

Abstract
Identifying the direct physiological targets of drugs and chemical probes remains challenging. Here we describe how resistance can be used to achieve ‘gold standard’ validation of a chemical inhibitor’s direct target in human cells. This involves demonstrating that a silent mutation in the target that suppresses inhibitor activity in cell-based assays can also reduce inhibitor potency in biochemical assays. Further, phenotypes due to target inhibition can be identified as those observed in the inhibitor-sensitive cells, across a range of inhibitor concentrations, but not in genetically matched cells with a silent resistance-conferring mutation in the target. We propose that chemotype-specific resistance, which is generally considered to be a limitation of molecularly-targeted agents, can be leveraged to deconvolve the mechanism of action of drugs and to properly use chemical probes.
Keywords: Drug resistance, chemical probes, target identification, chemical biology
Challenges in uncovering physiological targets of chemical inhibitors
Cell-permeable chemical inhibitors can have several advantages over other commonly used methods (e.g. RNAi, genetic knockout) to dissect cellular mechanisms[1, 2]. In particular, chemical inhibitors can block the function of their targets within minutes, allowing the timescale of the perturbation to match that of a dynamic cellular process being studied. In addition, if the chemical probes are reversible, relief from inhibitor treatment can allow the target protein’s functions to be turned on in cellular contexts within minutes. Importantly, chemical probes can also provide valuable starting points for the development of new chemotherapeutics.
In the past few decades, the discovery of chemical probes has relied largely on two types of approaches. One approach involves the selection of a target protein that has a critical role in a cellular process of interest or dysfunction of which has been linked to disease[3, 4]. Screens of chemical libraries or protein structure-guided methods are used to identify initial ‘hits’ that block the target protein’s function. These ‘hits’ are then optimized and their activity is analyzed in cellular contexts. The second approach, which mimics forward genetics, involves selection of a cellular process of interest and phenotype-based readouts are used to identify chemical inhibitors that perturb this process[5, 6]. Secondary cell-based screens are then employed to stratify initial ‘hits’. Biochemical and cell biological data are then used to deconvolve the inhibitor’s protein target. This approach can be attractive as it often yields new and unexpected ‘druggable’ targets involved in the cellular process.
For either of these approaches, firmly establishing that the target protein is indeed inhibited in cellular contexts remains challenging. Too often, physiological target inhibition is established by correlating loss-of-function (e.g. by RNAi) phenotypes with those due to chemical inhibition. This correlation can often fail for at least three reasons[1]. First, the resolution of the analysis of cellular phenotypes is often not high enough to tease apart subtle differences between on-target and off-target effects, particularly when inhibition of multiple different targets can lead to similar phenotypes (e.g. cell cycle arrest, cytoskeleton disruption, activation of the DNA damage response). Second, the mismatch in the timescales of the inhibition of target function following chemical inhibitor treatments or protein knockdown can lead to distinct phenotypes. Protein knockdown (e.g., by RNAi) can take several hours and thereby may result in the accumulation of cellular phenotypes that do not directly involve the target protein. This is less likely to be the case when phenotypes are analyzed immediately, within minutes, following chemical inhibitor treatments. Third, protein loss need not match activity inhibition. For example, the target may be part of a complex with other proteins and loss of the target may lead to the concomitant destabilization of associated proteins. In this case, the observed protein knockdown phenotypes may be related to the disruption of distinct functions of these other proteins. By contrast, chemical inhibition of the protein activity per se is less likely to destabilize the complex.
This review highlights research that suggests how resistance to chemical inhibitors can be analyzed to identify their direct physiological target. We focus on analyses of compounds active in eukaryotic cells. While resistance is typically considered to be a major disadvantage of molecularly targeted chemotherapeutics[7], we believe it can be used as an advantage for the unbiased analysis of cellular targets and to dissect the mechanism of action of chemical inhibitors. We propose that the highest standard, what we call ‘gold standard,’ proof of a chemical inhibitor’s direct physiological target is obtained when a point mutation that does not alter protein function can confer resistance to the chemical inhibitor both in biochemical assays and in cellular contexts. We also discuss how resistance can help with the proper use of chemical inhibitors as probes of cellular mechanisms. Finally, we suggest that multiple resistance-conferring mutations clustering at a region in a protein target can be used to model can be used to model interactions between inhibitor and target.
Cell-based Chemical Inhibitor Resistance
In cultured cells there are two major mechanisms that can confer resistance to chemical inhibitors[8]. First, multidrug resistance (MDR), which reduces the efficacy of multiple compounds, depends on reducing of the compound in the cell. This typically involves ATP binding cassette (ABC) transporter proteins that couple ATP hydrolysis to compounds efflux[9]. An important example of this type of resistance comes from clinical data for the antimitotic drug paclitaxel (Taxol), for which resistance observed in patients has been linked to ABC transporters[8]. Second, resistance specific for a chemical inhibitor, but not to other unrelated compounds causing distinct phenotypes, can arise. This type of resistance, which we refer to as ‘chemotype-specific resistance,’ can be the result of a mutation in the protein target that suppresses inhibitor binding. We note that such mutations must be otherwise ‘silent’ and not alter protein function to cause a phenotype.
A well-studied example of chemotype-specific resistance comes from studies of the kinase inhibitor imatinib mesylate (Gleevec®)[10]. Resistance to this drug can arise through a mutation in the binding pocket of the target kinase BCR-Abl. This point mutation does not substantially alter kinase activity but suppresses binding to the chemical inhibitor. Chemotype-specific resistance may also involve more complex cellular responses, such as the rewiring of a signaling network to circumvent the targeted pathway. In addition, the chemical inhibitor may be metabolized by cellular enzymes and effective compound concentrations may not be reached. However, the relative frequencies of these different modes of resistance in different human cell types is not well characterized and can be difficult to predict.
Leveraging Resistance Mutations for Target Identification in Genetically Tractable Organisms
Genetically tractable organisms, such as Saccharomyces cerevisiae (budding yeast), have played a key role in the study of chemical inhibitor resistance and analysis of physiological targets (reviewed in[11]) (Figure 1A). We consider a seminal study to be the identification of the target of rapamycin, a macrocyclic immunosuppressant compound that had been shown to block T cell activation[12]. Rapamycin, like the natural product FK506, had been shown to interact with and inhibit the activity of FK506 binding protein (FKBP) in vitro. However, rapamycin and FK506 blocked T cell activation at different points in the signaling pathway and antagonized each other’s inhibitory effect in T cells, suggesting that these compounds have distinct mechanisms of action.
FIGURE 1.
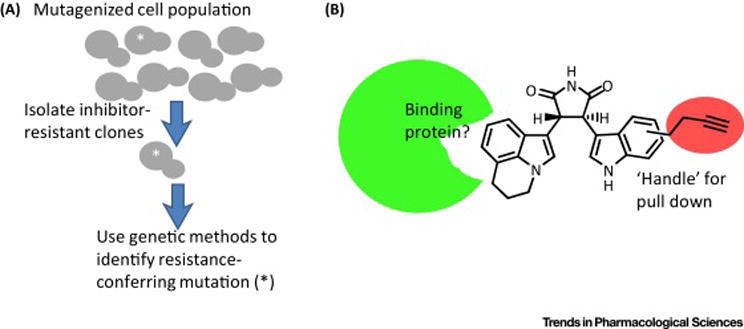
Common methods for methods for elucidation of the cellular targets of chemical inhibitors. (A) When compounds are active in genetically tractable organisms such as budding yeast, the mutations that confer resistance to the inhibitor can be analyzed. If the identified mutation is sufficient to confer chemotype-specific resistance, it can guide further studies that help establish the chemical inhibitor’s direct target. (B) A ‘handle’ can be introduced into the chemical inhibitor for ‘pull-down’ experiments. In the example shown, an alkyne is introduced into the parent compound. The modified analog should cause phenotypes that are similar to those due to treatments with the parent compound. Addition of the modified analog to cells is generally followed by the preparation of lysates. CLICK chemistry can then be used to attach a tag (e.g. biotin) for affinity-based isolation of the compound and associated proteins. Control experiments typically involve adding the unmodified parent compound to the pull-downs so that specific and non-specific interactions can be distinguished.
Rapamycin was found to be toxic to S. cerevisiae and strains lacking functional FKBP, expressed by the FPR1 gene, were resistant. As it was known that the FPR1 gene is not essential in budding yeast, these data were consistent with rapamycin having another target. Remarkably, a strain that was resistant to FK506, but sensitive to rapamycin, could be used to show that FK506 reversed the toxicity of rapamycin, suggesting that both compounds bind to FKBP in a competitive manner in cells. Dissociation of the rapamycin-FKBP complex in cells by FK506 could suppress rapamycin’s toxicity. Further analysis of resistance-conferring mutations in budding yeast led to the identification of tor mutants (for target of rapamycin), which were shown to be genetically unrelated to loss of FKBP function. The finding that the proteins expressed by two TOR genes are needed for the FKBP-dependent toxicity of rapamycin in budding yeast was a critical step that has led to a large body of work from many researchers that has now unraveled how these conserved proteins regulate cell growth and nutrient sensing[13].
Like budding yeast, fission yeast (Schizosaccharomyces pombe) has also played a central role in dissecting fundamental cellular mechanisms, such as regulation of the cell cycle[14]. Notably, fission yeast has certain cellular mechanisms and pathways, such as RNAi, that are not present in budding yeast but are present in humans. However, the use of fission yeast for chemical biology and inhibitor discovery has been restricted, as many compounds that are active in other organisms are not active in these cells. Analyses of drug sensitivity indicated that MDR mechanisms play a key role in the reduced sensitivity of fission yeast to chemical inhibitors[15]. These studies led to the design of yeast strains, called ‘MDR-suppressed’ (MDR-sup), with the key efflux pumps and MDR response genes deleted. These engineered strains are viable and can be used for chemical screens that take advantage of the powerful genetics of this system to identify new chemical probes[16, 17]. These MDR-sup fission yeast strains are also particularly useful for deciphering mechanisms of chemical inhibitor action as resistance can be analyzed in the absence of a robust MDR response.
To highlight the use of these fission yeast strains for chemical inhibitor target identification, we summarize the discovery of ribozinoindoles, the first potent and selective cell-permeable chemical inhibitors of ribosome biogenesis[16]. A key step that helped identify the targets of these compounds was the systematic analysis of inhibitor resistance. Sequencing multiple ribozinoindole-resistant clones identified mutations in the gene mdn1 encoding the ~540-KD protein, which is a member of the AAA+ ATPase family of proteins and is required for ribosome assembly. Critically, the introduction of these resistance mutations in cells was sufficient to confer resistance to ribozinoindole but not to cycloheximide, a chemically unrelated inhibitor that blocks protein synthesis. The full-length wild-type Mdn1 protein was generated in recombinant form and shown to be an active ATPase and this activity was suppressed by ribozinoindole. Importantly, a recombinant full-length Mdn1 containing the same point mutation that protected cells from this chemical inhibitor also suppressed inhibition of the ATPase activity by the compound in vitro. Together, these data provide the gold standard evidence that Mdn1 is the direct physiological target of ribozinoindoles[16]. There are many compounds that are known to be active in human cells but are not active in budding or fission yeast. Analyses in cells lacking MDR pathways suggest that this lack of chemical inhibitor activity is likely to be a consequence of divergence of cellular pathways or between homologous proteins such that compound binding is altered[15, 17]. Therefore, analysis of drug mechanisms and targets for many compounds active in human cells has relied on approaches other than the analysis of resistance.
Leveraging Resistance for Target Identification in Human Cells
Several approaches have been developed to analyze chemical inhibitor targets in human cells (for recent reviews see [18, 19]). A commonly used method involves compound-affinity-based ‘pull-downs’ [20] (Figure 1B). In this approach, the first step is to generate an inhibitor analog with a ‘handle’ (e.g. alkyne) that can be used to isolate compound-bound proteins from cellular lysates. Quantitative mass spectrometry is then utilized to identify associated proteins and select potential drug targets. A major challenge in using this approach is to ensure that inhibitor analogs for affinity purification have the same mechanism of action as the parent compound in cells. This depends on the availability of structure-activity relationship (SAR) data and on high-resolution dose-dependent assays that can uncover subtle differences in cellular phenotypes. This is particularly important when the potency of analogs with handles is lower than that of the unmodified parent compound. For this approach, as well as other approaches that involve genome-scale knockdown (e.g., by RNAi) to analyze chemical inhibitor mechanisms of action in human cells, establishing the direct inhibitor target in a cellular context relies on often unreliable correlations between candidate target protein knockdown and chemical inhibitor treatment phenotypes.
The use of resistance to analyze chemical inhibitor targets in human cells has not been pursued until recently. This was likely due to three reasons. First, while inhibitor-resistant human cells can be generated, it is difficult to separate mutations that cause resistance from those that are bystanders, as genetic methods such as backcrossing are not accessible. Second, the human genome is much larger and more complex in structure and organization than of widely used genetically tractable organisms such as yeast. Third, it has been unclear how frequently chemotype-specific resistance would arise through genetic changes in the direct protein target of a chemical inhibitor.
Recent advances in genome sequencing methodology encouraged us to reexamine the use of resistance in the identification of physiological targets of chemical inhibitors. In a pilot study, we focused on two inhibitors with known targets: BI-2536, a Polo-like kinase inhibitor, and velcade, a proteasome inhibitor[21]. We used HCT-116 cells, which are an adherent human colon cancer cell line, with relatively low expression of MDR efflux pumps, and are DNA-mismatch-repair deficient. These features were considered valuable as single clones could be readily isolated, MDR-based resistance was less likely, and diverse genetic changes (e.g., point mutations) would exist in the cell population. A key finding from these studies was that mutation in the protein target was likely to arise at a high frequency (>30% of clones) in chemotype-specific inhibitor-resistant clones. This is likely as these proteins have evolved to functions (e.g., modify substrate) rather than to bind chemical inhibitors. This observation had the practical implication that only a handful (six to eight) of inhibitor-resistant clones needed to be analyzed.
These findings, along with other studies, led to the development of a method that we have named DrugTargetSeqR (Figure 2)[21, 22]. Briefly, HCT116 cells are treated with the chemical inhibitor of interest at concentrations that kill most cells but at which a few drug-resistant clones can survive. These clones are isolated and expanded separately, keeping track of and limiting the total number of cell passages to so as avoid the accumulation of additional mutations. Resistance to the chemical inhibitor, which is typically a greater-than-fourfold decrease in potency, is confirmed. Clones that are resistant due to MDR mechanisms, which arise even in HCT116 cells, are excluded by testing chemically unrelated compounds that are known drug efflux pump substrates and cause phenotypes distinct from those due to the inhibitor of interest.
FIGURE 2.
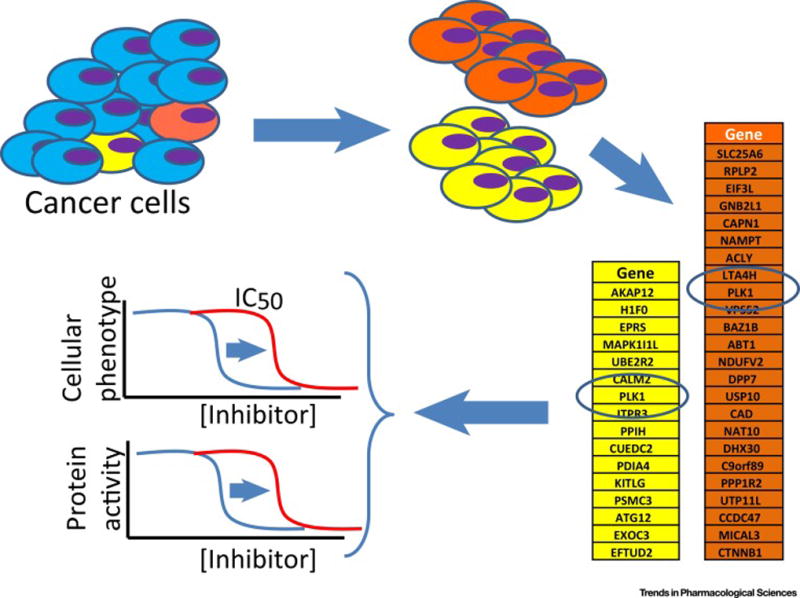
Schematic for DrugTargetSeqR. A genetically heterogeneous population of HCT116 cells is treated with the chemical inhibitor of interest. Clones that have reduced sensitivity to the inhibitor are isolated and expanded separately. Clones resistant to multidrug resistance (MDR) substrates are excluded from further analyses. Transcriptomes of six to eight clones and the parental cells are sequenced. Genetic differences between each clone and the parental cell population are identified. Genes that are altered (e.g. mutated) in multiple independent clones are selected. These mutations are introduced in other drug sensitive cell lines and those mutations sufficient to confer resistance are considered to be in the gene likely encoding the physiological target of the inhibitor. Biochemical assays are then used to test inhibition of the target protein’s activity or direct inhibitor binding. ‘Gold standard’ proof of target is established when the same mutation reduces the chemical inhibitor sensitivity in both cell-based and biochemical assays.
The transcriptomes of the chemotype-specific resistant clones are sequenced to facilitate unbiased analyses of changes in protein-coding sequences. This sequencing approach is favored to due to its lower cost compared with whole-genome sequencing and the fact that most chemical inhibitors target proteins. The genomes of these inhibitor-resistant clones are compared with that of the heterogeneous parental cell population to identify differences such as point mutations, insertions and deletions. Such comparisons typically identify a manageable number (typically on the order of hundreds) of genetic changes. We combine data for clones that are highly related at the level of genome sequence and are likely to share resistance mechanisms. Genetic changes (e.g. point mutations) that recur across multiple clones are then prioritized for further analyses. CRISPR-Cas9-based genome editing can be used to introduce these recurring mutations into another cell line that is known to be sensitive to the chemical inhibitor. If a mutation is sufficient to confer resistance to the chemical inhibitor, we then determine whether examine if the product of the gene carrying this mutation is a direct target of the inhibitor. This involves expressing the protein and establishing an activity assay (e.g., ATPase). A recombinant form of the protein carrying the genetic change (e.g., point mutation) is also generated. If the protein’s activity is inhibited by the compound and the mutant form is less sensitive, we can establish the gold-standard proof of the chemical inhibitor’s direct physiological target.
In developing and using DrugTargetSeqR, we have confirmed the direct physiological targets for BI-2536, velcade, and ispinesib, an inhibitor of a microtubule-based motor protein required for cell division[21, 22]. We also analyzed the mechanism of action of YM155, a cytotoxic compound that entered clinical trials as an anti-cancer drug[22]. Its mechanism of action was originally proposed to be suppression of survivin, a protein in the Aurora B kinase complex. Our analyses indicated that YM155 acts by triggering the DNA damage response rather than through altering survivin expression.
Other groups have employed approaches closely related to DrugTargetSeqR. Of note is work from a group at a pharmaceutical company that analyzed the mechanisms of action 6-thioguanine and triptolide, two cytotoxic compounds[23]. In this report resistance to these inhibitors was analyzed using KBM7 cells, a nearly haploid chronic myelogenous leukemia cell line. The advantage of using this cell line was nicely revealed by analyses of mutations in the protein ERCC that conferred resistance to triptolide. The point mutations in ERCC3 were found to be recessive; that is, they did not confer resistance to triptolide in the presence of the wild-type protein as would be the case in typical diploid human cell lines. However, these mutations were able to confer resistance in the human haploid cells. Importantly, 58% of the mutations identified in triptolide-resistant clones were in the target protein, consistent with our studies in HCT116 cells suggesting that chemotype-specific resistance arises at high frequency by mutations in the direct target.
Analyses of chemotype-specific resistance-conferring mutations in HCT116 cells has led to the identification of the target of indisulam, an aryl sulfonamide that entered clinical trials as an anti-cancer drug although its mechanism of action was unknown[24]. The exomes of six indisulam-resistant clones were analyzed and only three genes were mutated in at least three different clones. RBM39 was prioritized for further analyses as the different mutations mapped to one codon. Coimmunoprecipitation experiments demonstrated that RBM39, a protein involved in RNA splicing, is recruited to CUL4-DCAF15, an E3 ubiquitin ligase, in an indisulam-dependent manner. In the proposed model, indisulam induces the degradation of RBM39, which results in RNA splicing defects that ultimately cause toxicity. Additional analyses indicated that other sulfonamides, such as tasisulam and chloroquinoxaline sulfonamide (CQS), have a similar mechanism of action. Another study dissecting the mechanisms of these aryl sulfonamides also showed, using multiple lines of evidence that also included a resistance-conferring mutation, that these aryl sulfonamides selectively target splicing factors for degradation[25]. Together, these studies indicate that the analysis of resistance mutations can not only identify targets of enzyme inhibitors but also the mechanisms of action of compounds that promote protein-protein interactions.
Other examples of the use of HCT116 cells to probe mechanisms of action of an inhibitor include studies of mycolactone, an immunosuppressant that selectively blocks the translocation of a subset of the secreted proteome[26], and of CB-5083, an orally bioavailable inhibitor of the AAA+ protein VCP/p97, which regulates protein homeostasis pathways[27].
While DrugTargetSeqR and related methods have mainly been used to analyze cytotoxic compounds, as cell growth-based selection of resistant clones is relatively straightforward, resistance-based analyses can also be used to examine mechanisms of action of non-cytotoxic compounds. For these analyses, a hypothesis for the pathway targeted by the compound is needed. An illustrative example is the identification of the target of a compound called ISRIB that suppresses the integrated stress response (ISR)[28]. A cell line that expresses a fluorescent reporter for the activation of the ISR pathway was employed. Chemical mutagenesis was used and cells in which the ISRIB-induced suppression of the reporter was reversed were isolated and candidate genes were analyzed for mutations. Most of the ISRIB-resistant cells had one or more mutations that clustered at the N terminus of the guanine exchange factor eIF2B. These mutations were shown to be sufficient to confer resistance to ISRIB and, along with additional biochemical data, established eIF2b as the direct physiological target of this inhibitor in the context of ISR. Such an approach can be used to analyze physiological targets of other chemical inhibitors whose action is linked to specific cellular pathway but are not toxic to cells.
Additional uses of drug resistance-conferring mutations
A limitation to using chemical inhibitors as probes or drugs is that it is often unclear whether selective inhibition of the protein target is maintained across a range of compound concentrations. Dose-dependent phenotypes in cellular contexts are desired but it is likely, simply by mass action, that at higher inhibitor concentrations additional undesired targets are engaged (Figure 3). It is very difficult to establish how many cellular off targets are inhibited at doses needed to achieve complete on-target inhibition in cells.
FIGURE 3.
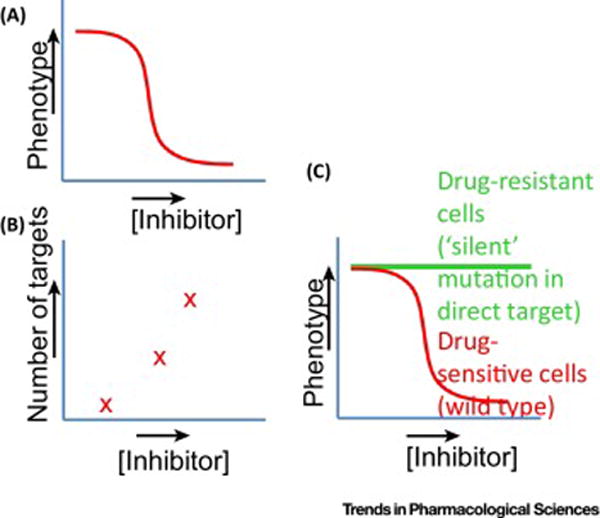
Using chemotype-specific resistance for chemical biology. (A) Dose-dependent effects of inhibitors are typically analyzed. As the inhibitor concentration increases, the more complete inhibition of the target’s activity in the cellular context is expected. (B) As inhibitor concentration increases the number of potential targets is also likely to be higher. Inhibition of different targets can make interpretation of phenotypes using chemical inhibitors difficult. (C) One solution is to examine dose-dependent responses to chemical inhibitors using two matched cell lines. One cell line is drug sensitive and the other is genetically identical but has a drug-resistance-conferring mutation in the target protein. It is critical that this mutation alone does not cause a phenotype (i.e., is a silent mutation). DrugTargetSeqR and related methods readily yield these genetically matched cell line pairs.
We propose that mutations in the direct target of the chemical inhibitor can be particularly useful to in addressing this limitation on the use of chemical inhibitors as probes. Chemical inhibitors should be used in parallel experiments with two matched cell lines, one that is inhibitor sensitive and another that is genetically identical but carries a silent resistance-conferring mutation in the direct target. Dose-dependent phenotypes that are observed in the wildtype cells but not the drug-resistant cells will be due to the activity of the target protein. By contrast, off-target effects would be observed in both cell lines. We and others have used such an approach to study the functions of cell cycle kinases and ribosome assembly mechanisms[16, 17]. In particular, we have used an inhibitor called Arkin-1 to analyze phenotypes associated with different levels of Aurora kinase (Akr1) activity in fission yeast. These studies reveal that high activity of this kinase is needed for proper chromosome compaction during cell division, but lower kinase activity is sufficient for regulation of the cell cycle checkpoint or chromosome-microtubule attachments.
This approach is not only useful for cell biological studies but also valuable for testing target engagement during drug development. The analogs generated during SAR campaigns to improve inhibitor properties can be tested in these matched cell line pairs and analogs that maintain specific on-target activity would be those that are active only in the wild-type cells and not in the cells carrying the resistance-conferring mutation in the direct target. Importantly, DrugTargetSeqR directly yields such matched human cell line pairs.
Our analyses suggest another important use of chemotype-specific resistance. In many cases multiple mutations at different positions in the target protein are identified. For cases where structural data is available, we find that these mutations cluster around the inhibitor-binding site in the target. For example, two of the mutations (G63S and R136G) in Polo-like kinase-1 that conferred resistance to the inhibitor BI-2536 were located in the inhibitor-binding pocket[21]. Therefore, we propose that when multiple resistance-conferring mutations in a protein are identified, they can help generate hypotheses for a drug’s binding site, even in the absence of a ligand-bound structure. For example, in the case of ribozinoindoles many of the resistance-conferring mutations that we identified in the mdn1 gene are proximal to the predicted AAA3-AAA4-domain interface, suggesting that the compound may bind this large protein at this site[16].
In other studies, cotransin (cyclic depsipeptide natural product derivatives) and Apratoxin A (a structurally unrelated cyclic natural product) resistant clones were identified using HCT116 cells[29]. Clustered mutations suggest putative binding sites for these inhibitors within the target protein, the Sec61 translocon, a transmembrane channel that interacts directly with the ribosome. When mapped onto a homology model of the human Sec61 channel, the resistance-conferring mutations for both compounds clustered in two partially overlapping regions near the lumenal end of the translocation channel. The authors surmised that this partial overlap of putative binding sites may explain how these two classes of compounds are able to block different subsets of secreted substrates while engaging the same cellular target.
These studies suggest how analyses of multiple resistance mutations across several inhibitors, even in the absence of compound-bound high-resolution structural data, can help with analyses of inhibitor binding modes and protein functions.
Concluding Remarks
Identification of the direct physiological targets of chemical inhibitors is a crucial step before these compounds can be used as probes or drugs. The resistance analysis-based methods for unbiased target identification in human cells described here are likely to continue to improve with new advances in technology, such as genomics and genome-editing approaches (see Outstanding Questions). Resistance-based methods will also be a valuable complement to the other approaches used to analyze chemical inhibitor targets in human cells. We believe that even when a potential direct target is identified using other methods (e.g., affinity-based approaches), mutant forms of the target should be generated and then tested to determine whether the same mutations suppress inhibitor activity in cell-based and biochemical assays. Genome editing, along with any available structural data for the candidate protein target, can be used to introduce specific mutations in human cells.
Outstanding questions.
To date, resistance selection has been conducted in only a handful of cell lines. Can other cell types can be used for DrugTargetSeqR and related methods? This is important because not all compounds are active in all human cell types.
Can this approach be applied to compounds that have multiple targets? It is likely that some compounds’ efficacies depend on engaging multiple proteins.
Proteins such as actin and tubulin are highly conserved and silent mutations may not arise frequently. Can resistance mutations be used to validate these highly conserved inhibitor targets?
Resistant populations in the presence of non-cytotoxic compounds have been isolated using reporter-based methods when a specific pathway is being studied. Can similar methods be developed for the unbiased identification of non-cytotoxic compounds when we lack a strong hypothesis for a pathway that may be inhibited?
In the past few decades, there have been major advances in the development of molecularly targeted therapeutics. It is also now clear that, while immediate benefits for patients can be achieved, long-term cures are rare. This is in large part due to resistance. While the clinical situation is complex, we believe that application of DrugTargetSeqR and related approaches for the systematic unbiased analysis of recurring genetic alterations that can suppress chemical inhibitor activity in cultured cells is likely to be very valuable. These data could help us better anticipate resistance and then address it by chemical design or the judicious selection of drug combinations before the start of treatments.
Trends Box.
-
-
Resistance-conferring mutations can help identify cellular targets of chemical probes and drugs.
-
-
Recent advances in genome sequencing and editing allow analyses of chemotype-specific resistance in human cells in culture.
-
-
Analysis of recurring chemotype-specific resistance-conferring mutations can help identify and validate a chemical inhibitor’s direct target in human cells.
-
-
Genetically-matched inhibitor-resistant and inhibitor-sensitive cell-lines can be used to dissect dose-dependent, on-target effects of a chemical inhibitor.
-
-
Multiple clustered, resistance-conferring mutations in the target protein can help identify an inhibitor’s binding site.
Acknowledgments
T.M.K. is grateful to the NIH/NIGMS for funding (GM98579) and R.M.M. acknowledges support from the Damon Runyon Cancer Research Foundation (DRG 2222-15).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest
The authors are unaware of any conflicts of interest.
References
- 1.Weiss WA, et al. Recognizing and exploiting differences between RNAi and small-molecule inhibitors. Nat Chem Biol. 2007;3(12):739–44. doi: 10.1038/nchembio1207-739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lampson MA, Kapoor TM. Unraveling cell division mechanisms with small-molecule inhibitors. Nat Chem Biol. 2006;2(1):19–27. doi: 10.1038/nchembio757. [DOI] [PubMed] [Google Scholar]
- 3.Drews J. Drug discovery: a historical perspective. Science. 2000;287(5460):1960–4. doi: 10.1126/science.287.5460.1960. [DOI] [PubMed] [Google Scholar]
- 4.Gibbs JB. Mechanism-based target identification and drug discovery in cancer research. Science. 2000;287(5460):1969–73. doi: 10.1126/science.287.5460.1969. [DOI] [PubMed] [Google Scholar]
- 5.Mitchison TJ. Probing cell division with “chemical genetics”. Harvey Lect. 2002;98:19–40. [PubMed] [Google Scholar]
- 6.Schreiber SL. Chemical genetics resulting from a passion for synthetic organic chemistry. Bioorg Med Chem. 1998;6(8):1127–52. doi: 10.1016/s0968-0896(98)00126-6. [DOI] [PubMed] [Google Scholar]
- 7.Holohan C, et al. Cancer drug resistance: an evolving paradigm. Nat Rev Cancer. 2013;13(10):714–26. doi: 10.1038/nrc3599. [DOI] [PubMed] [Google Scholar]
- 8.Gottesman MM, et al. Toward a Better Understanding of the Complexity of Cancer Drug Resistance. Annu Rev Pharmacol Toxicol. 2016;56:85–102. doi: 10.1146/annurev-pharmtox-010715-103111. [DOI] [PubMed] [Google Scholar]
- 9.Locher KP. Mechanistic diversity in ATP-binding cassette (ABC) transporters. Nat Struct Mol Biol. 2016;23(6):487–93. doi: 10.1038/nsmb.3216. [DOI] [PubMed] [Google Scholar]
- 10.Gorre ME, Sawyers CL. Molecular mechanisms of resistance to STI571 in chronic myeloid leukemia. Curr Opin Hematol. 2002;9(4):303–7. doi: 10.1097/00062752-200207000-00007. [DOI] [PubMed] [Google Scholar]
- 11.Zheng XS, et al. Genetic and genomic approaches to identify and study the targets of bioactive small molecules. Chem Biol. 2004;11(5):609–18. doi: 10.1016/j.chembiol.2003.08.011. [DOI] [PubMed] [Google Scholar]
- 12.Heitman J, et al. Targets for cell cycle arrest by the immunosuppressant rapamycin in yeast. Science. 1991;253(5022):905–9. doi: 10.1126/science.1715094. [DOI] [PubMed] [Google Scholar]
- 13.Gonzalez A, Hall MN. Nutrient sensing and TOR signaling in yeast and mammals. EMBO J. 2017;36(4):397–408. doi: 10.15252/embj.201696010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hayles J, Nurse P. Genetics of the fission yeast Schizosaccharomyces pombe. Annu Rev Genet. 1992;26:373–402. doi: 10.1146/annurev.ge.26.120192.002105. [DOI] [PubMed] [Google Scholar]
- 15.Kawashima SA, et al. Analyzing fission yeast multidrug resistance mechanisms to develop a genetically tractable model system for chemical biology. Chem Biol. 2012;19(7):893–901. doi: 10.1016/j.chembiol.2012.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kawashima SA, et al. Potent, Reversible, and Specific Chemical Inhibitors of Eukaryotic Ribosome Biogenesis. Cell. 2016;167(2):512–524 e14. doi: 10.1016/j.cell.2016.08.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kawashima SA, et al. A chemical biology strategy to analyze rheostat-like protein kinase-dependent regulation. Chem Biol. 2013;20(2):262–71. doi: 10.1016/j.chembiol.2013.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Feng Y, et al. Multi-parameter phenotypic profiling: using cellular effects to characterize small-molecule compounds. Nat Rev Drug Discov. 2009;8(7):567–78. doi: 10.1038/nrd2876. [DOI] [PubMed] [Google Scholar]
- 19.Schenone M, et al. Target identification and mechanism of action in chemical biology and drug discovery. Nat Chem Biol. 2013;9(4):232–40. doi: 10.1038/nchembio.1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Futamura Y, et al. Target identification of small molecules based on chemical biology approaches. Mol Biosyst. 2013;9(5):897–914. doi: 10.1039/c2mb25468a. [DOI] [PubMed] [Google Scholar]
- 21.Wacker SA, et al. Using transcriptome sequencing to identify mechanisms of drug action and resistance. Nat Chem Biol. 2012;8(3):235–7. doi: 10.1038/nchembio.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kasap C, et al. DrugTargetSeqR: a genomics- and CRISPR-Cas9-based method to analyze drug targets. Nat Chem Biol. 2014;10(8):626–8. doi: 10.1038/nchembio.1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Smurnyy Y, et al. DNA sequencing and CRISPR-Cas9 gene editing for target validation in mammalian cells. Nat Chem Biol. 2014;10(8):623–5. doi: 10.1038/nchembio.1550. [DOI] [PubMed] [Google Scholar]
- 24.Han T, et al. Anticancer sulfonamides target splicing by inducing RBM39 degradation via recruitment to DCAF15. Science. 2017;356(6336) doi: 10.1126/science.aal3755. [DOI] [PubMed] [Google Scholar]
- 25.Uehara T, et al. Selective degradation of splicing factor CAPERalpha by anticancer sulfonamides. Nat Chem Biol. 2017;13(6):675–680. doi: 10.1038/nchembio.2363. [DOI] [PubMed] [Google Scholar]
- 26.Baron L, et al. Mycolactone subverts immunity by selectively blocking the Sec61 translocon. J Exp Med. 2016;213(13):2885–2896. doi: 10.1084/jem.20160662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Anderson DJ, et al. Targeting the AAA ATPase p97 as an Approach to Treat Cancer through Disruption of Protein Homeostasis. Cancer Cell. 2015;28(5):653–65. doi: 10.1016/j.ccell.2015.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sekine Y, et al. Stress responses. Mutations in a translation initiation factor identify the target of a memory-enhancing compound. Science. 2015;348(6238):1027–30. doi: 10.1126/science.aaa6986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Paatero AO, et al. Apratoxin Kills Cells by Direct Blockade of the Sec61 Protein Translocation Channel. Cell Chem Biol. 2016;23(5):561–6. doi: 10.1016/j.chembiol.2016.04.008. [DOI] [PubMed] [Google Scholar]