

Abstract
The CRISPR-Cas revolution is taking place in virtually all fields of life sciences. Harnessing DNA cleavage with the CRISPR-Cas9 system of Streptococcus pyogenes has proven to be extraordinarily simple and efficient, relying only on the design of a synthetic single guide RNA (sgRNA) and its co-expression with Cas9. Here, we review the progress in the design of sgRNA from the original dual RNA guide for S. pyogenes and Staphylococcus aureus Cas9 (SpCas9 and SaCas9). New assays for genome-wide identification of off-targets have provided important insights into the issue of cleavage specificity in vivo. At the same time, the on-target activity of thousands of guides has been determined. These data have led to numerous online tools that facilitate the selection of guide RNAs in target sequences. It appears that for most basic research applications, cleavage activity can be maximized and off-targets minimized by carefully choosing guide RNAs based on computational predictions. Moreover, recent studies of Cas proteins have further improved the flexibility and precision of the CRISPR-Cas toolkit for genome editing. Inspired by the crystal structure of the complex of sgRNA-SpCas9 bound to target DNA, several variants of SpCas9 have recently been engineered, either with novel protospacer adjacent motifs (PAMs) or with drastically reduced off-targets. Novel Cas9 and Cas9-like proteins called Cpf1 have also been characterized from other bacteria and will benefit from the insights obtained from SpCas9. Genome editing with CRISPR-Cas9 may also progress with better understanding and control of cellular DNA repair pathways activated after Cas9-induced DNA cleavage.
Keywords: genome editing, CRISPR-Cas9 system, guide RNA
1. INTRODUCTION
The CRISPR (clustered regularly interspaced short palindromic repeats)-Cas (CRISPR-associated proteins) revolution is on its way (Jinek et al., 2012; Barrangou, 2014). It is now possible to perform genome editing in virtually any living organism accessible to experimental manipulation. More than 40 species have been modified in proof-of-principle experiments and many research papers have already been published that include evidence based on CRISPR-Cas-mediated genome editing. For example, in basic research, gene knockout can be performed by genome editing to test the role of a given gene in a biological process, which in most cases allows for definitive proof of a gene’s importance. Applications ranging from DNA imaging to genome-wide screens and control of transcription will also benefit from the CRISPR-Cas system and recent reviews have discussed these as well as many other exciting developments that are underway (Hsu et al., 2014; Mei et al., 2016).
The basic principles of genome editing were laid down more than 20 years ago when it was demonstrated that gene targeting is stimulated by several orders of magnitude if a DNA double strand break (DSB) is made at the target locus (Jasin, 1996). Zinc finger nucleases (ZFNs) and transcription activator-like effector nucleases (TALENs) were the first generations of programmable sequence-specific nucleases that paved the way of genome editing with CRISPR-Cas9. Compared to previous types of sequence-specific nucleases, the CRISPR-Cas9 system is relatively simple to use and very efficient. In this review, we will discuss optimization of the two components, the guide RNA and the Cas9 protein.
The guide RNA directs DNA cleavage by Cas9 to the target sequence (Jinek et al., 2012) and careful selection of the guide RNA is therefore the cornerstone of any experiment with the CRISPR-Cas9 system. First, we review the different types of guide RNAs that have been tested for Streptococcus pyogenes and Staphylococcus aureus Cas9 (SpCas9 and SaCas9) leading to the novel, more efficient designs. When using SpCas9, a great number of potential guide RNAs are usually available in the target sequence given the frequency of its protospacer adjacent motif (PAM; NGG) (Hwang et al., 2013). We then discuss the main criteria of guide RNA selection: the initial choice of target sequence, which depends on the goal of the genome editing experiment, efficiency and specificity. Finally, we review many different Cas9 proteins that are now available: SpCas9 variants that have been engineered to have a different PAM or higher specificity as well as Cas9 and Cpf1 (CRISPR from Prevotella and Francisella 1) proteins from different bacteria. Taken together, these recent developments greatly improve the flexibility and precision of genome editing with CRISPR-Cas systems.
2. Design of single guide RNAs
In their groundbreaking report of sequence-specific DNA cleavage by the crRNA (CRISPR RNA):tracrRNA (trans-activating crRNA)-Cas9 complex, Jinek et al. (2012) showed that changing the 20-nt target-specific sequence in the crRNA is sufficient to redirect DNA cleavage to any DNA sequence of interest, provided the latter is immediately followed by a PAM (NGG) for SpCas9. This paper triggered the CRISPR-Cas revolution of genome editing. Several highly influential experiments were also reported. In particular, they showed that the dual crRNA:tracrRNA could be replaced by a single “guide” RNA (sgRNA) where the sequence portions of crRNA and tracrRNA that hybridize together (the repeat and antirepeat sequences, respectively) are fused by a GAAA tetraloop (Fig. 1A and B).
Figure 1.
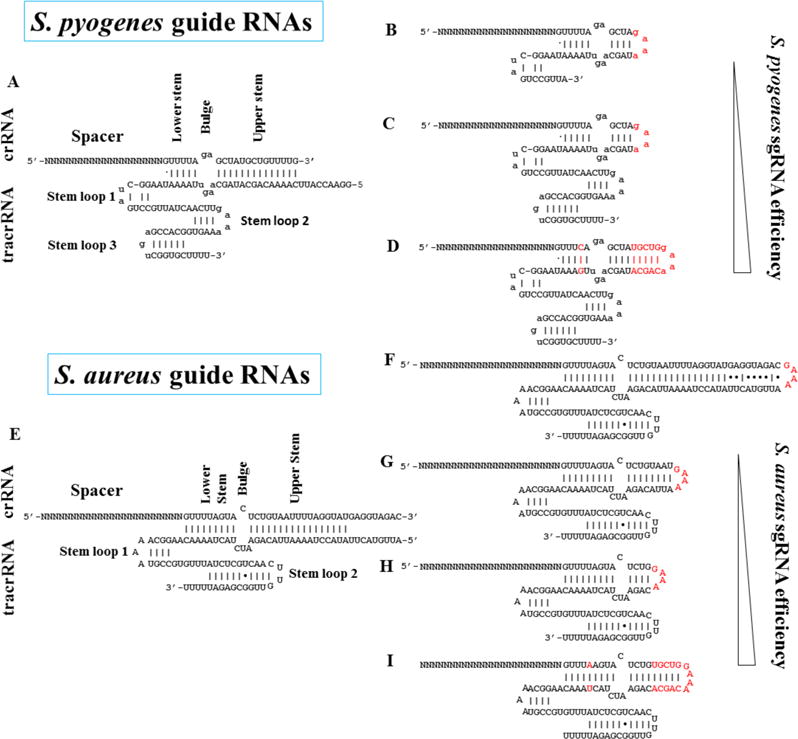
Design of synthetic sgRNA for S. pyogenes and S. aureus Cas9
A: The natural S. pyogenes guide RNA composed of crRNA and tracrRNA. B–D: Original design of Jinek et al. (2012) (B), standard, commonly used (C) and optimized (D) designs of S. pyogenes sgRNA. E: The natural S. aureus guide RNA. F–I: Original (F–H) and optimized (I) designs of S. aureus sgRNA. Red indicates the different sequences introduced to connect crRNA and tracrRNA in synthetic S. pyogenes and S. Aureus sgRNA. Mutations in the TTTT stretch of crRNA and the corresponding AAAA stretch of tracrRNA, which were made to improve sgRNA expressim from U6 promoter, are also indicated in red. sgRNA design impacts cleavage efficiency as indicated.
The tracrRNA sequence used in the first generation of sgRNAs was truncated, lacking 3′ tail (Fig. 1B) (Jinek et al., 2012; Cong et al., 2013) (Cong et al., 2013), whereas the cleavage efficiency was found to be much higher in cultured cells when the full-length 3′ tail of tracrRNA was included (Hsu et al., 2013; Mali et al., 2013; Pattanayak et al., 2013). Therefore, the standard sgRNA now used in experiments with Cas9 is a fusion of crRNA and full-length 3′-tail containing tracrRNA (Fig. 1C). When CRISPR-Cas9 system was used as a platform for dynamic imaging of repeated DNA sequences in living cells (Chen et al., 2013), two small modifications were introduced into the standard sgRNA: 1) a TTTT stretch was changed into TTTA to limit potential inhibition of sgRNA transcription from the U6 promoter, and 2) the sgRNA upper stem was extended to better mimic the portions of crRNA and tracrRNA that anneal together (Fig. 1D), both of which enabled much easier detection of telomeric DNA targeted by a GFP-tagged catalytically-inactive Cas9. More recently, similar modifications were found to enhance the knockout efficiency of CRISPR-Cas9-mediated genome editing (Dang et al., 2015). Although for most guides, the benefit was only modest, efficiency of a minority of guides increased up to eight-fold. As a result, pairs of guide RNAs used to induce genomic deletions were consistently 5- to 10-fold more efficient (Dang et al., 2015). Interestingly, several designs have also been tested for sgRNA from S. aureus. In this case, sgRNA with an upper stem extension that fully mimicks the repeat-antirepeat of the crRNA-tracrRNA induced much lower cleavage activity compared to sgRNA without an upper stem (Kleinstiver et al., 2015a) (Fig. 1E–H). More recently, modification of the TTTT stretch and an upper stem extension identical to that in optimized S. pyogenes sgRNAs (Fig. 1I) were found to further increase S. aureus sgRNA activity, thus allowing to efficiently restore dystrophin expression from the mutant Duchenne muscular dystrophy (DMD) gene of mdX mice (Tabebordbar et al., 2016). S. aureus sgRNA design was therefore critical to achieve high efficiency.
The studies mentioned above underscore the importance of sgRNA design when engineering novel CRISPR-Cas systems for genome editing. It is important to note that a powerful alternative to sgRNA is now to use dual guide RNAs produced by chemical synthesis. The full-length 3′-tail containing tracrRNA is constant and each target-specific crRNA is custom-synthesized. Because of their great simplicity, synthetic guide RNAs are becoming increasingly popular with researchers doing sgRNA/Cas9 injections in model organisms (Kim et al., 2014; Aida et al., 2015; Bhattacharya et al., 2015; Kotani et al., 2015). When used in co-transfection experiments with Cas9 mRNA or protein, synthetic dual guide RNAs are more efficient if they carry chemical modification that protect them from degradation (Rahdar et al., 2015). Compared to inexpensive plasmid-based solutions, the current costs of synthetic guide RNAs and Cas9 mRNA or protein from commercial suppliers still considerably limits their use in cell culture experiments. In the context of gene therapy, however, delivery methods of CRISPR-Cas reagents that do not require exogenous DNA are essential.
3. Selecting guide RNAs for knockout and knockin experiments
Many considerations may guide the choice of target DNA sequence and design of the genome edit. We briefly discuss here how to select guide RNAs to optimize genome editing efficiency in knockout and knockin experiments.
3.1. Selecting guide RNAs for knockout experiments
When investigating gene function, generation of constitutive loss-of-function mutants is an important step. For this purpose, guide target sequences are generally chosen in the 5′ exons of genes, because full-gene inactivation is expected from mutations that disrupt the reading frame and lead to highly truncated protein, if any. Interestingly, when investigating the inactivation of genes for chromatin-regulatory proteins, Shi et al. (2015) found that targeting functional domains was more efficient than targeting 5′ exons. Sequencing of the resulting mutations revealed that in-frame mutations in functional domains had led to function disruption. This type of guide RNA design, however, is only possible for proteins with well-characterized functional domains.
For gene knockout with a sgRNA, the mutations introduced are expected to be small insertions or deletions of random size that will therefore disrupt the open reading frame and yield knockouts in two cases out of three. Extensive sequencing revealed that frequency of out-of-frame mutations can in fact deviate significantly from the expected frequency (Bae et al., 2014). Bae et al. (2014) noted that some predominant mutations explained this unexpected finding. Importantly, predominant mutations occurred between short homologous sequence stretches and were therefore related to repair of the DSB by micro-homology-mediated end-joining (MMEJ). MMEJ involves annealing of short 2–8-nt sequences present on both sides of the cleavage site, hence the name. The molecular mechanisms involved are not fully understood, but both DNA ends are resected by exonucleases and the complementary single stranded sequences anneal, resulting in the final deletion. Based on the analysis of the sequences flanking the cleavage site, Bae et al. (2014) therefore proposed an “out-of-frame” score to quantify the probability that a given guide RNA will induce out-of-frame mutations. However, a recent analysis found no correlation between gene knockout and out-of-frame score (Fusi et al., 2015). This apparent discrepancy is most likely due to difference in cell types used, each having different levels of MMEJ repair pathway activity. When developing genome editing in a moth species, Lepideptora Littoralis, Koutroumpa et al. obtained an in-frame deletion that represented 60% of mutations (unpublished data). This in-frame deletion is indeed present between two micro-homology sequence stretches of 4 nt which are present on each side of the DSB and retrospectively, by using the out-of-frame score the corresponding guide RNA could have been avoided to obtain a higher rate of out-of-frame mutations!
To improve the yield of knockouts during genome editing, two interesting designs have been proposed that use more than one guide. In order to improve the yield of knockouts in mouse, Sunagawa et al. (2016) (Sunagawa et al., 2016) showed that instead of injecting a single guide into oocytes at the maximum possible concentration, it was more efficient to coinject three guides with each at 1/3 the maximum possible concentration. They tested three series of guides and obtained more than 95% mice with biallelic knockout mutations in each case. Such high efficiencies are very useful because they allow phenotypic analysis directly in F0 mice born from the injections. Reliable success with this promising method, however, requires three very efficient guides, which may be facilitated by prediction scores, as discussed below. A second interesting design is to use two guide RNAs to delete an entire exon or the whole coding sequence (Chen et al., 2014; Gratz et al., 2014; Paix et al., 2015) (Paix et al., 2015). In order to delete a common 5′ exon, two guide RNAs can be designed to target flanking intron sequences. Deletion of the target exon will generate mRNAs with fusion of flanking exons that should disrupt the reading frame in order to ensure loss-of-function. Whole-gene deletion was demonstrated in the fly Drosophila melanogaster where two guide RNAs were used to induce cleavage at positions flanking the target gene in order to stimulate replacement by an expression cassette of a fluorescent protein marker which facilitates the isolation of flies with the resulting null allele (Gratz et al., 2014). This approach has also been successful in the nematode Caenorhabditis elegans (Paix et al., 2015). The advantage of eliminating the whole coding sequence is that it creates a genuine null allele of the gene of interest, without any possibility of alternative protein expression. However, one limitation is that regulatory sequences of adjacent genes may be present in the target locus, potentially making it more difficult to attribute the phenotype to the loss-of-function of the target gene only.
3.2. Selecting guide RNAs for knockin experiments
Insertion of reporter sequences may be performed for many purposes, including easy tracing of gene expression, protein tagging and selection of modified cells. In this type of experiments, only guide RNAs within 20 bp from the knockin position can be used (Yang et al., 2013; Findlay et al., 2014;) because more distant cleavage will not stimulate efficient recombination with the donor plasmid during repair. In pioneer experiments with I-SceI nuclease, it was shown that stimulation of gene targeting required cleavage to take place less than 50 bp away from the point mutation (Elliott et al., 1998). In practice, with SpCas9, several PAMs can usually be found next to the insertion position and the corresponding guide RNAs can be tested. To drive cleavage closer to the knockin position, Cas9 proteins from different bacteria or variants of SpCas9 with alternative PAMs, which are listed in Fig. 2 and Table 2 and described further below, can also be considered. The preferred method for knockin is through homology-directed repair (HDR) with donor DNA in which the insert is flanked by sequences adjacent to the cleavage site. This allows for precise, predetermined sequence modification.
Figure 2.

SpCas9 variants with altered PAM or reduced off-target cleavage.
Several important SpCas9 variants have been engineered, either to modify the PAM or to reduce off-target cleavage. Functional changes in SpCas9 were achieved by mutation of the indicated key residues. VRQR-Cas9 specific to PAM NGA and VRER-Cas9 specific to PAM NGCG were obtained by mutating R1335 that directly contacts the PAM and other residues as indicated (Kleinstiver et al., 2015b). SpCas9 variants with reduced off-target cleavage, HF-Cas9 (Kleinstiver et al., 2016) and eCas9 1.1 (Slaymaker et al., 2016), were obtained by mutating residues that are involved in non-sequence-specific interaction with target DNA. The schematic structure of the SpCas9 is adapted from (Jiang et al., 2016). Structural domains of SpCas9 are delimited by the indicated amino acid positions. BH, bridge helix; Lk-I and Lk-II, linker peptides involved in conformational changes that elicit concerted DNA cleavage by RuvC and HNH nuclease domains.
Table 2.
Cas proteins and cognate PAMs available for genome editing in eukaryotic cells
| Cas protein | PAM | Origin | Reference |
|---|---|---|---|
| SpCas9 | NGG | S. pyogenes | Jinek et al., 2012 |
| VRQR-SpCas9 | NGA | Engineered from SpCas9 | Kleinstiver et al., 2015b |
| VRER-SpCas9 | NGCG | Engineered from SpCas9 | Kleinstiver et al., 2015b |
| SthCas9-CRISPR1 | N2AGAAW (W = A or T) | S. thermophilus | Muller et al., 2015 |
| SthCas9-CRISPR3 | NGGNG | S. thermophilus | Glemzaite et al., 2015 |
| NmCas9 | N4GMTT (M = A or C) | Neisseria meningitidis | Hou et al., 2013 |
| SaCas9 | N2GRRT (R = A or G) | S. aureus | Ran et al., 2015 |
| KKH-SaCas9 | N2NRRT (R = A or G) | Engineered from SaCas9 | Kleinstiver et al., 2015a |
| BlatCas9 | N4CND (D = A, G or T) | B. laterosporus | Karvelis et al., 2015 |
| AsCpf1 | TTTN | A. sp. BV3L6 | Zetsche et al., 2015 |
| LbCpf1 | TTTN | L. bacterium ND206 | Zetsche et al., 2015 |
N = A, T, C or G.
In addition to guide RNA position and activity, many factors determine knockin efficiency including length of homology arms and relative importance of DSB repair pathways. In gene drive experiments in mosquito (Gantz et al., 2015), genome editing is driven by transgenic expression of Cas9 in meiotic cells where homologous recombination (HR) prevails over non-homologous end-joining (NHEJ). Consequently, only knockin events take place and local mutations at the cleavage site due to NHEJ repair are hardly ever detected. In contrast, in most common experimental settings, NHEJ repair prevails and knockin events are much less frequent. Selection and/or screening strategies are therefore generally needed to identify knockin cells. Increasing knockin efficiency may be achieved by pharmacological inhibition of NHEJ or stimulation of HR to tip the balance between competing repair pathways in favor of knockin events(Song et al., 2016) (Maruyama et al., 2015) ((Chu et al., 2015); Maruyama et al., 2015; Song et al., 2016). Although no overt toxicity was observed during treatments, non-specific mutations may be induced during NHEJ inhibition. It would thus be of great interest to have better targeted methods, with NHEJ inhibition or other modulations of DNA repair restricted to the DSB induced by Cas9.
4. Lessons learned from genome editing in mouse embryonic stem (ES) cells
Problems with gene targeting observed in mouse knockout and knockin experiments over the last 30 years remain potential concerns when selecting guide RNAs and designing experiments of genome editing with CRISPR-Cas9. For example, inserting a sequence of 1.7–2.5 kb into human cell division cycle 14A (hCDC14A) and cell division cycle 14B (hCDC14B) genes resulted in unexpected exon skipping, making it impossible to exploit the modified cells as originally planned (Uddin et al., 2015). The following pitfalls have all been previously documented in the mouse (Müller, 1999): (i) As a result of small insertions or deletions in 5′ exons that disrupt the open reading frame, it is expected that protein expression will be abolished. However, protein expression may persist due to initiation of translation at alternative downstream ATG codons or simply due to the existence of alternative first coding exons. (ii) The regulatory sequences in the selection cassette used to identify the recombinant cells or organisms may perturb expression of the nearby genes and lead to a confounding phenotype. (iii) Deletion of whole genes may remove important regulatory sequences of distinct adjacent genes. (iv) Insertion of large artificial exons may not necessarily inactivate gene function and protein expression must be carefully monitored. (v) Careful genotyping is also important to rule out any complex rearrangements during genome editing that may affect neighboring genes in addition to the targeted sequences. Careful selection of guide RNAs and design of DNA sequence modification is therefore important to preclude uncertainties in analysis of edited genomes. Another lesson from the preCRISPR-age of gene targeting experiments in mouse ES cells is that the efficiency of genome editing may vary dramatically among loci. For some genes, huge screening efforts were required to find positive clones with the desired integration (Koller et al., 1991) and it was recommended to change the design of the gene targeting construct (Skarnes et al., 2011).
5. Selecting highly efficient guide RNAs
Guide RNA efficiency is crucial for genome editing and can vary considerably from one sequence to another. For example, Liu et al. (2016) recently tested a series of 218 guides and found that 89 were inactive as detected with the Surveyor assay. As detailed further below, it remains difficult to reliably predict cleavage efficiency from the guide sequence and, in practice, the best option is to test three guide RNAs and select the most efficient one (Ran et al., 2013). This can be done by examining the rate of mutations induced by the guides with the standard T7 assay (Larcher et al., 2014; Vouillot et al., 2015). Direct Sanger sequencing of PCR products of the target sequence can also be performed (Brinkman et al., 2014), which is not very sensitive but useful to detect the most efficient guide RNAs needed for knockin experiments. Other procedures are possible but require specific electrophoresis equipment or are more costly. For example, DNA capillary electrophoresis of PCR products can detect mutations (Dahlem et al., 2012) and the sensitivity will be increased by introducing a fluorescent primer and laser detection (Yang et al., 2015). Finally, fluorescent reporters, where fluorescent protein expression depends on cleavage and intramolecular recombination, can also been used but additional cloning of the target sequence into the reporter plasmid is required (Kim et al., 2011).
Many studies have identified sequence criteria or scores that correlate with guide efficiency. These are useful to limit the number of guide RNAs to be tested for genome editing. For instance, prediction scores are crucial in genome-wide screens that involve thousands of guides and indeed, second generation guide RNA libraries taking into prediction scores are proving much more efficient and reliable (Wang et al., 2015a; Doench et al., 2016). Table 1 provides a list of web resources for predicting sgRNA efficiency.
Table 1.
Web resources for predicting sgRNA efficiency
| Web resource | Experimental system | Reference |
|---|---|---|
| crispor.org | U6 promoter/mammalian cultured cells | Wang et al., 2014b |
| sgrna-design-v1 | U6 promoter/mammalian cultured cells | Doench et al., 2014 |
| cistrome.org/SSC | U6 promoter/mammalian cultured cells | Xu et al., 2015 |
| sgRNAscorer | U6 promoter/mammalian cultured cells | Chari et al., 2015 |
| crisprscan.org | Injected sgRNA/zebrafish embryos | Moreno-Mateos et al., 2015 |
| Wu-CRISPR | U6 promoter/mammalian cultured cells | Wong et al., 2015 |
| sgrna-design | U6 promoter/mammalian cultured cells | Fusi et al., 2015 |
| crispor.org | U6 promoter/D. melanogaster | Ren et al., 2014 |
| crispor.org | U6 promoter/C. elegans | Farboud and Meyer, 2015 |
| evaluateCRISPR | U6 promoter/fly cultured cells | Housden et al., 2015 |
Prediction scores for guide activity appear to depend on the experimental system considered. It was previously observed that a popular prediction score, based on data from guides tested in cell culture (Doench et al., 2014), was not reliable in zebrafish injection experiments (Varshney et al., 2015). Moreno-Mateos et al. (2015) collected the guide RNA activity of more than a thousand guides injected into zebrafish embryos by high throughput sequencing and devised a score that, in contrast, was shown to be very helpful in selecting guide sequences for injections into zebrafish. These findings suggest that guide RNA efficiency is strongly influenced by the experimental system. For example, Moreno-Mateos et al. (2015) showed that in their setup, the most efficient guide RNAs proved to be those that were the most stable after injections, including G-rich sequences known to form structures called G-quadruplexes. Such guide RNA stability may not be necessary for high activity when the guide is continuously expressed at high levels from a U6 promoter after lentiviral transduction. In the specific case of G-rich guides, it was in fact recently shown that they are generally inefficient in cultured mammalian cells (Malina et al., 2015). It therefore appears that the Moreno-Mateos score is particularly useful in zebrafish experiments for selecting guide RNAs produced by in vitro transcription.
All other scores available are based on datasets where guide RNA is expressed from a U6 promoter. The first score was devised by Wang et al. (2014a) from data of genome-wide guide RNA screens. The activity of guide RNAs targeting essential ribosomal genes was inferred by measuring the decrease of the corresponding guide RNA sequences during growth of the cells treated with the lentiviral guide RNA library. The second score was devised from a dataset of guides targeting cell surface proteins (Doench et al., 2014). The activity of 1841 guide RNAs was measured by quantifying the guide sequences in treated cells after sorting for the corresponding cell surface marker by FACS (fluorescence-activated cell sorting). These and other published datasets of guide RNA activity from the U6 promoter were analyzed with different machine learning algorithms to obtain novel scores (Wong et al., 2015; Xu et al., 2015). Compared to other datasets, Chari et al. (2015) measured guide activity on randomly integrated DNA sequences, thereby excluding potential bias due to the local chromatin context of genomic targets in previous assays, and proposed a score based on regression analysis of the data (Chari et al., 2015). Finally, Fusi et al. (2015) and Doench et al. (2016) extensively compared algorithm performance, used raw activity data rather than ranking, and devised a novel score.
Recent scores based on datasets with guide RNAs expressed from the U6 promoter were all shown to be superior to the first Doench score (Chari et al., 2015; Fusi et al., 2015; Wong et al., 2015; Xu et al., 2015). Among these, the score by Fusi et al. (2015) and Doench et al. (2016) appears to rely on the most exhaustive data analysis and is our current recommendation for the design of guides expressed from the U6 promoter in mammalian cells. However, it will be important to perform an independent cross-comparison of scores and systematically examine how they perform on datasets available to validate this recommendation. Currently, all the scores available have been established from datasets using Cas9 expressed from plasmid DNA or synthetic RNA, produced by in vitro transcription. It will therefore be important to know how the scores perform when guide RNAs are directly associated with recombinant Cas9 protein before injection or electroporation into the target cells (Kim et al., 2014; Bhattacharya et al., 2015; Kotani et al., 2015; Liang et al., 2015; Ménoret et al., 2015).
The molecular mechanisms underlying high guide RNA efficiency are not well understood. As mentioned above, guide activity depends on the expression system and several criteria associated with poor guide RNA activity are known. For instance, Wong et al. (2015) showed that guide RNAs that contain UUU in the seed region are often inefficient when expressed from the U6 promoter, possibly due to the inhibition of transcription by RNA polymerase III. Independently of the guide RNA expression system, specific sequence motifs that hinder the formation of the tracrRNA structure necessary for interaction with Cas9 may inhibit guide activity. In line with this idea, Wang et al. (2014a) and Hart et al. (2015) showed that guide RNAs that interact poorly with Cas9 exhibit lower activity and presence of U in the 4 nucleotides proximal to the PAM is anti-correlated to guide activity. Conversely, however, few guide sequence factors are known to correlate with higher guide RNA activity. Higher stability of the duplex between guide RNA and target DNA was shown to be favorable (Wong et al., 2015). Biophysical modeling and further insight into DNA cleavage by the guideRNA-Cas9 complex will be necessary and could be translated into better predictions of guide activity. For example, it has recently been reported that guide sequences still direct Cas9 DNA binding when their length is reduced from 20 to 14 nt but they no longer promote DNA cleavage (Dahlman et al., 2015). This unexpected finding, in addition to providing an elegant means for orthogonal transactivation and DNA cleavage, suggests a specific role for the 5′ end of the guide in cleavage activity. Finally, in addition to guide sequence, the target genomic environment could play an important role. So far, such a high proportion of guides tested are active that tight chromatin regulation of Cas9 access to its target sequences is unlikely. In genome-wide studies though, chromatin inaccessibility was reported to decrease binding of Cas9 (Wu et al., 2014) and possibly, at the target DNA sequence level, the presence of transcription factors, nucleosomes or specific epigenetic modifications could influence binding and cleavage. Recent in vitro studies indicated that binding of Cas9 to nucleosomal DNA may in fact be conditioned by accessibility of the PAM (Hinz et al., 2015).
To make it possible to easily compare activity scores of guide RNAs, we have recently integrated them into a web tool called CRISPOR (Haeussler et al.; available at crispor.org). In particular, it includes the Wang score that is only available otherwise in the form of computer code from the author. The Wang score has been experimentally demonstrated to be useful when designing a second generation library of human guides that proved to be much more efficient, demonstrating the usefulness of the Wang score (Wang et al., 2015a). Crispor.org also includes the Chari score with a computer code optimization that makes it much faster to run than the web site version made available by the authors (Chari et al., 2015). As discussed above, the Moreno-Mateos score is currently recommended for experiments in zebrafish using guide RNAs produced by in vitro transcription while the Fusi score is recommended in mammalian cells where the guide is expressed from the U6 promoter. Nevertheless, as a second option, guides that have high scores with other models may be taken into consideration.
6. Selecting guide RNAs with the fewest potential off-target sites
In Jinek et al. (2012)’s highly influential report, it has been demonstrated that for DNA cleavage in vitro, annealing of the guide to target DNA could tolerate up to five mismatches. Such a low specificity raised major concern for genome editing with CRISPR-Cas in living cells. The issue still deserves full and thorough investigation when considering therapeutic and agronomy applications. However, off-target cleavage events have now been extensively examined and some simple rules have emerged to minimize off-target effects for basic research.
Various methods have been used to examine off-targets. Potential off-targets sites were first identified through bioinformatics analysis, by searching the genome for sequences with mismatches to the target that were followed by a PAM motif (Hsu et al., 2013; Cho et al., 2014;). Assays that differ in sensitivity, labor involved and cost have been used to study induced mutations. The T7 assay is very easy and cheap to perform but only detects a minimum rate of 2%–5% mutant sequences. Conversely, PCR amplicon sequencing offers the highest sensitivity but is more expensive. Amongst several initial studies of CRISPR-Cas9 off-targets, the report by the Joung and Sander labs was alarming to many investigators (Fu et al., 2013). Of the six guide RNAs investigated, four had off-targets that were extensively modified, in some instances at higher rates than the bona fide target sequence. Next, the genome of edited cells was sequenced to search for potential mutations in an unbiased way. No off-target mutations could be reliably detected for the guide RNAs tested (Cho et al., 2014; Essletzbichler et al., 2014; Iyer et al., 2015). The results were encouraging but a firm conclusion cannot be drawn because the sensitivity from 20–25× genome coverage is relatively low and only a few treated cells were analyzed.
Finally, more sensitive methods were reported for detecting off-target mutations in an unbiased manner, that all involve high-throughput sequencing: GUIDE-seq (genome-wide, unbiased identification of DSBs enabled by sequencing), BLESS (direct in situ breaks labelling, enrichment on streptavidin and next-generation sequencing), HTGTS (high-throughput, genome-wide, translocation sequencing), IDLV (integrase-defective lentiviral vector) and Digenome-seq (in vitro Cas9-digested whole-genome sequencing). Of these methods, GUIDE-seq seems to be the most straightforward and cost-effective. It relies on capture of double-stranded oligonucleotides (dsODN) at DSBs (Tsai et al., 2015). When cotransfected with the guide RNA and Cas9 expression plasmids, a dsODN was shown to be captured at cleavage sites at a rate corresponding to one third of the targeted mutations. Using a primer to the known oligonucleotide sequence, it is thus possible to PCR-amplify and identify integration sites resulting from on- and off-target cleavage. The number of PCR sequence reads obtained is related to the cleavage rate by the guideRNA-Cas9 complex. GUIDE-seq is highly efficient in U2OS and HEK293 cells but may be difficult to carry out in other cell lines. We believe that this could be due to a particularly high rate of dsODN capture in U2OS and HEK293 relative to other cell lines. HTGTS and IDLV are two methods that also indirectly detect off-target cleavage sites, taking advantage of repair events that will join a known sequence to the DSBs and facilitate their identification (Frock et al., 2015; Wang et al., 2015b). HTGTS quantifies translocations between a known cleavage site and the DSBs while IDLV relies on capture of an integration-defective lentiviral vector. HTGTS and IDLV depend on much less frequent repair events than GUIDE-seq and are of more limited sensitivity. In contrast to the latter methods, BLESS and Digenome-seq directly detect DSBs, respectively in fixed cells or in the test tube (Kim et al., 2015; Ran et al., 2015). Digenome-seq is performed on genomic DNA treated with nuclease in vitro and appears to be the most sensitive method. When compared to GUIDE-seq and HTGTS, it detected more off-targets (especially off-targets with mutation rates below 0.1%). The initial sequencing step in Digenome-seq, however, generates a lot of false positives and candidate off-target sites need to be confirmed by next generation sequencing.
A fair comparison of unbiased methods for detecting off-targets requires using identical DNA samples resulting from identical expression levels of guide RNA and Cas9, and it may be difficult because sensitivity will be largely determined by the depth of high throughput sequencing. Some off-targets were identified by one method only (Kim et al., 2016), suggesting that exhaustive experimental identification of off-targets remains challenging. These findings suggest that it will now be important, depending on the genome editing context, to define thresholds on the nature and rate of off-targets that can be tolerated.
Taken together, a basic conclusion of studies that performed unbiased detection of off-targets is that in every case the off-targets detected are homologous to the guide. GUIDE-seq studies identified DSBs at unrelated sequences but these were also detected in control cells and proved to be sites that are prone to break in normal physiological conditions. Importantly, in most cases, only off-targets with up to three mismatches and a canonical PAM motif have a significant modification frequency (Kim et al., 2016). In a very few cases where a noncanonical PAM is found, usually NGA or NAG and the corresponding modification frequency is very low (Tsai et al., 2015). Off-targets with up to six mismatches or with gaps or bulges in the alignment are also possible but the modification frequencies are always very low ((Lin et al., 2014); Tsai et al., 2015). As a result, guide RNAs that have off-targets with less than three mismatches should generally be excluded. Importantly though, the possibility of off-target cleavage at sites with two or three mismatches detected by Tsai et al. (2015) could be reliably predicted using a novel off-target prediction method developed by Doench et al. (2016). They systematically analyzed the effects of different types of mismatches on DNA cleavage. In particular, all possible mismatches at each position were tested and it was found that mismatch identity could have a strong impact. The complete mutagenesis data was used to elaborate a novel score for predicting off-target cleavage, which was called CFD for cutting frequency determination. CFD correlated better with measured cleavage than previously available scores from Hsu and CCTop (CRISPR/Cas9 Target online predictor) (Hsu et al., 2013; Stemmer et al., 2015). For off-targets detected by Tsai et al. (2015) with more than three mismatches, no score is very reliable (Doench et al., 2016) and most likely, factors other than the off-target sequence itself, such as the chromatin environment, play a pivotal role in determining the rare off-targets out of the large number of related sequences with more than three mismatches. Finally, although prediction of off-targets has been improved by the CFD, it is still unclear how to translate all the predicted off-target scores into a reliable method that ranks guides according to their specificity.
Numerous online tools are now available to minimize off-targets in selection of guide RNAs. Some of the first tools developed were shown to be defective because they miss many off-targets (Tsai et al., 2015; Doench et al., 2016), possibly due to the selection of parameters of genome scanning algorithms that favor speed over precision. Unfortunately, this includes the very commonly used MIT and e-CRISP websites (Hsu et al., 2013; Heigwer et al., 2014). In contrast, many web tools are reliable, including the recent Cas-Off finder, sgRNAdesigner (Doench et al., 2016) and our CRISPOR tool (Haeussler et al.; available at crispor.org). Cas-Off finder is very fast and allows identifying off-targets with up to six mismatches (Bae et al., 2014b). sgRNAdesigner takes into account the CFD score and automatically ranks guides by predicted activity and specificity using arbitrarily defined criteria, making it the best suited for the design of thousands of guide RNAs in genome-wide screens (Doench et al., 2016). Crispor.org searches for off-targets for each guide and lists them with their off-target score and genomic annotations (Haeussler et al.; available at crispor.org). It is therefore possible to examine the results in detail and carefully exclude unwanted guides. For example, in cell culture experiments, off-targets in protein-coding sequences should be avoided. In model organisms, only potential off-targets residing on the chromosome of the target need to be considered, as chromosomal segregation during transmission will remove the others. This can drastically reduce the number of potential off-targets.
7. Novel Cas9 proteins and engineered SpCas9 provide additional genome editing flexibility and improved specificity
The best known Cas9 protein to date is SpCas9. Cas9 proteins from other bacteria have also been characterized (Table 2). In particular, several have been tested because they are smaller than SpCas9 (in the range of 1000 amino acids compared to 1368 for SpCas9) which facilitates packaging of the corresponding cDNAs into viral vectors for therapeutic applications (Ran et al., 2015). Cas9 from other bacteria than S. pyogenes generally have more complex PAMs, which provides them with higher specificity (Ran et al., 2015) but also limits the range of potential target sequences. Streptococcus thermophilus CRISPR1 Cas9 (SthCas9-CRISPR1) is an extreme case which requires a N2AGAAW (W = A or T) PAM (Müller et al., 2015). An interesting exception is that of Brevibacillus laterosporus Cas9 (BlatCas9) that has recently been shown to work in plants and only requires a simple N4CND PAM (D = A, G or T) (Karvelis et al., 2015). This target sequence flexibility will make BlatCas9 a very useful addition to the CRISPR-Cas toolkit.
Other types of RNA-guided endonucleases have also been recently discovered to be single effectors in CRISPR-based bacterial defense. Cpf1 is a protein family whose prototypical member was characterized from F. novicida, named FnCpf1 (Zetsche et al., 2015). FnCpf1 has a TTN PAM but unfortunately in the tests performed so far, it is inefficient in mammalian cells. In fact, of 16 Cpf1-family proteins from diverse bacteria tested, only two Cpf1 proteins derived from Acidaminococcus sp. BV3L6 (AsCpf1) and Lachnospiraceae bacterium ND2006 (LbCpf1) were active in mammalian cells, but they have a more restricted TTTN PAM than that of FnCpf1. All were shown to be active in vitro and the reason for inefficient cleavage by most Cpf1-family proteins in mammalian cells is unclear. Future engineering of FnCpf1 may improve its activity. Features such as the ability to interact with nucleosomal DNA, nuclear localization or optimal sgRNA design and expression may be involved. Cpf1 proteins have been suggested to facilitate HDR and precise genome editing because they produce staggered DNA ends (Zetsche et al., 2015). We note, however, that previous generations of programmable nucleases such as TALENs also produce staggered DNA ends but are not known to favor HDR over NHEJ compared to SpCas9.
Although the number of RNA-guided endonucleases is rapidly expanding, SpCas9 is likely to remain the workhorse of genome editing in most standard applications. Many genomes have already been successfully edited with it and the NGG PAM is sufficient for most applications. In addition, elucidation of SpCas9 structure has provided opportunities to rationally design variant SpCas9 with novel exciting features (Anders et al., 2014; Jinek et al., 2014; Nishimasu et al., 2014). By modifying amino acids involved in contacting the PAM sequence, it has been possible to engineer SpCas9 variants with novel PAM specificities (Fig. 2). Previously, mutation of residues that directly interact with the major groove of the PAM had not been sufficient to redirect PAM specificity (Anders et al., 2014), and Kleinstiver et al. (2015b) therefore devised a powerful screening strategy to select Cas9 variants carrying additional random mutations in the PAM interacting domain for novel PAM specificity. This allowed to isolate the variants VRQR-SpCas9, VRER-SpCas9 and KKH-SaCas9, that bind to NGA, NGCG and N2NRRT PAMs, respectively (Table 2; Fig. 2). VRQR-SpCas9 will be useful in many situations where precise targeting by SpCas9 is not possible, in particular in A/T rich sequences such as in the Plasmodium falciparum genome. KKH-SaCas9 significantly expands the range of sequences that can be targeted compared to SaCas9 and will be useful for gene therapy applications.
Furthermore, by modifying residues involved in non-sequence-specific contacts with DNA, it has also been possible to engineer variant SpCas9 proteins that drastically reduce off-targets observed with some guides. These “high precision” variants will be very important for therapeutic and agronomy applications (Kleinstiver et al., 2016; Slaymaker et al., 2016). Kleinstiver et al. (2016) developed one variant named HF-SpCas9 by mutating residues that contact the DNA strand annealing to guide RNA, while Slaymaker et al. (2016) developed another, named eCas9, by changing residues that contact the free complementary single strand DNA (Fig. 2).
Although mutations were designed to destabilize binding of SpCas9 to DNA, Kleinstiver et al. (2016) also raised the alternative possibility that conformation changes necessary for Cas9 activity could be inhibited. In particular, the Q926A mutation of HF-SpCas9 is located next to a peptide linker involved in the RuvC domain relocation that precedes concerted cleavage by the RuvC and HNH catalytic domains (Jiang et al., 2016). Interestingly, Slaymaker et al. (2016) screened mutations based on the hypothesis that progressive unwinding of DNA and 3′ to 5′ annealing of guide RNA is facilitated by non-specific interactions of Cas9 with the non-targeted DNA strand that inhibit DNA duplex formation. One major drawback of high precision variants is lower on-target activity. Although activity of most guides tested did not seem to be affected, several exhibited much lower efficiency with high precision variants of SpCas9 (Kleinstiver et al., 2016; Slaymaker et al., 2016). Dose–response studies may help to more finely address whether guide activity is compromised. Concerning the repertoire of target sequences, it was found that a G nucleotide is required at position 1 of the guide RNA sequence for efficient guide activity in cultured cells and this may be an additional limitation of high precision Cas9 variants (Kleinstiver et al., 2016).
8. Perspectives
The basic features of genome editing with the CRISPR-Cas9 system are now well established. Usually a guide RNA of suitable efficiency and specificity to drive efficient genome editing with SpCas9 can be easily selected. If not, SpCas9 variants engineered to work with different PAMs or Cas9 from different bacteria are alternative solutions that should allow targeting the sequence of interest. Further insights into the molecular steps of DNA cleavage, from PAM binding and initial unwinding of target DNA to concerted DNA cleavage by the two catalytic domains, may open up new opportunities of increasing the activity of specific guide RNA and/or Cas9 proteins. We can also expect future progress in our ability to control DNA sequence editing during repair of DSBs made by Cas9. Perhaps, through further engineering of Cas9 and of the molecular pathways involved in homology-directed repair, we can hope to obtain Cas9 variants that will perform programmed DNA sequence editing with the cut-and-paste precision of the transposases from which Cas9 proteins originally evolved to become the guardians of bacterial genomes (Shmakov et al., 2015).
Acknowledgments
We are grateful to our colleagues and collaborators for discussion and to Romain Domart for help with Figure 1. Studies in the lab of JPC were funded by ANR-II-INBS-0014. MH is funded by grants NIH/NHGRI5U41HG002371-15, NIH/NCI5U54HG007990-02 and by a grant from the California Institute of Regenerative Medicine, CIRM GC1R-06673C.
References
- Aida T, Chiyo K, Usami T, Ishikubo H, Imahashi R, Wada Y, Tanaka KF, Sakuma T, Yamamoto T, Tanaka K. Cloning-free CRISPR/Cas system facilitates functional cassette knock-in in mice. Genome Biol. 2015;16:87. doi: 10.1186/s13059-015-0653-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders C, Niewoehner O, Duerst A, Jinek M. Structural basis of PAM-dependent target DNA recognition by the Cas9 endonuclease. Nature. 2014;513:569–573. doi: 10.1038/nature13579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae S, Kweon J, Kim HS, Kim JS. Microhomology-based choice of Cas9 nuclease target sites. Nat Methods. 2014;11:705–706. doi: 10.1038/nmeth.3015. [DOI] [PubMed] [Google Scholar]
- Barrangou R. RNA events. Cas9 targeting and the CRISPR revolution. Science. 2014;344:707–708. doi: 10.1126/science.1252964. [DOI] [PubMed] [Google Scholar]
- Bhattacharya D, Marfo CA, Li D, Lane M, Khokha MK. CRISPR/Cas9: An inexpensive, efficient loss of function tool to screen human disease genes in Xenopus. Dev Biol. 2015;408:196–204. doi: 10.1016/j.ydbio.2015.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinkman EK, Chen T, Amendola M, van Steensel B. Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res. 2014;42:e168. doi: 10.1093/nar/gku936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chari R, Mali P, Moosburner M, Church GM. Unraveling CRISPR-Cas9 genome engineering parameters via a library-on-library approach. Nat Methods. 2015a doi: 10.1038/nmeth.3473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chari R, Mali P, Moosburner M, Church GM. Unraveling CRISPR-Cas9 genome engineering parameters via a library-on-library approach. Nat Methods. 2015b;12:823–826. doi: 10.1038/nmeth.3473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B, Gilbert LA, Cimini BA, Schnitzbauer J, Zhang W, Li GW, Park J, Blackburn EH, Weissman JS, Qi LS, Huang B. Dynamic imaging of genomic loci in living human cells by an optimized CRISPR/Cas system. Cell. 2013;155:1479–1491. doi: 10.1016/j.cell.2013.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Xu F, Zhu C, Ji J, Zhou X, Feng X, Guang S. Dual sgRNA-directed gene knockout using CRISPR/Cas9 technology in Caenorhabditis elegans. Sci Rep. 2014;4:7581. doi: 10.1038/srep07581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho SW, Kim S, Kim Y, Kweon J, Kim HS, Bae S, Kim JS. Analysis of off-target effects of CRISPR/Cas-derived RNA-guided endonucleases and nickases. Genome Res. 2014;24:132–141. doi: 10.1101/gr.162339.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu VT, Weber T, Wefers B, Wurst W, Sander S, Rajewsky K, Kühn R. Increasing the efficiency of homology-directed repair for CRISPR-Cas9-induced precise gene editing in mammalian cells. Nat Biotechnol. 2015;33:543–548. doi: 10.1038/nbt.3198. [DOI] [PubMed] [Google Scholar]
- Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, Zhang F. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahlem TJ, Hoshijima K, Jurynec MJ, Gunther D, Starker CG, Locke AS, Weis AM, Voytas DF, Grunwald DJ. Simple methods for generating and detecting locus-specific mutations induced with TALENs in the zebrafish genome. PLoS Genet. 2012;8:e1002861. doi: 10.1371/journal.pgen.1002861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahlman JE, Abudayyeh OO, Joung J, Gootenberg JS, Zhang F, Konermann S. Orthogonal gene knockout and activation with a catalytically active Cas9 nuclease. Nat Biotechnol. 2015 doi: 10.1038/nbt.3390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang Y, Jia G, Choi J, Ma H, Anaya E, Ye C, Shankar P, Wu H. Optimizing sgRNA structure to improve CRISPR-Cas9 knockout efficiency. Genome Biol. 2015;16:280. doi: 10.1186/s13059-015-0846-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doench JG, Fusi N, Sullender M, Hegde M, Vaimberg EW, Donovan KF, Smith I, Tothova Z, Wilen C, Orchard R, Virgin HW, Listgarten J, Root DE. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat Biotechnol. 2016 doi: 10.1038/nbt.3437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doench JG, Hartenian E, Graham DB, Tothova Z, Hegde M, Smith I, Sullender M, Ebert BL, Xavier RJ, Root DE. Rational design of highly active sgRNAs for CRISPR-Cas9-mediated gene inactivation. Nat Biotechnol. 2014;32:1262–1267. doi: 10.1038/nbt.3026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott B, Richardson C, Winderbaum J, Nickoloff JA, Jasin M. Gene conversion tracts from double-strand break repair in mammalian cells. Mol Cell Biol. 1998;18:93–101. doi: 10.1128/mcb.18.1.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Essletzbichler P, Konopka T, Santoro F, Chen D, Gapp BV, Kralovics R, Brummelkamp TR, Nijman SMB, Bürckstümmer T. Megabase-scale deletion using CRISPR/Cas9 to generate a fully haploid human cell line. Genome Res. 2014;24:2059–2065. doi: 10.1101/gr.177220.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farboud B, Meyer BJ. Dramatic enhancement of genome editing by CRISPR/Cas9 through improved guide RNA design. Genetics. 2015;199:959–971. doi: 10.1534/genetics.115.175166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Findlay GM, Boyle EA, Hause RJ, Klein JC, Shendure J. Saturation editing of genomic regions by multiplex homology-directed repair. Nature. 2014;513:120–123. doi: 10.1038/nature13695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frock RL, Hu J, Meyers RM, Ho YJ, Kii E, Alt FW. Genome-wide detection of DNA double-stranded breaks induced by engineered nucleases. Nat Biotechnol. 2015;33:179–186. doi: 10.1038/nbt.3101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fusi N, Smith I, Doench J, Listgarten J. In Silico Predictive Modeling of CRISPR/Cas9 guide efficiency. 2015 No. biorxiv;021568v1. [Google Scholar]
- Fu Y, Foden JA, Khayter C, Maeder ML, Reyon D, Joung JK, Sander JD. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat Biotechnol. 2013;31:822–826. doi: 10.1038/nbt.2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gantz VM, Jasinskiene N, Tatarenkova O, Fazekas A, Macias VM, Bier E, James AA. Highly efficient Cas9-mediated gene drive for population modification of the malaria vector mosquito Anopheles stephensi. Proc Natl Acad Sci U S A. 2015;112:E6736–6743. doi: 10.1073/pnas.1521077112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glemzaite M, Balciunaite E, Karvelis T, Gasiunas G, Grusyte MM, Alzbutas G, Jurcyte A, Anderson EM, Maksimova E, Smith AJ, Lubys A, Zaliauskiene L, Siksnys V. Targeted gene editing by transfection of in vitro reconstituted Streptococcus thermophilus Cas9 nuclease complex. RNA Biol. 2015;12:1–4. doi: 10.1080/15476286.2015.1017209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gratz SJ, Ukken FP, Rubinstein CD, Thiede G, Donohue LK, Cummings AM, O’Connor-Giles KM. Highly specific and efficient CRISPR/Cas9-catalyzed homology-directed repair in Drosophila. Genetics. 2014;196:961–971. doi: 10.1534/genetics.113.160713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heigwer F, Kerr G, Boutros M. E-CRISP: fast CRISPR target site identification. Nat Methods. 2014;11:122–123. doi: 10.1038/nmeth.2812. [DOI] [PubMed] [Google Scholar]
- Hinz JM, Laughery MF, Wyrick JJ. Nucleosomes Inhibit Cas9 Endonuclease Activity in Vitro. Biochemistry (Mosc) 2015;54:7063–7066. doi: 10.1021/acs.biochem.5b01108. [DOI] [PubMed] [Google Scholar]
- Housden BE, Valvezan AJ, Kelley C, Sopko R, Hu Y, Roesel C, Lin S, Buckner M, Tao R, Yilmazel B, Mohr SE, Manning BD, Perrimon N. Identification of potential drug targets for tuberous sclerosis complex by synthetic screens combining CRISPR-based knockouts with RNAi. Sci Signal. 2015;8:rs9. doi: 10.1126/scisignal.aab3729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou Z, Zhang Y, Propson NE, Howden SE, Chu LF, Sontheimer EJ, Thomson JA. Efficient genome engineering in human pluripotent stem cells using Cas9 from Neisseria meningitidis. Proc Natl Acad Sci U S A. 2013;110:15644–15649. doi: 10.1073/pnas.1313587110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu PD, Lander ES, Zhang F. Development and applications of CRISPR-Cas9 for genome engineering. Cell. 2014;157:1262–1278. doi: 10.1016/j.cell.2014.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu PD, Scott DA, Weinstein JA, Ran FA, Konermann S, Agarwala V, Li Y, Fine EJ, Wu X, Shalem O, Cradick TJ, Marraffini LA, Bao G, Zhang F. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol. 2013;31:827–832. doi: 10.1038/nbt.2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang WY, Fu Y, Reyon D, Maeder ML, Tsai SQ, Sander JD, Peterson RT, Yeh JRJ, Joung JK. Efficient genome editing in zebrafish using a CRISPR-Cas system. Nat Biotechnol. 2013;31:227–229. doi: 10.1038/nbt.2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer V, Shen B, Zhang W, Hodgkins A, Keane T, Huang X, Skarnes WC. Off-target mutations are rare in Cas9-modified mice. Nat Methods. 2015;12:479. doi: 10.1038/nmeth.3408. [DOI] [PubMed] [Google Scholar]
- Jasin M. Genetic manipulation of genomes with rare-cutting endonucleases. Trends Genet TIG. 1996;12:224–228. doi: 10.1016/0168-9525(96)10019-6. [DOI] [PubMed] [Google Scholar]
- Jiang F, Taylor DW, Chen JS, Kornfeld JE, Zhou K, Thompson AJ, Nogales E, Doudna JA. Structures of a CRISPR-Cas9 R-loop complex primed for DNA cleavage. Science. 2016 doi: 10.1126/science.aad8282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M, Jiang F, Taylor DW, Sternberg SH, Kaya E, Ma E, Anders C, Hauer M, Zhou K, Lin S, Kaplan M, Iavarone AT, Charpentier E, Nogales E, Doudna JA. Structures of Cas9 endonucleases reveal RNA-mediated conformational activation. Science. 2014;343:1247997. doi: 10.1126/science.1247997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karvelis T, Gasiunas G, Young J, Bigelyte G, Silanskas A, Cigan M, Siksnys V. Rapid characterization of CRISPR-Cas9 protospacer adjacent motif sequence elements. Genome Biol. 2015;16:253. doi: 10.1186/s13059-015-0818-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Bae S, Park J, Kim E, Kim S, Yu HR, Hwang J, Kim JI, Kim JS. Digenome-seq: genome-wide profiling of CRISPR-Cas9 off-target effects in human cells. Nat Methods. 2015;12:237–243. doi: 10.1038/nmeth.3284. 1 p following 243. [DOI] [PubMed] [Google Scholar]
- Kim D, Kim S, Kim S, Park J, Kim JS. Genome-wide target specificities of CRISPR-Cas9 nucleases revealed by multiplex Digenome-seq. Genome Res. 2016;26:406–4015. doi: 10.1101/gr.199588.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, Um E, Cho SR, Jung C, Kim H, Kim JS. Surrogate reporters for enrichment of cells with nuclease-induced mutations. Nat Methods. 2011;8:941–943. doi: 10.1038/nmeth.1733. [DOI] [PubMed] [Google Scholar]
- Kim S, Kim D, Cho SW, Kim J, Kim JS. Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Res. 2014;24:1012–1019. doi: 10.1101/gr.171322.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinstiver BP, Pattanayak V, Prew MS, Tsai SQ, Nguyen NT, Zheng Z, Joung JK. High-fidelity CRISPR-Cas9 nucleases with no detectable genome-wide off-target effects. Nature. 2016 doi: 10.1038/nature16526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinstiver BP, Prew MS, Tsai SQ, Nguyen NT, Topkar VV, Zheng Z, Joung JK. Broadening the targeting range of Staphylococcus aureus CRISPR-Cas9 by modifying PAM recognition. Nat Biotechnol. 2015a;33:1293–1298. doi: 10.1038/nbt.3404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinstiver BP, Prew MS, Tsai SQ, Topkar VV, Nguyen NT, Zheng Z, Gonzales APW, Li Z, Peterson RT, Yeh JRJ, Aryee MJ, Joung JK. Engineered CRISPR-Cas9 nucleases with altered PAM specificities. Nature. 2015b;523:481–485. doi: 10.1038/nature14592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koller BH, Kim HS, Latour AM, Brigman K, Boucher RC, Scambler P, Wainwright B, Smithies O. Toward an animal model of cystic fibrosis: targeted interruption of exon 10 of the cystic fibrosis transmembrane regulator gene in embryonic stem cells. Proc Natl Acad Sci U S A. 1991;88:10730–10734. doi: 10.1073/pnas.88.23.10730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotani H, Taimatsu K, Ohga R, Ota S, Kawahara A. Efficient Multiple Genome Modifications Induced by the crRNAs, tracrRNA and Cas9 Protein Complex in Zebrafish. PloS One. 2015;10:e0128319. doi: 10.1371/journal.pone.0128319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larcher T, Lafoux A, Tesson L, Remy S, Thepenier V, François V, Le Guiner C, Goubin H, Dutilleul M, Guigand L, Toumaniantz G, De Cian A, Boix C, Renaud JB, Cherel Y, Giovannangeli C, Concordet JP, Anegon I, Huchet C. Characterization of dystrophin deficient rats: a new model for Duchenne muscular dystrophy. PloS One. 2014;9:e110371. doi: 10.1371/journal.pone.0110371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemmens B, van Schendel R, Tijsterman M. Mutagenic consequences of a single G-quadruplex demonstrate mitotic inheritance of DNA replication fork barriers. Nat Commun. 2015;6:8909. doi: 10.1038/ncomms9909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang X, Potter J, Kumar S, Zou Y, Quintanilla R, Sridharan M, Carte J, Chen W, Roark N, Ranganathan S, Ravinder N, Chesnut JD. Rapid and highly efficient mammalian cell engineering via Cas9 protein transfection. J Biotechnol. 2015;208:44–53. doi: 10.1016/j.jbiotec.2015.04.024. [DOI] [PubMed] [Google Scholar]
- Lin Y, Cradick TJ, Brown MT, Deshmukh H, Ranjan P, Sarode N, Wile BM, Vertino PM, Stewart FJ, Bao G. CRISPR/Cas9 systems have off-target activity with insertions or deletions between target DNA and guide RNA sequences. Nucleic Acids Res. 2014;42:7473–7485. doi: 10.1093/nar/gku402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malina A, Cameron CJF, Robert F, Blanchette M, Dostie J, Pelletier J. PAM multiplicity marks genomic target sites as inhibitory to CRISPR-Cas9 editing. Nat Commun. 2015;6:10124. doi: 10.1038/ncomms10124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM. RNA-guided human genome engineering via Cas9. Science. 2013;339:823–826. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maruyama T, Dougan SK, Truttmann MC, Bilate AM, Ingram JR, Ploegh HL. Increasing the efficiency of precise genome editing with CRISPR-Cas9 by inhibition of nonhomologous end joining. Nat Biotechnol. 2015;33:538–542. doi: 10.1038/nbt.3190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mei Y, Wang Y, Chen H, Sun ZS, Ju XD. Recent Progress in CRISPR/Cas9 Technology. J Genet Genomics Yi Chuan Xue Bao. 2016;43:63–75. doi: 10.1016/j.jgg.2016.01.001. [DOI] [PubMed] [Google Scholar]
- Ménoret S, De Cian A, Tesson L, Remy S, Usal C, Boulé JB, Boix C, Fontanière S, Crénéguy A, Nguyen TH, Brusselle L, Thinard R, Gauguier D, Concordet JP, Cherifi Y, Fraichard A, Giovannangeli C, Anegon I. Homology-directed repair in rodent zygotes using Cas9 and TALEN engineered proteins. Sci Rep. 2015;5:14410. doi: 10.1038/srep14410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno-Mateos MA, Vejnar CE, Beaudoin JD, Fernandez JP, Mis EK, Khokha MK, Giraldez AJ. CRISPRscan: designing highly efficient sgRNAs for CRISPR-Cas9 targeting in vivo. Nat Methods. 2015;12:982–988. doi: 10.1038/nmeth.3543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller M, Lee CM, Gasiunas G, Davis TH, Cradick TJ, Siksnys V, Bao G, Cathomen T, Mussolino C. Streptococcus thermophilus CRISPR-Cas9 Systems Enable Specific Editing of the Human Genome. Mol Ther J Am Soc Gene Ther. 2015 doi: 10.1038/mt.2015.218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller U. Ten years of gene targeting: targeted mouse mutants, from vector design to phenotype analysis. Mech Dev. 1999;82:3–21. doi: 10.1016/s0925-4773(99)00021-0. [DOI] [PubMed] [Google Scholar]
- Nishimasu H, Ran FA, Hsu PD, Konermann S, Shehata SI, Dohmae N, Ishitani R, Zhang F, Nureki O. Crystal structure of Cas9 in complex with guide RNA and target DNA. Cell. 2014;156:935–949. doi: 10.1016/j.cell.2014.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paix A, Folkmann A, Rasoloson D, Seydoux G. High Efficiency, Homology-Directed Genome Editing in Caenorhabditis elegans Using CRISPR-Cas9 Ribonucleoprotein Complexes. Genetics. 2015;201:47–54. doi: 10.1534/genetics.115.179382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pattanayak V, Lin S, Guilinger JP, Ma E, Doudna JA, Liu DR. High-throughput profiling of off-target DNA cleavage reveals RNA-programmed Cas9 nuclease specificity. Nat Biotechnol. 2013;31:839–843. doi: 10.1038/nbt.2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahdar M, McMahon MA, Prakash TP, Swayze EE, Bennett CF, Cleveland DW. Synthetic CRISPR RNA-Cas9-guided genome editing in human cells. Proc Natl Acad Sci U S A. 2015;112:E7110–7117. doi: 10.1073/pnas.1520883112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Cong L, Yan WX, Scott DA, Gootenberg JS, Kriz AJ, Zetsche B, Shalem O, Wu X, Makarova KS, Koonin EV, Sharp PA, Zhang F. In vivo genome editing using Staphylococcus aureus Cas9. Nature. 2015;520:186–191. doi: 10.1038/nature14299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat Protoc. 2013;8:2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren X, Yang Z, Xu J, Sun J, Mao D, Hu Y, Yang SJ, Qiao HH, Wang X, Hu Q, Deng P, Liu LP, Ji JY, Li JB, Ni JQ. Enhanced specificity and efficiency of the CRISPR/Cas9 system with optimized sgRNA parameters in Drosophila. Cell Rep. 2014;9:1151–1162. doi: 10.1016/j.celrep.2014.09.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shmakov S, Abudayyeh OO, Makarova KS, Wolf YI, Gootenberg JS, Semenova E, Minakhin L, Joung J, Konermann S, Severinov K, Zhang F, Koonin EV. Discovery and Functional Characterization of Diverse Class 2 CRISPR-Cas Systems. Mol Cell. 2015;60:385–397. doi: 10.1016/j.molcel.2015.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skarnes WC, Rosen B, West AP, Koutsourakis M, Bushell W, Iyer V, Mujica AO, Thomas M, Harrow J, Cox T, Jackson D, Severin J, Biggs P, Fu J, Nefedov M, de Jong PJ, Stewart AF, Bradley A. A conditional knockout resource for the genome-wide study of mouse gene function. Nature. 2011;474:337–342. doi: 10.1038/nature10163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slaymaker IM, Gao L, Zetsche B, Scott DA, Yan WX, Zhang F. Rationally engineered Cas9 nucleases with improved specificity. Science. 2016;351:84–88. doi: 10.1126/science.aad5227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song J, Yang D, Xu J, Zhu T, Chen YE, Zhang J. RS-1 enhances CRISPR/Cas9-and TALEN-mediated knock-in efficiency. Nat Commun. 2016;7:10548. doi: 10.1038/ncomms10548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stemmer M, Thumberger T, Del Sol Keyer M, Wittbrodt J, Mateo JL. CCTop: An Intuitive, Flexible and Reliable CRISPR/Cas9 Target Prediction Tool. PloS One. 2015;10:e0124633. doi: 10.1371/journal.pone.0124633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunagawa GA, Sumiyama K, Ukai-Tadenuma M, Perrin D, Fujishima H, Ukai H, Nishimura O, Shi S, Ohno RI, Narumi R, Shimizu Y, Tone D, Ode KL, Kuraku S, Ueda HR. Mammalian Reverse Genetics without Crossing Reveals Nr3a as a Short-Sleeper Gene. Cell Rep. 2016;14:662–677. doi: 10.1016/j.celrep.2015.12.052. [DOI] [PubMed] [Google Scholar]
- Tabebordbar M, Zhu K, Cheng JKW, Chew WL, Widrick JJ, Yan WX, Maesner C, Wu EY, Xiao R, Ran FA, Cong L, Zhang F, Vandenberghe LH, Church GM, Wagers AJ. In vivo gene editing in dystrophic mouse muscle and muscle stem cells. Science. 2016;351:407–411. doi: 10.1126/science.aad5177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai SQ, Zheng Z, Nguyen NT, Liebers M, Topkar VV, Thapar V, Wyvekens N, Khayter C, Iafrate AJ, Le LP, Aryee MJ, Joung JK. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat Biotechnol. 2015;33:187–197. doi: 10.1038/nbt.3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uddin B, Chen NP, Panic M, Schiebel E. Genome editing through large insertion leads to the skipping of targeted exon. BMC Genomics. 2015;16:1082. doi: 10.1186/s12864-015-2284-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varshney GK, Pei W, LaFave MC, Idol J, Xu L, Gallardo V, Carrington B, Bishop K, Jones M, Li M, Harper U, Huang SC, Prakash A, Chen W, Sood R, Ledin J, Burgess SM. High-throughput gene targeting and phenotyping in zebrafish using CRISPR/Cas9. Genome Res. 2015;25:1030–1042. doi: 10.1101/gr.186379.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vouillot L, Thélie A, Pollet N. Comparison of T7E1 and surveyor mismatch cleavage assays to detect mutations triggered by engineered nucleases. G3 Bethesda Md. 2015;5:407–415. doi: 10.1534/g3.114.015834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Birsoy K, Hughes NW, Krupczak KM, Post Y, Wei JJ, Lander ES, Sabatini DM. Identification and characterization of essential genes in the human genome. Science. 2015;350:1096–1101. doi: 10.1126/science.aac7041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Wei JJ, Sabatini DM, Lander ES. Genetic screens in human cells using the CRISPR-Cas9 system. Science. 2014;343:80–84. doi: 10.1126/science.1246981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Wang Y, Wu X, Wang J, Wang Y, Qiu Z, Chang T, Huang H, Lin RJ, Yee JK. Unbiased detection of off-target cleavage by CRISPR-Cas9 and TALENs using integrase-defective lentiviral vectors. Nat Biotechnol. 2015;33:175–178. doi: 10.1038/nbt.3127. [DOI] [PubMed] [Google Scholar]
- Wong N, Liu W, Wang X. WU-CRISPR: characteristics of functional guide RNAs for the CRISPR/Cas9 system. Genome Biol. 2015;16:218. doi: 10.1186/s13059-015-0784-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Scott DA, Kriz AJ, Chiu AC, Hsu PD, Dadon DB, Cheng AW, Trevino AE, Konermann S, Chen S, Jaenisch R, Zhang F, Sharp PA. Genome-wide binding of the CRISPR endonuclease Cas9 in mammalian cells. Nat Biotechnol. 2014;32:670–676. doi: 10.1038/nbt.2889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Xiao T, Chen CH, Li W, Meyer CA, Wu Q, Wu D, Cong L, Zhang F, Liu JS, Brown M, Liu XS. Sequence determinants of improved CRISPR sgRNA design. Genome Res. 2015;25:1147–1157. doi: 10.1101/gr.191452.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Guell M, Byrne S, Yang JL, De Los Angeles A, Mali P, Aach J, Kim-Kiselak C, Briggs AW, Rios X, Huang PY, Daley G, Church G. Optimization of scarless human stem cell genome editing. Nucleic Acids Res. 2013;41:9049–9061. doi: 10.1093/nar/gkt555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Steentoft C, Hauge C, Hansen L, Thomsen AL, Niola F, Vester-Christensen MB, Frödin M, Clausen H, Wandall HH, Bennett EP. Fast and sensitive detection of indels induced by precise gene targeting. Nucleic Acids Res. 2015;43:e59. doi: 10.1093/nar/gkv126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zetsche B, Gootenberg JS, Abudayyeh OO, Slaymaker IM, Makarova KS, Essletzbichler P, Volz SE, Joung J, van der Oost J, Regev A, Koonin EV, Zhang F. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell. 2015;163:759–771. doi: 10.1016/j.cell.2015.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]