

Abstract
BACKGROUND
In X-linked adrenoleukodystrophy, mutations in ABCD1 lead to loss of function of the ALD protein. Cerebral adrenoleukodystrophy is characterized by demyelination and neurodegeneration. Disease progression, which leads to loss of neurologic function and death, can be halted only with allogeneic hematopoietic stem-cell transplantation.
METHODS
We enrolled boys with cerebral adrenoleukodystrophy in a single-group, open-label, phase 2–3 safety and efficacy study. Patients were required to have early-stage disease and gadolinium enhancement on magnetic resonance imaging (MRI) at screening. The investigational therapy involved infusion of autologous CD34+ cells transduced with the elivaldogene tavalentivec (Lenti-D) lentiviral vector. In this interim analysis, patients were assessed for the occurrence of graft-versus-host disease, death, and major functional disabilities, as well as changes in neurologic function and in the extent of lesions on MRI. The primary end point was being alive and having no major functional disability at 24 months after infusion.
RESULTS
A total of 17 boys received Lenti-D gene therapy. At the time of the interim analysis, the median follow-up was 29.4 months (range, 21.6 to 42.0). All the patients had gene-marked cells after engraftment, with no evidence of preferential integration near known oncogenes or clonal outgrowth. Measurable ALD protein was observed in all the patients. No treatment-related death or graft-versus-host disease had been reported; 15 of the 17 patients (88%) were alive and free of major functional disability, with minimal clinical symptoms. One patient, who had had rapid neurologic deterioration, had died from disease progression. Another patient, who had had evidence of disease progression on MRI, had withdrawn from the study to undergo allogeneic stem-cell transplantation and later died from transplantation-related complications.
CONCLUSIONS
Early results of this study suggest that Lenti-D gene therapy may be a safe and effective alternative to allogeneic stem-cell transplantation in boys with early-stage cerebral adrenoleukodystrophy. Additional follow-up is needed to fully assess the duration of response and long-term safety.
Adrenoleukodystrophy is an X-linked genetic disease that is caused by a defect in the gene ABCD1 (ATP-binding cassette, subfamily D, member 1), which encodes the per-oxisomal ABC half-transporter ALD protein.1 Mutations in ABCD1 result in abnormal breakdown of very-long-chain fatty acids, a process that predominantly affects adrenal and nervous-system tissues. The inflammatory cerebral phenotype known as cerebral adrenoleukodystrophy develops in approximately 35% of affected boys younger than 12 years of age and in a smaller percentage of affected patients 12 years of age or older.2,3 Learning and behavioral manifestations of cerebral adrenoleukodystrophy are often observed when affected patients are between the ages of 3 and 15 years (median age, 7 years) and are often followed by rapidly progressive loss of neurologic function. Most patients with cerebral adrenoleukodystrophy die within a decade after they receive the diagnosis if they are not treated with hematopoietic stem-cell transplantation.4
Allogeneic transplantation is the only effective therapy for cerebral adrenoleukodystrophy that has been identified to date, and it is more likely to be effective if it is performed at an early stage of neurodegeneration.5–8 Among symptomatic boys who have a high burden of cerebral white-matter disease at diagnosis, the outcome of transplantation is often unfavorable. In contrast, among boys who have undergone transplantation for presymptomatic disease that has been detected by means of early findings on imaging studies, good function and survival have been observed. Thus, rapid identification of potential highly matched hematopoietic stem-cell donors is important to ensure timely treatment. The long-term benefits of hematopoietic stem-cell transplantation in cerebral adrenoleukodystrophy are thought to be mediated by donor-derived replacement of myeloid-derived cells, possibly including microglial cells.2,9 There are notable limitations to allogeneic transplantation, including the risks of graft failure and graft-versus-host disease (GVHD). The most successful outcomes to date have been achieved with the use of cells from HLA-identical, unaffected related donors.10 Among patients who undergo transplantation with cells from such donors and do not have clinical evidence of disease, overall survival is good; treatment failure is usually due to transplantation-related complications or rapid disease progression during the engraftment of donor cells. In a small, single-center study, early transplantation with cells from matched related donors, which are available in a minority of cases, resulted in 100% survival.10
Gene therapy with autologous hematopoietic stem cells has been investigated as an alternative to allogeneic hematopoietic stem-cell transplantation.11–13 In the initial proof-of-principle, single-center study, four boys with cerebral adrenoleukodystrophy received autologous CD34+ hematopoietic stem cells transduced ex vivo with a lentiviral vector CG1711hALD that contained ABCD1 complementary DNA (cDNA). Results for two of those patients have been reported; both patients had functional expression of ALD protein and disease stabilization.14
We report the initial results of the STARBEAM study (ALD-102), an ongoing multicenter, single-group, open-label, phase 2–3 study of gene therapy with the Lenti-D drug product, which involves infusion of autologous CD34+ hematopoietic stem cells transduced ex vivo with the elivaldogene tavalentivec (Lenti-D) lentiviral vector that contains ABCD1 cDNA.
METHODS
STUDY DESIGN AND INVESTIGATIONAL THERAPY
The primary aim of the STARBEAM study is to assess the safety and efficacy of gene therapy with the Lenti-D drug product in male patients 17 years of age or younger who have cerebral adrenoleukodystrophy. The study protocols were approved by the requisite regulatory and human-protection committees. Patients were enrolled after a guardian provided consent and, when appropriate, the patient provided assent. Eligibility was restricted to patients who had gadolinium enhancement on magnetic resonance imaging (MRI) due to cerebral adrenoleukodystrophy and had the following signs of early-stage disease: a score on the cerebral adrenoleukodystrophy–specific neurologic function scale15 (which ranges from 0 to 25, with higher scores indicating more severe deficits) of 0 or 1, and a Loes score16 (which ranges from 0 to 34, with higher scores indicating an increased extent of lesions on MRI) of 0.5 to 9.0 (for further details, see the Supplementary Appendix, available with the full text of this article at NEJM.org). Patients who had an HLA-matched sibling who could donate cells for transplantation were excluded from the study.
A total of 18 patients were enrolled in the study; 1 of these patients did not meet the eligibility criteria. In 2016, after consultation with regulatory authorities, the target for enrollment was increased to 25 patients (but this target did not apply to the interim analysis).
CD34+ cells that were obtained from the enrolled patients by means of apheresis were transduced with the Lenti-D lentiviral vector. Cell trans-duction was performed in accordance with Good Manufacturing Practice conditions with the use of uniform and validated standard operating procedures. Patients received conditioning with busulfan and cyclophosphamide, after which the investigational drug product, which was made up of the transduced CD34+ cells, was infused. In the primary clinical study, the patients were followed for 2 years and then were offered enrollment in a 13-year long-term follow-up study. (For further details, see Fig. S1 and the Methods Section in the Supplementary Appendix.)
CLINICAL, IMAGING, AND LABORATORY ASSESSMENTS
Patients were assessed regularly for adverse events and disease progression (see the study protocol, available at NEJM.org). Gadolinium enhancement and lesion progression were evaluated by an experienced neuroradiologist who was unaware of patient identity, time since the infusion of the investigational drug product, and clinical status. The Loes score was used to assess the extent of lesions on MRI of the head; it is a commonly used assessment in patients with cerebral adrenoleukodystrophy.16 A score of less than 0.5 is considered to be normal. A stable Loes score was defined as an increase from baseline of less than 6 points or a score of 9 or less.
The cerebral adrenoleukodystrophy–specific neurologic function scale was used to evaluate the severity of gross neurologic dysfunction through an assessment for 15 disabilities across multiple domains.15 A score of 0 indicates the absence of clinical signs of cerebral disease. The 6 most severe disabilities included in the assessment are loss of communication, cortical blindness, tube feeding, total incontinence, wheelchair dependence, and complete loss of voluntary movement. These disabilities were judged to be of particular importance because they severely compromise a patient’s ability to function independently. Each of the 6 disabilities is classified in this study as a major functional disability (Table S1 in the Supplementary Appendix). The primary efficacy end point was being alive and having no major functional disabilities at 24 months.
Peripheral-blood mononuclear cells and the CD14+ cell fraction were assessed for expression of ALD protein with the use of flow cytometry. The vector copy number was determined with the use of a quantitative polymerasechain-reaction (PCR) assay. For analysis of vector integration sites, DNA was purified from peripheral-blood samples, and the total number of unique integration sites and the relative clonal contribution of the 10 most common integration sites were determined by means of linear amplification–mediated PCR, as described previously.17 All detectable integration sites were aggregated and analyzed. (For further details, see the Methods Section and Fig. S2 in the Supplementary Appendix.)
STUDY OVERSIGHT
The study was performed with approval from the Food and Drug Administration for an investigational new drug and with approval from Europe-an national authorities. The study was designed by six of the authors and the study sponsor, Bluebird Bio. The first two authors wrote the first draft of the manuscript, which was revised by the last author and substantively edited and approved by all the authors. All the authors made the decision to submit the manuscript for publication. Editorial assistance for an earlier version of the article was provided by employees of Bluebird Bio. The authors had access to the full data from the study, which were provided by Bluebird Bio. Laboratory data were generated by the authors and Bluebird Bio. Clinical data were collected by the authors, who performed the treatment and were responsible for all follow-up and clinical decisions. An independent data and safety monitoring board reviewed the safety data. The authors vouch for the completeness and accuracy of the data and of interpretation of the data and for adherence of the study to the protocol. Additional details regarding the study design are provided in the protocol.
RESULTS
PATIENTS AND TREATMENT
Between October 2013 and July 2015, a total of 17 patients, who were between 4 and 13 years of age at the time of enrollment, received the investigational gene therapy (Table 1). At baseline, the median Loes score was 2.0 (range, 1.0 to 7.5), and all the patients had a score of 0 on the neurologic function scale. Apheresis for manufacture of the Lenti-D drug product was accomplished with a single mobilization cycle and took one day for 7 of the patients and two days for 10 of the patients.
Table 1.
Baseline Characteristics of the Patients and the Drug Product.
| Characteristic | Value |
|---|---|
| Patients | |
| No. enrolled* | 17 |
| Age at enrollment (yr) | |
| Median | 6 |
| Range | 4–13 |
| Loes score† | |
| Median | 2.0 |
| Range | 1.0–7.5 |
| Score on neurologic function scale‡ | |
| Median | 0 |
| Range | 0 |
| Time from consent to infusion of drug product (days) | |
| Median | 67.0 |
| Range | 58.0–89.0 |
| Drug product | |
| Vector copy number (vector copies/diploid genome) | |
| Median | 1.0 |
| Range | 0.5–2.5 |
| Dose (CD34+ cells/kilogram of body weight) | |
| Median | 10,500,000 |
| Range | 6,000,000–19,400,000 |
All the patients were male.
Loes scores range from 0 to 34, with higher scores indicating an increased extent of lesions on magnetic resonance imaging.
Scores on the cerebral adrenoleukodystrophy–specific neurologic function scale range from 0 to 25, with higher scores indicating more severe deficits. The scale is used to evaluate the severity of gross neurologic dysfunction through an assessment for 15 disabilities across multiple domains; a score of 0 indicates the absence of clinical signs of cerebral disease.
Patients received the Lenti-D drug product, which contained 6.0 million to 19.4 million CD34+ cells per kilogram of body weight, with a vector copy number of 0.5 to 2.5 (Table 1). After the initial 4 patients were treated, the remaining 13 patients received granulocyte colony-stimulating factor (G-CSF) after the infusion to expedite engraftment. After the infusion, all the patients had neutrophil engraftment (median days to neutrophil engraftment, 31 [range, 20 to 39] without G-CSF and 12 [range, 11 to 20] with G-CSF), and all the patients had platelet engraftment (median days to platelet engraftment, 32 [range, 28 to 37] without G-CSF and 27 [range, 16 to 55] with G-CSF).
Data on the interim safety and efficacy assessments were available as of April 2017. At the time of data cutoff, the median follow-up after infusion was 29.4 months (range, 21.6 to 42.0), 16 of the 17 patients could be evaluated for the primary end point (i.e., had reached 24 months of follow-up), and 14 were enrolled in the long-term follow-up study.
SAFETY OUTCOMES
No toxic effects related to the infusion of the Lenti-D drug product were reported. Most adverse events associated with the treatment occurred during the conditioning phase or the first 2 weeks after the infusion, and they were generally consistent with the adverse events associated with myeloablative chemotherapy. One patient had hemorrhagic cystitis associated with BK virus on day 42. BK virus is a human polyomavirus that has been implicated as a common cause of late-onset hemorrhagic cystitis in patients who have undergone hematopoietic stem-cell transplantation. In this case, the cystitis resolved with conservative measures and was deemed to be possibly related to the drug product. No episodes of graft failure or GVHD were reported. There were no transplantation-related deaths. Serious adverse events and adverse events related to the drug product are listed in Table S2 in the Supplementary Appendix.
BIOMARKERS
Integration-site analysis revealed polyclonal reconstitution of peripheral blood. The total number of unique vector integration sites was between 251 and 5428. No integration site accounted for more than 30% of the total integration events on two consecutive tests. The vector copy number in peripheral blood ranged from 0.10 to 1.55 at month 24 (assessed in 14 patients) and from 0.36 to 1.83 at month 36 (assessed in 3 patients) (Fig. 1A); the number had generally stabilized by 2 months after infusion. No preferential integration was detected in or near genes that have previously been involved in serious adverse events associated with gene therapy (e.g., MDS1, EVI1, and LMO2). The 10 most common integration sites for each patient at the most recent follow-up are shown in Figure S2 in the Supplementary Appendix. Expression of ALD protein in peripheral-blood leukocytes was observed in all the patients at the most recent follow-up (Fig. 1B). The percentage of CD14+ cells that expressed ALD protein ranged from 5.4 to 71.4% at 6 months after infusion (median, 14.3%; assessed in 13 patients), from 6.9 to 54.5% at 12 months (median, 16.1%; assessed in 17 patients), from 4.9 to 54.8% at 18 months (median, 17.0%; assessed in 14 patients), and from 6.4 to 44.6% at 24 months (median, 19.0%; assessed in 13 patients) (Fig. 1B). Plasma levels of very-long-chain fatty acids were unaffected by treatment with Lenti-D. (For further details, see Figs. S2, S3, and S4 in the Supplementary Appendix.)
Figure 1. Vector Copy Number and Expression of ALD Protein.

Panel A shows the vector copy number in the Lenti-D drug product at infusion and in the peripheral blood for each of the 17 patients at 6 months after infusion. Panel B shows the vector copy number in the peripheral blood for each of the 17 patients at various time points after infusion. Panel C shows the expression of ALD protein in CD14+ cells in the peripheral blood at various time points after infusion; the horizontal lines in the boxes are median percentages, the top and bottom of the boxes are interquartile ranges, and the I bars are minimum and maximum percentages.
CLINICAL OUTCOMES
At the time of the interim analysis, 15 of the 17 patients (88%) were alive and free of major functional disabilities; these 15 patients maintained a score on the neurologic function scale of 0 or 1 (Fig. 2A). Two patients had neurologic disease progression. One of these patients (Patient 2016) withdrew from the study and later died from complications of allogeneic transplantation. In the other patient (Patient 2018), neurologic function deteriorated rapidly after treatment; a major functional disability (total incontinence) developed by month 9, and additional major functional disabilities continued to develop, including cortical blindness, loss of communication, and wheelchair dependence. Approximately 22 months after the infusion, the patient died from a viral infection complicated by rhabdomyolysis and acute kidney and liver failure; these complications and the immediate cause of death were judged to be not directly related to the investigational therapy. Shortly after data cutoff (at month 25), a patient (Patient 2013) had a score on the neurologic function scale of 2, as the result of a seizure, but had no other evidence of disease progression.
Figure 2. Neurologic Function and Clinical Outcomes Relative to Vector Copy Number in the Drug Product.

Panel A shows the scores on the cerebral adrenoleukodystrophy–specific neurologic function scale (which ranges from 0 to 25, with higher scores indicating more severe deficits) for each of the 17 patients at various time points after the infusion of the Lenti-D drug product; 12 patients had scores of 0 at all time points, with the last measurement obtained at month 36 (in 3 patients), month 30 (2), month 24 (5), or month 18 (2). The scale is used to evaluate the severity of gross neurologic dysfunction through an assessment for 15 disabilities across multiple domains; a score of 0 indicates the absence of clinical signs of cerebral disease. Panel B shows the clinical outcomes in the patients relative to the vector copy number in the drug product. Green circles represent patients with stable disease, and red triangles represent patients with a change in status. A change in neurologic function was defined as any change from baseline in the score on the neurologic function scale.
An exploratory analysis was designed to evaluate possible relationships between drug-product characteristics and clinical outcomes, including the occurrence of major functional disability or death and the occurrence of any change in neurologic function during the study. In the analysis, outcomes were plotted as a function of the vector copy number in the drug product. Worse clinical outcomes appeared to occur in patients who were treated with a drug product that had a lower vector copy number (Fig. 2B).
NEUROIMAGING
At the time of the interim analysis, lesion progression, as measured with the Loes score, had stabilized in 12 of the 17 patients (71%) (Fig. 3). Patient 2018, who had a relatively high Loes score of 7 at baseline, had rapid disease progression on MRI after treatment, during the early engraftment phase; the Loes score was 23 by month 12 and 25 by month 18. Patient 2016, who had new white-matter lesions by month 12, was withdrawn from the study by the treating physicians to undergo allogeneic stem-cell transplantation and later died from transplantation-related causes.
Figure 3. Extent of Lesions on MRI.
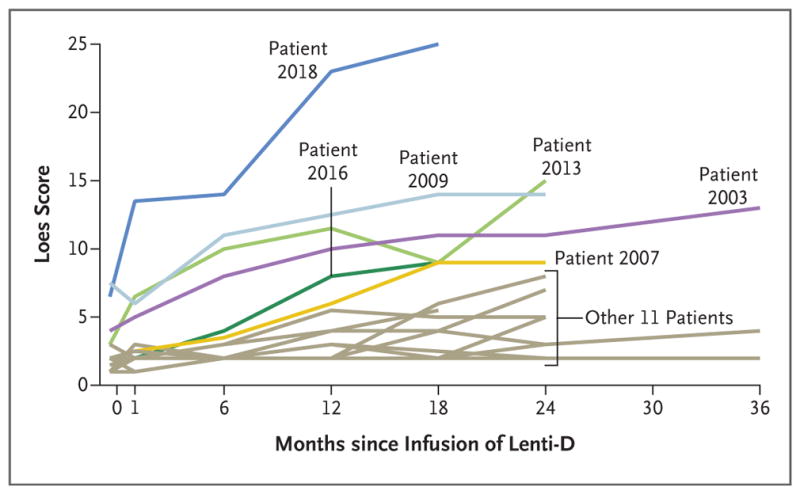
Shown are the Loes scores for each of the 17 patients at various time points after the infusion of the Lenti-D drug product. The Loes scores range from 0 to 34, with higher scores indicating an increased extent of lesions on magnetic resonance imaging (MRI). A score of 0.5 or less is considered to be normal.
Gadolinium enhancement, which was present at baseline in all the patients, had resolved by month 6 in the 16 patients who could be evaluated (Fig. 4). In 6 patients, diffuse gadolinium enhancement reemerged at month 12; the enhancement resolved again in 5 of these patients for whom results of later MRIs were available. Diffuse gadolinium enhancement reemerged in 2 of these patients at month 24; in addition, 1 patient had reemergence of enhancement at months 18 and 24, and Patient 2010 had reemergence of enhancement at month 24. In all the patients who had reemergence of gadolinium enhancement, the enhancement was less extensive than the enhancement seen at baseline. Representative MRIs are shown in Figure S5 in the Supplementary Appendix.
Figure 4. Gadolinium Enhancement on MRI.
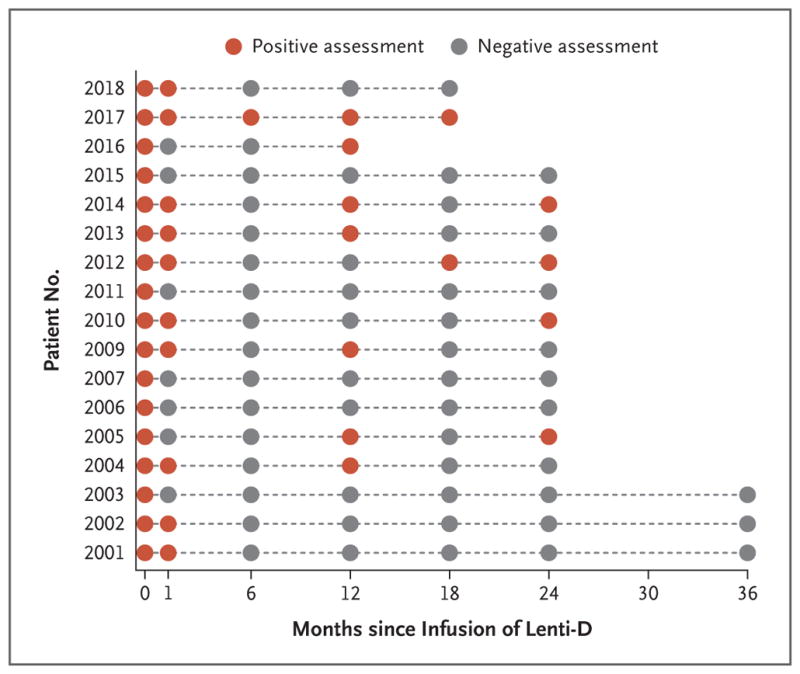
Shown are the results of assessments for gadolinium enhancement on MRI for each of the 17 patients at various time points after the infusion of the Lenti-D drug product. Gadolinium enhancement on reemergence after initial resolution was uniformly more diffuse than the enhancement seen at baseline but was still assessed as positive. Patient 2016 withdrew from the study at month 13. To review the MRIs, see Figure S5 in the Supplementary Appendix.
DISCUSSION
The goal of treatment of cerebral adrenoleuko-dystrophy is to halt progression of the disease as quickly and as safely as possible, thereby preventing the development of irreversible impairments, which compromise the ability to function independently.8 Hematopoietic stem-cell transplantation performed in the early stages of progressive cerebral adrenoleukodystrophy is known to be beneficial.4 Allogeneic transplantation has been shown to eventually halt disease progression in many patients, although some white-matter disease progression on MRI is commonly observed during the first 12 to 18 months after transplantation.6,7,18
Early results suggest that Lenti-D treatment is safe and has led to clinical stabilization in a large proportion of patients in this study. At the time of data cutoff, gene therapy was considered to be effective in 15 of the 17 patients who received treatment; 1 patient had multiple major functional disabilities, and 1 patient withdrew from the study. Most patients had stable neurologic function after Lenti-D treatment; 14 of the 17 patients remained free of major functional disabilities and had a score on the neurologic function scale of 0 or 1, indicating no or minimal clinical symptoms. Aside from the patient who withdrew from the study, 1 patient had a score on the neurologic function scale of 2 at 25 months after infusion (after a reported seizure), and 1 patient had multiple major functional disabilities, the earliest of which was evident at month 9. This patient had a rapid increase in both the Loes score and the score on the neurologic function scale as early as 2 weeks after treatment, findings that suggested that he may have had marked disease progression before treatment. The patient died from a viral infection complicated by rhabdomyolysis and acute kidney and liver failure. Similar to allogeneic transplantation,19 hematopoietic stem-cell gene therapy is not expected to have an effect on the phenotypes of adrenomyeloneuropathy or adrenal insufficiency.
At the time of the interim analysis, boys with cerebral adrenoleukodystrophy who had received Lenti-D therapy had generally stable disease or limited progression of disease on MRI of the head, as compared with the known rates of lesion progression among untreated boys (mean [±SD] increase in Loes score, 2.2±0.55 and 2.3±0.75 points per year for the posterior and anterior patterns, respectively).20 Neither hematopoietic stem-cell transplantation nor gene therapy appears to prevent white-matter lesion progression during the initial 12 to 18 months after treatment.6,7 Microglial cell death appears to play an important role in the pathophysiology of cerebral adrenoleukodystrophy,2 and therefore, early disease progression may occur during the time before replacement of microglial cells. Our data and the results of studies of allogeneic transplantation show that some disease progression on MRI during the first year after transplantation is common and therefore reinforce the urgency in identifying cerebral disease early and treating it swiftly. Infusion of autologous stem cells may have an advantage over allogeneic transplantation in this sense, because of the time saved by not needing to find a donor.
Although the presence of gadolinium enhancement before treatment has been clearly correlated with rapid disease progression, the kinetics of gadolinium enhancement after clinically successful allogeneic transplantation are not well understood. More data are needed to determine the importance of an evaluation of gadolinium enhancement after treatment in the assessment of the treatment response. Early results suggest that gadolinium enhancement does not appear to correlate with clinical outcomes after treatment.
Patients who have very rapidly progressive disease at the time of infusion may go on to have severe clinical symptoms. The quality of the drug product for autologous gene therapy, including the number of engraftable transduced stem cells and the level of expression of the transgene, may be critical to the prevention of neurologic disease progression. Although the sample size in this study is too small to allow statistical inferences, data from patients who had major functional disabilities or deterioration of neurologic function appeared to cluster at the lower end of the range for vector copy number.
We found no correlation between outcome and plasma level of very-long-chain fatty acids. It has been established that the plasma level of very-long-chain fatty acids does not correlate with the clinical severity or phenotype of ALD.21 Thus, since expression of ALD protein is probably related in part to vector copy number, a remaining issue that is not addressed by the current study is the effect of higher expression of the trans-gene in the progeny of transduced cells.
During the peritransplantation period, allogeneic transplantation requires immunosuppression, which has the potential to increase the risk of life-threatening infections. Among patients with cerebral adrenoleukodystrophy and other inherited metabolic disorders, allogeneic transplantation has been associated with a 100-day mortality rate of 8%, a 1-year mortality rate of 18%, a rate of graft failure of 6 to 14%, a rate of serious infection of 29%, a rate of acute grade 2, 3, or 4 GVHD of 8 to 45%, and a rate of chronic GVHD of up to 21%.5,10,22–24 Among patients in our study, who underwent gene therapy with an autologous approach, no GVHD or life-threatening infections were noted.
There have been concerns about mutagenesis related to viral integration after the addition of a gene to hematopoietic stem cells ever since leukemia and the myelodysplastic syndrome developed in patients who were treated with gamma-retro-viral vector gene therapy for several immunodeficiency diseases.25–28 Unlike the retroviral vectors used in those studies, the Lenti-D lentiviral vector (as well as other lentiviral and gamma-retro-viral vectors that are enhancer-deleted with the use of a self-inactivating vector design) so far appears to reduce the risk of mutagenesis.29–32 A longer follow-up and a larger sample size are required to confirm the low potential for genotoxic effects and the clinical efficacy and safety of gene therapy with the Lenti-D lentiviral vector.
Supplementary Material
Acknowledgments
Supported by Bluebird Bio; a grant awarded to the Harvard Clinical and Translational Science Center from the National Center for Research Resources and the National Center for Advancing Translational Sciences, National Institutes of Health (UL1 TR001102); a Patient-Powered Research Network Award from the Patient-Centered Outcomes Research Institute; and the Great Ormond Street Hospital and University College London Great Ormond Street Institute of Child Health Biomedical Research Centre.
We thank the patients and families who are participating in this study; ALD Connect for communication to families about the trial; the clinical and laboratory staff at our institutions for patient care and data collection (Catherine Becker, N.P., Razina Aziz-Bose, Brenda MacKinnon, R.N., Matthew Cavanaugh, and Stephanie Patriarca at Massachusetts General Hospital and Boston Children’s Hospital; André Baruchel, M.D., Jean-Hugues Dalle, M.D., and Jerome Larghero, M.D., at Hôpital Bicetre; Dayna Terrazas, R.N., Michael Linetsky, M.D., and Donald B. Kohn, M.D., at the University of California, Los Angeles; and Katie Snell at the Great Ormond Street Institute of Child Health); Dr. Daniel Loes for serving as the central MRI reader for the study and providing important guidance on interpretation of MRI data; John Baalser, Ph.D., and Susan Paadre, Ph.D., at Veristat for statistical support; Katherine Lewis, M.A., and Joseph Biedenkapp, Ph.D., for providing editorial assistance for an earlier version of the manuscript; and the following employees of Bluebird Bio for contributions to the design and execution of the study: Mohammed Asmal, M.D., Ph.D., for medical monitoring and data analysis; Lilian Yengi, Ph.D., for assay development and interpretation; Gabor Veres, Ph.D., for data analysis and critical review; Robert Ross, M.D., for medical advice; Christopher Horvath, M.D., for study design and data analysis; Philip Gregory, Ph.D., for data analysis and critical review; Kendrick Goss, Ph.D., for assay development; and Elizabeth Graf for data management.
Funded by Bluebird Bio and others; STARBEAM ClinicalTrials.gov number, NCT01896102; ClinicalTrialsRegister.eu number, 2011-001953-10.
Footnotes
Disclosure forms provided by the authors are available with the full text of this article at NEJM.org.
References
- 1.Moser HW. Adrenoleukodystrophy: phenotype, genetics, pathogenesis and therapy. Brain. 1997;120:1485–508. doi: 10.1093/brain/120.8.1485. [DOI] [PubMed] [Google Scholar]
- 2.Eichler FS, Ren JQ, Cossoy M, et al. Is microglial apoptosis an early pathogenic change in cerebral X-linked adrenoleukodystrophy? Ann Neurol. 2008;63:729–42. doi: 10.1002/ana.21391. [DOI] [PubMed] [Google Scholar]
- 3.Powers JM, Liu Y, Moser AB, Moser HW. The inflammatory myelinopathy of adrenoleukodystrophy: cells, effector molecules, and pathogenetic implications. J Neuropathol Exp Neurol. 1992;51:630–43. doi: 10.1097/00005072-199211000-00007. [DOI] [PubMed] [Google Scholar]
- 4.Mahmood A, Raymond GV, Dubey P, Peters C, Moser HW. Survival analysis of haematopoietic cell transplantation for childhood cerebral X-linked adrenoleukodystrophy: a comparison study. Lancet Neurol. 2007;6:687–92. doi: 10.1016/S1474-4422(07)70177-1. [DOI] [PubMed] [Google Scholar]
- 5.Peters C, Charnas LR, Tan Y, et al. Cerebral X-linked adrenoleukodystrophy: the international hematopoietic cell transplantation experience from 1982 to 1999. Blood. 2004;104:881–8. doi: 10.1182/blood-2003-10-3402. [DOI] [PubMed] [Google Scholar]
- 6.Polgreen LE, Chahla S, Miller W, et al. Early diagnosis of cerebral X-linked adrenoleukodystrophy in boys with Addison’s disease improves survival and neurological outcomes. Eur J Pediatr. 2011;170:1049–54. doi: 10.1007/s00431-011-1401-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baumann M, Korenke GC, Weddige-Diedrichs A, et al. Haematopoietic stem cell transplantation in 12 patients with cerebral X-linked adrenoleukodystrophy. Eur J Pediatr. 2003;162:6–14. doi: 10.1007/s00431-002-1097-3. [DOI] [PubMed] [Google Scholar]
- 8.Engelen M, Kemp S, de Visser M, et al. X-linked adrenoleukodystrophy (X-ALD): clinical presentation and guidelines for diagnosis, follow-up and management. Orphanet J Rare Dis. 2012;7:51. doi: 10.1186/1750-1172-7-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shapiro E, Krivit W, Lockman L, et al. Long-term effect of bone-marrow transplantation for childhood-onset cerebral X-linked adrenoleukodystrophy. Lancet. 2000;356:713–8. doi: 10.1016/S0140-6736(00)02629-5. [DOI] [PubMed] [Google Scholar]
- 10.Miller WP, Rothman SM, Nascene D, et al. Outcomes after allogeneic hematopoietic cell transplantation for childhood cerebral adrenoleukodystrophy: the largest single-institution cohort report. Blood. 2011;118:1971–8. doi: 10.1182/blood-2011-01-329235. [DOI] [PubMed] [Google Scholar]
- 11.Naldini L. Ex vivo gene transfer and correction for cell-based therapies. Nat Rev Genet. 2011;12:301–15. doi: 10.1038/nrg2985. [DOI] [PubMed] [Google Scholar]
- 12.Naldini L. Gene therapy returns to centre stage. Nature. 2015;526:351–60. doi: 10.1038/nature15818. [DOI] [PubMed] [Google Scholar]
- 13.Williams DA, Thrasher AJ. Concise review: lessons learned from clinical trials of gene therapy in monogenic immunodeficiency diseases. Stem Cells Transl Med. 2014;3:636–42. doi: 10.5966/sctm.2013-0206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cartier N, Hacein-Bey-Abina S, Bartholomae CC, et al. Hematopoietic stem cell gene therapy with a lentiviral vector in X-linked adrenoleukodystrophy. Science. 2009;326:818–23. doi: 10.1126/science.1171242. [DOI] [PubMed] [Google Scholar]
- 15.Moser HW, Loes DJ, Melhem ER, et al. X-linked adrenoleukodystrophy: overview and prognosis as a function of age and brain magnetic resonance imaging abnormality: a study involving 372 patients. Neuropediatrics. 2000;31:227–39. doi: 10.1055/s-2000-9236. [DOI] [PubMed] [Google Scholar]
- 16.Loes DJ, Hite S, Moser H, et al. Adrenoleukodystrophy: a scoring method for brain MR observations. AJNR Am J Neuroradiol. 1994;15:1761–6. [PMC free article] [PubMed] [Google Scholar]
- 17.Schmidt M, Schwarzwaelder K, Bartholomae C, et al. High-resolution insertion-site analysis by linear amplification-mediated PCR (LAM-PCR) Nat Methods. 2007;4:1051–7. doi: 10.1038/nmeth1103. [DOI] [PubMed] [Google Scholar]
- 18.Saute JA, Souza CF, Poswar FO, et al. Neurological outcomes after hematopoietic stem cell transplantation for cerebral X-linked adrenoleukodystrophy, late onset metachromatic leukodystrophy and Hurler syndrome. Arq Neuropsiquiatr. 2016;74:953–66. doi: 10.1590/0004-282X20160155. [DOI] [PubMed] [Google Scholar]
- 19.van Geel BM, Poll-The BT, Verrips A, Boelens JJ, Kemp S, Engelen M. Hematopoietic cell transplantation does not prevent myelopathy in X-linked adrenoleukodystrophy: a retrospective study. J Inherit Metab Dis. 2015;38:359–61. doi: 10.1007/s10545-014-9797-1. [DOI] [PubMed] [Google Scholar]
- 20.Loes DJ, Fatemi A, Melhem ER, et al. Analysis of MRI patterns aids prediction of progression in X-linked adrenoleuko-dystrophy. Neurology. 2003;61:369–74. doi: 10.1212/01.wnl.0000079050.91337.83. [DOI] [PubMed] [Google Scholar]
- 21.Moser AB, Kreiter N, Bezman L, et al. Plasma very long chain fatty acids in 3,000 peroxisome disease patients and 29,000 controls. Ann Neurol. 1999;45:100–10. doi: 10.1002/1531-8249(199901)45:1<100::aid-art16>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 22.Mitchell R, Nivison-Smith I, Anazodo A, et al. Outcomes of haematopoietic stem cell transplantation for inherited metabolic disorders: a report from the Australian and New Zealand Children’s Haematology Oncology Group and the Australasian Bone Marrow Transplant Recipient Registry. Pediatr Transplant. 2013;17:582–8. doi: 10.1111/petr.12109. [DOI] [PubMed] [Google Scholar]
- 23.Beam D, Poe MD, Provenzale JM, et al. Outcomes of unrelated umbilical cord blood transplantation for X-linked adrenoleukodystrophy. Biol Blood Marrow Transplant. 2007;13:665–74. doi: 10.1016/j.bbmt.2007.01.082. [DOI] [PubMed] [Google Scholar]
- 24.Amartino H, Eichler F, Duncan C, et al. Expanding our understanding of cerebral adrenoleukodystrophy and interim phase 2/3 results of an autologous hematopoietic stem cell gene therapy. J Inborn Errors Metab Screen; Abstracts presented at the 13th International Congress of Inborn Errors of Metabolism — ICIEM 2017; 2017. pp. 1–413. abstract. [Google Scholar]
- 25.Hacein-Bey-Abina S, Garrigue A, Wang GP, et al. Insertional oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1. J Clin Invest. 2008;118:3132–42. doi: 10.1172/JCI35700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Howe SJ, Mansour MR, Schwarz-waelder K, et al. Insertional mutagenesis combined with acquired somatic mutations causes leukemogenesis following gene therapy of SCID-X1 patients. J Clin Invest. 2008;118:3143–50. doi: 10.1172/JCI35798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stein S, Ott MG, Schultze-Strasser S, et al. Genomic instability and myelodysplasia with monosomy 7 consequent to EVI1 activation after gene therapy for chronic granulomatous disease. Nat Med. 2010;16:198–204. doi: 10.1038/nm.2088. [DOI] [PubMed] [Google Scholar]
- 28.Avedillo Díez I, Zychlinski D, Coci EG, et al. Development of novel efficient SIN vectors with improved safety features for Wiskott-Aldrich syndrome stem cell based gene therapy. Mol Pharm. 2011;8:1525–37. doi: 10.1021/mp200132u. [DOI] [PubMed] [Google Scholar]
- 29.Hacein-Bey-Abina S, Pai S-Y, Gaspar HB, et al. A modified γ-retrovirus vector for X-linked severe combined immunodeficiency. N Engl J Med. 2014;371:1407–17. doi: 10.1056/NEJMoa1404588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Aiuti A, Biasco L, Scaramuzza S, et al. Lentiviral hematopoietic stem cell gene therapy in patients with Wiskott-Aldrich syndrome. Science. 2013;341:1233151. doi: 10.1126/science.1233151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Biffi A, Montini E, Lorioli L, et al. Lentiviral hematopoietic stem cell gene therapy benefits metachromatic leuko-dystrophy. Science. 2013;341:1233158. doi: 10.1126/science.1233158. [DOI] [PubMed] [Google Scholar]
- 32.Hacein-Bey Abina S, Gaspar HB, Blondeau J, et al. Outcomes following gene therapy in patients with severe Wiskott-Aldrich syndrome. JAMA. 2015;313:1550–63. doi: 10.1001/jama.2015.3253. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.