

Abstract
Rationale:
Linear nevus sebaceous syndrome (LNSS) is a rare neurocutaneous syndrome, characterized by nevus sebaceous,central nervous system (CNS), ocular and skeletal abnormalities. The present study describes KRAS somatic mosaic mutation in a case of LNSS with lymphatic malformations (LMs).
Patient concerns:
A 4-month-old female with a clinical diagnosis of LNSS presented with infantile spasms, mental retardation, skull dysplasia, ocular abnormalities, congenital atrial septal defect, and LMs.
Diagnosis:
Cervical ultrasonography revealed a 4.6 × 4.6 × 2.2cm no echo packet with clear boundary in the subcutaneous tissues of the right neck. The neck MRI indicated a cyst in the subcutaneous tissues of the right neck. Whole-exome sequencing revealed a low-level heterozygous mutation of the KRAS gene (c.35C > T; p.G12D, 19%) in the skin lesion sample. This mutation was not present in the blood samples of the patient and her parents.
Interventions:
The patient received sclerotherapy with paicibanil (OK-432) injection for the cyst.
Outcomes:
Following 1 year of treatment, the patient exhibited fewer seizures. The mental and motor development was significantly improved. The patient can currently walk with assistance and speak simple words.
Lessons:
LNSS is a rare, congenital neurocutaneous syndrome consisting of a spectrum of abnormalities involving the skin, central nervous system, eyes, LMs and other systems. LNSS can be caused by postzygotic somatic mutation in the RAS family of genes. Multidisciplinary evaluation and treatment is needed.
Keywords: infantile spasm, KRAS, linear nevus sebaceous syndrome (LNSS), lymphatic malformations, somatic mutation
1. Introduction
Linear nevus sebaceous syndrome (LNSS) is a rare, congenital neurocutaneous syndrome characterized by midline facial skin lesions (linear nevus sebaceous), seizures, and mental retardation.[1] The incidence of this disease is approximately 1 per 10,000 live births.[1] Nevus sebaceous (NS) is a hallmark of LNSS.[2] Previous studies have reported multiple epileptic syndromes in association with LNSS, such as infantile spasms, Lnennox-gastaut syndrome, and Othahara syndrome.[1,3] Multisystem disorders namely, ocular, skeletal, cardiovascular, and urinary defects were also frequently reported in LNSS.[1,4]
LNSS occurs sporadically with no sexual predilection, and is not associated with chromosomal abnormalities. The condition is believed to be a result of somatic mutations.[5] It has been demonstrated that activating RAS mutations can lead to constitutive activation of the RAF-MEK-ERK signaling pathway and result in increased cellular proliferation.[6] Postzygotic somatic mutations in the HRAS, KRAS, and NRAS genes are associated with the incidence of LNSS.[5,7–10] The commonest mutation in LNSS is the c.37G > C (p.Gly13Arg) in the HRAS gene that is present in more than 90% of the reported disease cases.[10]
We report a case of LNSS patient with NS in the scalp and the facial region, infantile spasms, mental retardation, ocular abnormalities, congenital heart disease, and skull dysplasia. The patient exhibited lymphatic malformations (LMs) in the right neck, which has never been documented before, to the best of our knowledge. Whole-exome sequencing analysis detected a somatic mutation in the KRAS gene in exon 1 (c.35C > T; p.G12D).
2. Ethics and consent
Written informed consent was obtained from the patient's guardian (father), who agreed to publication of the patient's condition. The use of clinical specimens and clinical data was reviewed and approved by the ethics committee of the Children's Hospital of Zhejiang University.
3. Methods
Clinical data and imaging analysis were collected by the electronic medical record system of the Children's Hospital of Zhejiang University. A small piece of lesion skin that was obtained from the forehead and peripheral blood samples of the child and her parents were collected. Genomic DNA was extracted from the skin and blood samples according to the manufacturer's protocols (Qiagen Inc, Valencia).
Whole exome sequencing was conducted by Precision MD Shanghai (Shanghai Genome Institute, Shanghai, China). A total of 4676 genes (all known gene of genetic disease, designed by Beijing JinZhun Gene Technology Co., Ltd., Beijing, China) were targeted for capture and deep sequencing, including candidate genes associated with LNSS. Targeted sequence enrichment was carried out using the Agilent SureSelect Target Enrichment Kit (Agilent Technologies Inc., San Francisco). Genomic DNA was prepared for sequencing using a Covaris S1 Ultrasonicator (Covaris, MA). Adaptor-ligated libraries were constructed using Paired-End Genomic DNA kits (Illumina, CA). High throughput sequencing was conducted on a HiSeq2500 sequencer (Illumina Inc., San Diego, CA). Sequencing data were aligned to hg19 using the Burrows–Wheeler Aligner (BWA) software. Polymerase chain reaction (PCR) duplicates were removed with the “samtools” software and variants were identified using the Genome Analysis Toolkit (GATK, version 3.4) software from the (Broad Institute, Cambridge).
4. Results
4.1. Case presentation
A female 4-month-old infant was admitted in the pediatric neurology department of our hospital with new-onset seizures. The seizures were characterized by forward head dropping, rapid arms, and legs jerk. These disorders occurred frequently in clusters, and lasted up to 5 seconds. The patient's development was delayed and was associated with a concomitant inability to raise the head and gaze the target. Physical examination demonstrated well-demarcated, raised, hairless, pink plaques on the left side of the scalp, face, ear, and the neck. An additional plaque was observed in the midline of the face (Fig. 1). Multiple, protuberated, merged masses were noted on the binocular conjunctiva and corneal, notably in the left eye. An ocular pterygium resulted in left eyeball side protrusion (Fig. 2). Grade 2 soft systolic murmurs were recorded in precordia by heart auscultation. The video electroencephalogram (EEG) examination indicated hypsarrhythmia. Magnetic resonance imaging (MRI) of the head demonstrated that the corpus callosum was thin with minimal cortical atrophy (Fig. 3). A 3-dimensional (3D) reconstruction of the head by computerized tomography (CT) demonstrated signs of dysplasia of the brain. The bony part of the left temporal parietal bone was thinner and a multiple calcification of the posterior wall of eyeball was observed (Fig. 4). Echocardiography demonstrated that the atrial septal defect was approximately healed. The X-rays of the chest and limbs were normal. Abdominal ultrasound examination indicated the liver, pancreas, spleen, kidneys, and retroperitoneum were normal. Fundoscopy indicated no obvious turbidity in the anterior chamber and crystal. Flat retina was evident with the exception of a small mass on the left temporal retina. Routine blood tests demonstrated that the main biochemical parameters, the electrolyte levels, the thyroid and parathyroid function, as well as the calcium and phosphorus levels were normal.
Figure 1.
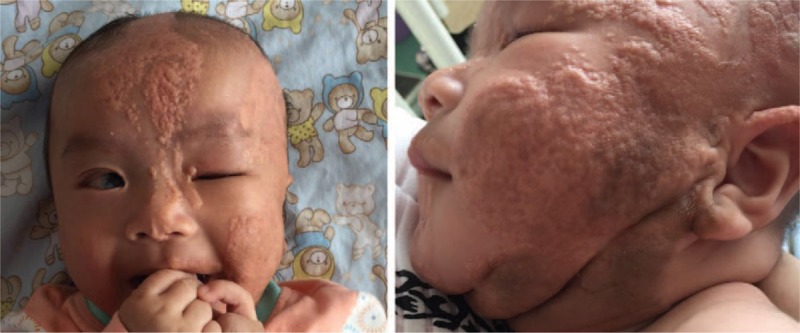
Nevus sebaceous on the left side of scalp, face, ear and neck, and midline of face.
Figure 2.
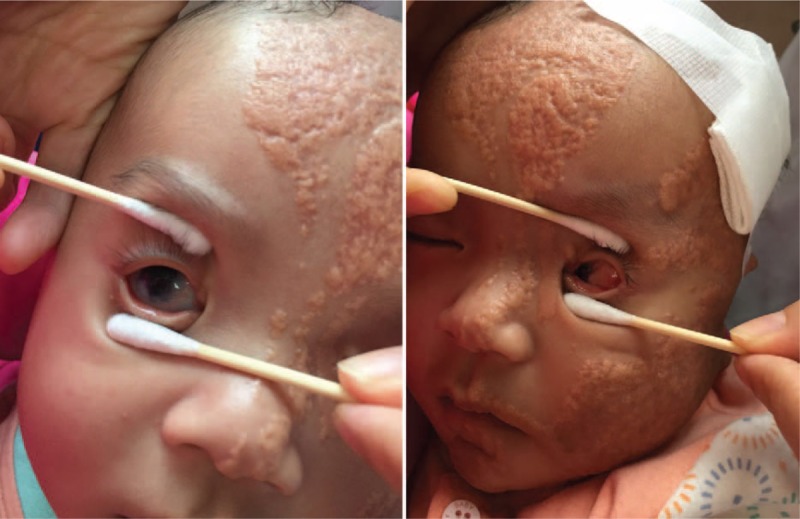
Binocular conjunctiva and corneal exhibit multiple uplift, mutually connected mass, notably in the left eye. An ocular pterygium causes left eyeball side protrusion.
Figure 3.
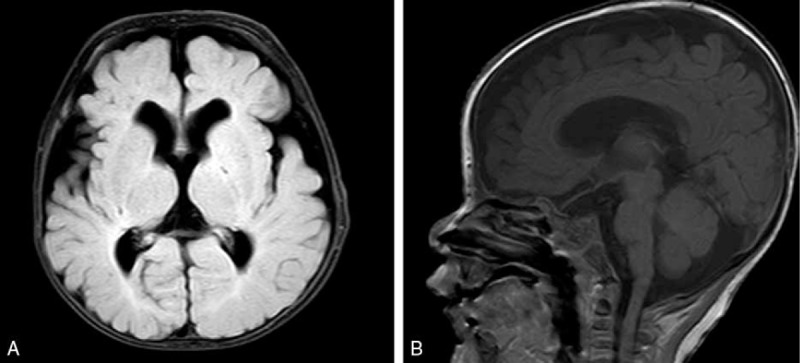
Brain MRI indicates the thinning of the corpus callosum and cortical atrophy. (A) FLAIR sequence, axial. (B) T1-weighted, sagittal.
Figure 4.
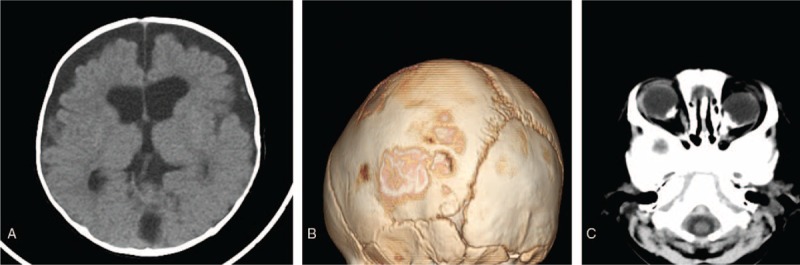
CT-scan and 3D reconstruction of the head indicate cerebral dysplasia (A), maldevelopment of the left temporal parietal bone (B), and multiple calcification of the posterior wall of eyeballs (C).
The infant was born at 40+ 1 weeks of gestation by cesarean section due to intrauterine hypoxia. The Apgar score at birth was normal. The birth weight was 3300 g. Her parents were unrelated, and her mother experienced a normal pregnancy. Following birth, abnormal lesions of the skin and ocular regions were noted. The right side of the neck exhibited a 5 × 5 cm size mass that fluctuated during palpation. A trial septal defect (0.49 cm) was detected by echocardiography. Head MRI indicated a wide anterior temporal space and no abnormal signal in the brain parenchyma. Cervical ultrasonography revealed a 4.6 × 4.6 × 2.2 cm no echo packet with clear boundary in the subcutaneous tissues of the right neck. The neck MRI indicated a cyst with long T1 and T2 signal intensities in the subcutaneous tissues of the right neck. The cyst exhibited dimensions of approximately 29 × 34 × 38 mm (Fig. 5). LMs of the right neck were diagnosed. One month following birth, the patient received sclerotherapy with one dose of paicibanil (OK-432) injection for the cyst every month. The cervical mass disappeared following 2 months of therapy. The patient underwent a skin lesion biopsy in the temporal scalp. The pathological findings were in accordance with the changes noted in case of NS (Fig. 6).
Figure 5.
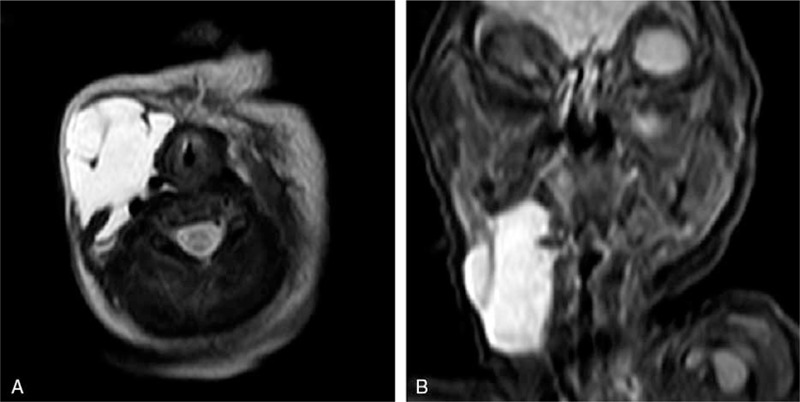
Cervical MRI demonstrates the presence of lymphatic malformation on the right side of the neck. (A) T2-weighted, axial. (B) T2-weighted, coronal.
Figure 6.
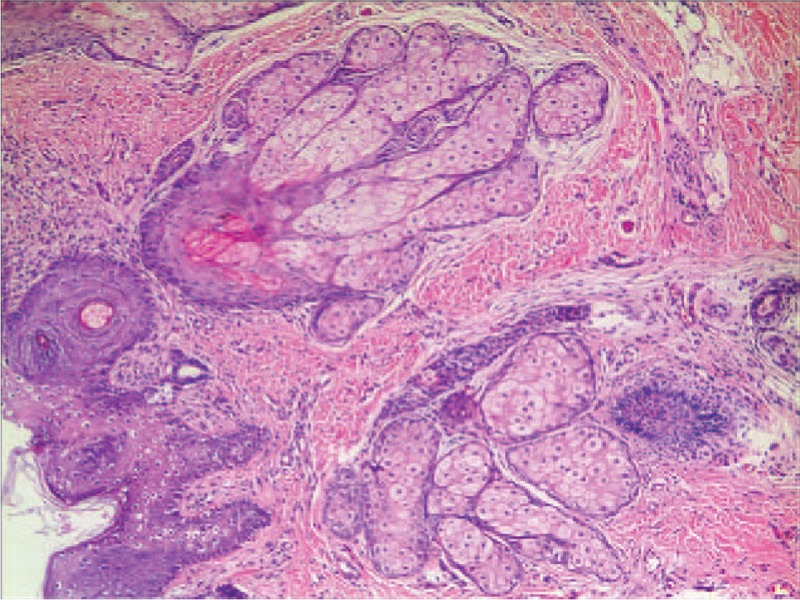
Histopathology of nevus sebaceous reveals acanthosis and a large number of mature/nearly mature sebaceous glands in the dermis (HE staining, magnification × 50).
LNSS complicated with infantile spasms was diagnosed. The seizures were controlled by administration of adrenocorticotropic hormone (ACTH), topiramate, and clobazam. Following 1 year of treatment, the patient exhibited fewer seizures. The mental and motor development was significantly improved. The patient can currently walk with assistance and speak simple words.
4.2. Genetic analysis
Whole-exome sequencing analysis revealed a mutation in the KRAS gene in exon 1 (c.35C > T; p.G12D) in the skin lesion sample. The allele frequency of the altered nucleotide in the skin lesion was 19% (sequencing depth: 35/183) according to the amplicon-based library (Fig. 7). Deep sequencing of the DNA extracted from the patient's blood samples and her parents demonstrated the absence of KRAS mutations. Sanger-based sequencing was not conducted due to the low level of the mosaic mutation.
Figure 7.
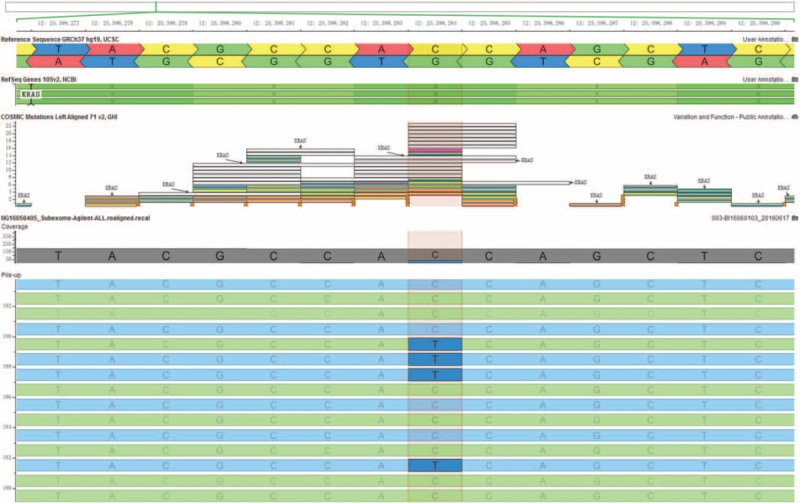
A low-level heterozygous mutation in the KRAS gene (c.35C > T; p.G12D, 19%) was detected in a skin lesion sample.
5. Discussion
LNSS is a subtype of the epidermal nevus syndrome. It has been traditionally defined as the triad of NS, seizures, and mental retardation.[1] Several reports have shown that this syndrome encompasses a broad spectrum of neurologic, ophthalmic, skeletal, cardiovascular, and urologic defects.[1,3,4] The syndrome has been reported in the past under various names, such as linear nevus sebaceous of Jadassohn (NSJ), Schimmelpenning–Feufstein–Mims syndrome (SFM), and Jadassohn nevus Phakomatosis (JNP).
NS is a hamartoma that manifests in the epidermis, hair follicles, and sebaceous and apocrine glands.[1] It usually presents at birth as a yellow-orange to pink, finely papillomatous, alopecic plaque that is often oval and/or linear. The lesions vary in size, but are unusually large and exophytic. This disorder commonly appears in the scalp and face, whereas it occasionally occurs in the neck and trunk.[1,4] NS exhibits 3 dermatological stages: stage 1 lasts from birth to puberty and is associated with a small and hairless lesion; stage 2 occurs during puberty, and is characterized by massive development of the sebaceous glands, papillomatous epidermal hyperplasia, and maturation of the apocrine glands due to androgen stimulation; stage 3 presents with a possible malignant transformation of the lesion and the most common pathology is basal cell epithelioma.[4] Giant sebaceous nevi exhibit a 5% to 20% chance of cutaneous neoplastic development, notably the development of basal cell carcinoma (more than 90%).[4] Malignant transformation usually occurs in the third stage, rarely before the age of 30 years. Surgical excision of the skin lesion remains controversial to date, although many reports suggest complete excision and reconstruction for purposes of cosmetic purposes and reducing the risk of developing malignancy.[11] The present patient exhibited a large NS in the scalp and face, and the dermatologist recommended surgical excision before puberty.
More than 60% of patients with LNSS demonstrate associated neurologic abnormalities.[1,3] Seizures and mental retardation are present in 67% and 61% of LNSS cases, respectively.[3] Seizures usually occur in the first year following birth, which is usually resistant to conventional antiepileptic drugs.[12] Hemimegalencephaly, as noted in the present case, is reported in approximately 72% of the patients.[3] The patient of the present study was 4 months old and exhibited nodding spasms, hypsarrhythmia on EEG, and mental retardation, which could be diagnosed as infantile spasms. Her head MRI only showed a thin region of corpus callosum and minimal cortical atrophy, in the absence of hemimegalencephaly. She received ACTH, topiramate, and clobazam treatment. Following 1 year of treatment, the patient demonstrated a full recovery.
LNSS is often complicated with ocular abnormalities, which have an incidence of approximately 50% to 59%.[13] These ocular abnormalities include colobomas of the lid, strabismus, cataracts, corneal vascularization, ocular hemangiomas, generalized retinal degeneration, bulbar dermoids with pannus formation, iris and chorioretinal colobomas, hamartomas of the eyelid, asymmetry of orbital bones, and esotropia.[1,13,14] The present case exhibited corneal vascularization on bilateral eyes, and a vascularized epibullar mass in the left cornea suggesting a diagnosis of choristoma. Fundus examination revealed a small mass on the left eye temporal retina. The ophthalmologist considered that her eyesight might be normal, as the patient exhibited the ability to see objects and distinguish colors. However, the left eye corneal pannus and mass may grow afterwards, covering the pupil, leading to vision problem. Therefore, surgical removal of the mass is recommended.
LMs are traditionally called lymphangiomas. These disorders comprise low-flow vascular anomalies of the lymphatic system that occur in 1 of 2000 to 4000 live births.[15,16] LMs can be divided into 3 types: macrocystic (diameter > 1 cm), microcystic (diameter <1 cm), and mixed.[15] More than 70% of LMs are observed in the head and neck regions. In case of the disease manifesting in the aerodigestive tract, life-threatening airway obstruction may occur, which requires immediate surgery.[16] Ultrasound and MRI are important diagnostic approaches for LMs in order to identify the location size and extent of the lesions.[15] Various treatment methods have been described in the literature, such as surgery, sclerotherapy, laser therapy, sildenafil, propranolol, sirolimus, and vascularized lymph node transfer.[15] In this case, the LMs disappeared and no recurrence was noted during the 1-year follow-up following 2 treatment cycles of sclerotherapy with paicibanil. To the best of our knowledge, this is the first report of LNSS associated with LMs.
In the present study, a KRAS mutation (c.35C > T; p.G12D) was identified in the skin lesion sample of the patient with LNSS, but not in the peripheral blood samples of the patient and her parents. This finding is consistent with the previous reports by Groesser et al[5] and Wang et al.[5,9] Discordance for LNSS from monozygotic twins supported the concept of a postzygotic somatic mutation in the etiology of the syndrome.[17,18] Several reports suggested that LNSS is associated with oncogenic mutations of the RAS family genes.[5,7,9,19–21] These mutations were only present in the cells of NS and were not found in the normal skin cells and/or blood. Groesser et al[5] analyzed the tissues from 2 unrelated LNSS patients with different RAS mutations: one carried a mutation in the HRAS gene (G13R) and another in the KRAS gene (G12D). Lim et al[8] identified a somatic NRAS mutation (Q61R) in a 7-year-old Caucasian female with LNSS. Mutant cells carrying these mutations demonstrated constitutive activation of the RAF-MEK-ERK signaling pathway and resulted in increased cellular proliferation.[6,22] Mutations in the RAS signaling pathway can cause lymphedema, chylothorax, and/or chylous ascites. It is postulated that the RAS pathway may play a role in the maintenance of the lymphatic system.[23] In the present case, LNSS associated with LMs was possibly caused by the somatic mutation of the KRAS gene in the early embryo. It is widely accepted that KRAS gene mutations play important roles in the regulation of the intracellular signaling pathways involved in lung cancer and the clinical response of the patients to the medical drug treatment for lung cancer.[24] Currently, no successful anti-KRAS therapies exist for lung cancer; however, RAF/MEK/ERK inhibitors have shown preliminary exciting activity for a wide range of RAS-mutated malignancies.[25,26] Thus, we propose that pharmacological targeting of the RAF/MEK/ERK pathway with MEK inhibitors may provide novel future medical therapies for the treatment of LNSS.
In conclusion, LNSS is a rare, congenital neurocutaneous syndrome that can include LMs. LNSS can be caused by postzygotic somatic mutation in the RAS family of genes. In the present study, we described the first case of LNSS with LMs, for which a somatic mosaic KRAS mutation (c.35C > T; p.G12D) was identified in the skin lesion of the patient.
Footnotes
Abbreviations: 3D = three-dimensional, ACTH = adrenocorticotropic hormone, BWA = Burrows–Wheeler Aligner, CT = computed tomography, EEG = electroencephalogram, JNP = Jadassohn nevus Phakomatosis, LMs = lymphatic malformations, LNSS = linear nevus sebaceous syndrome, MRI = magnetic resonance imaging, NS = nevus sebaceous, NSJ = nevus sebaceus of Jadassohn, PCR = polymerase chain reaction, SFM = Schimmelpenning–Feufstein–Mims syndrome.
The authors disclose no conflicts of interest.
References
- [1].Eisen DB, Michael DJ. Sebaceous lesions and their associated syndromes: part II. J Am Acad Dermatol 2009;61:563–78. quiz 579–580. [DOI] [PubMed] [Google Scholar]
- [2].Herman TE, Siegel MJ. Hemimegalencephaly and linear nevus sebaceous syndrome. J Perinatol 2001;21:336–8. [DOI] [PubMed] [Google Scholar]
- [3].van de Warrenburg BP, van Gulik S, Renier WO, et al. The linear naevus sebaceus syndrome. Clin Neurol Neurosurg 1998;100:126–32. [DOI] [PubMed] [Google Scholar]
- [4].Yu KC, Lalwani AK. Inner ear malformations and hearing loss in linear nevus sebaceous syndrome. Int J Pediatr Otorhinolaryngol 2000;56:211–6. [DOI] [PubMed] [Google Scholar]
- [5].Groesser L, Herschberger E, Ruetten A, et al. Postzygotic HRAS and KRAS mutations cause nevus sebaceous and Schimmelpenning syndrome. Nat Genet 2012;44:783–7. [DOI] [PubMed] [Google Scholar]
- [6].Aoki Y, Niihori T, Narumi Y, et al. The RAS/MAPK syndromes: novel roles of the RAS pathway in human genetic disorders. Hum Mutat 2008;29:992–1006. [DOI] [PubMed] [Google Scholar]
- [7].Levinsohn JL, Tian LC, Boyden LM, et al. Whole-exome sequencing reveals somatic mutations in HRAS and KRAS, which cause nevus sebaceus. J Invest Dermatol 2013;133:827–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Lim YH, Ovejero D, Sugarman JS, et al. Multilineage somatic activating mutations in HRAS and NRAS cause mosaic cutaneous and skeletal lesions, elevated FGF23 and hypophosphatemia. Hum Mol Genet 2014;23:397–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Wang H, Qian Y, Wu B, et al. KRAS G12D mosaic mutation in a Chinese linear nevus sebaceous syndrome infant. BMC Med Genet 2015;16:101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Aslam A, Salam A, Griffiths CE, et al. Naevus sebaceus: a mosaic RASopathy. Clin Exp Dermatol 2014;39:1–6. [DOI] [PubMed] [Google Scholar]
- [11].Margulis A, Bauer BS, Corcoran JF. Surgical management of the cutaneous manifestations of linear nevus sebaceus syndrome. Plast Reconstr Surg 2003;111:1043–50. [DOI] [PubMed] [Google Scholar]
- [12].Pavone L, Curatolo P, Rizzo R, et al. Epidermal nevus syndrome: a neurologic variant with hemimegalencephaly, gyral malformation, mental retardation, seizures, and facial hemihypertrophy. Neurology 1991;41((Pt 1)):266–71. [DOI] [PubMed] [Google Scholar]
- [13].Park JM, Kim DS, Kim J, et al. Epibulbar complex choristoma and hemimegalencephaly in linear sebaceous naevus syndrome. Clin Exp Dermatol 2009;34:e686–9. [DOI] [PubMed] [Google Scholar]
- [14].Kausar A, Zafar SN, Altaf S, et al. Ophthalmic manifestations of linear nevus sebaceous/organoid nevus syndrome. J Coll Phys Surg Pak 2015;25:220–2. [PubMed] [Google Scholar]
- [15].Bagrodia N, Defnet AM, Kandel JJ. Management of lymphatic malformations in children. Curr Opin Pediatr 2015;27:356–63. [DOI] [PubMed] [Google Scholar]
- [16].Zhou Q, Zheng JW, Mai HM, et al. Treatment guidelines of lymphatic malformations of the head and neck. Oral Oncol 2011;47:1105–9. [DOI] [PubMed] [Google Scholar]
- [17].Schworm HD, Jedele KB, Holinski E, et al. Discordant monozygotic twins with the Schimmelpenning-Feuerstein-Mims syndrome. Clin Genet 1996;50:393–7. [DOI] [PubMed] [Google Scholar]
- [18].Rijntjes-Jacobs EG, Lopriore E, Steggerda SJ, et al. Discordance for Schimmelpenning-Feuerstein-Mims syndrome in monochorionic twins supports the concept of a postzygotic mutation. Am J Med Genet A 2010;152a:2816–9. [DOI] [PubMed] [Google Scholar]
- [19].Bourdeaut F, Herault A, Gentien D, et al. Mosaicism for oncogenic G12D KRAS mutation associated with epidermal nevus, polycystic kidneys and rhabdomyosarcoma. J Med Genet 2010;47:859–62. [DOI] [PubMed] [Google Scholar]
- [20].Kuroda Y, Ohashi I, Enomoto Y, et al. A postzygotic NRAS mutation in a patient with Schimmelpenning syndrome. Am J Med Genet A 2015;167a:2223–5. [DOI] [PubMed] [Google Scholar]
- [21].Sun BK, Saggini A, Sarin KY, et al. Mosaic activating RAS mutations in nevus sebaceus and nevus sebaceus syndrome. J Invest dermatol 2013;133:824–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Dereure O. MAP-kinase pathway activation in nevus sebaceous and Schimmelpenning syndrome. Ann Dermatol Venereol 2013;140:326–7. [DOI] [PubMed] [Google Scholar]
- [23].Brouillard P, Boon L, Vikkula M. Genetics of lymphatic anomalies. J Clin Invest 2014;124:898–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Luo SY, Lam DC. Oncogenic driver mutations in lung cancer. Transl Respir Med 2013;1:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Li BT, Janku F, Patel MR, et al. First-in-class oral ERK1/2 inhibitor Ulixertinib (BVD-523) in patients with advanced solid tumors: final results of a phase I dose escalation and expansion study. J Clin Oncol 2017;35(15 suppl):2508–12508. [Google Scholar]
- [26].Chenard-Poirier M, Kaiser M, Boyd K, et al. Results from the biomarker-driven basket trial of RO5126766 (CH5127566), a potent RAF/MEK inhibitor, in RAS- or RAF-mutated malignancies including multiple myeloma. J Clin Oncol 2017;35(15 suppl):2506–12506. [Google Scholar]