

Graphical Abstract
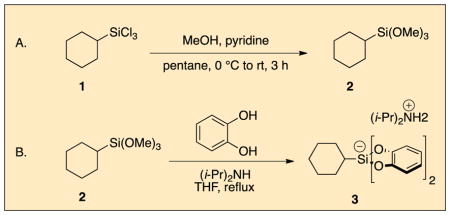
Procedure
A. Cyclohexyltrimethoxysilane (2)
A 250 mL, oven-dried, two-necked, round-bottomed flask is charged with a 3.2 cm, Teflon-coated magnetic oval stir bar and coupled with a 50 mL dropping funnel. Both the dropping funnel and the round-bottomed flask are sealed with a rubber septum. The system is evacuated for 10 min and back-filled with nitrogen. This process is repeated twice more, and then, the flask is charged by syringe with pentane (180 mL), anhydrous pyridine (21.0 mL, 20.5 g, 260 mmol, 4 equiv), and anhydrous methanol (10.5 mL, 8.3 g, 260 mmol, 4 equiv) (Notes 1–3).
Additionally, a solution of cyclohexyltrichlorosilane 1 (14.14 g, 65.0 mmol, 1.0 equiv) in pentane (37 mL) is prepared in an addition funnel. (Figure 1, left) The stirred, homogeneous solution (Note 4) is cooled to 0 °C (external temperature) via an ice-water bath while under a nitrogen atmosphere (Figure 1). After cooling for 10 min the solution in the dropping funnel is added dropwise to the flask over 35 min (Notes 5, 6, and 7). A voluminous white precipitate (pyridinium hydrochloride, Figure 1, right) forms upon addition of 1. Following complete addition of 1, the reaction mixture is stirred at 0 °C for 5 min. The ice bath is removed, and the heterogeneous solution is stirred for 3 h at room temperature.
Figure 1.

Reaction setup for synthesis of 2
Upon confirmation that the reaction is complete by crude 1H NMR, the stirring is stopped, and the solids are allowed to settle (Figure 2) (Note 8). The reaction mixture is decanted from the solid pyridinium hydrochloride and transferred to a 1000 mL separatory funnel (Note 9). The white pyridinium salt is washed with pentane (100 mL) to assist in transferring all of the product to the funnel. The separatory funnel is charged with deionized H2O (250 mL). The layers are separated, and the aqueous layer is extracted with pentane (2 × 125 mL). The combined organic layers are washed with 2 M aqueous HCl (2 × 100 mL), saturated aqueous NaHCO3 (150 mL), deionized water (150 mL), and then saturated aqueous NaCl (150 mL). The organic layer is then dried over sodium sulfate (25 g), and after filtration, the solvent is removed in vacuo by rotary evaporation (Note 10) to furnish pure 2 (12.49 g, 94%) (Notes 11 and 12) as a clear, colorless oil (Figure 3).
Figure 2.

Pyridinium hydrochloride produced during synthesis of 2
Figure 3.

Sample of 2 isolated after workup
B. Diisopropylammonium bis(catecholato)cyclohexylsilicate (3)
A 250 mL, oven-dried, single-necked, round-bottomed flask is charged with a 3.2 cm, Teflon-coated, magnetic oval stir bar and catechol (10.74 g, 97.5 mmol, 1.95 equiv) (Notes 13, 14, and 15). The flask is sealed with a rubber septum and flushed with nitrogen via a nitrogen line inlet. The flask is then charged via syringe with anhydrous tetrahydrofuran (60 mL) and anhydrous diisopropylamine (8.40 mL, 6.07 g, 60.0 mmol, 1.2 equiv) (Note 13) (Figure 4). The homogeneous solution is stirred at room temperature for 2 min.
Figure 4.

a) Catechol in tetrahydrofuran; b) After addition of 1 drop of diisopropylamine; c) After addition of all diisopropylamine
The septum is removed and the flask is charged with cyclohexyltrimethoxysilane, 2, (10.20 g, 50.0 mmol, 1 equiv) followed immediately by additional anhydrous tetrahydrofuran (40 mL). The flask is equipped with a septum-sealed 25.5 cm reflux condenser (with a nitrogen line inlet). The flask is then placed in a mineral oil bath and heated to reflux (Note 16) for 16 h (Figure 5). After briefly cooling the flask, a crude sample is assessed by 1H NMR and judged to be incomplete at this time (Notes 17 and 18). The reaction is restarted by first removing the solvent from the flask by rotary evaporation (Notes 19 and 20) yielding a sticky, voluminous, off-pink solid. The flask is recharged with anhydrous tetrahydrofuran (50 mL), and then sonicated for 5 min at 27 °C in a water bath in order to separate the solid from the walls of the flask. The flask is resealed with a rubber septum and flushed with nitrogen via a nitrogen line inlet. The flask is then recharged with anhydrous diisopropylamine (4.20 mL, 3.04 g, 30.0 mmol, 0.6 equiv). The flask is reequipped with a septum-sealed 25.5 cm reflux condenser (with a nitrogen line inlet). The flask is then placed in a mineral oil bath and heated to reflux (Note 16) for 16 h. After briefly cooling the flask, a crude sample is assessed by 1H NMR and judged to be incomplete at this time. The process is repeated three more times, after which time the reaction is judged as complete. The solvent is removed from the flask by rotary evaporation (Note 20) giving a dry, voluminous, off-pink solid. The flask is diluted with diethyl ether (200 mL) and is sonicated for 5 min at 27 °C in a water bath. The solid is collected by vacuum filtration through a 500 mL, D4 porosity fritted funnel.
Figure 5.

Reaction setup for synthesis of 3
The flask is rinsed with additional diethyl ether (50 mL) and the slurry is added to the funnel. The combined solids are washed with diethyl ether (100 mL) and are allowed to dry on vacuum for 10 min (Note 21). The solid is placed under vacuum for 30 min (Note 22), giving pure 3 (20.24 g, 96%) (Note 23, and 24) as a white, free flowing powder (Figure 6).
Figure 6.

Sample of 3 isolated after workup
Working with Hazardous Chemicals
The procedures in Organic Syntheses are intended for use only by persons with proper training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as “Prudent Practices in the Laboratory” (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at http://www.nap.edu/catalog.php?record_id=12654). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices.
In some articles in Organic Syntheses, chemical-specific hazards are highlighted in red “Caution Notes” within a procedure. It is important to recognize that the absence of a caution note does not imply that no significant hazards are associated with the chemicals involved in that procedure. Prior to performing a reaction, a thorough risk assessment should be carried out that includes a review of the potential hazards associated with each chemical and experimental operation on the scale that is planned for the procedure. Guidelines for carrying out a risk assessment and for analyzing the hazards associated with chemicals can be found in Chapter 4 of Prudent Practices.
The procedures described in Organic Syntheses are provided as published and are conducted at one’s own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein.
Discussion
The rich history of organosilanes intersecting with organic chemistry to fill otherwise impossible-to-meet gaps is unparalleled. Not only do organosilanes serve as some of the most utilized protecting groups, but the unique hypervalency of silicon allows it to serve as much more than a simple blocking group.2 Organosilanes serve as popular Lewis acids that can enable asymmetric processes, act as coupling partners in transition metal-catalyzed cross-coupling reactions, facilitate the perfluoroalkylation of various functional groups, and as mild stoichiometric reductants. Several of these transformations hinge on the intermediacy of pentavalent silicon. In addition to being a common intermediate, certain pentavalent silicon species are quite stable and can be readily prepared.3 One such species, organobis(catecholato)silicates, drew our attention as a potential cross coupling partner for the recently described Ni/photoredox dual catalytic paradigm.4 First prepared by Frye in 1964,5 these hypervalent silicate species have had limited synthetic applications. Apart from the elegant reports by DeShong6 and Hosomi,7 examples of alkylsilicates as cross-coupling partners for Csp2-Csp2 bond formation were rare and, to the best of our knowledge, their use in Csp3-C sp2bond construction was unknown prior to 2014. That year and again in 2015, Nishigaichi reported the first examples of Csp3-Csp2 bond construction using alkyl bis(catecholato)silicates and 1,4-dicyanoarenes via radical-mediated decyanation.8 In lieu of these reports, we envisioned that organobis(catecholato)silicates could serve as unmatched precursors for alkyl radicals under photoredox conditions. Indeed, we quickly found that alkylbis(catecholato)silicates readily undergo oxidation via a SET to the excited state of various inorganic and organic photocatalysts. In part, this susceptibility to photooxidation can be attributed to their low oxidation potentials (E° ~ +0.40 V to +0.75 V vs SCE for all silicates examined to date). The homogeneity in redox values is a consequence of a leveling effect educed by the catecholate moiety. That is, the electrochemical potentials of each derivative are more or less equivalent because the redox reaction is occurring at a site that is virtually unchanged between substrates and is distal to the homolysis event.
We and others have recently found that alkylbis(catecholato)silicates are particularly well-suited for Ni/photoredox dual catalysis (Figure 7). 9,10 Using this paradigm, Csp3-Csp2 bonds can be constructed under remarkably mild, base-free conditions with only innocuous byproducts generated during the reaction. Utilization of this powerful reaction manifold has enabled the cross-coupling of benzylic/allylic, 2° and 1° Csp3-hybridized radicals with aryl and/or alkenyl electrophiles. In addition, alkylsilicates can be employed as H-atom abstractors of thiols to facilitate thioetherification via the integration of thiyl radicals with Ni/photoredox dual catalysis.
Figure 7.

Applications of alkylbis(catecholato)silicates in Ni/photoredox dual catalysis
During the course of these investigations, a robust method for preparation of alkylsilicates was needed. We identified two key impasses where optimization was needed. First, the ideal starting material needed to be selected. Alkyltrichlorosilanes and trialkoxyalkylsilanes were both viable candidates. Whereas the former offered more diversity in substrate scope because of the easy of hydrosilylation with trichlorosilane, they are far more sensitive to hydrolysis.11 Moreover, difficulties encountered in the direct synthesis of alkylsilicates from alkyltrichlorosilanes (e.g., difficulties in purification, production of voluminous HCl salts, and decomposition of the product during extended reaction times) prompted us to examine the viability of trialkoxyalkylsilanes. Ultimately, these proved to be the optimal starting materials. Specifically, we found that the best results were achieved using trimethoxyalkylsilanes, likely because of the lack of steric encumbrance at silicon when attempting nucleophilic displacement. To improve the diversity of available trimethoxyalkylsilanes, we devised a reliable and user-friendly method for converting alkyltrichlorosilanes into their trimethoxy analogues. Thus, we proceeded with these species as ideal starting materials for silicate synthesis.
Secondly, we examined a number of counter-ions, evaluating each based on solubility, synthetic ease, and bench-top stability. Ultimately, alkylammonium cations were recognized as the ideal counter-ion for our purposes because : i) the amine needed for the reaction to proceed served as both a base and the cation for the resulting ammonium silicate; ii) because the amine is protonated, it is inherently inert to redox processes; iii) the solubility properties of the alkylsilicate can be tuned by the structure of the amine base; iv) internal salts can be formed (i.e., if an amine-containing trimethoxyalkylsilane is used), alleviating the need for exogenous base; v) the ammonium salt is easily removed during workup following reactions in which the alkylsilicate is used. Although triethylamine was utilized during our earlier studies, we have come to favor diisopropylamine as the amine base because the resulting diisopropylammonium alkylsilicates readily solidify, making isolation and purification much more facile.
A number of alkylsilicates can be prepared in the manner outlined in this protocol. In many cases, the requisite trimethoxyalkoxysilanes are commercially available. Given that there were cases where only the alkyltrichlorosilanes were available, we have included the aforementioned procedure for converting these chlorinated species to their corresponding trimethoxyalkylsilanes. Table 1 provides some representative alkylsilicates that can be accessed using this protocol. In cases where the alkylsilicate could not be prepared by this method or was accompanied by significant side reactions, an alternative protocol is available and described in one of our recent publications.9b
Table 1.
Structural Diversity of ammonium alkylsilicates

|
Acknowledgments
We thank NIGMS (RO1 GM113878) for financial support of this research. C.B.K. is grateful for an NIH NRSA postdoctoral fellowship (F32GM117634-01).
Biographies

Professor Gary Molander completed his undergraduate studies at Iowa State University under the tutelage of Professor Richard Larock. He earned his Ph.D. at Purdue University under the direction of Professor Herbert Brown and undertook postdoctoral training with Professor Barry Trost at the University of Wisconsin, Madison. He began his academic career at the University of Colorado, Boulder, moving to the University of Pennsylvania in 1999, where he is currently the Hirschmann–Makineni Professor of Chemistry. His research interests focus on the development of new synthetic methods for organic synthesis using organotrifluoroborates and organobis(catecholato)silicates.

Mr. Kingson Lin is an undergraduate student at the University of Pennsylvania. He will be graduating in 2017 with a B.A in Biochemistry and a M.S. in Chemistry. He has recently been named a Novartis undergraduate scholar and is a recipient of the College Alumni Society Research Grant. He joined the Molander group in September of 2015 and studies organo(biscatecholato)silicates and their application in photoredox/Ni dual catalysis.

Dr. Christopher B. Kelly studied at Stonehill College in Easton, MA, where he received his B.S. in Biochemistry in 2010. That same year, he joined the University of Connecticut (UConn) where he performed his doctoral studies under the supervision of Dr. Nicholas Leadbeater. While at UConn, he developed new synthetic methods in organofluorine and oxoammonium salt chemistry. After earning his Ph.D. in organic chemistry in 2015, he joined Prof. Molander’s group at the University of Pennsylvania as a National Institutes of Health postdoctoral fellow. His research focuses on new synthetic methods involving organobis(catecholato)silicates and photoredox catalysis.

Dr. Matthieu Jouffroy received his M.Sc. in Molecular and Supramolecular Chemistry from the University of Strasbourg in 2011. He obtained his Ph.D. from the same university in 2014 on the synthesis of confining ligands built upon cyclodextrin scaffolds under Dr. Dominique Matt and Professor Dominique Armspach. He then worked at Tokyo Tech under Professor Kohtaro Osakada and Dr. Daisuke Takeuchi on monophosphine Pd(II) complexes in styrene/CO copolymerization via a JSPS postdoctoral fellowship. He is currently a postdoctoral researcher at the University of Pennsylvania under Prof. Gary Molander. His current research interests are focused on Ni/photoredox cross-coupling with organosilicates.

Andrés García-Domínguez obtained his B.Sc. in Chemistry from Autónoma University in Madrid (Spain) in 2011. In 2013, he received his M.Sc. in Organic Chemistry from the same university under the supervision of Prof. Dr. Diego J. Cárdenas and moved to University of Zurich (Switzerland) to carry out his Ph.D. work under the supervision of Prof. Dr. Cristina Nevado. His research interests are currently focused on the development of new transition metal-catalyzed methodologies for dicarbofunctionalizations of multiple bonds.

Estíbaliz Merino obtained her Ph.D. degree from the Autónoma University (Madrid-Spain). After a postdoctoral stay with Prof. Magnus Rueping at Goethe University Frankfurt and RWTH-Aachen University in Germany, she worked with Prof. Avelino Corma in Instituto de Tecnología Química-CSIC (Valencia) and Prof. Félix Sánchez in Instituto de Química Orgánica General-CSIC (Madrid) in Spain. At present, she is research associate in Prof. Cristina Nevado’s group in University of Zürich. She is interested in the development of new catalytic methods for the synthesis of natural products and in the development of new materials with application in heterogeneous catalysis.
Appendix. Chemical Abstracts Nomenclature (Registry Number)
Cyclohexyltrichlorosilane (98-12-4)
Methanol (67-56-1)
Pyridine (110-86-1)
Cyclohexyltrimethoxysilane (17865-54-2)
Catechol: 1,2-Benzenediol (120-80-9)
N,N-Diisopropylamine: 2-Propanamine, N-(1-methylethyl)-; (108-18-9)
Footnotes
The following reagents in this section were purchased from commercial sources and used without further purification: Pentane (≥99%, Acros Organics) and methanol (extra dry over 4 Å molecular sieves, 99.8%, Acros Organics).
Pyridine was purchased from Fisher scientific (Certified ACS grade, ≥99%) and stored over KOH pellets.
Although pentane is used in this protocol, a number of other solvents can be used (e.g., n-hexane, n-heptane, diethyl ether, tetrahydrofuran) with similar results.. For simple aliphatic alkyltrichlorosilanes hydrocarbon solvents are favored because of ease of product isolation.
The solution is stirred at 500 rpm throughout the reaction.
The solution in the funnel has a milky color.
Cyclohexyltrichlorosilane was purchased from TCI Europe (>98.0%) and used without further purification
After the addition, the funnel was rinsed with 2 mL of pentane and added into the reaction mixture.
An aliquot (0.1 mL) was taken via 1 mL syringe through the septum and transferred into a vial. The solvent was evaporated at 375 mmHg at 25 °C for 15 minutes and CDCl3 (0.7 mL) was added.
Alternatively, filtration through a medium porosity fritted funnel and washing the pyridinium salt with multiple small washes of pentane can be performed (~ 3 × 25 mL)
Bath temperature: 40 °C. First evaporation at pressure >550 mmHg. To dry further the compound, it was submitted to 250 mmHg for 1 h. Due to the volatility of the product 2 (bp = 207–209 °C), very long evaporation times may lead to reduced yields.
A second reaction on identical scale provided 12.72 g (96%) of the same product.
The product has been characterized as follows: 1H NMR (CDCl3, 400 MHz) δ 0.87 (tt, J = 12.4, 3.0 Hz, 1 H), 1.18 – 1.31 (m, 5 H), 1.70 – 1.78 (m, 5 H), 3.58 (s, 9 H). 13C NMR (CDCl3, 101 MHz) δ 22.3, 26.7 (2 × C) 27.6 (2 × C), 50.8 (3 × C). FT-IR (neat, ATR) 2923, 2841, 1447, 1196, 1090, 851, 827, 797, 754 cm−1. HRMS (CI+) calcd for C9H21O3Si [M+H]+: 205.1255, found: 205.1255. The purity of the compound was calculated by qNMR using 12.8 mg of dimethyl fumarate (DF, purity: 97%) and 19.4 mg of the compound 2. Two signals for the product 2 were selected (0.87 and 1.70–1.78 ppm) and the normalized integrals values per proton equivalent were calculated (dividing each integral by the corresponding number of protons). The integral of the product 2 (Int2) was calculated as the average of both normalized integrals. The number of protons (n2) was set to one. The purity was determined using the following equation:
- nDF = number of protons giving rise to a given NMR signal (the total number of protons is set to one because an average of all normalized integrals is carried out).
- IntDF = Average of normalized integrals values (for the two signals of DF).
- MWDF = Molecular weight of DF.
- mDF = mass of DF.
- PDF = Purity of DF (as percent value).
- n2 = number of protons giving rise to a given NMR signal (the total number of protons is set to one because an average of all normalized integrals is carried out).
- Int2 = Average of normalized integrals values (for the two signals of compound 2 at 0.87 and 170–1.78 ppm).
- MW2 = Molecular weight of compound 2.
- m2 = mass of compound 2.
The following reagents in this section were purchased from commercial sources: tetrahydrofuran (Certified, ≥99%, Fisher Scientific) catechol (99%, Alfa Aesar), and diisopropylamine (99+%, Alfa Aesar). Tetrahydrofuran was dried using a solvent purification system from Inert Technology co. (Content of water: 20–50 ppm). Diisopropylamine was distilled from calcium hydride before use. Catechol was purified as described in Note 15.
The mixture is stirred at 750 rpm throughout the reaction.
Catechol is recrystallized from hot heptane. In a single-necked 500 mL round-bottomed flask, catechol (20 g) is suspended in heptane (400 mL). The reaction mixture is refluxed over 15 min and after removing the heating source the solids are allowed to settle. The hot liquid phase is transferred to a beaker and allowed to cool to room temperature, where it was held for 1 h. The white solid is filtered, washed with pentane and dried under vacuum.

Alternatively, catechol can be sublimed. Ultimately, both purification methods are useful to ensure the resulting silicate is near colorless. The color of alkylsilicates does not have any bearing on the Ni/photoredox cross-coupling developed by our laboratory.
Oil bath temperature maintained at 85 °C using an Heidolph MR Hei Tec hot plate equipped with a Pt 1000 temperature probe dipped into the oil bath.
Although on smaller scale (<20 mmol) preparations, restarting the reaction is not necessary, larger scale reactions tend to stall after this time period. This may relate to the methanol content in solution. By restarting the reaction in the described manner one time, the product was obtained in 89% yield. The reaction reaches completion (total time: 64 h) by restarting three times more. Another reaction provided 16.34 g (78%) with three restarts.
When the reaction is cooled at rt, the reflux condenser is removed and 0.1 mL of the suspension taken, transferred to a 10 mL vial and the solvent evaporated (see Note 10). Residual solvent signals for THF and MeOH can be found and the reaction is judged to be complete when no catechol (1H NMR (DMSO-d6) δ = 6.60 and 6.70 ppm) is observed.
Care should be used when removing the solvent because the solid has a tendency to bump.
Bath temperature: 40 °C. Pressure >190 mmHg. Because of the volatility of 2 (bp = 207–209 °C), it is recommended that very low pressures be avoided.
Although 3 (and most diisopropylammonium alkylsilicates) are mostly insoluble in ether, they do have a slight solubility. As such it is recommended that only the suggested amount of ether be used when washing to avoid diminished yield.
In a rotatory evaporator at 40 °C, 30 mmHg or in a vacuum line (3 × 10−2 mmHg). Care should be used because the solid has a tendency to bump.
The product (3) has been characterized as follows: 1H NMR (DMSO-d6, 400 MHz) δ 0.61–0.68 (m, 1 H), 0.94–1.12 (m, 5 H), 1.20 (d, J = 6.5 Hz, 12 H), 1.46–1.49 (m, 5 H), 3.35 (sept, 2 H), 6.40 (d, J = 5.5 Hz, 4 H), 6.41 (d, J = 5.5 Hz, 2H), 6.49 (d, J = 5.5 Hz, 2 H), 6.50 (d, J = 5.6 Hz, 2H), 8.00 (br s, 2 H). 13C NMR (DMSO-d6, 101 MHz) δ 18.9, 26.8, 27.7, 28.3, 30.2, 46.3, 109.2, 116.9, 151.0. FT-IR (cm−1, neat, ATR) 3034, 2847, 1484, 1354, 1239, 1197, 1147, 1102, 1016, 887, 819, 743, 729, 661, 595, 519, 429, 419. HRMS (ES−) calcd for [M-iPr2NH2]−: 327.1058 C18H19O4Si, found: 327.1059 Melting Point: 209–210 °C (uncorrected).
Further purification can be accomplished by dissolving the alkylsilicate in a minimal amount of dichloromethane followed by adding a minimum amount of pentane as an antisolvent. The resulting solid is then filtered and washed with a 50:50 mixture by volume of pentane to ether. The purity of the compound 3 was calculated by qNMR using 4.7 mg of dimethyl fumarate (DF, purity: 97%) and 5.8 mg of the compound 3 (See details in Note 10).
References
- 2.(a) Greene TW, Wuts PGM. Protective Groups In Organic Synthesis. 3. John Wiley & Sons; New Jersey: 2007. [Google Scholar]; (b) Dilman AD, Ioffe SL. Chem Rev. 2003;103:733–772. doi: 10.1021/cr020003p. [DOI] [PubMed] [Google Scholar]; (c) Chang WTT, Smith RC, Regens CS, Bailey AD, Werner NS, Denmark SE. Org React. 2011;75:213–746. [Google Scholar]; (d) Chatgilialoglu C. Acc Chem Res. 1992;25:188–194. [Google Scholar]; (e) Prakash GKS, Yudin AK. Chem Rev. 1997;97:757–786. doi: 10.1021/cr9408991. [DOI] [PubMed] [Google Scholar]
- 3.(a) Rendler S, Oestreich M. Synthesis. 2005;11:1727–1747. [Google Scholar]; (b) Chuit C, Corriu RJP, Reye C, Young JC. Chem Rev. 1993;93:1371–1448. [Google Scholar]
- 4.Seminal reports:Tellis JC, Primer DN, Molander GA. Science. 2014;345:433–436. doi: 10.1126/science.1253647.Zuo Z, Ahneman DT, Chu L, Terret JA, Doyle AG, MacMillan DWC. Science. 2014;345:437–440. doi: 10.1126/science.1255525.
- 5.Frye CL. J Am Chem Soc. 1964;86:3170–3171. [Google Scholar]
- 6.Seganish WM, DeShong P. J Org Chem. 2004;69:1137–1143. doi: 10.1021/jo035309q. [DOI] [PubMed] [Google Scholar]
- 7.Hosomi A, Kohra S, Tominaga Y. Chem Pharm Bull. 1988;36:4622–4625. [Google Scholar]
- 8.(a) Matsuoka D, Nishigaichi Y. Chem Lett. 2014;43:559–561. [Google Scholar]; (b) Matsuoka D, Nishigaichi Y. Chem Lett. 2015;44:163–165. [Google Scholar]
- 9.(a) Jouffroy M, Primer DN, Molander GA. J Am Chem Soc. 2016;138:475–478. doi: 10.1021/jacs.5b10963. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Patel NR, Kelly CB, Jouffroy M, Molander GA. Org Lett. 2016;18:764–767. doi: 10.1021/acs.orglett.6b00024. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Jouffroy M, Kelly CB, Molander GA. Org Lett. 2016;18:876–879. doi: 10.1021/acs.orglett.6b00208. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Jouffroy M, Davies GHM, Molander GA. Org Lett. 2016;18:1606–1609. doi: 10.1021/acs.orglett.6b00466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.(a) Corcé V, Chamoreau LM, Derat E, Goddard JP, Ollivier C, Fensterbank L. Angew Chem, Int Ed. 2015;54:11414–11418. doi: 10.1002/anie.201504963. [DOI] [PubMed] [Google Scholar]; (b) Chenneberg L, Lévêque C, Corcé V, Baralle A, Goddard JP, Ollivier C, Fensterbank L. Synlett. 2016;27:731–735. [Google Scholar]; (c) Lévêque C, Chenneberg L, Corcé V, Goddard JP, Ollivier C, Fensterbank L. Org Chem Front. 2016;3:462–465. [Google Scholar]
- 11.Troegel D, Stohrer J. Coord Chem Rev. 2011;255:1440–1459. [Google Scholar]