

Abstract
Rationale:
Immunoglobulin G4-related disease (IgG4-RD) is characterized by tumor-like lesions, a dense lymphoplasmacytic infiltrate rich in IgG4-positive plasma cells, storiform fibrosis, and obliterative phlebitis. IgG4-RD has been described in a variety of organ systems; however, it rarely involves the central nervous system.
Patient concerns:
A 17-year-old woman visited our clinic with a complaint of blurred vision for the past 5 months. She also reported a painless right submandibular mass that had been present for 1 year. Her best-corrected visual acuity (BCVA) was 2.0 LogMAR, with an almost total visual field defect in the right eye.
Diagnoses:
Magnetic resonance imaging (MRI) revealed lobulated parasellar tumors with perineural spreading along branches of the trigeminal nerves causing right optic nerve compression. A craniotomy with tumor removal and submandibular gland biopsy was performed. Histopathological analysis of the tumor revealed stromal fibrosis with atypical lymphoid infiltrations. Histopathological and immunohistochemical analysis of the submandibular gland confirmed the diagnosis of IgG4-RD.
Interventions:
The patient was administered 500mg/d of pulse methylprednisolone for 3 days, 500mg of intravenous rituximab every 2 weeks (for a total of 2 doses), and 500mg of intravenous pulse cyclophosphamide every month (for a total of 3 doses).
Outcomes:
Two months after the initiation of immunosuppressive therapy, the patient's BCVA returned to 0.1 LogMAR with visual field defect recovery. The follow-up MRI showed the almost complete disappearance of the previously contrast-enhanced lesions.
Lessons:
Herein, we report a rare case of IgG4-RD presenting as a parasellar tumor and present a review of the related literature. Based on the case report, we propose that aggressive therapy with glucocorticoid, rituximab, and cyclophosphamide may potentially be useful for treating such cases.
Keywords: cerebral pseudotumor, histopathology, IgG4-related disease, optic neuropathy
1. Introduction
Immunoglobulin G4-related disease (IgG4-RD) is a recently recognized fibroinflammatory condition characterized by tumor-like lesions, a dense lymphoplasmacytic infiltrate rich in IgG4-positive plasma cells, storiform fibrosis, and obliterative phlebitis.[1] IgG4-RD has been described in a variety of organ systems: the pancreas, biliary tree, liver, salivary glands, lacrimal glands, periorbital tissues, lymph nodes, thyroid, retroperitoneum, kidneys, aorta, lungs, prostate, meninges, and pituitary gland.[2] The fibroinflammatory lesions frequently form a mass that may destroy the involved organ, mimicking malignancy.[3] Histopathological and immunohistochemical analyses of biopsy specimens remain the cornerstone in the diagnosis of IgG4-RD. Elevated concentrations of IgG4 in serum are also helpful in diagnosing IgG4-RD, but approximately 30% of patients with IgG4-RD have normal serum IgG4 levels.[4] Patients often respond well to corticosteroid therapy.[5] Herein, we report a rare case of IgG4-RD presenting as a parasellar tumor that showed a good response to corticosteroid and immunosuppressive therapy.
2. Methods and results
2.1. Case report
2.1.1. Patient information and clinical findings
A 17-year-old adolescent girl with a 9-year history of atopic dermatitis visited our clinic with a complaint of blurred vision for the past 5 months. She also reported a painless right submandibular mass that had been present for 1 year. Physical examination revealed enlargement of bilateral submandibular glands with right side predominance; moreover, an enlarged lymph node, about 1.2 cm in size, was noted on the right side of the neck. The neurologic examination disclosed decreased sensation to pin-prick over the right perinasal area. Her best-corrected visual acuity (BCVA) was 2.0 LogMAR, with an almost total visual field defect in the right eye (Fig. 1 A). The visual evoked potentials showed absent response in the right eye and prolonged P100 latency (130 ms) in the left eye, which was suggestive of functional perturbation of the bilateral prechiasmatic optic pathway, with the right side being worse. Magnetic resonance imaging (MRI) revealed lobulated good contrast-enhancing tumors in the bilateral parasellar regions with extracranial extension along the ophthalmic (V1), maxillary (V2), and mandibular (V3) branches of the bilateral trigeminal nerves (Fig. 2A–F). The tumor had a larger component on the right and extended into the right orbital apex through the superior orbital fissure. There was a contrast-enhancing soft tissue in the right orbital apex suggesting perineural spreading of the tumor along the nasociliary branch of the ophthalmic nerve (V1) with compression of the right optic nerve. The perineural spreading of the parasellar tumors was also evident with erosion and widening of the right superior orbital fissure and the bilateral foramen rotundum and the foramen ovale visible on the post-surgical follow-up Computed Tomography (not shown).
Figure 1.

Right eye visual field (A) before and (B) after treatment with glucocorticoid, rituximab, and cyclophosphamide. (A) The patient initially had an almost total visual field defect. (B) The visual field defect exhibited dramatic recovery 2 months after the initiation of immunosuppressive therapy.
Figure 1 (Continued).
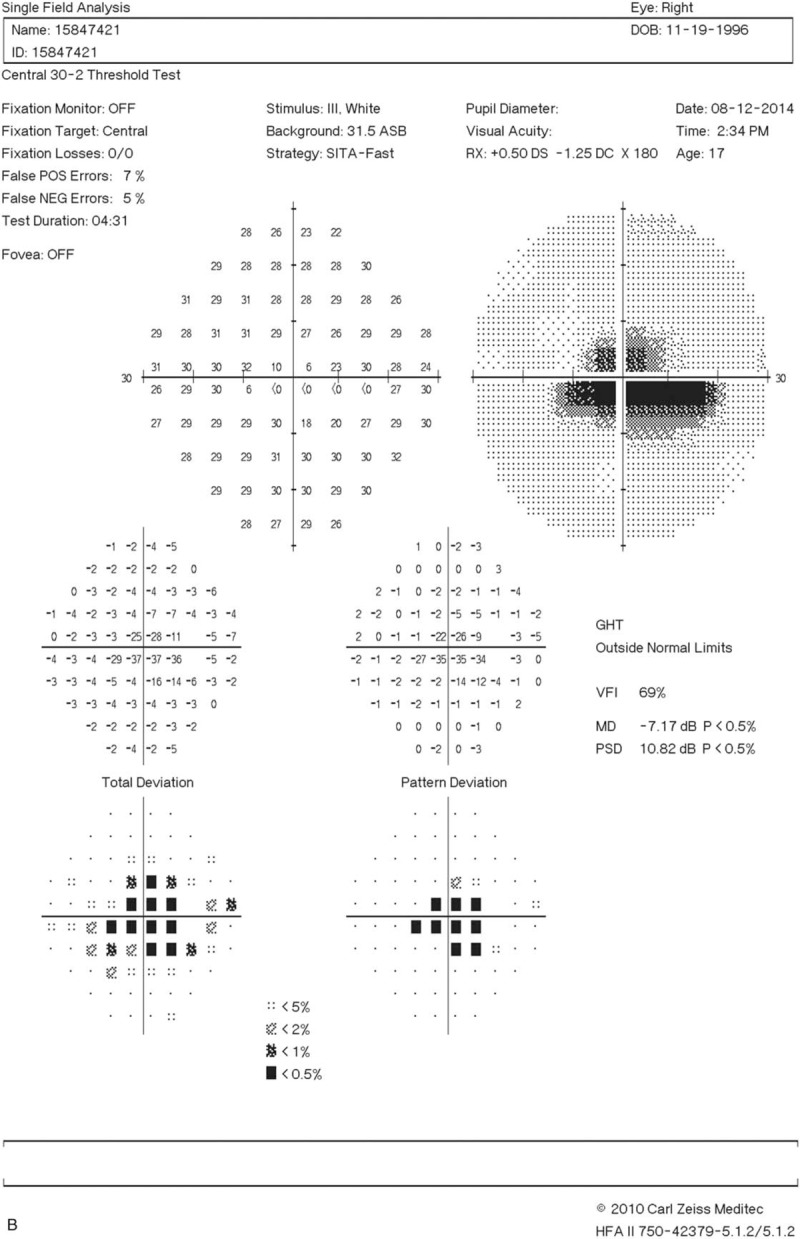
Right eye visual field (A) before and (B) after treatment with glucocorticoid, rituximab, and cyclophosphamide. (A) The patient initially had an almost total visual field defect. (B) The visual field defect exhibited dramatic recovery 2 months after the initiation of immunosuppressive therapy.
2.1.2. Diagnostic assessment
A craniotomy with tumor removal and submandibular gland biopsy was performed.
Histopathological examination of the parasellar tumor revealed stromal fibrosis with atypical lymphoid infiltration (Fig. 3). Abundant plasma cells and focal eosinophils infiltration were noted as well. Those atypical lymphoid infiltrates consisted largely of CD3-positive T cells with focal CD20-positive B cells (data not shown). The immunohistochemical (IHC) study for kappa and lambda light chains did not reveal a monoclonal B-cell population. An IHC stain for anaplastic lymphoma kinase-1 (ALK-1) was negative. Histopathological examination of the submandibular gland showed dense infiltration of lymphocytes and plasma cells with storiform fibrosis; in addition, phlebitis without obliteration of the lumen was noted (Fig. 4). An IHC study of the submandibular gland revealed CD138-positive plasma cells with mixed CD3 and CD20-positive small lymphocyte infiltration. Increased IgG4-positive plasma cell (>50 IgG4+ cells/HPF with IgG4+/IgG+ cell ratio >40%) infiltration was also noted. According to the above histopathological features and immunophenotypic findings of the submandibular gland, a diagnosis of immunoglobulin G4-related sialadenitis was considered. However, the patient's visual acuity did not recover after tumor removal and, moreover, elevated serum IgG4 (IgG4: 511 mg/dL, IgG: 1770 mg/dL) was noted. Hence, a diagnosis of IgG4-related cerebral pseudotumor with compressive optic neuropathy was considered.
Figure 2.
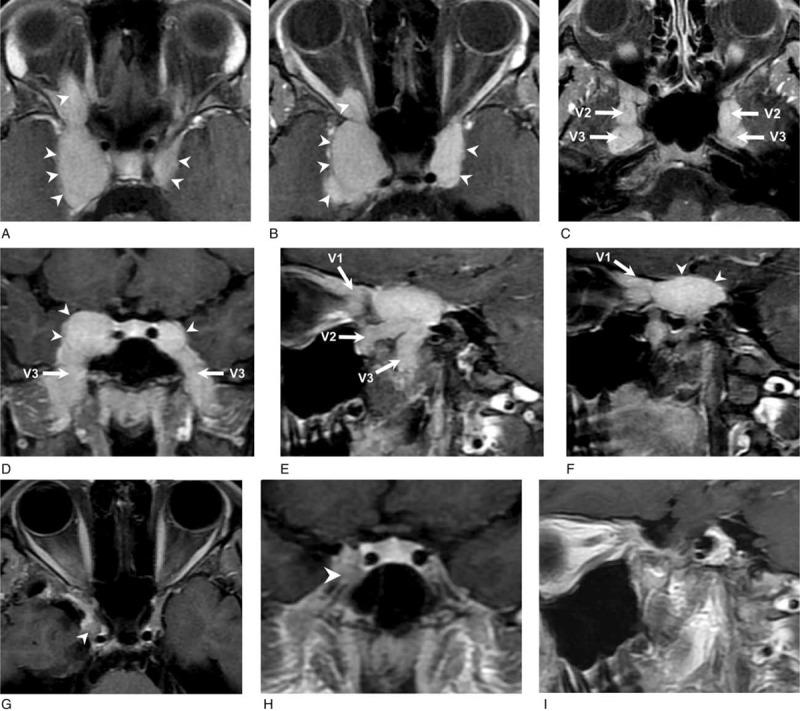
Brain MRI gadolinium-enhanced T1-weighted images with fat-saturation before (A–F) and after treatment with glucocorticoid, rituximab, and cyclophosphamide (G–I). (A–C) Axial images at about the sellar floor level from superior to inferior. Lobulated tumors with homogeneously good contrast-enhancement in the bilateral parasellar regions (arrow head) were found. The tumors extended extracranially along the ophthalmic (V1), maxillary (V2, arrow), and mandibular (V3, arrow) branches of the bilateral trigeminal nerves. The tumor had a larger component on the right; moreover, the right superior orbital fissure was eroded and widened. There was a contrast-enhancing soft tissue in the right orbital apex suggesting perineural spreading of the tumor along the nasociliary branch of the ophthalmic nerve (V1) with compression of the right optic nerve. (D) Coronal image at the pituitary gland level. Lobulated tumors with homogeneously good contrast-enhancement in the bilateral parasellar regions were found (arrow head). The tumors extended extracranially through the bilateral foramens rotundum and foramens ovale along the maxillary (V2) and mandibular (V3, arrow) branches of the trigeminal nerve, respectively. (E, F) Sagittal images at the right parasellar level from lateral to medial. The contrast-enhancing tumor extended extracranially through the superior orbital fissure, foramen rotundum, and foramen ovale along the ophthalmic (V1, arrow), maxillary (V2, arrow), and mandibular (V3, arrow) branches of the right trigeminal nerve, respectively. (G) Axial image at the sellar floor level and (H) coronal image at the pituitary gland level. The contrast-enhancing parasellar tumors mostly disappeared, with a small, residual, and less contrast-enhancing nodule in the lateral part of the right cavernous sinus (arrow head). (I) Sagittal image at the right parasellar level. The extracranially extended tumor resolved. MRI = magnetic resonance imaging.
Figure 3.
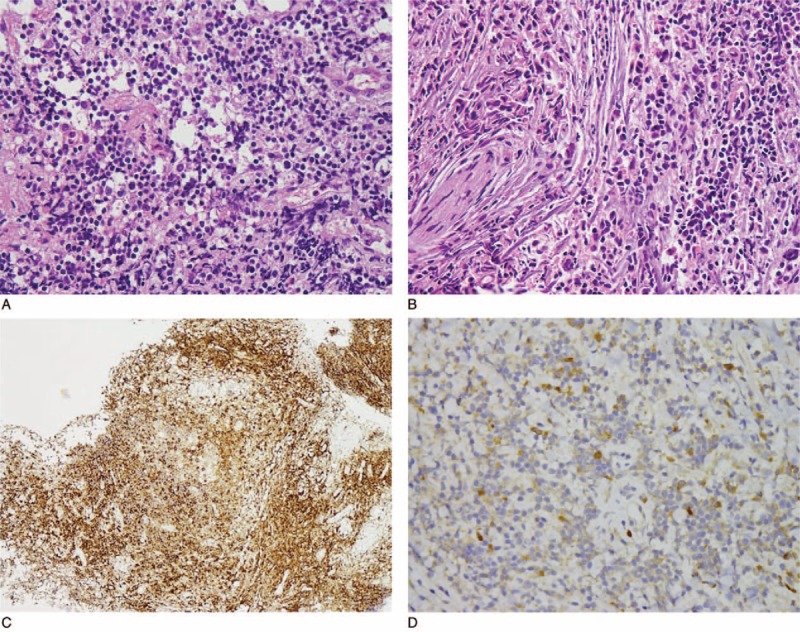
Atypical lymphoid infiltrate at parasellar lesion, revealing (A) mixed population and including (B) some plasma cells in fibrous stroma (hematoxylin and eosin, ×400). (C) Abundant plasma cells were highlighted by CD138 immunostain (×100). (D) Immunohistochemistry revealed a few IgG4-positive cells (×400).
2.1.3. Therapeutic intervention, follow-up, and outcomes
The patient was then administered 500 mg/d of pulse methylprednisolone for 3 days, 500 mg of intravenous rituximab every 2 weeks (for a total of 2 doses), and 500 mg of intravenous pulse cyclophosphamide every month (for a total of 3 doses). The dose of prednisolone was kept at 10 mg/d for maintenance therapy. Two months after the initiation of immunosuppressive therapy, the patient's BCVA returned to 0.1 LogMAR (Fig. 5) with visual field defect recovery (Fig. 1 B). The follow-up MRI showed the almost complete disappearance of the previously contrast-enhanced lesions with only a small amount of residual soft tissue in the lateral part of the right cavernous sinus (Fig. 2G–I).
Figure 4.
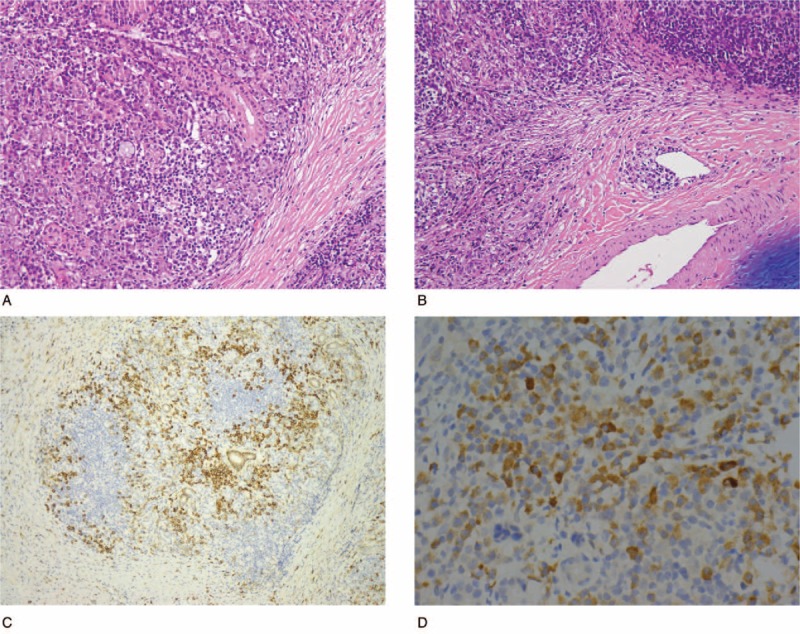
Histopathology of the submandibular gland. Dense lymphoplasmacytic infiltrate with storiform fibrosis was shown (A), as was the presence of phlebitis without obliteration (B) (hematoxylin and eosin, ×200). (C) Plasma cells were highlighted by CD138 immunostain (×100). (D) Immunohistochemistry revealed increased IgG4-positive cells (>50 IgG4+ cells/HPF) (×400).
Figure 5.
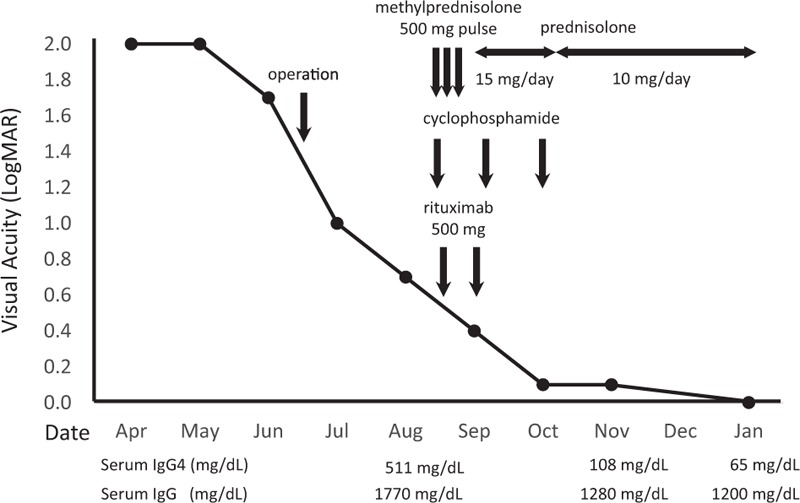
Visual acuity changes during the treatment course. The patient underwent craniotomy with tumor removal in June 2014. Her visual acuity only improved to 0.7 LogMAR in the following 2 months. She was then administered pulse methylprednisolone at 500 mg/d for 3 days, 500 mg of intravenous rituximab every 2 weeks (for a total of 2 doses), and 500 mg of intravenous pulse cyclophosphamide every month (for a total of 3 doses). The dose of prednisolone was kept at 10 mg/d for maintenance therapy. Two months after the initiation of immunosuppressive therapy, her BCVA returned to 0.1 LogMAR. The serum IgG4 also decreased after the initiation of immunosuppressive therapy.
3. Discussion and conclusions
Orbital inflammatory pseudotumors (OIPTs) often manifest as unilateral extraocular myositis,[6,7] causing periorbital pain, restriction of extraocular muscle movement, proptosis, or diplopia. Such OIPTs are commonly restricted to the orbit; however, the extraorbital extension of pseudotumors with dural-based infiltrate involving the cranial fossa and cavernous sinus has been reported.[6–9] In recent years, it has been suggested that some cases of OIPTs may be related to the entity known as IgG4-RD.[8,9] Moreover, IgG4-RD has previously been described in the central nervous system, with hypophysitis[10] and pachymeningitis[11] being the most common manifestations. Here we present the first case, to our knowledge, of an IgG4-related cerebral pseudotumor with perineural spreading along branches of the trigeminal nerves causing compressive optic neuropathy. The histopathology of the parasellar tumor revealed atypical lymphoid infiltration, mimicking lymphoid neoplasm. This atypical lymphocytic infiltrate was composed predominantly of T cells, with scattered aggregates of B-cells; moreover, no immunoglobulin light chain restriction was identified. Hence, the possibility of lymphoid malignancy was excluded. There was abundant plasma cell infiltration with stromal fibrosis and scattered eosinophils were also noted, observations which were compatible with the histopathological findings indicating IgG4-RD. To date, no international consensus criteria have been established for the diagnosis of extra-pancreatic IgG4-RD. A variety of cutoff points, ranging from >10 to >50 IgG4-positive plasma cells per high-power field (cells/HPF), have been proposed with regard to different organ sites.[1] Some studies advocate using the ratio of IgG4-positive plasma cells[5] to IgG-positive plasma cells to assist in making the diagnosis of extra-pancreatic IgG4-RD, where a ratio >30% to 50% is suggestive of a diagnosis.[12–16] One study, which analyzed the histopathological features of IgG4-related meningeal disease, demonstrated an increased number of IgG4-positive cells ranging from 11.8 to 54.2 cells/HPF.[17] The authors recommend the use of ≥10 IgG4-positive cells/HPF as a minimum criterion for the diagnosis of IgG4-related meningeal disease.[17] In the case presented herein, the immunohistochemical examination of the parasellar tumor revealed 20 IgG4-positive cells/HPF, with the ratio of IgG4/IgG-positive cells being around 10%. Although the lobulated parasellar tumor extended along the ophthalmic (V1), maxillary (V2), and mandibular (V3) branches of the bilateral trigeminal nerves, it did not cause excruciating trigeminal neurological symptoms except for decreased sensitivity to pin-pricks over the right perinasal area, which was supplied by the external nasal branch of the right anterior ethmoidal nerve (from the nasociliary branch of the right ophthalmic nerve). In addition, the patient in this case initially presented with compressive optic neuropathy. At the same time, right submandibular gland swelling was noted along with elevated serum IgG4. The histopathological examination of the submandibular gland suggested the diagnosis of IgG4-RD. Even though the patient had blurred vision for as long as 5 months, with an almost total visual field defect, after corticosteroid and immunosuppressive therapy, the patient's BCVA returned to 0.1 LogMAR with visual field defect recovery. Furthermore, the follow-up MRI showed almost complete remission of the brain tumor. Since autoimmune pancreatitis (AIP) is considered to be the pancreatic manifestation of an IgG4-related systemic disease, the international consensus of diagnostic criteria (ICDC) for AIP has included elevated serum IgG4 levels and the histological changes of other organ involvement (OOI) as findings that can assist in making the diagnosis.[18] Our case also indicates that elevated serum IgG4 levels and OOI can assist in making the diagnosis of IgG4-RD in atypical presentations. Moreover, we propose that aggressive therapy with glucocorticoid, rituximab, and cyclophosphamide may be a potentially beneficial treatment for IgG4-related cerebral pseudotumor.
In summary, we report herein a unique case of IgG4-related cerebral pseudotumor with perineural spreading along branches of the trigeminal nerves causing compressive optic neuropathy. We also identify the histopathological and MRI features of IgG4-related cerebral pseudotumor. Moreover, we propose that aggressive therapy with glucocorticoid, rituximab, and cyclophosphamide may potentially be useful for treating such cases.
Footnotes
Abbreviations: BCVA = best-corrected visual acuity, IgG4-RD = immunoglobulin G4-related disease, IHC = immunohistochemical, MRI = magnetic resonance imaging.
PCW and PTT contributed equally to this work.
Informed consent: Informed consent was obtained from the patient.
The authors do not have any source of funding.
The authors report no conflicts of interest.
References
- [1].Deshpande V, Zen Y, Chan JK, et al. Consensus statement on the pathology of IgG4-related disease. Mod Pathol 2012;25:1181–92. [DOI] [PubMed] [Google Scholar]
- [2].Cheuk W, Chan JK. IgG4-related sclerosing disease: a critical appraisal of an evolving clinicopathologic entity. Adv Anat Pathol 2010;17:303–32. [DOI] [PubMed] [Google Scholar]
- [3].Deshpande V. The pathology of IgG4-related disease: critical issues and challenges. Semin Diagn Pathol 2012;29:191–6. [DOI] [PubMed] [Google Scholar]
- [4].Sah RP, Chari ST. Serologic issues in IgG4-related systemic disease and autoimmune pancreatitis. Curr Opin Rheumatol 2011;23:108–13. [DOI] [PubMed] [Google Scholar]
- [5].Stone JH, Zen Y, Deshpande V. IgG4-related disease. N Engl J Med 2012;366:539–51. [DOI] [PubMed] [Google Scholar]
- [6].Tedeschi E, Ugga L, Caranci F, et al. Intracranial extension of orbital inflammatory pseudotumor: a case report and literature review. BMC Neurol 2016;16:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Kaushik C, Ramakrishnaiah R, Relia N, et al. Radiological case: orbital pseudotumor with cavernous sinus and sellar extension. Appl Radiol 2015;44:46–8. [Google Scholar]
- [8].Haraguchi A, Ando T, Ueki I, et al. A case of compressive optic neuropathy putatively caused by IgG4-related idiopathic orbital inflammation. Acta Med Nagasaki 2012;57:29–32. [Google Scholar]
- [9].Tiegs-Heiden CA, Eckel LJ, Hunt CH, et al. Immunoglobulin G4-related disease of the orbit: imaging features in 27 patients. AJNR Am J Neuroradiol 2014;35:1393–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Shimatsu A, Oki Y, Fujisawa I, et al. Pituitary stalk lesions (infundibulo-hypophysitis) associated with immunoglobulin G4-related systemic disease: an emerging clinical entity. Endocr J 2009;56:1033–41. [DOI] [PubMed] [Google Scholar]
- [11].Joshi D, Jager R, Hurel S, et al. Cerebral involvement in IgG4-related disease. Clin Med (Lond) 2015;15:130–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Cheuk W, Yuen HK, Chu SY, et al. Lymphadenopathy of IgG4-related sclerosing disease. Am J Surg Pathol 2008;32:671–81. [DOI] [PubMed] [Google Scholar]
- [13].Kitagawa S, Zen Y, Harada K, et al. Abundant IgG4-positive plasma cell infiltration characterizes chronic sclerosing sialadenitis (Kuttner's tumor). Am J Surg Pathol 2005;29:783–91. [DOI] [PubMed] [Google Scholar]
- [14].Masaki Y, Dong L, Kurose N, et al. Proposal for a new clinical entity, IgG4-positive multiorgan lymphoproliferative syndrome: analysis of 64 cases of IgG4-related disorders. Ann Rheum Dis 2009;68:1310–5. [DOI] [PubMed] [Google Scholar]
- [15].Zen Y, Inoue D, Kitao A, et al. IgG4-related lung and pleural disease: a clinicopathologic study of 21 cases. Am J Surg Pathol 2009;33:1886–93. [DOI] [PubMed] [Google Scholar]
- [16].Zen Y, Onodera M, Inoue D, et al. Retroperitoneal fibrosis: a clinicopathologic study with respect to immunoglobulin G4. Am J Surg Pathol 2009;33:1833–9. [DOI] [PubMed] [Google Scholar]
- [17].Lindstrom KM, Cousar JB, Lopes MB. IgG4-related meningeal disease: clinico-pathological features and proposal for diagnostic criteria. Acta Neuropathol 2010;120:765–76. [DOI] [PubMed] [Google Scholar]
- [18].Shimosegawa T, Chari ST, Frulloni L, et al. International consensus diagnostic criteria for autoimmune pancreatitis: guidelines of the International Association of Pancreatology. Pancreas 2011;40:352–8. [DOI] [PubMed] [Google Scholar]