

Summary
Zika virus (ZIKV) infection causes devastating congenital abnormities and Guillain-Barré syndrome. The ZIKV envelope (E) protein is responsible for viral entry and represents a major determinant for viral pathogenesis. Like other flaviviruses, the ZIKV E protein is glycosylated at amino acid N154. To study the function of E glycosylation, we generated a recombinant N154Q ZIKV that lacks the E glycosylation, and analyzed the mutant virus in mammalian and mosquito hosts. In mouse models, the mutant was attenuated, as evidenced by lower viremia, decreased weight loss, and no mortality; however, knockout of E glycosylation did not significantly affect neurovirulence. Mice immunized with the mutant virus developed a robust neutralizing antibody response and were completely protected from wild-type ZIKV challenge. In mosquitoes, the mutant virus exhibited diminished oral infectivity for the Aedes aegypti vector. Collectively, the results demonstrate that the E glycosylation is critical for ZIKV infection of mammalian and mosquito hosts.
Keywords: Zika virus, glycosylation, flavivirus entry, mosquito transmission, vaccine
eTOC Blurb
Zika virus (ZIKV) causes devastating congenital abnormities and Guillain-Barré syndrome. Fontes-Garfias et al. showed that the glycosylation of ZIKV envelope protein plays an important role in infecting mosquito vectors and in pathogenesis in mouse.
Introduction
Zika virus (ZIKV) was originally isolated in 1947 from the Zika Forest of Uganda, and belongs to genus Flavivirus from family Flaviviridae (Weaver et al., 2016). Others flaviviruses, such as dengue (DENV), Yellow fever (YFV), West Nile (WNV), and Japanese encephalitis viruses (JEV), are medically important pathogens. About 80% of ZIKV infections are asymptomatic (Fauci and Morens, 2016). The recent epidemics have documented that ZIKV infections are associated with a broad range of devastating disorders comprising congenital Zika syndrome (Costello et al., 2016) as well as Guillain-Barré syndrome (Fauci and Morens, 2016). ZIKV is mainly transmitted by Aedes mosquitoes, and is also transmitted through sexual and blood transfusion routes (Aliota et al., 2017). It is a public health priority to develop effective vaccines and therapeutics of ZIKV (Shan et al., 2016a).
ZIKV has a positive, single-strand RNA genome of about 11,000 nucleotides that encode three structural proteins (capsid [C], precursor membrane [prM], and envelope [E]) and seven non-structural proteins (NS1, NS2A, NS2B, NS3, NS4A, NS4B, and NS5). The structural proteins along with genomic RNA form viral particles, while the nonstructural proteins are involved in replication, assembly, and evasion of the host immune system (Pierson and Diamond). Flaviviruses enter the host cell via receptor-mediated endocytosis. The viral E protein first interacts with cellular surface attachment factors and receptors (Hamel et al., 2015). The virions are then internalized through the endocytic pathway (Rey et al., 2017). In the late endosome, low pH triggers a series of conformational changes of the E protein, leading to the fusion of viral and cellular membranes. After uncoating from nucleocapsids, the viral genome is released into the cytoplasm where translation occurs. Genomic replication takes place in the replication complexes in vesicle packets (VPs) formed in the endoplasmic reticulum (ER). Virus assembly takes place on the ER surface at sites juxtaposed to the VPs, where structural proteins packages the newly synthesized viral RNA to form immature virions that bud into the ER lumen (Welsch et al., 2009). Mature infectious virions form as they transit through the Golgi network where the pr segment is cleaved from the prM by host protease Furin. The mature virions are released from infected cells via exocytosis (Acosta et al., 2014).
The flavivirus E protein is a major surface glycoprotein involved in modulating the viral infection cycle and eliciting antibody response (Pierson and Diamond). The E protein of most flaviviruses is post-translationally modified by N-linked glycosylation at amino acid 153/154 within a highly conserved glycosylation motif of N-X-T/S at positions154-156, indicating the biological importance of this modification; however, some flaviviruses isolates lack E glycosylation, suggesting that the function of E can be achieved without the N-linked glycan (Adams et al., 1995; Beasley et al., 2004; Berthet et al., 1997; Post et al., 1992; Vorndam et al., 1993). For the WNV E protein, glycosylation plays a critical role in neuroinvasiveness in mice (Beasley et al., 2005; Shirato et al., 2004), but does not show any effect on infection of mosquitos; in cell culture, the E glycosylation knockout reduces WNV replication in mammalian BHK and avian QT6 cells, but does not affect viral replication in mosquito C6/36 cells (Murata et al., 2010). For DENV E protein, there are two glycosylation sites (at residues N67 and N153). Mutagenesis studies showed that N153Q mutation reduces viral replication in BHK and C6/36 cells, but does not affect viral replication in intrathoraxially injected mosquitoes (Bryant et al., 2007). Depletion of N67 glycosylation of E protein abolishes the ability of DENV-2 to produce infectious virus in mammalian cells, but not in mosquito cells (Bryant et al., 2007; Mondotte et al., 2007). For ZIKV, phylogenetic analysis showed that the prototype African stain MR776a contains a four-amino acid deletion spanning the N154 glycosylation site (Fig. 1A); this deletion might represent an adaptation when the original MR776a isolate was passaged in mouse brains for about 148 rounds (Dick et al., 1952). Several other historical strains (e.g., Malaysian strain P6-740 in Fig. 1A) also lack the glycosylation motif, whereas all the contemporary epidemic strains contained the E N154 glycosylation site. The biological function of ZIKV E glycosylation remains to be determined. The goal of this study was to characterize the role of E glycosylation in ZIKV replication in cell culture, virulence in mouse models, and infection of mosquito vector.
Figure 1.
Characterization of N154Q mutant. (A) Amino acid sequence aligment. The sequences of the E glycosylation site (positions 154-156) were compared among ZIKV strains FSS13025 (GeneBank number KU955593.1), MR776a (GeneBank number AY632535), P6-740 (GeneBank number KX377336.1), and 41525-DAK (GeneBank number KU955591.1), YFV strain Asibi (GeneBank number AY640589), WNV strain NY99 (GeneBank number DQ211652), JEV strain SA14 (GeneBank number D90194), DENV-1 strain SG/07K3640DK1/2008 (GeneBank number GQ398255), DENV-2 strain 16681 (GeneBank number NC_001474), DENV-3 strain SG/05K863DK1/2005 (GeneBank number EU081190), and DENV-4 strain SG/06K2270DK1/2005 (GeneBank number GQ398256). The conserved glycosylation site at Asn 154 is highlighted in green. The glycosylation motif N-X-S/T is underlined. It should be noted that ZIKV P6-740 strain does not contain the conserved glycosylation motif of N-X-T/S. (B) 3-D dimer structure of ZIKV envelope protein. E protein dimer is shown in ribbon form; E protein monomers are colored in light and dark green and the transmembrane regions are colored in blue and purple. The N154 glycans on each monomer are labeled and shown projecting on the E protein surface (Protein Data Bank accession codes: 5IRE). (C) IFA of viral protein expression in cells transfected with WT and N154Q RNAs. Vero cells were electroporated with 10 µg of genome-length WT and N154Q RNA. On day 3 p.t., IFA was performed to examine viral E protein expression using a mouse mAb (4G2). Green and blue represent E protein and nuclei (stained with DAPI), respectively. (D) Plaque morphologies of WT and N154Q viruses. (E) Sequencing traces of E gene of WT and N154Q viruses. (F) Endoglycosidase analyses of E protein. Proteins from WT and N154Q virus-infected Vero cells (MOI of 0.01) were analyzed by Western blot at 5 days post-infection. Lysates were treated with PNGase F for 1 h at 37°C. Western blot analysis of enzyme digested E was assessed by 12% SDS-PAGE under reducing condition using rabbit anti-E IgG as primary antibody. Symbols “Ο” and “∆” indicate E proteins with and without glycosylation, respectively. (G-I) Comparison of growth kinetics of WT and N154Q viruses in Vero (G), BHK (H) and C6/36 (I) cells. Vero, BHK and C3/36 cells were infected with WT or N154Q virus at an MOI of 0.01. Viral titers were measured at indicated time points using plaque assays on Vero cells. Means and SDs from three independent replicates are shown. Statistics were performed using unpaired Student’s t test; **very significant (p < 0.01).
Results
Characterization of E glycosylation-knockout ZIKV in cell culture
Sequence alignment showed that, except for the high passage strain MR776a and several other historical strains (e.g., Malaysian P6-740), the E proteins from almost all other ZIKV isolates (including all the contemporary epidemic strains) have a conserved N-linked glycosylation motif (N-X-T/S; Fig. 1A). The glycan, linked to residue N154 (Fig. 1B), is exposed on the surface of domain I of the E protein (Sirohi et al., 2016). To study the function of E glycosylation, we engineered a mutation encoding N154Q in E protein of Cambodian ZIKV strain FSS13025, a close relative of all strains from the Americas. The N154Q mutation was elected because the side chains of glutamine and asparagine have the same polarity and differ by only one carbon. Upon transfection into Vero cells, the WT and mutant genomic RNAs generated similar amounts of E-expressing cells on day 3 post-transfection (Fig. 1C). Similar plaque morphologies were observed for the WT and N154Q viruses derived from the transfected cells (Fig. 1D). Sequencing of the full genome of the N154Q virus confirmed the engineered mutation without reversion or other mutations (Fig. 1E). Continuous passaging of the mutant virus for three rounds (3 days per round) on Vero cells did not change the engineered N154Q substitution.
To demonstrate the glycosylation status of the viral E protein, we treated the infected cell lysates with peptide N-glycosidase F (PNGase F), an enzyme that removes high mannose and complex carbohydrates. Western blotting showed that the PNGaseF treatment increased the mobility of the WT E protein; in contrast, the PNGaseF treatment did not change the mobility of the N154Q E protein, which migrated slightly faster than the WT E protein (Fig. 1F). The result indicated that the E protein expressed from the N154Q virus was not glycosylated.
We compared the replication kinetics of the WT and mutant viruses on three cell lines derived from the African green monkey (Vero), baby hamster kidney (BHK), and Ae. albopictus mosquito (C6/36). The two viruses showed comparable replication kinetics on Vero and BHK cells (Fig. 1G&H). In contrast, the mutant virus reproducibly generated about 10-fold more infectious progeny virus on C6/36 cells than the WT virus on day 1 post-infection (p.i.), whereas the WT virus generated equivalent and slightly higher viral titers than the mutant virus on days 2 and 3 p.i. (Fig. 1I). Overall, the data suggested that depletion of E glycosylation affects the initial cycle of ZIKV replication in mosquito C6/36, but not in mammalian Vero and BHK cells. We currently don’t know how the WT virus caught up with the mutant virus in C6/36 cells on day 2 and 3 p.i., whereas the yield of mutant virus was about 10-fold higher than the WT virus on day 1 post-infection.
Depletion of E glycosylation improves ZIKV attachment, virion assembly, and the infectivity of progeny virus in C6/36 cells
Since the E glycosylation mutant reproducibly generated more infectious virus on C6/36 cells on day 1 p.i. (Fig. 1I), we examined the effect of E glycosylation on the early steps (cell attachment and entry) and late steps (virion assembly and release) of an infection cycle. For analyzing viral attachment and entry, C6/36 cells were incubated with equal amounts of infectious WT or E N154Q virus (MOI of 1) at 4°C for 1 h, allowing the viruses to attach to the cell surface without entering (Fig. 2A). After 1 h incubation at 4°C, the cells from group I (Fig. 2A) were washed with PBS to remove unattached virus, and the amounts of viruses that had attached to the cell surface were measured by quantitative RT-PCR; the mutant attached 1.8-fold more virus to cells than the WT virus did (Fig. 2B). The cells from group II (Fig. 2A) were further incubated at 30°C to initiate viral entry. At different time points, the infected cells were stringently washed with an alkaline high-salt solution to remove free virus as well as cell surface-associated virus, and the intracellular viral RNA was quantified using RT-PCR. In agreement with the attachment result, significantly higher levels of mutant viral RNA were detected than those of the WT viral RNA at 1.5 to 7 h .p.i. (Fig. 2B). The results indicated that the E N154Q mutation enhanced virus attachment and/or entry on C6/36 cells.
Figure 2.
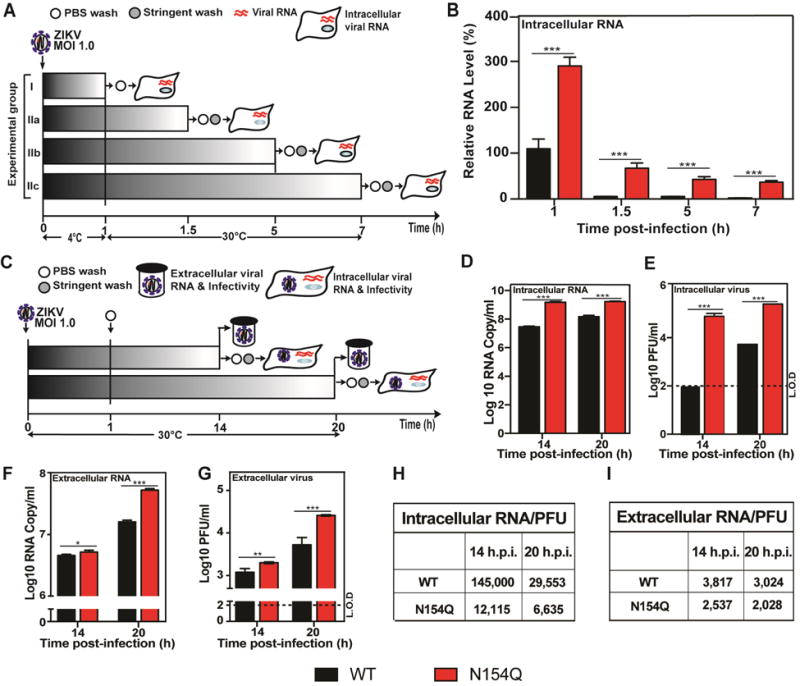
Effects of N154Q mutation on ZIKV life cycle in C6/36 cells. (A) Overview of the experimental design to examine the virus attachment and entry. (B) Intracellular viral RNAs quantified at given time points from intracellular viral RNAs and GAPDH RNAs by qRT-PCR accordingly. The relative viral RNA levels were calculated by normalizing the viral RNAs at each time point to that of 1 h post-infection (set at 100%). Each data point represents the averaged relative RNA of three independent experiments. Statistics were performed using unpaired Student’s t test; *significant (p < 0.05); **very significant (p < 0.01); ***extremely significant (p < 0.001). (C) Overview of the experimental design to monitor a single cycle of ZIKV infection. (D-E) C3/36 cells were infected with WT or N154Q virus at an MOI of 1.0. After infection, virus inoculums were removed and cells were washed three times with PBS. To quantify intracellular viral RNAs at 14 and 20 h p.i., cells were further stringently washed with alkaline high-salt solution at the time the sample was harvested. Intracellular viral RNAs were measured by qRT-PCR and normalized using the cellular GAPDH RNA levels. Extracellular viral RNAs were determined by qRT-PCR, and infectious viruses were quantified by plaque assay. (D) Intracellular viral RNA. (E) Intracellular virus infectivity. (F) Extracellular RNA. (G) Extracellular virus infectivity. Each data point represents the average and standard deviations of three independent experiments. Statistics were performed using unpaired Student’s t test; *significant (p < 0.05); **very significant (p < 0.01); ***extremely significant (p < 0.001). (H-I) Intracellular and extracellular RNA Copy/PFU ratio.
For analyzing the effect of E N154Q mutation on virion assembly and progeny virus infectivity, C6/36 cells were infected with WT or mutant viruses at an MOI of 1 (Fig. 2C). The unattached viruses in inocula were removed at 1 h p.i. by three times washing with PBS. After incubating the infected cells at 30°C for 14 and 20 h, we measured intracellular and extracellular levels of viral RNAs and infectious viruses (Fig. 2D–G). In all cases, the mutant virus generated more viral RNA and infectious virions than the WT virus. To measure the efficiency of intracellular virion assembly, we calculated the intracellular viral RNA/PFU ratios; the results showed that the intracellular RNA/PFU ratios derived from the WT virus were 12- and 4.5-fold higher than those derived from the mutant virus at 14 and 20 h p.i., respectively (Fig. 2H), suggesting that the mutant virus was more efficient in intracellular virion assembly in C6/36 cells.
Next, we calculated the extracellular viral RNA/PFU ratios to estimate the infectivity of progeny viruses derived from the C6/36 cells. As summarized in Fig. 2I, the extracellular RNA/PFU ratios derived from the WT virus were 1.5-fold greater than those derived from the mutant at both 14 and 20 h p.i., suggesting that the infectivity of WT progeny virus was lower. Altogether, the results indicated that glycosylation of E protein decreased viral attachment, virion assembly, and the infectivity of progeny virus in C6/36 cells. In DENV-2, depletion of E glycosylation was also shown to increase virus entry, but reduce virion release (Lee et al., 2010).
E N154Q mutation attenuates ZIKV in A129 mice
We evaluated the virulence and immunogenicity of the E N154Q mutant virus in the A129 mice, an immune compromised ifnar−/− model for ZIKV infection (Rossi et al., 2016). Fig. 3A outlines the experimental scheme. Three-week-old mice were infected with 104 PFU of WT or mutant virus via the subcutaneous route. The infected mice were measured for weight loss (Fig. 3B), viremia (Fig. 3C), and mortality (Fig. 3D). Compared with the PBS-inoculated control animals, no significant weight loss was observed in the mutant virus-infected mice, whereas the WT-infected animals exhibited significant weight loss (Fig. 3B). Mice infected with the mutant generated 3,734- and 4,311-fold lower peak viremia on day 2 and 3 p.i., respectively, than those infected with the WT virus (Fig. 3C). No mortality was observed in the mutant-infected mice, whereas 40% mortality was observed in the WT virus-infected animals (Fig. 3D). Collectively, the results demonstrated that the E glycosylation knockout attenuated ZIKV in the A129 mice. The observed attenuation was not due to the differences in thermostability or temperature sensitivity of the two viruses, as they exhibited similar thermostability and temperature sensitivity on Vero cells (Supplementary Fig. 1).
Figure 3.
Comparison of virulence between WT and N154Q in the A129 mice. Three-week old mice (5 mice per group) were infected with 104 PFU of WT or N154Q virus or PBS via subcutaneous injection. (A) Scheme of vaccination and challenge. (B) Mouse weight loss after infection with WT or N154Q virus. Mock or infected mice (n = 5 per group) were monitored for weight loss over the course of 9 days post-infection. A two-way ANOVA test was performed to evaluate the statistical significance of weight differences among WT and N154Q infected mice with mock group at each point. Error bar represent standard deviation. Symbols *** indicate P values <0.001. (C) Mouse viremia after infection with WT and N154Q viruses. Viremia were quantified using plaque assay. The limit of detection (L.O.D.) for viremia is 100 PFU/ml. (D) Mortality for WT, N154Q and mock group. (E) Pre-challenge neutralization antibody titers. On day 28 p.i., mouse sera were measured for antibody neutralizing titers using a mCherry ZIKV infection assay. The expression of mCherry in infected Vero cells was analyzed by a fluorescent microscopy at 28 days post-infection. (F) Post-challenge viremia. On day 28 post-infection, mice were challenged with 1×105 PFU parental virus (ZIKV strain FSS13025) via the I.P. route. Viremia on day 2 post-challenge was quantified using plaque assay. Error bar represent standard deviation.
To test if immunization with E N154Q virus could protect mice from WT ZIKV infection, we measured the neutralizing antibody titers of the infected mice on day 28 p.i. and then challenged the animals with 105 PFU of ZIKV strain FSS13025 via the subcutaneous route. Comparable high levels of neutralizing titers were detected from the WT and mutant virus-infected animals (Fig. 3E). No infectious virus was detected from either the WT or mutant virus-infected animals post challenge; in contrast, viremia of 106 PFU/ml was detected in the PBS-immunized control mice (Fig. 3F). These results indicated that the E glycosylation knockout virus could elicit robust antibody response and protected mice from ZIKV challenge.
E glycosylation mutant ZIKV induces higher innate cytokine responses in dendritic cells (DCs)
Mammalian DCs are one of the cell types that are first infected after an infectious mosquito bite. We compared the abilities of WT and E N154Q viruses to replicate and induce innate cytokine responses in primary DCs. Bone marrow-derived DCs from A129 mice were infected with WT or mutant viruses at an MOI of 1. Extracellular viruses, intracellular viral RNAs, and intracellular mRNA levels of different cytokines were measured post-infection. The mutant virus produced more infectious virus and intracellular viral RNA than the WT virus on days 3 and 4 p.i. (Fig. 4A&B). The mutant virus also induced significantly higher cytokine expression, including IFN-β, IL-1β, and IL-6 (Fig. 4C). These results suggested that the E N154Q ZIKV induced higher primary cytokine responses, which may in turn suppress viral replication in infected animals.
Figure 4.
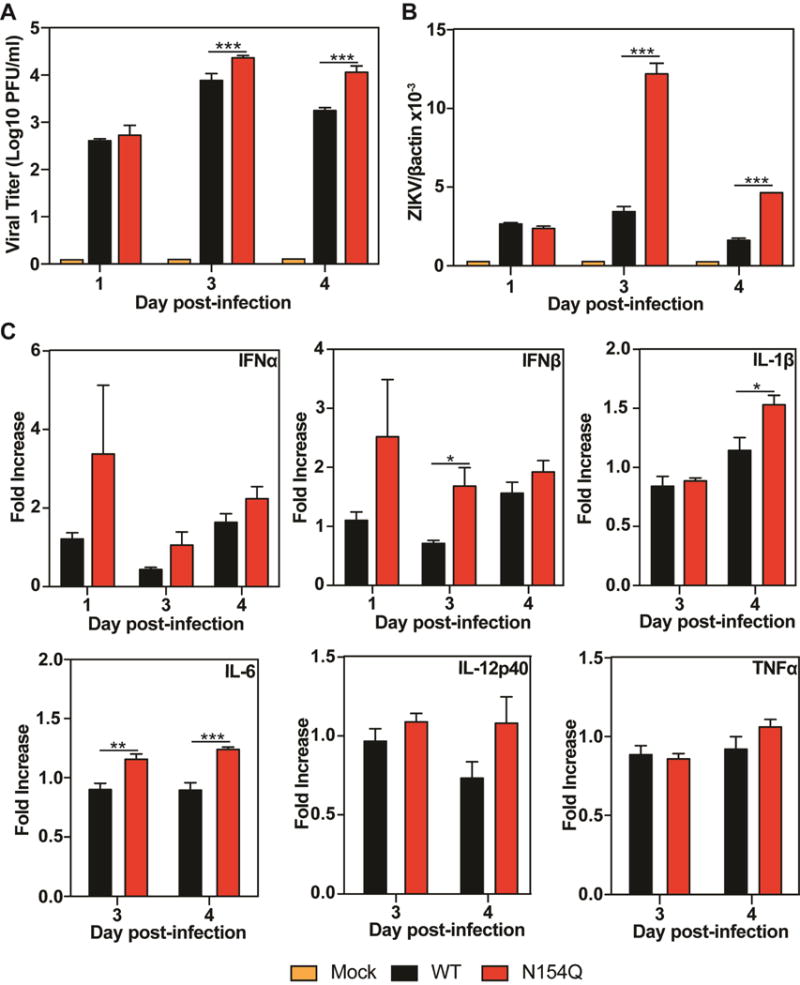
Comparison of viral replication and innate cytokine responses in mouse DCs. Bone marrow-derived DCs from A129 mice were infected with WT or E N154Q mutant virus at an MOI of 1. At days 1, 3, and 4 p.i., extracellular infectious viruses (A) and intracellular viral RNAs (B) were measured by plaque assay and quantitative RT-PCR, respectively. In addition, quantitative RT-PCR was used to quantify intracellular mRNA levels of different cytokines, including IFNα, IFNβ, IL-1β, IL-6, IL-12p40, and TNFα (C). Data are presented as means ± SEM, n = 6. Statistical significance is presented as P < 0.05 (*), P< 0.01 (**), and P < 0.001 (***) when compared to the WT ZIKV-infected group using unpaired t test.
E glycosylation does not significantly affect ZIKV neurovirulence
We compared the neurovirulence of WT and E N154Q viruses through intracranial injection of 1-day-old CD-1 outbred mice. As reported previously (Shan et al., 2017), neonates succumbed to WT ZIKV infection in a dose-responsive manner (Fig. 5). Mice infected with E N154Q virus showed mortality rates similar to those infected with the WT virus. As a control, no death was observed in PBS-inoculated mice (Fig. 5). The result suggested that E glycosylation did not significantly contribute to ZIKV neurovirulence.
Figure 5.
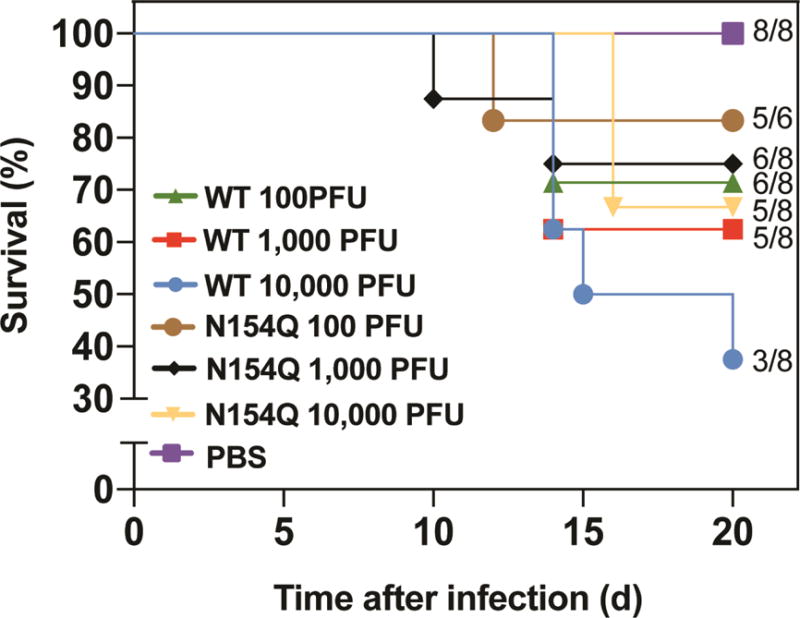
Comparison of neurovirulence between WT and N154Q in CD-1 mice. One-day old mice (6 or 8 mice per group) were infected with 100 to 10,000 PFU of WT or N154Q virus via intra-cranial injection. Survival numbers and total number of infected mice were calculated. Survival percentages of infected mice are presented. No statistical significant difference was observed after performing Log-rank (Mantel-Cox) test to analyze the differences in survival between WT and N154Q.
E N154Q mutation diminishes the ability of ZIKV to infect Aedes aegypti mosquitoes
We examined the effect of E glycosylation on ZIKV infection of Ae. aegypti, the main urban vector. Mosquitoes derived from Galveston, Texas were fed on artificial human blood meals containing 106 PFU/ml of WT or mutant virus. On day 7 post-feeding, engorged mosquitos with similar blood meal sizes were analyzed for the presence of virus in the bodies to estimate viral infection rates. Consistent with previous findings (Roundy et al., 2017), WT virus showed an infection rate of 48% (Fig. 6). In contrast, only 4% of the mosquitoes fed on the mutant virus were infected. The results indicated that glycosylation of E protein was important for ZIKV to infect Ae. aegypti.
Figure 6.
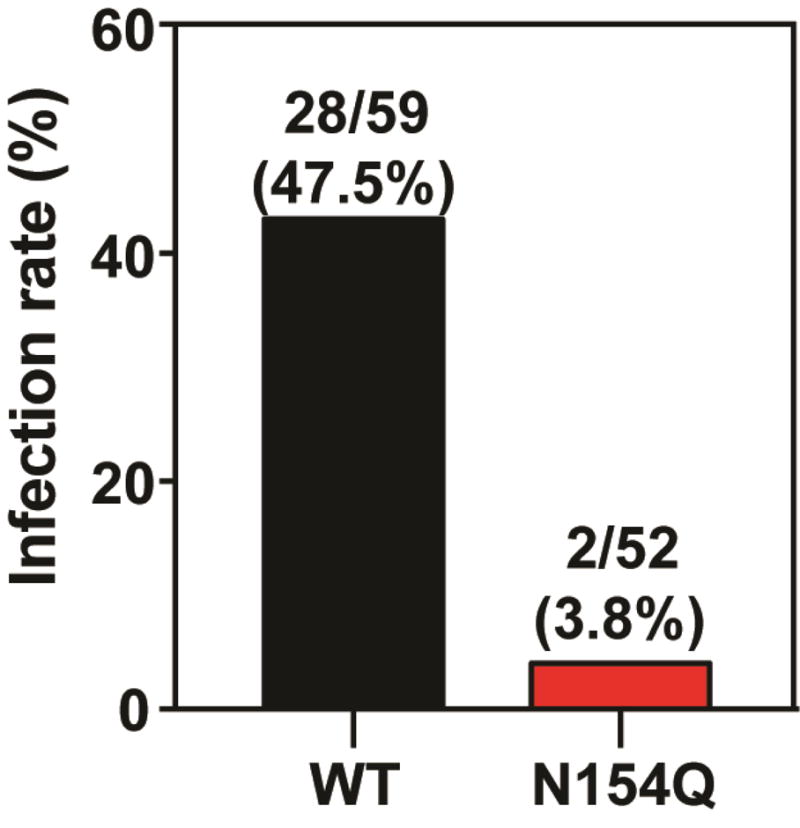
Infection of WT and N154Q in Aedes aegypti. A. aegypti mosquitoes were fed with WT or N154Q virus on artificial blood meals for 30 minutes. On day 7 post-feeding, individual engorged, incubated mosquitoes were homogenized and infection was assayed by immunostaining of viral protein expression on inoculated Vero cells. The number of infected mosquitos and total number of engorged mosquitoes are indicated.
Discussion
Like other flaviviruses, the E protein from almost all ZIKV strains is glycosylated at amino acid N154. Beside the N154 site, the E protein from some flaviviruses, such as DENV, has a second glycosylation site at N67 (Mukhopadhyay et al., 2003). Because the life cycle of ZIKV alternates between mammalian and mosquito hosts, we examined the role of E N154 glycosylation in viral replication in A129 mice and Ae. aegypti mosquitos. We also compared the viral replication of WT and E glycosylation mutant viruses in mammalian and mosquito cell lines. Discrepant results were observed from the in vitro and in vivo experiments in both hosts. In mosquito host, when infecting C6/36 cells, ZIKV E glycosylation negatively regulated viral attachment, virion assembly, and infectivity of progeny virus (Fig. 2); in contrast, when Ae. aegypti imbibed blood meals spiked with a high level of virus, the glycosylation was found to be critical for ZIKV infection (Fig. 6). In the in vitro mammalian models, the E glycosylation did not affect viral replication in BHK and Vero cells; however, knockout of E glycosylation significantly attenuated ZIKV in the A129 mouse model, as evidenced by reduced viremia, less weight loss, and no mortality (Fig. 3B–D). Interestingly, E glycosylation depletion did not significantly affect the neurovirulence when the mutant virus was directly injected into the brains of newborn CD1 mice (Fig. 5). Such in vitro and in vivo discrepancies were also observed previously when analyzing the function of E glycosylation in other flaviviruses. In the case of DENV-2, knockout of N153 glycosylation of E protein reduced viral replication in C6/36 cells (Lee et al., 2010; Mondotte et al., 2007), but did not affect viral replication in intrathoracically inoculated Ae. aegypti mosquitoes (Bryant et al., 2007). In WNV, knockout of E N154 glycosylation did not affect viral replication in C6/36 cells, but significantly reduced viral transmission in Culex mosquitoes (Moudy et al., 2009). These in vitro and in vivo discrepancies are likely due to the lack of cellular factor(s) and complex immune systems in cell lines. Such cellular factor(s) and immune systems could play important roles in interacting with glycosylated E protein on virion surface and in launching robust antiviral activities, respectively. In the case of ZIKV, it remains to be determined the cell types and their contributions to the neurovirulence in the infected CD1 mice.
Flavivirus enters host cells via receptor-mediated endocytosis (Pierson and Diamond). The carbohydrates on viral E protein function as an initial attachment molecule to host cells through C-type lectins, a family of host proteins with carbohydrate-binding activity. Previous studies showed that lectins from mammalian and mosquito hosts engage flavivirus E protein through two distinct mechanisms. In mammalian cells, lectin molecules such as DC-SIGN are associated with cytoplasmic membrane through their transmembrane domains (Bernhard et al., 2004). When DENV infects DCs, it binds to DC-SIGN on cell surface, leading to subsequent interactions with cellular receptor(s) and viral entry (Lozach et al., 2005). Cryo-electron microscope structure show that DC-SIGN binds to two glycosylation sites at N67 of two neighboring E proteins in each icosahedral asymmetric unit (Pokidysheva et al., 2006). It is conceivable that the E glycosylation-mediated flaviviral attachment may be dependent on the expression level of lectins on mammalian cell surface. This might explain the discrepancy of ZIKV replication in mammalian cell lines and in A129 mice. In support this of this hypothesis, transient expression of DC-SIGN in HEK-293 cells enhanced WT ZIKV infection (Hamel et al., 2015). However, our result from DC infection (Fig. 4) argues against the above hypothesis. Compared with the WT virus, the glycosylation mutant replicated more efficiently and induced greater innate cytokine responses in the DCs derived from the A129 mice (Fig. 4). The greater initial cytokine response may in return restrict viral replication, leading to attenuated viremia and no mortality in the mutant virus-infected A129 mice; in addition, the higher innate cytokine response in DCs could boost stronger adaptive immunity in mice, resulting in high levels of neutralizing antibodies and complete protection upon WT ZIKV challenge (Fig. 3). However, it should be noted that our current results do not exclude other possibilities that may also contribute to the in vitro and in vivo discrepancy in mammalian hosts.
In the mosquito host, distinct C-type lectins modulate different flavivirus infections. C-type lectins mosGCTL-1 and mosGCTL-3 are critical for the attachment of WNV and DENV-2, respectively, to cellular surface in mosquitoes (Cheng et al., 2010; Liu et al., 2014). In contrast to mammalian lectin DC-SIGN that are presented on cellular surface through their transmembrane domains, mosquito lectins are not directly associated with the cell surface. These soluble, cell-free lectins could interact with the glycan of flaviviral E proteins to form a lectin-virus complex, which could then bind to specific lectin-binding proteins located on the mosquito cell surface, leading to viral attachment and entry. For example, WNV binds to cell-free lectin mosGCTL-1 which subsequently interacts with cell surface protein mosPTP-1 in mosquito (Cheng et al., 2010). Intriguingly, mosPTP-1 cannot serve as a cellular surface receptor to bind the mosGCTL-3/DENV-2 complex, suggesting that, similar to the specificity of mosGCTLs for different flaviviruses, DENV-2 may employ a mosPTP-1 analog to mediate viral entry into mosquito cells (Liu et al., 2014). The distinct mechanism of mosquito lectin-mediated viral attachment may also explain the discrepancy of our in vitro and in vivo results. The amounts of cell-free lectins and their cell surface receptors may be different between C6/36 cell line and live mosquitoes.
Since the E glycosylation mutation attenuated ZIKV, we explored its potential use for live-attenuated vaccine development. A single-shot immunization of A129 mice with 104 PFU E N154Q virus elicited a robust neutralizing antibody response and fully prevented viremia upon WT ZIKV challenge. Although E N154Q virus was attenuated in A129 mice and in infecting Ae. aegypti mosquitoes, this safety profile is not as good as two recently reported, live-attenuated vaccine candidates: a 3′UTR deletion virus and NS1 glycosylation knockout virus (Richner et al., 2017; Shan et al., 2017). The latter two vaccine candidates did not cause any death after 103 PFU of vaccine viruses were injected intracranially into 1-day-old CD-1 mice, and no mosquitoes were infected after feeding on blood meals containing 106 PFU/ml of the vaccine candidate viruses. In contrast, we observed mortality in CD-1 mice infected with 102 PFU of E N154Q virus (Fig. 5), and 4% of engorged mosquitoes were infected after blood meal feeding (Fig. 6). These results indicated that ZIKV with the E N154Q mutation alone is not a safe vaccine candidate. However, the E glycosylation mutation could be combined with other attenuation approaches for live-attenuated vaccine development.
In summary, our results demonstrate the importance of E glycosylation for ZIKV infection in A129 mice and Ae. aegypti mosquitoes. Together with previous studies, our data support the conclusion that E glycosylation of various flaviviruses may function differently when the viruses alternate their infections between mammalian and mosquito hosts. Future studies are needed to further understand the selective advantages of the E glycosylation, particular in mammalian hosts. Finally, the discrepant results derived from cell culture, mouse model, and mosquito underscore the importance in performing experiments both in vitro and in vivo to confirm the findings of from different experimental flavivirus systems.
Experimental Procedures
Cells and Antibodies
Vero and C6/36 cells were cultured as previously reported (Yang et al., 2017). The following antibodies were used in this study: a mouse monoclonal antibody (mAb) 4G2 cross-reactive with flavivirus E protein (ATCC), Rabbit Anti-Zika (African) Envelop DIII IgG (Alpha Diagnostic International), ZIKV-specific HMAF [hyper-immune ascitic fluid obtained from the World Reference Center of Emerging Viruses and Arboviruses (WRCEVA) at the University of Texas Medical Branch], anti-mouse IgG antibody labeled with horseradish peroxidase (KPL, Gaithersburg, MD),Anti-Rabbit IgG (H+L) Antibody Horseradish Peroxidase-labeled (KPL, Gaithersburg, MD), and goat anti-mouse IgG conjugated with Alexa Fluor®488 (Thermo Fisher Scientific). The ZIKV Cambodian strain FSS13025 (GenBank number KU955593.1) was generated from an infectious cDNA clone pFLZIKV as described previously (Shan et al., 2016b).
Plasmid construction
The E N154Q mutations were introduced to the ZIKV full length cDNA infectious clone pFLZIKV (Shan et al., 2016b). A shuttle vector approach was used to amplify the DNA fragment between unique restriction sites NotI and AvrII using corresponding primers. Mutations were introduced to a fragment using QuickChange II XL Site-Directed Mutagenesis Kit (Agilent Technologies), then the fragment was digested and assembled to pFLZIKV. E. coli strain Top 10 (Invitrogen) was used to propagate the plasmids. The fragment and full length plasmids were validated by Sanger DNA. All restriction enzymes were purchased from New England BioLabs (Ipswitch, MA).
RNA Transcription, transfection, and indirect immunofluorescence assay (IFA)
RNA transcription and transfection were performed as previously described (Shan et al., 2016b). For IFA, Vero cells transfected with viral RNA were seeded in 8-well Lab-Tek II chamber slide (Thermo Fisher Scientific, Waltham, MA). At indicated time points, cells were fixed in 100% methanol at −20°C for 15 min. After 1 h incubation in blocking buffer containing 1% FBS and 0.05% Tween-20 in PBS, the cells were treated with mouse monoclonal antibody (mAb) 4G2 for 1 h and washed three times with PBS (5 min for each wash). The cells were then incubated with Alexa Fluor® 488 goat anti-moue IgG (Thermo Fisher Scientific) for 1 h in blocking buffer, and washed three times with PBS. The cells were mounted in Vectashield® mounting medium with DAPI (4’, 6-diamidino-2-phenylindole; Vector Laboratories, Inc.). Fluorescence images were observed under a fluorescence microscope (Olympus).
Plaque assay
Twenty h prior infection, Vero cells were seeded into a 24 well plate cells (4 × 105 cells/well). Viral samples were 10-fold serially diluted six times in DMEM with 2% FBS and 1% penicillin/streptomycin. For each dilution, 100 l sample was added to the Vero cells seeded in a 24 well plate; infections were executed in triplicates. After 1 hour incubation at 37°C, 0.5 ml methyl cellulose overlay containing 2% FBS and 1% penicillin/streptomycin was added to each well, and the plate was incubated at 37°C for 4 days. Following incubation, the methyl cellulose overlay, and the cells were fixed with 3.7% formaldehyde for 30 min at room temperature. After removing the fixative, the plate was stained with 1% crystal violet for 10 min. Visible plaques were counted, and viral titers (PFU/ml) were calculated.
Virus replication kinetics
Vero (8 × 105 cells/well) and C6/36 cells (1.2 × 106 cells/well) were seeded in 6-well plates 24 h prior infection. Cells were inoculated with WT or N154Q virus at an MOI of 0.01; infections were executed in triplicates. Virus stocks were diluted in DMEM containing 2% FBS and 1% penicillin/streptomycin. One hundred l of virus was added to each well of the 6-well plates. Cells were incubated for 1 h (5% CO2 at 37°C for Vero cells and at 30°C for C6/36 cells), the inocul a were removed, and cells were washed three times with PBS. Subsequently, 3 ml of fresh media containing 2% FBS and 1% penicillin/streptomycin, was added to each well. The plates were incubated for 5 days and supernatants were collected daily. Standard plaque assay on Vero cells was used to determine the replication curves following a protocol mentioned above.
Western blots and glycosidase treatment
Vero cells were seeded in T-175 flask (1.75 × 107 cells/flask), inoculated with virus at an MOI of 0.01, and incubated at 37°C until the cytopathogenic effect was observed. The infected cells were harvested, washed with cold PBS, and lysed with RIPA buffer. The lysed cells were placed on a Fisher Scientific™ Mini-Tube Rotator for a gentle agitation for 1 h at 4°C. The lysates were then centrifuged for 10 min at 15,000 rpm at 4°C, to remove cell debris. Lysate aliquots were treated with Peptide N-Glycosidase F (PGNase F) in accordance with the manufacturer’s instructions (New England BioLabs, Inc.). Proteins we analyzed under denaturing conditions in 12% SDS-polyacrylamide gel electrophoresis (SDS-PAGE), and transferred using a Trans-Blot® Turbo™ Blotting System (Bio-Rad Laboratories) onto a polyvinylidene difluoride (PVDF) membrane. Blots were then blocked in TBST buffer (10 nM Tris-HCl, pH 7.5, 150 nM NaCl, and 0.1% Tween 20) supplemented with 5% skim milk for 1 h, followed by probing with primary antibodies (1:2000 dilution) for 1 h at room temperature. After three washes with TBST buffer, the blots were incubated with goat anti-rabbit conjugated to HRP (1:5000 dilution) in TBST buffer with 5% milk for 1 h, followed by three washed with TBST buffer. Amersham™ ECL™ Prime Western Blotting detection reagent (GE Healthcare, Chicago, IL) was utilized to detect the antibodies.
Mice experiments
All animal work was performed as approved by the UTMB Institutional Animal Care and Use Committee (IACUC). Virulence was determined by using three-week-old A129 mice, a model susceptible to ZIKV infection (Rossi et al., 2016). A129 mice were subcutaneously injected with 104 PFU of WT or E N154Q virus, five mice per group. PBS was used to dilute the virus stocks to the desired concentration. The inoculum was back-titrated to verify the viral dose. Mock-infected mice were given PBS by the same route. Mice were monitored for weight loss and signs of disease daily. Mice were bled via retro-orbital sinus (RO), after being anesthetized every other day, to quantify the viremia using plaque assay on Vero cells. On day 28 post-immunization, mice were anesthetized and bled to measure neutralization antibody titers using a mCherry ZIKV infection assay. Mice were challenged on day 28 post infection with ZIKV WT FSS13025 with 1 × 105 via intraperitoneal (I.P.) injection. On day 2 post-challenge, the mice were bled to measure viremia. Viral titers of sera and inoculum were determined by plaque assay on Vero cells as described above.
Infection and cytokine response in DCs
Bone-marrow derived DCs were generated as described previously (Daffis et al., 2008). Briefly, bone marrow cells from A129 mice were isolated and cultured for 6 days in RPMI-1640 supplemented with granulocyte-macrophage-colony stimulating factor, and interleukin-4 (Peprotech) to generate myeloid DC. Day 6-cultured DCs were infected with ZIKV at an MOI of 1. At day 1, 3, and 4 p.i., culture fluids were measured for infectious virus using plaque assays on Vero cells (Xie et al., 2017). The infected cells were extracted for total intracellular RNA using Trizol (Invitrogen). The amounts of intracellular viral RNA and various cytokine RNAs were quantified using quantitative RT-PCR as reported previously (Wang et al., 2004).
Neurovirulence on newborn CD-1 mice
Groups of 1 day-old outbred CD-1 mice (n=6 or 8) were injected with WT or N154Q viruses with tenfold dilutions from 10,000 PFU to 100 PFU by the intracranial route. Mice were monitored daily for morbidity and mortality.
Antibody neutralization assay
Neutralizing activity of mouse sera was assessed using a mCherry ZIKV infection assay. Sera were 2-fold serially diluted starting at 1:100 in DMEM with 2% FBS and 1% penicillin/streptomycin, then incubated with mCherry ZIKV at 37°C for 2 h. Antibody-virus complexes were added to pre-seeded Vero cells in 96-well plates. After 48 h post-infection, cells were visualized by fluorescence microscopy using Cytation 5 Cell Imaging Multi-Mode Reader (Biotek) to quantify the mCherry fluorescence-positive cells. The percentage of fluorescence-positive cells in the non-treatment controls was set at 100%. The fluorescence-positive cells from serum-treated wells were normalized to those of non-treatment controls. A four-parameter sigmoidal (logistic) model in the software GraphPad Prism 7 was used to calculate the neutralization titers (NT50).
Infection of Mosquitoes with ZIKV
Aedes aegypti colony mosquitoes derived from Galveston, TX, were exposed for 30 min to artificial blood meals consisting of 1% (weight/vol) sucrose, 20% (vol/vol) FBS, 5 mM ATP, 33% (vol/vol) PBS-washed human blood cells (UTMB Blood Bank), and 33% (vol/vol) DMEM medium. The 1 ml-blood meal was combined with 1 ml virus and offered to cartons of Ae. aegypti in Hemotek 2-ml heated reservoirs (Discovery Workshops) covered with a mouse skin. The final viral load in the blood meals was 1×106 PFU/ml. Engorged mosquitoes were incubated at 28°C, 80% relative humidity on a 12:12 h light:dark cycle with ad libitum access to 10% sucrose solution for 7 days and then frozen at −20°C overnight. To assess infection, the whole body of each individual mosquito was homogenized (Retsch MM300 homogenizer, Retsch Inc., Newton, PA) in DMEM with 20% FBS and 250 µg/ml amphotericin B. The samples were then centrifuged for 10 min at 5,000 × rpm. Afterward, 50 µl supernatants were inoculated into 96-well plates containing Vero cells at 37°C and 5% CO2 for 3 days. Cells were fixed with a mixture of ice-cold acetone and methanol (1:1) solution and immunostained as described previously (Shan et al., 2016b). The infection rate was calculated using the number of virus-positive mosquito bodies divided by the total number of engorged mosquitoes.
Quantitative reverse transcription PCR (qRT-PCR)
Viral RNAs were extracted from the supernatant using QIAamp® viral RNA Mini Kit (Qiagen®), and intracellular total RNAs were purified using RNeasy® Mini Kit (Qiagen®). Extracted RNAs were eluted in 50 l RNase-free water. ZIKV RNA copies were determined using a specific probe (5′-FAM/AGCCTACCT/ZEN/TGACAAGCAATCAGACACTCAA/3IABkFQ-3′) and a primer set (ZIKV_1193F: 5′-CCGCTGCCCAACACAAG-3′; ZIKV_1269R: 5′- CCACTAACGTTCTTTTGCAGACAT-3′). The probe contains a 5′-FAM reporter dye, 3′ IBFQ quencher, and internal ZEN quencher. Following manufacturer’s instructions, 15- l reactions of the QuantiTect Probe RT-PCR Kit (QIAGEN) and 1.5 l RNA templates were used to performed qRT-PCR assays using the LightCycler® 480 System (Roche). In vitro transcribed full-length ZIKV RNA were used as RNA standard for RT-PCR quantification. The mRNA level of the housekeeping gene glyceraldehyde-3-phophate dehydrogenase (GAPDH) was measured using an iTaq™ Universal SYBR® Green One-Step Kit (Bio-Rad) and primers: H_GAPDH-F (5′-TGTTGCCATCAATGACCCCTT-3′), H_GAPDH-R (5′-CTCCACGACGTACTCAGCG-3′), AD_GADPH-F (5′-GGTATGGCTTTCCGTGTCCC-3′), and AD_GADPH-R (5′-GCGGCTTCCTTGACCTTCTG-3′).
Quantification of extra- and intracellular infectious virions
At given time points, about 1 ml culture fluids were harvested and centrifuged at 500 × g for 5 min to remove cell debris prior to storage at −80°C. Infected cells w ere washed three times with cold PBS to remove unbound virions. Cell surface-associated viruses were removed by a 3 min wash with cold alkaline-high-salt solution (1 M NaCl and 50 mM sodium bicarbonate, pH 9.5). After twice cold-PBS washes, the cells were detached using 0.25% Trypsin-EDTA (ThermoFisher Scientific), and suspended in 3 ml DMEM medium containing 2% FBS. Total cells were collected by centrifugation at 1,000 × g for 5 min. The cell pellets were resuspended in 250 l DMEM medium with 2% FBS. One hundred microliters of the cell suspensions were centrifuged at 1,000 × g for 5 min to pellet the cells; the pelleted cells were then used for intracellular viral RNA extraction. The remaining 150 l of cell suspensions were lysed using a single freeze-thaw cycle (frozen at −80°C and thawed at 37°C). Afterwa rd, cellular debris was removed by centrifugation at 3,200 × g for 5 min at 4°C, and t he supernatant was harvested for plaque assay to determine the intracellular infectivity.
Data analysis
GraphPad Prism v7.02 software were used to analyze all data. Results were expressed as the mean ± standard deviation (SD). Comparisons of groups were performed using Mann-Whitney test or one-way ANOVA with a multiple comparisons correction. A P value of <0.05 indicates statistically significant.
Supplementary Material
Highlights.
Viral envelope glycosylation is critical for Zika virus virulence in A129 mice
Glycosylation-deficient Zika virus can protect mice from wild-type virus challenge
Envelope glycosylation is required for Zika virus infection of Aedes aegypti vector
Acknowledgments
We thank colleagues at University of Texas Medical Branch for helpful discussions and support during the course of this study. P.-Y.S. lab was supported by University of Texas Medical Branch (UTMB) startup award, University of Texas STARs Award, CDC grant for the Western Gulf Center of Excellence for Vector-Borne Diseases, Pan American Health Organization grant SCON2016-01353, the Kleberg Foundation Award, UTMB CTSA UL1TR-001439, and NIH grant AI127744. This research was also partially supported by NIH grants AI120942 and AI099123 to S.C.W and T.W., respectively.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions
C.R.F., C.S., H.L., A.E.M., D.B.A.M., X.X., J.Z., C.M.R., and M.W. performed experiments and data analysis. C.R.F., C.S., A.E.M., S.L.R., S.C.W., T.W., and P.-Y.S. designed the experiments and interpreted the results. C.R.F., C.S., S.L.R., S.C.W., T.W., and P.-Y.S. wrote the manuscript.
References
- Acosta EG, Kumar A, Bartenschlager R. Revisiting dengue virus-host cell interaction: new insights into molecular and cellular virology. Adv Virus Res. 2014;88:1–109. doi: 10.1016/B978-0-12-800098-4.00001-5. [DOI] [PubMed] [Google Scholar]
- Adams SC, Broom AK, Sammels LM, Hartnett AC, Howard MJ, Coelen RJ, Mackenzie JS, Hall RA. Glycosylation and antigenic variation among Kunjin virus isolates. Virology. 1995;206:49–56. doi: 10.1016/s0042-6822(95)80018-2. [DOI] [PubMed] [Google Scholar]
- Aliota MT, Bassit L, Bradrick SS, Cox B, Garcia-Blanco MA, Gavegnano C, Friedrich TC, Golos TG, Griffin DE, Haddow AD, et al. Zika in the Americas, year 2: What have we learned? What gaps remain? A report from the Global Virus Network. Antiviral Res. 2017;144:223–246. doi: 10.1016/j.antiviral.2017.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beasley DW, Davis CT, Estrada-Franco J, Navarro-Lopez R, Campomanes-Cortes A, Tesh RB, Weaver SC, Barrett AD. Genome sequence and attenuating mutations in West Nile virus isolate from Mexico. Emerg Infect Dis. 2004;10:2221–2224. doi: 10.3201/eid1012.040647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beasley DW, Whiteman MC, Zhang S, Huang CY, Schneider BS, Smith DR, Gromowski GD, Higgs S, Kinney RM, Barrett AD. Envelope protein glycosylation status influences mouse neuroinvasion phenotype of genetic lineage 1 West Nile virus strains. J Virol. 2005;79:8339–8347. doi: 10.1128/JVI.79.13.8339-8347.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernhard OK, Lai J, Wilkinson J, Sheil MM, Cunningham AL. Proteomic analysis of DC-SIGN on dendritic cells detects tetramers required for ligand binding but no association with CD4. J Biol Chem. 2004;279:51828–51835. doi: 10.1074/jbc.M402741200. [DOI] [PubMed] [Google Scholar]
- Berthet FX, Zeller HG, Drouet MT, Rauzier J, Digoutte JP, Deubel V. Extensive nucleotide changes and deletions within the envelope glycoprotein gene of Euro-African West Nile viruses. J Gen Virol. 1997;78(Pt 9):2293–2297. doi: 10.1099/0022-1317-78-9-2293. [DOI] [PubMed] [Google Scholar]
- Bryant JE, Calvert AE, Mesesan K, Crabtree MB, Volpe KE, Silengo S, Kinney RM, Huang CY, Miller BR, Roehrig JT. Glycosylation of the dengue 2 virus E protein at N67 is critical for virus growth in vitro but not for growth in intrathoracically inoculated Aedes aegypti mosquitoes. Virology. 2007;366:415–423. doi: 10.1016/j.virol.2007.05.007. [DOI] [PubMed] [Google Scholar]
- Cheng G, Cox J, Wang P, Krishnan MN, Dai J, Qian F, Anderson JF, Fikrig E. A C-type lectin collaborates with a CD45 phosphatase homolog to facilitate West Nile virus infection of mosquitoes. Cell. 2010;142:714–725. doi: 10.1016/j.cell.2010.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costello A, Dua T, Duran P, Gulmezoglu M, Oladapo OT, Perea W, Pires J, Ramon-Pardo P, Rollins N, Saxena S. Defining the syndrome associated with congenital Zika virus infection. Bull World Health Organ. 2016;94:406–406A. doi: 10.2471/BLT.16.176990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daffis S, Samuel MA, Suthar MS, Keller BC, Gale M, Jr, Diamond MS. Interferon regulatory factor IRF-7 induces the antiviral alpha interferon response and protects against lethal West Nile virus infection. J Virol. 2008;82:8465–8475. doi: 10.1128/JVI.00918-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dick GW, Kitchen SF, Haddow AJ. Zika virus. I. Isolations and serological specificity. Transactions of the Royal Society of Tropical Medicine and Hygiene. 1952;46:509–520. doi: 10.1016/0035-9203(52)90042-4. [DOI] [PubMed] [Google Scholar]
- Fauci AS, Morens DM. Zika Virus in the Americas - Yet Another Arbovirus Threat. N Engl J Med. 2016 doi: 10.1056/NEJMp1600297. [DOI] [PubMed] [Google Scholar]
- Hamel R, Dejarnac O, Wichit S, Ekchariyawat P, Neyret A, Luplertlop N, Perera-Lecoin M, Surasombatpattana P, Talignani L, Thomas F, et al. Biology of Zika Virus Infection in Human Skin Cells. J Virol. 2015;89:8880–8896. doi: 10.1128/JVI.00354-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee E, Leang SK, Davidson A, Lobigs M. Both E protein glycans adversely affect dengue virus infectivity but are beneficial for virion release. J Virol. 2010;84:5171–5180. doi: 10.1128/JVI.01900-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Zhang F, Liu J, Xiao X, Zhang S, Qin C, Xiang Y, Wang P, Cheng G. Transmission-blocking antibodies against mosquito C-type lectins for dengue prevention. PLoS Pathog. 2014;10:e1003931. doi: 10.1371/journal.ppat.1003931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozach PY, Burleigh L, Staropoli I, Navarro-Sanchez E, Harriague J, Virelizier JL, Rey FA, Despres P, Arenzana-Seisdedos F, Amara A. Dendritic cell-specific intercellular adhesion molecule 3-grabbing non-integrin (DC-SIGN)-mediated enhancement of dengue virus infection is independent of DC-SIGN internalization signals. J Biol Chem. 2005;280:23698–23708. doi: 10.1074/jbc.M504337200. [DOI] [PubMed] [Google Scholar]
- Mondotte JA, Lozach PY, Amara A, Gamarnik AV. Essential role of dengue virus envelope protein N glycosylation at asparagine-67 during viral propagation. J Virol. 2007;81:7136–7148. doi: 10.1128/JVI.00116-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moudy RM, Zhang B, Shi PY, Kramer LD. West Nile virus envelope protein glycosylation is required for efficient viral transmission by Culex vectors. Virology. 2009;387:222–228. doi: 10.1016/j.virol.2009.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay S, Kim BS, Chipman PR, Rossmann MG, Kuhn RJ. Structure of West Nile virus. Science. 2003;302:248. doi: 10.1126/science.1089316. [DOI] [PubMed] [Google Scholar]
- Murata R, Eshita Y, Maeda A, Maeda J, Akita S, Tanaka T, Yoshii K, Kariwa H, Umemura T, Takashima I. Glycosylation of the West Nile Virus envelope protein increases in vivo and in vitro viral multiplication in birds. Am J Trop Med Hyg. 2010;82:696–704. doi: 10.4269/ajtmh.2010.09-0262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierson TC, Diamond MS. Flaviviruses p 747–794. In: Knipe DM, Howley PM, editors. Fields virology. 6th. Vol. 1 [Google Scholar]
- Pokidysheva E, Zhang Y, Battisti AJ, Bator-Kelly CM, Chipman PR, Xiao C, Gregorio GG, Hendrickson WA, Kuhn RJ, Rossmann MG. Cryo-EM reconstruction of dengue virus in complex with the carbohydrate recognition domain of DC-SIGN. Cell. 2006;124:485–493. doi: 10.1016/j.cell.2005.11.042. [DOI] [PubMed] [Google Scholar]
- Post PR, Santos CN, Carvalho R, Cruz AC, Rice CM, Galler R. Heterogeneity in envelope protein sequence and N-linked glycosylation among yellow fever virus vaccine strains. Virology. 1992;188:160–167. doi: 10.1016/0042-6822(92)90745-b. [DOI] [PubMed] [Google Scholar]
- Rey FA, Stiasny K, Heinz FX. Flavivirus structural heterogeneity: implications for cell entry. Curr Opin Virol. 2017;24:132–139. doi: 10.1016/j.coviro.2017.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richner J, Jagger B, Shan C, Fontes C, Dowd K, Cao B, Himansu S, Caine E, Nunes B, Medeiros D, et al. Vaccine mediated protection against Zika virus induced congenital disease. Cell. 2017;170:273–283. doi: 10.1016/j.cell.2017.06.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi SL, Tesh RB, Azar SR, Muruato AE, Hanley KA, Auguste AJ, Langsjoen RM, Paessler S, Vasilakis N, Weaver SC. Characterization of a Novel Murine Model to Study Zika Virus. Am J Trop Med Hyg. 2016;94:1362–1369. doi: 10.4269/ajtmh.16-0111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roundy CM, Azar SR, Rossi SL, Huang JH, Leal G, Yun R, Fernandez-Salas I, Vitek CJ, Paploski IA, Kitron U, et al. Variation in Aedes aegypti Mosquito Competence for Zika Virus Transmission. Emerg Infect Dis. 2017;23:625–632. doi: 10.3201/eid2304.161484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan C, Muruato AE, Nunes BT, Luo H, Xie X, Medeiros DB, Wakamiya M, Tesh RB, Barrett AD, Wang T, et al. A live-attenuated Zika virus vaccine candidate induces sterilizing immunity in mouse models. Nat Med. 2017 doi: 10.1038/nm.4322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan C, Xie X, Barrett ADT, Garcia-Blanco MA, Tesh RB, Vasconcelos PFC, Vasilakis N, Weaver SC, Shi PY. Zika Virus: Diagnosis, Therapeutics, and Vaccine ACS Infectious Diseases. 2016a;2:170–172. doi: 10.1021/acsinfecdis.6b00030. [DOI] [PubMed] [Google Scholar]
- Shan C, Xie X, Muruato AE, Rossi SL, Roundy CM, Azar SR, Yang Y, Tesh RB, Bourne N, Barrett AD, et al. An Infectious cDNA Clone of Zika Virus to Study Viral Virulence, Mosquito Transmission, and Antiviral Inhibitors. Cell Host Microbe. 2016b;19:891–900. doi: 10.1016/j.chom.2016.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirato K, Miyoshi H, Goto A, Ako Y, Ueki T, Kariwa H, Takashima I. Viral envelope protein glycosylation is a molecular determinant of the neuroinvasiveness of the New York strain of West Nile virus. Journal of General Virology. 2004;85:3637–3645. doi: 10.1099/vir.0.80247-0. [DOI] [PubMed] [Google Scholar]
- Sirohi D, Chen Z, Sun L, Klose T, Pierson TC, Rossmann MG, Kuhn RJ. The 3.8 A resolution cryo-EM structure of Zika virus. Science. 2016;352:467–470. doi: 10.1126/science.aaf5316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vorndam V, Mathews JH, Barrett AD, Roehrig JT, Trent DW. Molecular and biological characterization of a non-glycosylated isolate of St Louis encephalitis virus. J Gen Virol. 1993;74(Pt 12):2653–2660. doi: 10.1099/0022-1317-74-12-2653. [DOI] [PubMed] [Google Scholar]
- Wang T, Town T, Alexopoulou L, Anderson JF, Fikrig E, Flavell RA. Toll-like receptor 3 mediates West Nile virus entry into the brain causing lethal encephalitis. Nat Med. 2004;10:1366–1373. doi: 10.1038/nm1140. [DOI] [PubMed] [Google Scholar]
- Weaver SC, Costa F, Garcia-Blanco MA, Ko AI, Ribeiro GS, Saade G, Shi PY, Vasilakis N. Zika virus: History, emergence, biology, and prospects for control. Antiviral Res. 2016;130:69–80. doi: 10.1016/j.antiviral.2016.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsch S, Miller S, Romero-Brey I, Merz A, Bleck CK, Walther P, Fuller SD, Antony C, Krijnse-Locker J, Bartenschlager R. Composition and three-dimensional architecture of the dengue virus replication and assembly sites. Cell Host Microbe. 2009;5:365–375. doi: 10.1016/j.chom.2009.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiteman MC, Wicker JA, Kinney RM, Huang CY, Solomon T, Barrett AD. Multiple amino acid changes at the first glycosylation motif in NS1 protein of West Nile virus are necessary for complete attenuation for mouse neuroinvasiveness. Vaccine. 2011;29:9702–9710. doi: 10.1016/j.vaccine.2011.09.036. [DOI] [PubMed] [Google Scholar]
- Xie X, Yang Y, Muruato AE, Zou J, Shan C, Nunes BT, Medeiros DB, Vasconcelos PF, Weaver SC, Rossi SL, et al. Understanding Zika Virus Stability and Developing a Chimeric Vaccine through Functional Analysis. MBio. 2017;8 doi: 10.1128/mBio.02134-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Shan C, Zou J, Muruato AE, Bruno DN, de Almeida Medeiros Daniele B, Vasconcelos PF, Rossi SL, Weaver SC, Xie X, et al. A cDNA Clone-Launched Platform for High-Yield Production of Inactivated Zika Vaccine. EBioMedicine. 2017;17:145–156. doi: 10.1016/j.ebiom.2017.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.