming the presence of the flavin cofactor.
To verify that the prepared fusion enzymes were fully functional, the activities of both fusion partners were measured (Table 1). The observed peroxidase activities for all fusion enzymes were in good agreement with the k cat. values determined for the isolated peroxidase. Accordingly, it can be concluded that the activity of the peroxidase was unaffected by fusing it to the oxidases. Also, the oxidases displayed activity when fused to SviDyP, although the activities were somewhat lower than the activities of the non‐fused enzymes. Oxidase activities of 15–43 % were observed for the fused oxidases ChitO*, EugO, HMFO, and HotAldO. This could be partly explained because the activity was measured at a fixed substrate concentration (k obs), which would yield lower rates when compared with k cat. values taken from the literature. Another explanation of the lower observed rates may lie in incomplete flavin cofactor incorporation. Yet, prolonged incubation of the fusion enzymes did not result in higher activities. The somewhat lowered oxidase activities may also be caused by structural effects of bringing the enzymes together. Nonetheless, it can be concluded that both fusion partners of the created fusion enzymes show significant activities. Therefore, we started to explore their use as bifunctional biocatalysts.
Table 1.
Peroxidase and oxidase activities of the fusion enzymes.[a]
| Fusion | k obs [s−1] | Fusion | k obs [s−1] | ||
|---|---|---|---|---|---|
| enzyme | Peroxidase | Oxidase | enzyme | Peroxidase | Oxidase |
| P‐ChitO* | 7.7 (6.6) | 1.0 (6.523) | P‐HMFO | 7.1 (6.6) | 9.0 (2121) |
| P‐EugO | 5.0 (6.6) | 2.5 (1222) | P‐HotAldO | 8.6 (6.6) | 0.43 (1.920) |
[a] The peroxidase activity was measured by using Reactive Blue 19 as a substrate at pH 4.0. The oxidase activities of P‐EugO and P‐HMFO towards vanillyl alcohol were measured at pH 7.5 and 8.0, respectively. The activity of P‐HotAldO towards xylitol was measured at pH 7.5, and the activity of P‐ChitO* towards cellobiose was measured at pH 7.6. The values in parentheses indicate the k cat. values of the separate enzymes, as determined for SviDyP (see the Supporting Information) or as reported in the literature.
There are numerous applications in which the combined use of a peroxidase and oxidase is exploited for detection purposes. One known application that uses such a P‐oxidase couple is the combined use of glucose oxidase and HRP in biosensors to determine the glucose level in blood.17 Glucose oxidase oxidizes glucose to gluconic acid in the presence of molecular oxygen, and the formed hydrogen peroxide is subsequently used to translate the oxidase activity into a readout. We explored SviDyP–oxidase fusion enzymes for their use in detecting sugars. SviDyP is a representative of a newly discovered class of peroxidases, the DyP‐type peroxidases, which have the advantage over HRP that they are typically easily overexpressed and purified from a heterologous host, such as E. coli.24, 27 First, we produced and probed native SviDyP for its performance, and found it to be mainly active at pH 3–7 at ambient temperature, with an optimum for activity towards Reactive Blue 19 at pH 4.0. SviDyP is active towards 4‐aminoantipyrine (AAP) and 3,5‐dichloro‐2‐hydroxybenzenesulfonic acid (DCHBS), which are commonly used as chromogenic substrates in peroxidase assays (AAP/DCHBS assay). The P‐ChitO* and P‐HotAldO fusion enzymes were tested with the AAP/DCHBS assay for their use in detecting sugars. ChitO is active towards mono‐, di‐, and oligosaccharides and is the only oxidase known to be able to oxidize N‐acetylated carbohydrates.23, 31 Various ChitO mutants have been engineered that display distinct preferences for different carbohydrates. This would allow the generation of dedicated P‐oxidase fusions for the detection of specific mono‐ and oligosaccharides. HotAldO is mainly active on alditols, such as xylitol and sorbitol, which would allow its use for xylitol or sorbitol sensing.20 To test the fusion enzymes, pH 6 was used because this was the value at which optima of the oxidases and peroxidase overlapped. With saturating concentrations of test sugars (24 mm cellobiose for P‐ChitO* and 1.4 mm xylitol for P‐HotAldO), a clear and rapid color developed at a rate of 0.3 s−1 for both sugars. The rate of color formation was close to the observed rate when native SviDyP was tested in the AAP/DCHBS assay (0.4 s−1). This indicates that under the employed conditions the peroxidase is rate determining in the assay. A more detailed analysis of the sensitivity of P‐ChitO* and P‐HotAldO revealed that the fusion enzymes were able to detect low levels of cellobiose (25 μm) and xylitol (10 μm; Figure S3 in the Supporting Information). When using Amplex Red as a fluorogenic peroxidase substrate, we could even lower the detection limit by one order of magnitude (Figure S4). This shows that such peroxidase–oxidase fusion enzymes are perfectly suited for sensing purposes by harboring the full catalytic arsenal for an oxygen‐driven biosensor.
For the generated P‐HMFO and P‐EugO fusion enzymes, we explored their use in fully linked cascade reactions. We imagined that, except for the use of the oxidase‐generated hydrogen peroxide, the aromatic product formed by the oxidases could also be used as a substrate for the fused peroxidase. DyP‐type peroxidases have been shown to act on various aromatic compounds, whereas HMFO and EugO are, among other substrates, active on monophenolic compounds.21, 22, 27, 28, 29, 30 This overlap in substrate/product scope is perfect for one‐pot cascade reactions. In earlier work, we showed that another DyP‐type peroxidase, TfuDyP, dimerized vanillyl alcohol, vanillin, and vanillyl acetone.28 Dimerization of phenolic compounds is a known reaction for peroxidases and laccases, and involves oxidative phenolic coupling and keto–enol tautomerization.32, 33 Divanillin is a desired taste/flavor enhancer and is reported to give an impression of creaminess to food and to mask the sense of bitterness.33 In this work, we examined whether P‐EugO and P‐HMFO could produce divanillin from vanillyl alcohol, through a cascade reaction in which vanillyl alcohol was oxidized to vanillin by an oxidase and subsequently dimerized to divanillin by SviDyP (Figure 1). P‐EugO and P‐HMFO were incubated with vanillyl alcohol at pH 5.5, and the reaction mixtures were subsequently analyzed by LC‐MS (Figures S5–S14). Both P‐EugO and P‐HMFO were found to convert vanillyl alcohol. After 21 h, P‐HMFO had oxidized 90 % of vanillyl alcohol into vanillin (69 %) and divanillin and related oligomers (21 %). Under the same conditions, P‐EugO converted 92 % of vanillyl alcohol into vanillin (53 %) and a higher amount of oligomers (39 %), of which the most dominant product was divanillin (see the Supporting Information). These results demonstrate that the fusion enzymes are suitable for the production of the taste enhancer divanillin.33 Apart from being recognized as flavors, vanillin and divanillin are also considered as renewable building blocks for the production of bio‐based plastics.34, 35, 36 Furthermore, divanillin and related phenolic dimers have an antimetastatic potential.37 Recently, we developed a one‐pot, two‐step cascade reaction in which EugO and HRP or SviDyP were combined to produce low‐molecular‐weight lignin‐like oligomers from eugenol.38 The created fusion enzyme P‐EugO simplifies this newly developed approach to synthesize lignin oligomer from eugenol. HPLC analysis revealed that incubation of eugenol with P‐EugO gave the same lignin products: phenyl coumaran, pinoresinol, coniferyl alcohol, dieugenol, and a lignin tetramer (Figure S15).
In conclusion, we made four active fusion enzymes of DyP‐type peroxidase, SviDyP, and four different oxidases that we termed P‐oxidases. All designed fusion enzymes could be overexpressed by E. coli as a soluble protein. SviDyP proved to be a good substitute for HRP in the HRP‐coupled assay and could be applied at an acidic pH. This SviDyP assay could be applied to explore the substrate scope of oxidases or as a biosensor for the detection of, for instance, sugars. SviDyP has an overlapping substrate/product scope with multiple oxidases, which is perfect for cascade reactions. Fusion enzymes P‐HMFO and P‐EugO were used in one‐pot, two‐step cascade reactions. P‐HMFO could be used to prepare divanillin as the main product, whereas P‐EugO could be used for the synthesis of lignin oligomers. For future work, it would be interesting to shift the pH optima of the oxidase and peroxidase closer together. The pH optima of several enzymes were previously shifted through site‐directed mutagenesis.39, 40, 41 By optimizing these artificial fusions of redox enzymes, novel, effective, bifunctional biocatalysts can be developed.
Experimental Section
Chemicals, reagents, and enzymes: Chemicals, media components, and reagents were obtained from Sigma(–Aldrich), Merck, BD, Acros Organics, TCI, Alfa Aesar, Thermo Fisher, and Fisher Scientific. Amplex Red (Amplisyn Red) was obtained from SynChem. Oligonucleotides and HRP were obtained from Sigma. Restriction enzyme HindIII was obtained from New England Biolabs. The Pfu‐ Ultra Hotstart PCR master mix was from Agilent Technologies, and the In‐Fusion HD EcoDry cloning kit was obtained from Clontech.
Cloning: The genes of oxidases ChitO* (ChitO triple mutant, Q268R/G270E/S410R), EugO, HMFO, and HotAldO were amplified and cloned C terminally to the SviDyP gene in vector pBAD His‐SviDyP;26 for original and new plasmids, see Table S1 in the Supporting Information. pBAD His‐SviDyP contains C terminally to the SviDyP gene, a stop codon, a HindIII restriction site, and another stop codon. The vector was linearized by using restriction enzyme HindIII. The gene of HotAldO (including a C‐terminal His6 tag, without the first codon for methionine) was cloned into pBAD His‐SviDyP by using restriction free cloning.42 The obtained plasmid contained a stop codon between the genes of SviDyP and HotAldO. The stop codon was mutated to serine by QuikChange PCR to yield vector pBAD His‐SviDyP‐HotAldO‐His. The above‐mentioned stop codon was mutated to serine before cloning of the other oxidase genes. The oxidase genes were subsequently amplified and cloned into the obtained plasmid by In‐Fusion cloning (In‐Fusion HD EcoDry cloning kit, Clontech). The HindIII restriction site was retained on both sides of the oxidase genes. E. coli strain TOP10 (Invitrogen) was transformed by the obtained plasmids.
Culture growth and enzyme purification: Precultures were grown on lysogeny broth medium (LB 5 mL) at 37 °C, 135 rpm, overnight. To inoculate 400 mL Terrific Broth (TB) medium, 1:100 preculture was added. These cultures were grown at 37 °C, with shaking at 135 rpm, until the OD600 reached about 0.4–0.6, after which they were induced by 0.02 % l‐arabinose and grown at 17 °C and 135 rpm for 70 h. All cultures were supplemented with ampicillin (50 μg mL−1). Cells were harvested by centrifugation at 6700 g and 4 °C for 20 min (Beckman Coulter, Avanti JE centrifuge, JLA 10.500 rotor). Pellets were washed with buffer A (50 mm potassium phosphate, 0.5 m NaCl, pH 8.0), harvested by centrifugation (3000 g, 4 °C, 40 min, Eppendorf centrifuge 5810R), and stored at −20 °C before use. Prior to enzyme purification, the pellets were thawed and resuspended in buffer A supplemented with phenylmethanesulfonyl fluoride (PMSF; 0.1 mm). Cells were disrupted by sonication (70 % amplitude, 5 min total on time with cycles of 5 s on and 10 s off) and the cell‐free extract was obtained by centrifugation at 16 000 g and 4 °C for 15 min (VWR, Micro Star 17R centrifuge). The enzymes were purified from the cell‐free extract by using a 5 mL His‐Trap HP column (GE Health care). The columns were washed with buffer A and buffer A supplemented with 6, 12, and 24 mm imidazole. The enzymes were eluted with 300 mm imidazole in buffer A. Subsequently, the buffer was exchanged to buffer B (20 mm potassium phosphate, 150 mm NaCl, pH 7.5) by using a 10 mL Econo‐Pac 10 DG desalting column (BioRad). The purified enzymes were flash frozen with liquid nitrogen and stored at −20 °C. UV/Vis absorbance spectra of the enzymes were recorded between λ=250 and 800 nm at ambient temperature (V‐660 spectrophotometer, Jasco). The protein concentrations were determined by using the Lambert–Beer law and the predicted molecular extinction coefficients (ExPASy ProtParam tool43) were as follows: ϵ 280 nm=48 470 m −1 cm−1 for SviDyP, ϵ 280 nm=124 915 m −1 cm−1 for P‐ChitO* (SviDyP‐ChitO*, in case it contained one disulfide bond), ϵ 280 nm=127 770 m −1 cm−1 for P‐EugO (SviDyP‐EugO), ϵ 280 nm=126 850 m −1 cm−1 for P‐HMFO (SviDyP‐HMFO), and ϵ 280 nm=116 880 m −1 cm−1 for P‐HotAldO (SviDyP‐HotAldO).
Steady‐state kinetic analysis of Svi DyP: The steady‐state kinetic parameters of SviDyP were determined for Reactive Blue 19 (ϵ 595 nm=10 mm −1 cm−1[26]) in sodium citrate buffer (50 mm, pH 4.0) with H2O2 (100 μm) and enzyme (20 nm). SviDyP was added to start the reaction. Oxidation of Reactive Blue 19 was followed spectrophotometrically (JASCO V‐660) at ambient temperature.
Oxidase and peroxidase activity of the fusion enzymes: The activities of both fusion partners were determined separately. For all reactions, a saturating substrate concentration of 20 times the K M value was used. For SviDyP, the same reaction conditions were used as described above, with a substrate concentration of 100 μm Reactive Blue 19 (K M=4.6 μm). For the oxidases, the same reaction mixtures and pH values were used as described before.20, 21, 22, 23 Prior to the reactions, the oxidases were incubated with 100 μm FAD for 1 h at ambient temperature. Vanillyl alcohol was used as a substrate for EugO22 and HMFO,21 d‐(+)‐cellobiose for ChitO*,23 and xylitol for HotAldO.20 The oxidation of vanillyl alcohol was followed spectrophotometrically at λ=340 nm (vanillin, ϵ 340 nm=14 mm −1 cm−1 at pH 7.522 and 8.021). The oxidation of d‐(+)‐cellobiose and xylitol were followed by means of a HRP‐coupled assay. In this assay, hydrogen peroxide was produced by the oxidases and used by HRP to couple DCHBS and AAP to a pink product (ϵ 515 nm=26 mm −1 cm−1).20, 23
Svi DyP coupled assay for the detection of oxidase substrates: This assay was a variant of the HRP coupled assay mentioned above and made use of the peroxidase activity of dye‐decolorizing peroxidase SviDyP instead of HRP. The coupled activity of fusion enzymes P‐ChitO* and P‐HotAldO were determined at pH 5 (50 mm sodium citrate buffer) and 6 (50 mm potassium phosphate buffer). The reaction mixtures contained 0.1 mm AAP, 1.0 mm DCHBS, and 23.8 mm (20×K M) d‐(+)‐cellobiose for ChitO* or 1.4 mm xylitol (20×K M) for HotAldO. The formation of the pink product was followed spectrophotometrically at ambient temperature (ϵ 515 nm=26 mm −1 cm−1). To determine whether the oxidase or peroxidase was the limiting factor in these reactions, the reactions were repeated in the presence of H2O2 (100 μm) to determine the optimal reaction rate of SviDyP.
Analysis of the sensitivity of the Svi DyP assay: The sensitivity of the coupled assay was studied by determining the lower concentration limit for substrate detection, as described before.44 Reaction mixtures (200 μL) contained 0.1 mm AAP, 1.0 mm DCHBS, 150 nm fusion enzyme, and varying substrate concentrations (0.5 μm–1 mm) in 50 mm potassium phosphate buffer pH 6.0. d‐(+)‐Cellobiose and xylitol were used as substrates for P‐ChitO* and P‐HotAldO, respectively. The enzymes were added to start the reaction. Reactions were performed in triplicate and the absorbance at λ=515 nm (pink product, ϵ 515 nm=26 mm −1 cm−1) was followed at ambient temperature for 15 min by using a SynergyMX (BioTek) plate reader. The obtained values after 15 min were corrected for both the path length and the blank. For comparison, the sensitivity of the coupled assay was also determined by using 60 μm Amplex Red (10‐acetyl‐3,7‐dihydroxyphenoxazine, Amplisyn Red) instead of AAP/DCHBS. A stock of 6.0 mm Amplex Red was prepared in DMSO. The oxidation of Amplex Red was followed by measuring the fluorescence of the product, resorufin (λ ex=530 nm, λ em=590 nm), for 15 min at ambient temperature.
One‐pot cascade reaction for the synthesis of divanillin and related dimers and oligomers: Vanillyl alcohol was dissolved in water at a concentration of 50 mm. Reaction mixtures (2.0 mL) contained 2 mm vanillyl alcohol and 1.0 μm SviDyP, P‐HMFO, or P‐EugO in sodium citrate buffer (50 mm, pH 5.5). In the case of SviDyP, 500 μm H2O2 was added. Reaction mixtures were incubated in 15 mL closed tubes at 30 °C and 100 rpm, for 21 h. Control reactions were prepared without enzyme. After 2, 3, and 21 h, samples were taken. Enzymes were heat‐inactivated at 95 °C for 10 min, after which time the samples were centrifuged for 5 min at 16,100×g. Reaction products were analyzed by reversed‐phase HPLC by using a Jasco HPLC system. Samples (10 μL) were injected onto a Grace Altima HP C18 column (5 μm, 2.1×150 mm, with 1.0 cm precolumn of the same material). Solvents used were as follows: A: water with 0.1 % formic acid; B: acetonitrile. HPLC method: 2 min 10 % B, 2–20 min gradient to 70 % B, 20–23 min 70 % B, 23 min 10 % B, followed by 7 min re‐equilibration. Detection by a UV detector at λ=280 nm and flow rate of 0.5 mL min−1. LC‐MS analysis was performed on a Surveyor HPLC‐DAD instrument coupled to an LCQ Fleet detector by using scanning for both positive and negative modes. Samples were injected onto a Grace Altima HP C18 column (3 μm, 2.1×100 mm, with 1.0 cm precolumn of the same material), flow rate 0.3 mL min−1. Solvents used were as follows: A: water with 0.1 % formic acid; B: acetonitrile with 0.08 % formic acid. LC‐MS method: 2 min 100 % A, 2–32 min gradient to 80 % B, 32–37 min 80 % B, 37–38 min 100 % A, 38–48 min 100 % A re‐equilibration.
One‐pot cascade reaction for the synthesis of lignin‐like oligomers from eugenol: The activity of P‐EugO towards eugenol was assayed as described before.38 Reaction mixtures (2.0 mL) contained 1.0 μm P‐EugO, 10 mm eugenol, and 5 % DMSO (v/v) in potassium phosphate buffer (20 mm, pH 6.0). A stock solution of 300 mm eugenol was prepared in DMSO. For comparison, a reaction mixture containing SviDyP (1.0 μm) and EugO (1.0 μm) was assayed. All reactions were performed in duplicate and compared with a reaction without enzyme. Reaction mixtures were incubated at 30 °C and 50 rpm in 20 mL Pyrex tubes with a headspace to volume ratio of 10:1. Samples (200 μL) were taken after 24 and 96 h. These samples were heat‐treated and analyzed by reversed‐phase HPLC, as described above, for the production of divanillin and related oligomers.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This work was supported by the NWO graduate program: synthetic biology for advanced metabolic engineering, project number 022.004.006, The Netherlands.
D. I. Colpa, N. Lončar, M. Schmidt, M. W. Fraaije, ChemBioChem 2017, 18, 2226.
References
- 1. Schweizer E., Hofmann J., Microbiol. Mol. Biol. Rev. 2004, 68, 501–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Patel M. S., Nemeria N. S., Furey W., Jordan F., J. Biol. Chem. 2014, 289, 16615–16623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Doi R. H., Kosugi A., Nat. Rev. Microbiol. 2004, 2, 541–551. [DOI] [PubMed] [Google Scholar]
- 4. Elleuche S., Appl. Microbiol. Biotechnol. 2015, 99, 1545–1556. [DOI] [PubMed] [Google Scholar]
- 5. Pérez-Arellano I., Carmona-Álvarez F., Martínez A. I., Rodríguez-Díaz J., Cervera J., Protein Sci. 2010, 19, 372–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Charles I. G., Keyte J. W., Brammar W. J., Smith M., Hawkins A. R., Nucleic Acids Res. 1986, 14, 2201–2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yourno J., Kohno T., Roth J. R., Nature 1970, 228, 820–824. [DOI] [PubMed] [Google Scholar]
- 8. Yu K., Liu C., Kim B.-G., Lee D.-Y., Biotechnol. Adv. 2015, 33, 155–164. [DOI] [PubMed] [Google Scholar]
- 9. Conrado R. J., Varner J. D., Delisa M. P., Curr. Opin. Biotechnol. 2008, 19, 492–499. [DOI] [PubMed] [Google Scholar]
- 10. Matthews S., Tee K. L., Rattray N. J., Mclean K. J., Leys D., Parker D. A., Blankley R. T., Munro A. W., FEBS Lett. 2017, 591, 737–750. [DOI] [PubMed] [Google Scholar]
- 11. Torres Pazmiño D. E., Snajdrova R., Baas B.-J., Ghobrial M., Mihovilovic M. D., Fraaije M. W., Angew. Chem. Int. Ed. 2008, 47, 2275–2278; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 2307–2310. [Google Scholar]
- 12. Beyer N., Kulig J. K., Bartsch A., Hayes M. A., Janssen D. B., Fraaije M. W., Appl. Microbiol. Biotechnol. 2017, 101, 2319–2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Meynial Salles I., Forchhammer N., Croux C., Girbal L., Soucaille P., Metab. Eng. 2007, 9, 152–159. [DOI] [PubMed] [Google Scholar]
- 14. Seo H. S., Koo Y. J., Lim J. Y., Song J. T., Kim C. H., Kim J. K., Lee J. S., Choi Y. D., Appl. Environ. Microbiol. 2000, 66, 2484–2490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Abdel-Hamid A. M., Solbiati J. O., Cann I. K. O., Adv. Appl. Microbiol. 2013, 82, 1–28. [DOI] [PubMed] [Google Scholar]
- 16. Ander P., Marzullo L., J. Biotechnol. 1997, 53, 115–131. [DOI] [PubMed] [Google Scholar]
- 17. Barham D., Trinder P., Analyst 1972, 97, 142–145. [DOI] [PubMed] [Google Scholar]
- 18. Chun H. J., Park Y. M., Han Y. D., Jang Y. H., Yoon H. C., Biochip J. 2014, 8, 218–226. [Google Scholar]
- 19. Mundaca-Uribe R., Bustos-Ramírez F., Zaror-Zaror C., Aranda-Bustos M., Neira-Hinojosa J., Peña-Farfal C., Sens. Actuators B 2014, 195, 58–62. [Google Scholar]
- 20. Winter R. T., Heuts D. P. H. M., Rijpkema E. M. A., van Bloois E., Wijma H. J., Fraaije M. W., Appl. Microbiol. Biotechnol. 2012, 95, 389–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dijkman W. P., Fraaije M. W., Appl. Environ. Microbiol. 2014, 80, 1082–1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jin J., Mazon H., van den Heuvel R. H. H., Janssen D. B., Fraaije M. W., FEBS J. 2007, 274, 2311–2321. [DOI] [PubMed] [Google Scholar]
- 23. Ferrari A. R., Lee M., Fraaije M. W., Biotechnol. Bioeng. 2015, 112, 1074–1080. [DOI] [PubMed] [Google Scholar]
- 24. Krainer F. W., Glieder A., Appl. Microbiol. Biotechnol. 2015, 99, 1611–1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yu W., Liu W., Huang H., Zheng F., Wang X., Wu Y., Li K., Xie X., Jin Y., PLoS One 2014, 9, e110319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Colpa D. I., Fraaije M. W., J. Mol. Catal. B 2016, 134, 372–377. [Google Scholar]
- 27. Colpa D. I., Fraaije M. W., van Bloois E., J. Ind. Microbiol. Biotechnol. 2014, 41, 1–7. [DOI] [PubMed] [Google Scholar]
- 28. Lončar N., Colpa D. I., Fraaije M. W., Tetrahedron 2016, 72, 7276–7281. [Google Scholar]
- 29. Ogola H. J. O., Kamiike T., Hashimoto N., Ashida H., Ishikawa T., Shibata H., Sawa Y., Appl. Environ. Microbiol. 2009, 75, 7509–7518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kim S. J., Shoda M., Appl. Environ. Microbiol. 1999, 65, 1029–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Heuts D. P. H. M., Janssen D. B., Fraaije M. W., FEBS Lett. 2007, 581, 4905–4909. [DOI] [PubMed] [Google Scholar]
- 32. Nishimura R. T., Giammanco C. H., Vosburg D. A., J. Chem. Educ. 2010, 87, 526–527. [Google Scholar]
- 33. Krings U., Esparan V., Berger R. G., Flavour Fragrance J. 2015, 30, 362–365. [Google Scholar]
- 34. Fache M., Boutevin B., Caillol S., Eur. Polym. J. 2015, 68, 488–502. [Google Scholar]
- 35. Llevot A., Grau E., Carlotti S., Grelier S., Cramail H., Polym. Chem. 2015, 6, 7693–7700. [Google Scholar]
- 36. Amarasekara A. S., Razzaq A., ISRN Polym. Sci. 2012, 2012, 1–5. [Google Scholar]
- 37. Jantaree P., Lirdprapamongkol K., Kaewsri W., Thongsornkleeb C., Choowongkomon K., Atjanasuppat K., Ruchirawat S., Svasti J., J. Agric. Food Chem. 2017, 65, 2299–2306. [DOI] [PubMed] [Google Scholar]
- 38.M. H. M. Habib, P. J. Deuss, N. Lončar, M. Trajkovic, M. W. Fraaije, Adv. Synth. Catal 2017, https://doi.org/10.1002/adsc.201700650.
- 39. Pokhrel S., Joo J. C., Yoo Y. J., Biotechnol. Bioprocess Eng. 2013, 18, 35–42. [Google Scholar]
- 40. Mendes S., Brissos V., Gabriel A., Catarino T., Turner D. L., Todorovic S., Martins L. O., Arch. Biochem. Biophys. 2015, 574, 99–107. [DOI] [PubMed] [Google Scholar]
- 41. Tomschy A., Brugger R., Lehmann M., Svendsen A., Vogel K., Kostrewa D., Lassen S. F., Burger D., Kronenberger A., van Loon A. P. G. M., et al., Appl. Environ. Microbiol. 2002, 68, 1907–1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. van den Ent F., Löwe J., J. Biochem. Biophys. Methods 2006, 67, 67–74. [DOI] [PubMed] [Google Scholar]
- 43. Gasteiger E., Hoogland C., Gattiker A., Duvaud S., Wilkins M. R., Appel R. D., Bairoch A. in Proteomics Protocols Handbook (Ed.: J. M. Walker), Humana, Totowa, 2005, pp. 571–607. [Google Scholar]
- 44. Ferrari A. R., Gaber Y., Fraaije M. W., Biotechnol. Biofuels 2014, 7, 37.24612932 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary