

Abstract
Kidneys have a major role in normal physiology and metabolic homeostasis. Loss or impairment of kidney function is a common occurrence in several metabolic disorders, including hypertension and diabetes. Chronic kidney disease (CKD) affect nearly 10%of the population worldwide; ranks 18th in the list of causes of death; and contributes to a significant proportion of healthcare costs. The tissue repair and regenerative potential of kidneys are limited and they decline during aging. Recent studies have demonstrated a key role for epigenetic processes and players, such as DNA methylation, histone modifications, noncoding (nc)RNA, and so on, in both kidney development and disease. In this review, we highlight these recent findings with an emphasis on aberrant epigenetic changes that accompany renal diseases, key targets, and their therapeutic value.
Introduction
Analyses of data from genome projects revealed that only a small portion of the transcribed genome encodes protein-coding RNA and most is noncoding [1–4]. ncRNA transcripts act alone or in concert with proteins to bring about gene regulation, which is central to the functional diversification of multiple cell types in higher eukaryotes [5–7]. Aberrant gene regulation is linked to various human pathologies in many organs and tissues during development as well as aging. Kidneys are important organs that help sustain the stable composition of blood, secrete important hormones, contribute to mineral and blood pressure homeostasis and so on. Recent evidence indicates that gene regulatory mechanisms involving both protein- and nonprotein-coding players are key for homeostatic renal function as well as disease [8–11]. Here, we summarize ncRNA as well as epigenetic modifications mediating gene regulation (Fig. 1) and mechanisms specific to mammalian kidneys during development, with a focus on processes that are crucial during the onset and/or progression of renal diseases.
Figure 1.
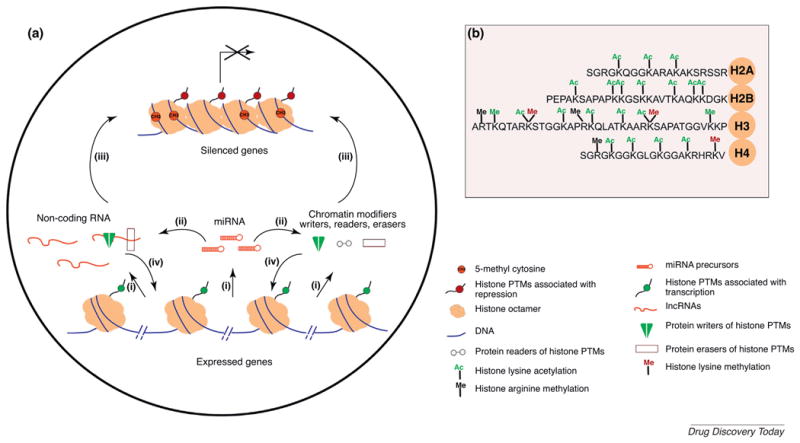
Gene regulation by non-coding RNA and epigenetic modifications.
(a) Mechanisms involving chromatin modifiers and ncRNAs. (i) miRNAs, long (l)ncRNA and mRNAs are transcribed in the nucleus by RNA polymerases. Based on sequence homology, (ii) miRNAs incorporated into miRISC can regulate the levels of mRNAs of chromatin modifier proteins and lncRNAs by either target degradation or translational repression, resulting in the silencing of target gene expression. lncRNAs and epigenetic and/or chromatin modifiers [writers, such as DNA methyl transferases (Dnmts), histone acetyl transferases (HATs), and histone methyl transferases KMTs; readers, such as Bromodomain-containing proteins (e.g., Brd4) and chromo domain-containing proteins (e.g., HP1); and erasers, such as lysine demethylases (Kdms), and histone deacetylases (HDACs)] can regulate the establishment or maintenance of gene repression (iii) or activation (iv) at specific loci alone or in combination, by selective incorporation or removal of post-translational modifications on histone substrates, thereby maintaining preferential access to transcriptional machinery. (b) Well-characterized post-translational modifications (PTMs) on N-terminal tails of core histones (H2A, H2B, H3, and H4). Acetylation (Ac) and methylation (Me) on lysine (K) and arginine (R). Modifications predominantly found at active genes are indicated in green and silenced genes in red.
Noncoding RNAs and gene regulation
Most of the transcribed genome comprises several classes of ncRNAs, including RNAs involved in protein translation and ribosomal biogenesis, spliceosomal RNAs, small nucleolar (sno) RNAs, and relatively newly described classes of ncRNA, such as endogenous small ncRNAs [i.e., miRNAs, small interfering (si) RNAs, and piwi-interacting (pi)RNAs], long ncRNAs (lncRNAs), and circular RNAs (circRNA)]. In this review, we focus on the impact of the latter class of RNAs and proteins that are involved in their biogenesis on renal gene regulation.
RNA interference (RNAi) and the impact of small RNAs on gene regulation have been detailed in several excellent reviews [12,13]. Whereas small ncRNAs have important roles in the modulation of homeostatic gene regulation, their levels are often perturbed during disease conditions and, thus, they have emerged as an important tool for diagnosis and/or therapeutic targeting [14–16]. Small ncRNAs are an expanding universe that includes the prototype, miRNAs [17], and several new classes, such as agotrons [18]. miRNAs are small endogenous RNAs that are produced from hairpin loop-containing long primary miRNAs in the nucleus [19]. These are then processed into precursor miRNAs and are eventually processed to mature miRNAs that inhibit protein synthesis, mainly by targeting the 3′ untranslated region (UTR) of mRNAs. Recent studies demonstrated that some miRNAs act via binding to the 5′ UTR as well as coding sequences (CDS) [20,21]. Imperfect sequence complementarity of miRNA binding to target mRNA sequence leads to translational repression and/or mRNA degradation via deadenylation and exonucleolytic decay. Canonical miRNA biogenesis requires the action of RNase III class enzymes, such as nuclear Drosha and nucleocytoplasmic Dicer. An alternative biogenesis pathway involves mirtrons that bypass Drosha and utilizes splicing to generate pre-miRNAs [22].
miRNAs in kidney development and homeostasis
Several independent studies have revealed the role of mature miRNAs in podocyte homeostasis [23–26]. Conditional deletion of Dicer in the podocytes of murine kidneys via NPSH2 (Podocin) promoter-driven Cre recombinase resulted in glomerular abnormalities, wasting, and proteinuria, ultimately leading to death resulting from kidney failure with associated glomerulosclerosis and fibrosis [23–25]. Several miRNAs belonging to the miR-30 family [23,25] as well as miR-23b, -24, and -26a [24], were found to be downregulated in Dicer-conditional knockout (cKO) glomeruli. Dicer deletion was accompanied by a concomitant upregulation of mRNAs with target sequences of the miR-30 family, such as Vimentin (Vim), Advanced glycosylation end-product-specific receptor (Ager), and Heat-shock protein 20 (Hsp20) [25]. Inducible loss of Drosha in postnatal kidneys recapitulated the phenotype of podocyte-specific Dicer-deficient kidneys, including proteinuria and renal failure [26].
The small RNA pathway appears to have an important role in the survival of the progenitor population during nephrogenesis [27,28]. Loss of Dicer in early metanephric mesenchyme resulted in severe renal dysgenesis partly because of the premature loss of Cbp/p300-interacting transactivator with Glu/Asp-rich carboxy-terminal domain 1 (Cited1), a key transcription factor involved in kidney development, as well as increased levels of the proapoptotic protein Bim [28]. By contrast, perturbation of miRNA biogenesis in the ureteric bud derivatives, via targeting either Dgrc8, a Drosha-associated protein component of the nuclear miRNA microprocessor complex, or Dicer, led to the development ofcystic and hydronephrotic phenotypes [27,29–31]. Deletion of Dicer expression in paired box 8 (Pax8)-expressing cells led to glomerulocystic disease. Downregulation of beta-catenin and upregulation of glycogen synthase kinase 3β (GSK3β) levels were also observed [32]. Two recent studies reported profound effects of Dicer deletion in Foxd1+ progenitor cells, which give rise to renal stroma during development [33,34]. Loss of Dicer in renal stromal progenitors resulted in the disruption of their patterning, nephrogenesis, and abnormal podocyte differentiation and vascular patterning [33]. Among a list of miRNAs that were downregulated in Dicer-deficient stromal cells, miR-214, -199a-5p, and -199a-3p had been previously shown to regulate stromal cell functions ex vivo, including WNT pathway activation, migration, and proliferation. Phua et al. [34] reported dysregulated apoptosis via increased proapoptotic proteins, such as Bim, in Dicer-deficient Foxd1+ progenitor cells, leading to defects in nephrogenesis and vascular patterning. Although Dicer function was linked to its miRNA maturation functions in most of these studies, miRNA-independent functions of Dicer might also have a role in the observed phenotypes [35]. A single study of the role of the miR-17∼92 cluster during kidney development indicated that, although nephron-lineage specification was unaltered in the absence of miR-17∼92, nephron progenitor proliferation was severely affected, resulting in a reduced number of nephrons [36]. As a result of nephron-specific loss of miR-17∼92 function, renal disease akin to chronic kidney disease (CKD) manifested as early as 6 weeks of age. Future studies are required to pinpoint the role of an array of small RNAs in kidney development and homeostasis using loss- and gain-of-function approaches.
Modulation of miRNAs in renal fibrosis
Several studies have demonstrated that levels of miRNAs are altered in various renal diseases and, in many cases, they have a causative role in the pathophysiology of the disease. Several key miRNAs have emerged as potential biomarkers (Table 1) and/or therapeutic targets (Table 2). CKD is common and is associated with high morbidity and mortality worldwide. In the USA in 2014, CKD had a prevalence of 14.8%, with a Medicare spending of more than US$50 billion for adults aged 65 years and older (www.usrds.org/adr.aspx). Common diseases, such as diabetes, hypertension, obesity, and cardiovascular syndrome, are important risk factors for CKD. Irrespective of the nature of the initial insult, renal fibrosis is a common occurrence in CKD and several decades of research have indicated that transforming growth factor beta (TGF-β) has a crucial role in this process. The expression of several miRNAs is altered during pathological renal fibrosis. miR-214 and miR-21 were found to be upregulated during renal damage in multiple models of renal injury [37]. Functional studies indicated that miR-214 and miR-21 are key players in renal fibrosis, as demonstrated by the finding that a deficiency of genes encoding miR-214 and miR-21 led to a 93% and 75% reduction in the fibrotic phenotype, respectively, in a mouse model of renal fibrosis following unilateral ureteral obstruction (UUO) [38]. Furthermore, subcutaneous delivery of anti-miR-214 recapitulated the effect of miR-214 deficiency during renal fibrosis (86% relative to control anti-miR). In another study, miR-21 was found to be highly enriched in fibrotic kidneys in mouse models of fibrosis as well as in human transplant kidneys with nephropathy [39]. In this study, ablation of miR-21 expression via either a gene KO strategy or antago-miR led to a reduction in kidney fibrosis. Peroxisome proliferator-activated receptor-α (PPAR-α) was found to be a direct key target of miR-21 in renal fibrosis models and effects of antagomiR-21 were abrogated upon PPAR-α deficiency, thereby linking miRNA-mediated repression of lipid metabolism to fibrosis [39]. This is in agreement with recent studies linking altered lipid metabolism to renal injury and fibrosis [40,41]. However, in a mouse model of diabetic nephropathy (DN), deletion of miR-21 exacerbated the disease phenotype via increased glomerular injury [42], suggesting that miR-21 has a distinct role in different cell types; thus, a more comprehensive understanding is required for effective therapeutic targeting of miR-21 in CKD.
Table 1. ncRNAs as biomarkers of kidney disease.
| RNA | Kidney disease | Source | Modulation | Species tested | Diagnostic/predictive power | Refs |
|---|---|---|---|---|---|---|
| Small RNA | ||||||
| miR-21, -210, -16, -320 | AKI | Plasma | Increased | Human | Predictive of mortality | [123] |
| miR-138-5p, -1971, -218-1-3p, -489 | AKI | Urine | Increased | Rat | Correlates with degree of renal injury | [124] |
| miR-494 | AKI | Urine | Increased | Human | Predictive of AKI | [71] |
| miR-188,-30a, -30e | Contrast-induced AKI | Plasma | Increased | Human | Early biomarker | [125] |
| miR-10a, -30d | Focal segmental glomerulosclerosis | Urine | Increased | Human | Correlates with kidney injury | [126] |
| miR-21, -216a | CKD | Urine | Increased | Human | Dialysis-free survival | [127] |
| miR-17 | IgAN | Urine | Increased | Human | Predictive of disease | [127] |
| miR-29b, -29c, -93 | IgAN | Urine | Decreased | Human | Correlates with renal fibrosis | [46] |
| miR-155, -146a | IgAN | Urine | Increased | Human | Correlates with renal fibrosis | [128] |
| miR-130a, -145 | DN | Urine exosomes | Increased | Human | Correlates with microalbuminuria in patients with type 1 diabetes mellitus | [129] |
| miR-155, -424 | DN | Urine exosomes | Decreased | Human | Correlates with microalbuminuria in patients with type 1 diabetes mellitus | [129] |
| miR-320c, -6068 | DN | Urine exosomes | Increased | Human | Correlates with disease progression | [130] |
| miR-371-5P, -423-5P, -1224-3p | Lupus nephritis | Peripheral blood mononuclear cells | Increased | Human | Predictive of disease | [131] |
| miR-29c | Lupus nephritis | Urine | Decreased | Human | Correlative of renal fibrosis | [45] |
| miR-125a, -629, -602, -628 | Renal transplant rejection | Renal biopsy | Increased | Human | Correlates with acute rejection | [132] |
| miR-142(3) | Renal transplant dysfunction | Urine | Increased | Human | Predictive of chronic allograft dysfunction, interstitial fibrosis, tubular atrophy | [133] |
| miR-200b, -125b,-99a | Renal transplant dysfunction | Urine | Decreased | Human | Help monitor allograft function; correlate with dysfunction | [134] |
| miR-143(2) | Autosomal dominant PKD | Urine | Increased | Human | Distinguishes from other CKD | [135] |
| Long ncRNA | ||||||
| TapSAKI | AKI | Plasma | Increased | Human | Predictive of 28-day survival | [93] |
| uc002rpc, uc001mng | Renal transplant rejection | Renal biopsy | Increased | Human | Predictive of acute rejection | [136] |
| RP11-354P17.15-001 | Renal transplant rejection | Urine | Increased | Human | Correlate with acute rejection and loss of kidney function | [92] |
Table 2. ncRNAs as therapeutic targets for kidney disease.
| RNA | Target cell type | Target genes | Kidney disease | Observed change from homeostasis | Application/Therapeutic strategy | Refs |
|---|---|---|---|---|---|---|
| Small RNA | ||||||
| miR-150 | Renal tubular epithelial cells, endothelial cells | c-MYB (direct), IGF1R (indirect) | AKI | Slight diminution during AKI | Genetic ablation conferred protection against AKI; treatment modality not tested | [69] |
| miR-24 | Renal tubular epithelial cells, endothelial cells | HMOX1 (direct), H2AX (direct) | AKI | Increased during AKI | Antagonism using LNA; preventative administration conferred protection against AKI in a mouse model | [70] |
| miR-21 | Renal tubular epithelial cells | PTEN (direct), AKT phosphorylation (indirect) | AKI | Increased during AKI; mediator of Xenon preconditioning effect | Exogenous administration might confer protection against AKI; not tested in in vivo models | [72] |
| miR-687 | Renal tubular epithelial cells | PTEN (direct) | AKI | Increased during AKI | Blockade using anti-miR LNA conferred protection during preventative regimen in mouse model of AKI | [73] |
| miR-494 | Renal tubular epithelial cells | ATF3 (direct) | AKI | Increased during AKI | Blockade using lentiviral mediated anti-miR administration conferred renoprotection in a mouse model of AKI | [71] |
| miR-382 | Renal tubular epithelial cells | Kallikrein 5 | CKD | Increased during kidney fibrosis | Antagonism using LNA conferred protection against fibrosis in a mouse model; preventative treatment | [137] |
| miR-21 | Inflammatory macrophages, pericytes | PPARa | CKD | Increased during kidney fibrosis | Antagonism using LNA conferred protection against fibrosis in a mouse model | [39] |
| miR-29 | Mesangial cells, Podocytes | COLLI and COLLIV | CKD | Decreased during kidney fibrosis; renoprotective agents, such as ROCK inhibitor, restored levels of miR-29 | Supplementation could prove beneficial; untested potential | [44] |
| miR-192 | Mesangial cells | ZEB1/2 | DN | Increased during DN | LNA-mediated antagonism alleviated proteinuria in mouse models | [138] |
| miR-23b | Renal tubular epithelial cells | G3BP2 | DN | Decreased in diabetic milieu | Exogenous supplementation of miR-23b or silencing of G3BP2 in diabetic mice alleviated features of fibrosis and albuminuria; similarly, administration of miR-23b antagomiR promoted renal fibrosis in healthy mice | [139] |
| miR-17∼92 | Renal tubular epithelial cells | PKD1, PKD2, HNF1b, PKHD1 | PKD | Increased in cystic kidneys | Genetic deletion suppressed cyst growth | [140] |
| miR-21 | Renal tubular epithelial cells | PDCD4 | PKD | Increased in cystic kidneys | Genetic deletion suppressed cyst growth | [141] |
| miR-148b | Peripheral blood mononuclear cells | C1GALT1 (core 1,β1,3-galactosyltransferase 1) | IgAN | Increased in PBMCs from patients with IgAN | Untested potential applications include reconstitution of miR-148b-silenced PBMCs/bone marrow chimeras during early stages of disease | [142] |
| Long ncRNA | ||||||
| lnc-MGC | Podocytes | ATF3, CUGBP2, PUM2, TNRC6B, HuR, PTEN | DN | Increased in diabetic milieu | LNA-mediated silencing of lnc-MGC ameliorated pathobiological features | [86] |
| Tug1 | Podocytes | PGC1a pathway | DN | Decreased in diabetic milieu | Untested potential applications include supplementation of Tug1 | [87] |
The expression of several miRNAs is downregulated by TGF-β, a potent profibrotic factor. In an UUO model of kidney fibrosis, miR-29 family members (miR-29a/b/c) were identified as key miRNAs that were downregulated by TGF-β signaling in a SMAD family member 3 (Smad3)-dependent manner [43]. In in vitro cultures of renal tubular epithelial cells, overexpression of miR-29 effectively inhibited TGF-β-induced expression of Collagen-I and -III. Similarly,Wang et al. [44] observed that TGF-β1 treatment downregulated miR-29a/b/c in renal proximal tubular epithelial cells (NRK52E), primary mesangial cells, as well as human conditionally immortalized podocytes, with a concomitant increase in the levels of extracellular matrix (ECM) proteins. Overexpression of miR-29a/b/c in both RPTEC cells and podocytes led to the downregulation of ECM genes, such as those encoding Collagen-I and Connective tissue growth factor (CTGF), at both the RNA and protein level. In support of this observation, administration of renoprotective agents, such as a Rho-associated protein C kinase (ROCK) inhibitor, restored the levels of miR-29 in a mouse model of renal fibrosis [44]. Levels of miR-29c in urinary exosomes negatively correlated with renal fibrosis in lupus nephritis, highlighting the potential of miR-29c as a non-invasive biomarker [45]. Lower urinary levels of miR-29 family members, miR-29b and miR-29c, correlated well with proteinuria and renal function in patients with IgA nephropathy (IgAN) [46].
TGF-β-induced tissue fibrosis often entails epithelial to mesenchymal transition (EMT) and several reports have suggested a key role for miRNAs in this process. In human cancer cell lines, miR-200 family members were downregulated during EMT induced by TGF-β [47], whereas overexpression of miR-200 family members as well as miR-205, another miRNA that is significantly downregulated by the TGF-β pathway, was sufficient to prevent EMT via modulation of the transcriptional repressors, ZEB1 and ZEB2 [48]. Similar downregulation of miR-200 family members was reported in animal models of renal fibrosis as well as in cultured renal epithelial cells. In NRK52E cells, treatment with either TGF-β1 or TGF-β2 reduced the expression of three members of the miR-200 family: miR-200a, miR-200b, and miR-200c. Overexpression of either miR-200a or miR-141 resulted in a decrease in the ECM genes encoding Collagen I (ColI), Fibronectin (FN), and alpha smooth muscle actin (aSMA), indicating that these miRNAs act as a cellular break against the fibrotic pathway [49]. miR-200a and miR-141 were also found to be downregulated in another widely used mouse model of renal fibrosis following UUO [50]. Similarly, miR-130b was downregulated by TGF-β [51] and its levels negatively correlated with tubulointerstitial fibrosis and Snail expression, as well as with serum creatinine and proteinuria [52]. Studies have also indicated that TGF-β can positively modulate the expression of miRNAs during renal fibrosis. Expression of miR-192 was upregulated in mesangial cells both in vitro, upon treatment with TGF-β [53], and in vivo in mice glomeruli in diabetic milieu [54]. Feedforward regulation of TGF-β expression by miR-192 was also observed [53]. In proximal tubular epithelial cells, the converse has been reported [55]. Thus, miR-192 has a role in the regulation of the TGF-β pathway and in a cell type and stage-specific manner in DN. In addition, miR-192 modulates several miRs, including miR-216a, -217, and -200b/c, as an amplifier of TGF-β action [56].
Detailed mechanisms and roles of the above miRNAs in renal fibrosis have been reviewed in detail elsewhere, including the vital role of TGF-β signaling in this process [57–59]. Recent studies have suggested that, akin to lower eukaryotes, there is evidence for cell to cell transfer of miRNA in vertebrates [60]. Whether similar interactions occur between different renal cell types during fibrosis and the cellular proteins that might be involved in this process remain to be studied. However, miRNAs have been discovered in urinary exosomes [61,62], one of the component pathways involved in intercellular communication. Detailed evaluation of these exosomes for miRNA and their mRNA targets will offer mechanistic insights into exosome biogenesis and sorting in renal pathologies. In addition to renal fibrosis in the context of DN, the role of ncRNAs in other chronic renal diseases, such as IgAN, lupus nephritis, polycystic kidney disease (PKD), hypertensive nephrosclerosis, and renal transplant nephropathy, is increasingly appreciated and detailed elsewhere [8,11,63–66].
miRNAs as biomarkers and therapeutic targets in acute kidney injury
The cellular abundance of several miRNAs is altered during acute kidney injury (AKI), a debilitating disorder associated with an increased rate of morbidity and mortality, especially in the intensive care setting. miRNAs are emerging as potential biomarkers and therapeutic targets for AKI. In a miRNA profiling study, miR-21, miR17-5p, and miR-106a were found to increase during renal ischemia-reperfusion injury (IRI)-mediated AKI [67]. Interestingly, these miRNAs also marked maintenance and repair phases following injury, suggesting their potential utility for AKI prognosis and risk stratification. ncRNAs that showed promise as AKI biomarkers have been summarized in Table 1.
miRNAs also modulated the outcomes of AKI in murine models, thereby presenting potential therapeutic avenues (Table 2). Postnatal loss of Dicer specifically in renal proximal tubular epithelial cells in mice showed no apparent pathology, but these mice were surprisingly resistant to IRI induced renal injury (IRI) [68]. In this mouse model, cKO of Dicer in proximal tubular cells using phosphenolpyruvate carboxy kinase (PEPCK) promoter-driven Cre recombinase led to protection from renal injury, with less apoptosis and tissue damage and better survival. The findings suggested a pathogenic role for Dicer and associated miRNAs that require Dicer-mediated processing during their biogenesis in the settings of AKI. Several subsequent studies involving the perturbation of individual miRNAs during AKI revealed distinct effects suggesting that miRNAs in general adversely affect the pathophysiology of the disease, corroborating the Dicer cKO study.
A recent study elucidated the protective effects of downregulating miR-150 in mouse models of AKI [69]. In this study, genetic deletion of miR-150 conferred renoprotection in both myocardial infarction-induced AKI as well as a bilateral renal IRI model, via restoration of RNA and protein levels of insulin-like growth factor 1R (IGF1R) and concomitant cellular survival following injury. In a detailed study by Lorenzen et al. [70], the level of miR-24 was found to increase in tubular epithelial as well as endothelial cells following AKI, leading to cellular death. The well-known tissue-protective protein heme-oxygenase (Hmox1) was identified as a direct target of miR-24. In a mouse model of AKI, antagnosim of miR-24 using locked nucleic acid (LNA) ameliorated renal injury when administered 24 h before the induction of injury.. miR-494 is another miRNA that impacts cell survival and death following AKI [71]. Activating transcription factor 3 (ATF3), a key molecule regulating cell survival following stress, is a direct target of miR-494 that is upregulated during AKI. Antagonism of miR-494 following lentiviral-mediated delivery of antisense miR-494 relieved ATF3 repression and conferred renoprotection in a mouse model of AKI.
Several miRNAs that target phosphatase and tensin homology (PTEN) protein have been implicated in the pathophysiology of AKI. Of these, miR-21 was upregulated by xenon preconditioning and conferred renoprotection against lipopolysaccharide-induced AKI via PTEN downregulation [72]. By contrast, in an ischemic AKI model, another PTEN modulator, miR-687, was upregulated in a hypoxia inducible factor-1 alpha (HIF-1α)-dependent manner and LNA-mediated blockade of miR-687 relieved PTEN repression and conferred renoprotection [73]. In light of the study on miR-687 mediated PTEN modulation revealing a protective role of PTEN [73] and the role of miR-21 in promoting renal fibrosis [39], the therapeutic effectiveness and duration of miR-21 supplementation either directly or via xenon pretreatment in AKI settings remain to be addressed. Together, the miRNA studies have furthered our understanding of the complex pathophysiology of AKI, especially identifying key signaling nodes for therapeutic targeting and potential non-invasive biomarkers. Further studies could provide a more complete understanding of the temporal effects of miRNA induction, including any protective effect during AKI.
Long noncoding RNAs in renal disease
lncRNAs are >200 nucleotides long, and are transcribed from a diverse array of genomic loci, including repeat regions, promoters, introns, and intergenic regions [74,75]. Recent large-scale genome-wide identification of lncRNA loci was aided by localizing intergenic regions that have chromatin features (i.e., high promoter H3K4me3 and H3K36me3 across the gene length, ‘K4–K36 domain’) similar to that of actively transcribed genes [76]. ncRNAs originating from these intergenic regions are referred to as long intergenic ncRNA (lincRNA). Although most lncRNAs are not highly conserved, they have been shown to have important cellular functions. Although this review focuses on lncRNAs that have roles in transcriptional and post-transcriptional roles, lncRNAs, such as nuclear-enriched autosomal transcript 1 (NEAT1), serve as structural determinants of subnuclear bodies, such as paraspeckles [77]. Well-characterized lncRNAs that function in transcriptional regulation include Xist, H19, KCNQ1ot1, Hotair, and Repeat A (RepA). Several studies have elucidated distinct mechanisms by which lincRNAs regulate gene expression, although a complete understanding is yet to be achieved [78]. Many lncRNAs were shown to interact with chromatin modifiers, such as Ezh2, a H3K27 methyltransferase, to bring about repressive chromatin modifications. RepA, a lncRNA involved in X chromosome inactivation (XCI),interacts with and recruits Ezh2 to the X chromosome during the initiation of XCI in the course of early embryonic development [79]. Apart from mediating Ezh2-dependent repressive chromatin modifications, a repressive function of lncRNA by direct inhibition of RNA polymerase elongation has also been demonstrated. During cellular response to heat-shock stress, lncRNA B2 SINE RNA directly binds RNA polymerase II, leading to a transcriptional pause [80]. A recent study reported that such a transcriptional pause brought about by lncRNA B2 was relieved at certain loci that encode stress-response genes in a histone methyltransferase-independent function of Ezh2 via modulation of B2 lncRNA stability [81]. Thus, lncRNAs appear to be a component of both positive and negative gene regulation mediated by Ezh2. Although lncRNA transcripts themselves have functional roles in gene regulation, recent evidence also indicates that transcription at lncRNA loci, enhancer activity, and splicing can also have important roles in the gene regulation of neighboring genes [82]. Transactivation functions of lncRNAs were observed in the case of Staufen1 (STAU1)-mediated mRNA decay (SMD) [83]. During SMD, translationally active mRNAs are degraded via target mRNA binding by the RNA-binding protein STAU1, and lncRNAs named ‘half-STAU1-binding site RNAs’ (1/2-sbsRNAs) were found to transactivate this process. Furthermore, some lncRNAs act as miRNA sponges, leading to derepression of miRNA target genes, as in the examples cited below. Following is a summary of the roles of a few lncRNAs in renal disease.
Besides regulating the expression of small RNAs, TGF-β also modulates the levels of lncRNAs in renal cell lineages. TGF-β upregulated the imprinted lncRNA, H19, in the renal tubular epithelial cell line, HK2, as well as in the kidneys in mouse models of renal fibrosis [84]. RNAi-mediated knockdown of lncRNA H19 effectively abrogated an increase in ECM gene expression both in vivo and in vitro. In these models, lncRNA H19 acts as a sponge and sequesters miR-17, thereby relieving the repression of miR-17 targets, such as Fibronectin and Fibronectin type-III domain containing 3A (FNDC3A) [84]. Genome-wide analysis of ncRNA expression in kidneys from Smad3-KO and wild-type mice during UUO and antiglomerular basement membrane glomerulonephritis revealed that 21 lncRNAs were co-expressed in both these models and were regulated by Smad3. Among these, the promoters of ncRNAs (np_17856 and np_5318) were found to have conserved binding sites and enrichment of Smad3. Furthermore, the expression of these RNAs was enhanced in experimental models of CKD and reliant on the presence of Smad3 [85].
lncRNAs are emerging as important players in the pathophysiology of DN. A recent study identified a lncRNA, lnc-MGC, that hosts a megacluster of miRNAs (miR-379–3072) and is differentially upregulated in diabetic kidneys via the endoplasmic reticulum (ER)-stress response pathway [86]. LNA-mediated silencing of lnc-MGC resulted in reduced levels of the host lncRNA as well as the associated miRNA cluster, leading to attenuation of profibrotic gene expression, ECM deposition, and hypertrophy. In another recent study, lncRNA taurine upregulated gene 1 (Tug1) was implicated in the pathobiological response of podocytes in diabetic milieu. Tug1 was repressed in podocytes from diabetic mice with concomitant downregulation of PGC-1α and its pathway genes and, consequently, mitochondrial bioenergetics [87]. Ex-pression of Tug1 appears to confer tissue protection because its overexpression prevented apoptosis and enhanced cellular function following liver injury in a transplant model [88].
In an unbiased profiling study to identify lncRNAs that are modulated during renal tubular epithelial cell responses to hypoxia and inflammatory stimuli, Lin et al. [89] identified several differentially expressed lncRNAs. Of these, modulation of MIR210HG, linc-ATP13A4-8, and linc-KIAA1737-2 abundance was confirmed in human tubular epithelial cells isolated from transplant biopsies as well as in epithelial cells exposed to plasma from patients with sepsis [89]. Although a specific role for these lncRNAs has not been attributed, pathway and loci analyses were indicative of their involvement in biological pathways related to cellular stress and inflammatory response. A recent study identified a lncRNA, Psoriasis susceptibility related-RNA gene induced by Stress (PRINS), as a promoter of regulated on activation, normal T cell expressed and secreted (RANTES) expression in hypoxic renal tubules [90]. Among several differentially modulated lncRNAs identified in a qPCR array methodology, PRINS was shown to transactivate RANTES expression. However, the in vivo role of PRINS in the kidney is yet to be demonstrated.
Several lncRNAs have been identified as potential biomarkers of renal transplant rejection [91–93]. The abundance of three intergenic lncRNAs (LNC-MYH13-3:1, RP11-395P13.3-001, and RP11-354P17.15-001) was strongly altered in the urine of patients with acute rejection [92]. Of these, RP11-354P17.15-001 was induced in epithelial cells by the proinflammatory cytokine, interleukin 6 (IL-6), whose level correlated with higher decline in renal filtration function 1 year post transplantation and level declined with anti-rejection therapies. The same group also identified an intronic lncRNA, Transcript predicting Survival in AKI (TapSAKI) as a predictor of mortality in critically ill patients with AKI [93]. In a study assessing the expression of inactive X-specific transcripts (XIST), a lncRNA involved in female X chromosome inactivation, XIST was found to be a potential urinary biomarker of membranous nephropathy [94]. Glomerular nephritis-associated change in XIST levels was observed only in female patients, whereas XIST transcripts were less abundant and unaltered in males. A recent study reported that the lncRNA, Activated by TGF-β (ATB), is a promising biomarker in predicting acute rejection of renal allografts [94]. Interestingly, lncRNA ATB had been previously shown to be a competing endogenous RNA for miR-200.
Understanding of the role of lncRNAs in other renal diseases, especially in renal cell carcinoma, is rapidly expanding and several excellent studies and reviews have been published on this topic [95–98].
Epigenetic alterations in kidney diseases
The precise and coordinated expression of genes in defined cellular niches is crucial during the development of various tissues, including kidneys, as exemplified by iterative reciprocal inductive morphogenesis patterns dictated by differential gene expression in distinct cell lineages in the developing mammalian kidneys. Gene regulation during organism development and homeostasis is a complex process that is precisely controlled by various stimuli and via access of transcriptional machinery and transcription factors to the genome in multiple cell types. This access is mediated mostly via covalent modifications of both DNA and histones, which tightly pack DNA through nucleosomes. Methylation of DNA at the fifth position of cytosine in CpG dinucleotides is one of the key regulators of gene repression during mammalian embryonic development via prevention of access to transcription factors to target promoters. Compared with lower eukaryotes, the human genome is relatively CpG depleted, with 2.8 ×107 CpGs. Of these, CpG islands that are generally associated with promoters, 3′UTRs, and first exons of protein-coding genes are unmethylated except at a few developmental and pluripotency associated genes. Most other CpGs that are spread across the genome (60-80%) are commonly found in a methylated state [99]. Availability of transcription factors and local chromatin structures are key determinants of cell type-specific gene expression programs, and the methylation of CpGs across the genome offers an efficient way for cell types to remember the transcriptional programs during replication, along with certain other factors. The mammalian DNA methyltransferases, Dnmt3a, Dnmt3b, and Dnmt1, establish and maintain DNA methylation at distinct regions of the genome and have nonoverlapping functions. Dnmt1 and Dnmt3b global KO mice die before term. Dnmt1 KO leads to early lethality at embryonic day (E)9.5 shortly after gastrulation and before the development of the kidney [100]. Dnmt3b KO in mice is embryonically lethal, with mortality occurring around E14.5–18.5 because of multiple developmental defects, including growth impairment and rostral neural tube defects [101]. Although Dnmt3a-KO mice had reduced DNA methylation, newborns were of normal appearance at birth, but died within the first month and were runted. In humans, mutations of DNMT3b cause Immunodeficiency, Centromeric instability, and Facial anomalies syndrome (ICF) [102].
Following genome-wide methylation analyses of fibroblasts from fibrotic human kidneys, Bechtel et al. [103] showed that epigenetic silencing via cytosine methylation at the CpG island of RAS protein activator-like 1 (RASAL1), a member of the RAS-GAP family, was a key molecular event in renal fibrosis. In a mouse model of renal fibrosis induced by folic acid administration, Rasal1 was hypermethylated in a Dnmt1-dependent manner and 5′-aza-cytidine, a DNA methylation inhibitor, effectively decreased fibroblast activation and accumulation of type I collagen (Col1A1) as well as α-SMA-positive myofibroblasts. Furthermore, production of α-SMA and Col1A1 as well as proliferation was attenuated in fibrotic fibroblasts upon treatment with 5′-azacytidine. RNAi-mediated silencing of Rasal1 in vitro in cultured fibroblasts also led to phenotypic effects similar to those observed by DNA methylation-mediated gene silencing as well as hyperactive Ras signaling. More recent studies point to the role of Ten-eleven translocation methylcytosine dioxygenase (Tet) family proteins, specifically Tet3 in DNA demethylation at the Rasal1 promoter, during the reversal of renal fibrosis mediated by bone morphogenetic protein 7 (Bmp7) [104]. In this study, Tet3 expression was downregulated in many mouse models of experimentally induced renal fibrosis, with the expression levels of other members of the Tet family, Tet1 and Tet2, remaining unchanged relative to the controls.These studies indicate a role for DNA demethylation at specific promoters and regulatory regions; specifically, changes in DNA methylation at the Rasal1 promoter might be useful as a biomarker of renal fibrosis and other chronic renal diseases, as well as reporters of effective therapy.
Another interesting epigenetic regulation of gene expression in kidneys has been demonstrated in the case of the antiaging gene Klotho, which is abundantly expressed in kidneys and is a major source of the circulating form of the protein. Azuma et al. [105] demonstrated that DNA methylation has a crucial role in conferring kidney-specific expression of Klotho. Interestingly, Klotho expression is attenuated during acute and chronic kidney disease and, in the latter, a role for DNA hypermethylation has been demonstrated. In a mouse model of CKD induced by administration of protein-bound uremic toxins, the Klotho promoter was hypermethylated, leading to decreased gene expression, and was reversed by the DNA methyltransferase inhibitor, 5′-azacytidine [106]. Furthermore, renoprotective agents against renal fibrosis and/or CKD, such as Rhein, a plant-derived anthraquinone, reversed DNA hypermethylation-mediated epigenetic silencing of Klotho during fibrosis [107]. In addition to DNA methylation-mediated regulation, the Klotho gene is subject to chromatin modification-mediated epigenetic silencing. An increase in H3K9 methylation, a repressive histone modification mark,was observed during Klotho silencing [108]. Renal fibrosis was accompanied by an increase in the expression of G9a, a key H3K9 methyltransferase, and by enrichment of H3K9 monomethylation marks at the Klotho promoter, which could be erased by BIX01294, an inhibitor of G9a [109]. Such erasure of H3K9 monomethyl marks restored Klotho expression and conferred protection in a mouse model of renal fibrosis. Besides methylation, repressive chromatin modification brought about by histone deacetylases is emerging as a potential target in the treatment of fibrosis. In a mouse model of DN, RNAi-mediated silencing of histone deacetylase 9 (HDAC9) attenuated glomerulosclerosis, inflammatory cytokine release, podocyte apoptosis, and renal injury [110]. Similarly, inhibitors of sirtuin 1 (Sirt1) and sirtuin 2 (Sirt2), members of the well-known sirtuin class of histone deacetylases, alleviated renal fibrosis and lessened ECM deposition [111].
There is increasing evidence for permissive chromatin alterations at the promoters of ECM genes during renal fibrosis. Treatment of rat mesangial cells with TGF-β led to enrichment of H3K4 mono-, di-, and trimethylation marks, with a concomitant decrease in repressive H3K9 di- and trimethylation levels in their promoters and an increase in mRNA levels of the H3K4 monomethyltransferase, Set7/9 [112]. Interestingly, TGF-β-modulated recruitment of Set7/9 to ECM gene promoters can be effectively blocked by TGF-β-specific antibodies. Furthermore, siRNA-mediated knockdown of Set7/9 resulted in reduced global levels of H3K4me1 and TGF- β-induced ECM gene levels. The role of histone modifications in regulating the expression of yet another modulator of renal fibrosis, thioredoxin-interacting protein (TxnIP), has been demonstrated. In DN, there is dysregulation of the antioxidant pathway mediated by TxnIP [113]. A recent study indicated that diabetic milieu-induced increased TxnIP expression in mesangial cells is associated with an increase in permissive histone marks, such as H3K9ac and H3K4me, and a decrease in H3K27me at the TxnIP promoter [114]. Together, these reports point to important roles of modulators of the epigenetic state in defining the outcome of kidney fibrosis and/or CKD in vitro and in vivo. In a study by Badal et al. [115], an intersection of miRNAs and chromatin remodeling was observed in podocytes. miR-93 was downregulated in podocytes under high glucose conditions, leading to elevated levels of its target, mitogen and stress-activated kinase 2 (Msk2), a histone kinase that phosphorylates H3 serine 10 [115]. Whereas exogenous supplementation of miR-93 showed therapeutic promise in slowing the progression of DN, further studies are required to fully understand the kinetics of H3S10 phosphorylation and its target genes that escape repression during DN.
In acutely injured kidneys, there are only a few studies of the role of DNA methylation in regulating gene expression, with initial reports arising from transplant kidneys that sustain ischemic injury. Mehta et al. [116] reported that the Calcitonin A (Calca) gene promoter was hypermethylated in cells obtained from the urine of transplant patients with biopsy-proven acute tubular necrosis compared with healthy controls. More recently, levels of the DNA demethylases, Tet1 and Tet2, were found to be downregulated in kidneys that underwent AKI, although a biological role for these enzymes in regulating gene expression during AKI has not yet been demonstrated [117]. We have just begun to understand the robust chromatin modifications that acutely injured kidneys undergo. One of the first studies of chromatin alterations during AKI showed the involvement of the chromatin remodeling factor, SWI/SNF-related, matrix-associated, actin-dependent regulator of chromatin, subfamily a, member 4 (BRG1), a transcriptional coregulator during stress-induced expression of proinflammatory genes, tumor necrosis factor alpha (TNF-α), and monocyte chemoattractant protein (MCP-1) in human tubular epithelial cells. Silencing of BRG1 attenuated RNA polymerase II promoter occupancy with a concomitant reduction in the expression of these proinflammatory genes [118]. In a study by Mar et al. [119], time-dependent changes in chromatin modifications at select gene loci following ischemic or endotoxemic AKI or a combination of both were elucidated. AKI-induced expression of proinflammatory genes, such as those encoding TNF-α, and of kidney injury markers, such as neutrophil gelatinase-associated lipocalin (Ngal) and kidney injury molecule 1 (Kim-1), was associated with broad enrichment of permissive histone modifications, RNA polymerase II recruitment, and diminution of repressive marks at these loci. Several studies have pointed to the impact of permissive histone acetylation marks and the protein players involved in maintaining the balance between open and closed chromatin structures by such marks in response to AKI. Activating transcription factor 3 (ATF3), a known transcriptional repressor that attenuates the expression of the proinflammatory genes encoding IL-6 and IL-12b (IL-12b) during AKI is shown to mediate its action via interacting with histone deacetylase 1 (HDAC1), resulting in gene repression via H4 deacetylation [120]. Recently, p300/CBP-associated factor-1 (PCAF-1), a histone acetyltransferase, was shown to be upregulated during endotoxemic AKI, mediating expression of proinflammatory genes, such as those encoding vascular adhesion molecule1 (VCAM-1) and MCP-1, via increased H3K18 acetylation [121]. Similarly, HDAC inhibitors, such as phenylthiobutanoic acid (PTBA), have proven to be beneficial in accelerating recovery following AKI and in reducing postinjury fibrosis in murine models of AKI [122]. Studies thus far indicate that AKI-induced alteration of histone acetylation is deleterious in general, because many of these acetylated loci tend to be on proinflammatory genes. However, a genome-wide profiling of chromatin modifications approach following ChIP-seq will help gain a better understanding of the complex dynamics of histone acetylation and methylation status in modulating the cellular response to kidney injury.
Concluding remarks and outlook
Our understanding of the epigenetic regulation of renal gene expression during homeostasis and disease is rapidly evolving. These studies have shed new light on the pathobiology of kidney diseases, increasing the plethora of avenues for early detection as well as therapeutic targeting. Encouraging findings from an increasing number of small animal studies that target aberrant epigenetic changes that are deleterious to the outcome of renal injury and/or causative of renal diseases point towards attractive future therapies. Significant promise is seen in the context of both small and long ncRNAs as potential biomarkers of kidney injury. A combination of protein and ncRNA biomarkers could improve the prognosis of renal disease, such AKI, which has not been possible thus far. Future promises and challenges lie in understanding and integrating the vast knowledge of temporal genome-wide chromatin changes that occur during renal homeostasis and disease as well as identifying precise roles of small and long ncRNAs and epigenetic protein players to devise effective therapies against several kidney diseases that currently lack targeted therapeutic avenues.
Acknowledgments
We apologize to researchers whose work could not be included because of length restrictions. We acknowledge research support from the NIH: R21AG047412 (NIA) to S.B.K and K01AR069197 (NIAMS) to S.K.K.
References
- 1.Eddy SR. Non-coding RNA genes and the modern RNA world. Nat Rev Genet. 2001;2:919–929. doi: 10.1038/35103511. [DOI] [PubMed] [Google Scholar]
- 2.Kapranov P, et al. The majority of total nuclear-encoded non-ribosomal RNA in a human cell is ‘dark matter’ un-annotated RNA. BMC Biol. 2010;8:149. doi: 10.1186/1741-7007-8-149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Djebali S, et al. Landscape of transcription in human cells. Nature. 2012;489:101–108. doi: 10.1038/nature11233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Guttman M, et al. Ribosome profiling provides evidence that large noncoding RNAs do not encode proteins. Cell. 2013;154:240–251. doi: 10.1016/j.cell.2013.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rinn JL, Chang HY. Genome regulation by long noncoding RNAs. Annu Rev Biochem. 2012;81:145–166. doi: 10.1146/annurev-biochem-051410-092902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Engreitz JM, et al. Long non-coding RNAs: spatial amplifiers that control nuclear structure and gene expression. Nat Rev Mol Cell Biol. 2016;17:756–770. doi: 10.1038/nrm.2016.126. [DOI] [PubMed] [Google Scholar]
- 7.Lee JT. Epigenetic regulation by long noncoding RNAs. Science. 2012;338:1435–1439. doi: 10.1126/science.1231776. [DOI] [PubMed] [Google Scholar]
- 8.Marques FZ, et al. The emerging role of non-coding RNA in essential hypertension and blood pressure regulation. J Hum Hypertens. 2015;29:459–467. doi: 10.1038/jhh.2014.99. [DOI] [PubMed] [Google Scholar]
- 9.Butterworth MB. MicroRNAs and the regulation of aldosterone signaling in the kidney. Am J Physiol Cell Physiol. 2015;308:C521–C527. doi: 10.1152/ajpcell.00026.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chandrasekaran K, et al. Role of microRNAs in kidney homeostasis and disease. Kidney Int. 2012;81:617–627. doi: 10.1038/ki.2011.448. [DOI] [PubMed] [Google Scholar]
- 11.Trionfini P, et al. MicroRNAs in kidney physiology and disease. Nat Rev Nephrol. 2015;11:23–33. doi: 10.1038/nrneph.2014.202. [DOI] [PubMed] [Google Scholar]
- 12.Rana TM. Illuminating the silence: understanding the structure and function of small RNAs. Nat Rev Mol Cell Biol. 2007;8:23–36. doi: 10.1038/nrm2085. [DOI] [PubMed] [Google Scholar]
- 13.Wilson RC, Doudna JA. Molecular mechanisms of RNA interference. Annu Rev Biophys. 2013;42:217–239. doi: 10.1146/annurev-biophys-083012-130404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li Z, Rana TM. Therapeutic targeting of microRNAs: current status and future challenges. Nat Rev Drug Discov. 2014;13:622–638. doi: 10.1038/nrd4359. [DOI] [PubMed] [Google Scholar]
- 15.van Rooij E, Olson EN. MicroRNA therapeutics for cardiovascular disease: opportunities and obstacles. Nat Rev Drug Discov. 2012;11:860–872. doi: 10.1038/nrd3864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kota SK, Balasubramanian S. Cancer therapy via modulation of micro RNA levels: a promising future. Drug Discov Today. 2010;15:733–740. doi: 10.1016/j.drudis.2010.07.003. [DOI] [PubMed] [Google Scholar]
- 17.Ghildiyal M, Zamore PD. Small silencing RNAs: an expanding universe. Nat Rev Genet. 2009;10:94–108. doi: 10.1038/nrg2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hansen TB, et al. Argonaute-associated short introns are a novel class of gene regulators. Nat Commun. 2016;7:11538. doi: 10.1038/ncomms11538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 20.Zhou H, Rigoutsos I. MiR-103a-3p targets the 5′ UTR of GPRC5A in pancreatic cells. RNA. 2014;20:1431–1439. doi: 10.1261/rna.045757.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hausser J, et al. Analysis of CDS-located miRNA target sites suggests that they can effectively inhibit translation. Genome Res. 2013;23:604–615. doi: 10.1101/gr.139758.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ruby JG, et al. Intronic microRNA precursors that bypass Drosha processing. Nature. 2007;448:83–86. doi: 10.1038/nature05983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Harvey SJ, et al. Podocyte-specific deletion of dicer alters cytoskeletal dynamics and causes glomerular disease. J Am Soc Nephrol. 2008;19:2150–2158. doi: 10.1681/ASN.2008020233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ho J, et al. Podocyte-specific loss of functional microRNAs leads to rapid glomerular and tubular injury. J Am Soc Nephrol. 2008;19:2069–2075. doi: 10.1681/ASN.2008020162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shi S, et al. Podocyte-selective deletion of dicer induces proteinuria and glomerulosclerosis. J Am Soc Nephrol. 2008;19:2159–2169. doi: 10.1681/ASN.2008030312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhdanova O, et al. The inducible deletion of Drosha and microRNAs in mature podocytes results in a collapsing glomerulopathy. Kidney Int. 2011;80:719–730. doi: 10.1038/ki.2011.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nagalakshmi VK, et al. Dicer regulates the development of nephrogenic and ureteric compartments in the mammalian kidney. Kidney Int. 2011;79:317–330. doi: 10.1038/ki.2010.385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ho J, et al. The pro-apoptotic protein Bim is a microRNA target in kidney progenitors. J Am Soc Nephrol. 2011;22:1053–1063. doi: 10.1681/ASN.2010080841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pastorelli LM, et al. Genetic analyses reveal a requirement for Dicer1 in the mouse urogenital tract. Mamm Genome. 2009;20:140–151. doi: 10.1007/s00335-008-9169-y. [DOI] [PubMed] [Google Scholar]
- 30.Bartram MP, et al. Conditional loss of kidney microRNAs results in congenital anomalies of the kidney and urinary tract (CAKUT) J Mol Med. 2013;91:739–748. doi: 10.1007/s00109-013-1000-x. [DOI] [PubMed] [Google Scholar]
- 31.Bartram MP, et al. Loss of Dgcr8-mediated microRNA expression in the kidney results in hydronephrosis and renal malformation. BMC Nephrol. 2015;16:55. doi: 10.1186/s12882-015-0053-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Iervolino A, et al. Selective dicer suppression in the kidney alters GSK3beta/beta-catenin pathways promoting a glomerulocystic disease. PLoS One. 2015;10:e0119142. doi: 10.1371/journal.pone.0119142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nakagawa N, et al. Dicer1 activity in the stromal compartment regulates nephron differentiation and vascular patterning during mammalian kidney organogenesis. Kidney Int. 2015;87:1125–1140. doi: 10.1038/ki.2014.406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Phua YL, et al. Renal stromal miRNAs are required for normal nephrogenesis and glomerular mesangial survival. Physiol Rep. 2015;3:e12537. doi: 10.14814/phy2.12537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Johanson TM, et al. MicroRNA-independent roles of the RNase III enzymes Drosha and Dicer. Open Biol. 2013;3:130144. doi: 10.1098/rsob.130144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Marrone AK, et al. MicroRNA-17∼92 is required for nephrogenesis and renal function. J Am Soc Nephrol. 2014;25:1440–1452. doi: 10.1681/ASN.2013040390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Denby L, et al. miR-21 andmiR-214 are consistently modulated during renal injury in rodent models. Am J Pathol. 2011;179:661–672. doi: 10.1016/j.ajpath.2011.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Denby L, et al. MicroRNA-214 antagonism protects against renal fibrosis. J Am Soc Nephrol. 2014;25:65–80. doi: 10.1681/ASN.2013010072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chau BN, et al. MicroRNA-21 promotes fibrosis of the kidney by silencing metabolic pathways. Sci Transl Med. 2012;4:121ra118. doi: 10.1126/scitranslmed.3003205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kume S, et al. Role of altered renal lipid metabolism in the development of renal injury induced by a high-fat diet. J Am Soc Nephrol. 2007;18:2715–2723. doi: 10.1681/ASN.2007010089. [DOI] [PubMed] [Google Scholar]
- 41.Kang HM, et al. Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nat Med. 2015;21:37–46. doi: 10.1038/nm.3762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lai JY, et al. MicroRNA-21 in glomerular injury. J Am Soc Nephrol. 2015;26:805–816. doi: 10.1681/ASN.2013121274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Qin W, et al. TGF-beta/Smad3 signaling promotes renal fibrosis by inhibiting miR-29. J Am Soc Nephrol. 2011;22:1462–1474. doi: 10.1681/ASN.2010121308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang B, et al. Suppression of microRNA-29 expression by TGF-beta1 promotes collagen expression and renal fibrosis. J Am Soc Nephrol. 2012;23:252–265. doi: 10.1681/ASN.2011010055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sole C, et al. miR-29c in urinary exosomes as predictor of early renal fibrosis in lupus nephritis. Nephrol Dial Transplant. 2015;30:1488–1496. doi: 10.1093/ndt/gfv128. [DOI] [PubMed] [Google Scholar]
- 46.Wang G, et al. Urinary miR-21, miR-29, and miR-93: novel biomarkers of fibrosis. Am J Nephrol. 2012;36:412–418. doi: 10.1159/000343452. [DOI] [PubMed] [Google Scholar]
- 47.Park SM, et al. The miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes Dev. 2008;22:894–907. doi: 10.1101/gad.1640608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gregory PA, et al. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat Cell Biol. 2008;10:593–601. doi: 10.1038/ncb1722. [DOI] [PubMed] [Google Scholar]
- 49.Wang B, et al. miR-200a Prevents renal fibrogenesis through repression of TGF-beta2 expression. Diabetes. 2011;60:280–287. doi: 10.2337/db10-0892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xiong M, et al. The miR-200 family regulates TGF-beta1-induced renal tubular epithelial to mesenchymal transition through Smad pathway by targeting ZEB1 and ZEB2 expression. Am J Physiol Renal Physiol. 2012;302:F369–F379. doi: 10.1152/ajprenal.00268.2011. [DOI] [PubMed] [Google Scholar]
- 51.Castro NE, et al. Transforming growth factor beta1 (TGF-beta1) enhances expressionofprofibrotic genes througha novel signaling cascade and microRNAs in renal mesangial cells. J Biol Chem. 2014;289:29001–29013. doi: 10.1074/jbc.M114.600783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bai X, et al. MicroRNA-130b improves renal tubulointerstitial fibrosis via repression of Snail-induced epithelial–mesenchymal transition in diabetic nephropathy. Sci Res. 2016;6:20475. doi: 10.1038/srep20475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kato M, et al. AmicroRNA circuit mediates transforming growth factor-beta1 autoregulation in renal glomerular mesangial cells. Kidney Int. 2011;80:358–368. doi: 10.1038/ki.2011.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kato M, et al. MicroRNA-192 indiabetic kidney glomeruli and its function in TGF-beta-induced collagen expression via inhibition of E-box repressors. Proc Natl Acad Sci USA. 2007;104:3432–3437. doi: 10.1073/pnas.0611192104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Krupa A, et al. Loss of MicroRNA-192 promotes fibrogenesis in diabetic nephropathy. J Am Soc Nephrol. 2010;21:438–447. doi: 10.1681/ASN.2009050530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kato M, et al. TGF-beta activates Akt kinase through a microRNA-dependent amplifying circuit targeting PTEN. Nat Cell Biol. 2009;11:881–889. doi: 10.1038/ncb1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chung AC, Lan HY. MicroRNAs in renal fibrosis. Front Physiol. 2015;6:50. doi: 10.3389/fphys.2015.00050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Srivastava SP, et al. MicroRNAs in kidney fibrosis and diabetic nephropathy: roles on EMT and EndMT. BioMed Res Int. 2013;2013:125469. doi: 10.1155/2013/125469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang B, Ricardo S. Role of microRNA machinery in kidney fibrosis. Clin Exp Pharmacol Physiol. 2014;41:543–550. doi: 10.1111/1440-1681.12249. [DOI] [PubMed] [Google Scholar]
- 60.Boon RA, Vickers KC. Intercellular transport of microRNAs. Arterioscler Thromb Vasc Biol. 2013;33:186–192. doi: 10.1161/ATVBAHA.112.300139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lv LL, et al. Isolation and quantification of microRNAs from urinary exosomes/microvesicles for biomarker discovery. Int J Biol Sci. 2013;9:1021–1031. doi: 10.7150/ijbs.6100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Perez-Hernandez J, et al. Increased urinary exosomal microRNAs in patients with systemic lupus erythematosus. PLoS One. 2015;10:e0138618. doi: 10.1371/journal.pone.0138618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Noureddine L, et al. MicroRNAs and polycystic kidney disease. Drug Discov Today Dis Models. 2013;10:e137–e1743. doi: 10.1016/j.ddmod.2013.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Szeto CC, Li PK. MicroRNAs inIgA nephropathy. Nat Rev Nephrol. 2014;10:249–256. doi: 10.1038/nrneph.2014.50. [DOI] [PubMed] [Google Scholar]
- 65.Zan H, et al. MicroRNAs in lupus. Autoimmunity. 2014;47:272–285. doi: 10.3109/08916934.2014.915955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Scian MJ, et al. MiRNAs in kidney transplantation: potential role as new biomarkers. Expert Rev Mol Diagn. 2013;13:93–104. doi: 10.1586/erm.12.131. [DOI] [PubMed] [Google Scholar]
- 67.Kaucsar T, et al. Activation of the miR-17 family and miR-21 during murine kidney ischemia-reperfusion injury. Nucleic Acid Ther. 2013;23:344–354. doi: 10.1089/nat.2013.0438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wei Q, et al. Targeted deletion of Dicer from proximal tubules protects against renal ischemia-reperfusion injury. J Am Soc Nephrol. 2010;21:756–761. doi: 10.1681/ASN.2009070718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ranganathan P, et al. MicroRNA-150 deletion in mice protects kidney from myocardial infarction-induced acute kidney injury. Am J Physiol Renal Physiol. 2015;309:F551–F558. doi: 10.1152/ajprenal.00076.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lorenzen JM, et al. MicroRNA-24 antagonism prevents renal ischemia reperfusion injury. J Am Soc Nephrol. 2014;25:2717–2729. doi: 10.1681/ASN.2013121329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lan YF, et al. MicroRNA-494 reduces ATF3 expression and promotes AKI. J Am Soc Nephrol. 2012;23:2012–2023. doi: 10.1681/ASN.2012050438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Jia P, et al. miR-21 contributes to xenon-conferred amelioration of renal ischemia-reperfusion injury in mice. Anesthesiology. 2013;119:621–630. doi: 10.1097/ALN.0b013e318298e5f1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bhatt K, et al. MicroRNA-687 induced by hypoxia-inducible factor-1 targets phosphatase and tensin homolog in renal ischemia-reperfusion injury. J Am Soc Nephrol. 2015;26:1588–1596. doi: 10.1681/ASN.2014050463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Esteller M. Non-coding RNAs inhuman disease. Nat Rev Genet. 2011;12:861–874. doi: 10.1038/nrg3074. [DOI] [PubMed] [Google Scholar]
- 75.Kung JT, et al. Long noncoding RNAs: past, present, and future. Genetics. 2013;193:651–669. doi: 10.1534/genetics.112.146704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Guttman M, et al. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature. 2009;458:223–227. doi: 10.1038/nature07672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Clemson CM, et al. An architectural role for a nuclear noncoding RNA: NEAT1 RNA is essential for the structure of paraspeckles. Mol Cell. 2009;33:717–726. doi: 10.1016/j.molcel.2009.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ulitsky I, Bartel DP. lincRNAs: genomics, evolution, and mechanisms. Cell. 2013;154:26–46. doi: 10.1016/j.cell.2013.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lee JT, Bartolomei MS. X-inactivation, imprinting, and long noncoding RNAs in health and disease. Cell. 2013;152:1308–1323. doi: 10.1016/j.cell.2013.02.016. [DOI] [PubMed] [Google Scholar]
- 80.Allen TA, et al. The SINE-encoded mouse B2 RNA represses mRNA transcription in response to heat shock. Nat Struct Mol Biol. 2004;11:816–821. doi: 10.1038/nsmb813. [DOI] [PubMed] [Google Scholar]
- 81.Zovoilis A, et al. Destabilization of B2 RNA by EZH2 activates the stress response. Cell. 2016;167:1788–1802. doi: 10.1016/j.cell.2016.11.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Paralkar VR, et al. Unlinking an lncRNA from its associated cis element. Mol Cell. 2016;62:104–110. doi: 10.1016/j.molcel.2016.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Gong C, Maquat LE. lncRNAs transactivate STAU1-mediated mRNA decay by duplexing with 3′ UTRs via Alu elements. Nature. 2011;470:284–288. doi: 10.1038/nature09701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Xie H, et al. Long non-coding RNA-H19 antagonism protects against renal fibrosis. Oncotarget. 2016;7:51473–51481. doi: 10.18632/oncotarget.10444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zhou Q, et al. Identification of novel long noncoding RNAs associated with TGF-beta/Smad3-mediated renal inflammation and fibrosis by RNA sequencing. Am J Pathol. 2014;184:409–417. doi: 10.1016/j.ajpath.2013.10.007. [DOI] [PubMed] [Google Scholar]
- 86.Kato M, et al. An endoplasmic reticulum stress-regulated lncRNA hosting a microRNA megacluster induces early features of diabetic nephropathy. Nat Commun. 2016;7:12864. doi: 10.1038/ncomms12864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Long J, et al. Long noncoding RNA Tug1 regulates mitochondrial bioenergetics in diabetic nephropathy. J Clin Invest. 2016;126:4205–4218. doi: 10.1172/JCI87927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Su S, et al. Overexpressionofthe long noncoding RNA TUG1 protects against cold-induced injuryofmouse liversbyinhibiting apoptosis and inflammation. FEBS J. 2016;283:1261–1274. doi: 10.1111/febs.13660. [DOI] [PubMed] [Google Scholar]
- 89.Lin J, et al. The long noncoding RNA landscape in hypoxic and inflammatory renal epithelial injury. Am J Physiol Renal Physiol. 2015;309:F901–F913. doi: 10.1152/ajprenal.00290.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Yu TM, et al. RANTES mediates kidney ischemia reperfusion injury through a possible role of HIF-1alpha and LncRNA PRINS. Sci Rep. 2016;6:18424. doi: 10.1038/srep18424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Chen W, et al. Microarray analysis of long non-coding RNA expression in human acute rejection biopsy samples following renal transplantation. Mol Med Rep. 2014;10:2210–2216. doi: 10.3892/mmr.2014.2420. [DOI] [PubMed] [Google Scholar]
- 92.Lorenzen JM, et al. Long noncoding RNAs in urine are detectable and may enable early detection of acute T cell-mediated rejection of renal allografts. Clin Chem. 2015;61:1505–1514. doi: 10.1373/clinchem.2015.243600. [DOI] [PubMed] [Google Scholar]
- 93.Lorenzen JM, et al. Circulating long noncoding RNA TapSaki is a predictor of mortality in critically ill patients with acute kidney injury. Clin Chem. 2015;61:191–201. doi: 10.1373/clinchem.2014.230359. [DOI] [PubMed] [Google Scholar]
- 94.Huang YS, et al. Urinary Xist is a potential biomarker for membranous nephropathy. Biochem Biophys Res Commun. 2014;452:415–421. doi: 10.1016/j.bbrc.2014.08.077. [DOI] [PubMed] [Google Scholar]
- 95.Lorenzen JM, Thum T. Long noncoding RNAs in kidney and cardiovascular diseases. Nat Rev Nephrol. 2016;12:360–373. doi: 10.1038/nrneph.2016.51. [DOI] [PubMed] [Google Scholar]
- 96.Qiao HP, et al. Long non-coding RNA GAS5 functions as a tumor suppressor in renal cell carcinoma. Asian Pac J Cancer Prev. 2013;14:1077–1082. doi: 10.7314/apjcp.2013.14.2.1077. [DOI] [PubMed] [Google Scholar]
- 97.Wu Y, et al. Suppressed expression oflong non-coding RNA HOTAIR inhibits proliferation and tumourigenicity of renal carcinoma cells. Tumour Biol. 2014;35:11887–11894. doi: 10.1007/s13277-014-2453-4. [DOI] [PubMed] [Google Scholar]
- 98.Seles M, et al. Current insights into long non-coding RNAs in renal cell carcinoma. Int J Mol Sci. 2016;17:573. doi: 10.3390/ijms17040573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Smith ZD, Meissner A. DNA methylation: roles in mammalian development. Nat Rev Genet. 2013;14:204–220. doi: 10.1038/nrg3354. [DOI] [PubMed] [Google Scholar]
- 100.Li E, et al. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell. 1992;69:915–926. doi: 10.1016/0092-8674(92)90611-f. [DOI] [PubMed] [Google Scholar]
- 101.Okano M, et al. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 1999;99:247–257. doi: 10.1016/s0092-8674(00)81656-6. [DOI] [PubMed] [Google Scholar]
- 102.Li E. Chromatin modification and epigenetic reprogramming in mammalian development. Nat Rev Genet. 2002;3:662–673. doi: 10.1038/nrg887. [DOI] [PubMed] [Google Scholar]
- 103.Bechtel W, et al. Methylation determines fibroblast activation and fibrogenesis in the kidney. Nat Med. 2010;16:544–550. doi: 10.1038/nm.2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Tampe B, et al. Tet3-mediated hydroxymethylation of epigenetically silenced genes contributes to bone morphogenic protein 7-induced reversal of kidney fibrosis. J Am Soc Nephrol. 2014;25:905–912. doi: 10.1681/ASN.2013070723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Azuma M, et al. Promoter methylation confers kidney-specific expression of the Klotho gene. FASEB J. 2012;26:4264–4274. doi: 10.1096/fj.12-211631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Sun CY, et al. Suppression of Klotho expression by protein-bound uremic toxins is associated with increased DNA methyltransferase expression and DNA hypermethylation. Kidney Int. 2012;81:640–650. doi: 10.1038/ki.2011.445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Zhang Q, et al. Rhein reversal of DNA hypermethylation-associated Klotho suppression ameliorates renal fibrosis in mice. Sci Rep. 2016;6:34597. doi: 10.1038/srep34597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Rubinek T, et al. Epigenetic silencing of the tumor suppressor klotho in human breast cancer. Breast Cancer Res Treat. 2012;133:649–657. doi: 10.1007/s10549-011-1824-4. [DOI] [PubMed] [Google Scholar]
- 109.Irifuku T, et al. Inhibition of H3K9 histone methyltransferase G9a attenuates renal fibrosis and retains klotho expression. Kidney Int. 2016;89:147–157. doi: 10.1038/ki.2015.291. [DOI] [PubMed] [Google Scholar]
- 110.Liu F, et al. Silencing of histone deacetylase 9 expression in podocytes attenuates kidney injury in diabetic nephropathy. Sci Rep. 2016;6:33676. doi: 10.1038/srep33676. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 111.Ponnusamy M, et al. Blocking sirtuin 1 and 2 inhibits renal interstitial fibroblast activation and attenuates renal interstitial fibrosis in obstructive nephropathy. J Pharmacol Exp Ther. 2014;350:243–256. doi: 10.1124/jpet.113.212076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Sun G, et al. Epigenetic histone methylation modulates fibrotic gene expression. J Am Soc Nephrol. 2010;21:2069–2080. doi: 10.1681/ASN.2010060633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Advani A, et al. Expression, localization, and function of the thioredoxin system in diabetic nephropathy. J Am Soc Nephrol. 2009;20:730–741. doi: 10.1681/ASN.2008020142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.De Marinis Y, et al. Epigenetic regulation of the thioredoxin-interacting protein (TXNIP) gene by hyperglycemia in kidney. Kidney Int. 2016;89:342–353. doi: 10.1016/j.kint.2015.12.018. [DOI] [PubMed] [Google Scholar]
- 115.Badal SS, et al. miR-93 regulates Msk2-mediated chromatin remodelling in diabetic nephropathy. Nat Commun. 2016;7:12076. doi: 10.1038/ncomms12076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Mehta TK, et al. Quantitative detection of promoter hypermethylationasa biomarker of acute kidney injury during transplantation. Transplant Proc. 2006;38:3420–3426. doi: 10.1016/j.transproceed.2006.10.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Huang N, et al. Reduction of DNA hydroxymethylationinthe mouse kidney insulted by ischemia reperfusion. Biochem Biophys Res Commun. 2012;422:697–702. doi: 10.1016/j.bbrc.2012.05.061. [DOI] [PubMed] [Google Scholar]
- 118.Naito M, et al. BRG1 increases transcription of proinflammatory genes in renal ischemia. J Am Soc Nephrol. 2009;20:1787–1796. doi: 10.1681/ASN.2009010118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Mar D, et al. Heterogeneity of epigenetic changes at ischemia/reperfusion-and endotoxin-induced acute kidney injury genes. Kidney Int. 2015;88:734–744. doi: 10.1038/ki.2015.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Li HF, et al. ATF3-mediated epigenetic regulation protects against acute kidney injury. J Am Soc Nephrol. 2010;21:1003–1013. doi: 10.1681/ASN.2009070690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Huang J, et al. Histone acetyltransferase PCAF regulates inflammatory molecules in the development of renal injury. Epigenetics. 2015;10:62–72. doi: 10.4161/15592294.2014.990780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Novitskaya T, et al. A PTBA small molecule enhances recovery and reduces postinjury fibrosis after aristolochic acid-induced kidney injury. Am J Physiol Renal Physiol. 2014;306:F496–F504. doi: 10.1152/ajprenal.00534.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Lorenzen JM, et al. Circulating miR-210 predicts survival in critically ill patients with acute kidney injury. Clin J Am Soc Nephrol. 2011;6:1540–1546. doi: 10.2215/CJN.00430111. [DOI] [PubMed] [Google Scholar]
- 124.Zhou X, et al. Identification of urinary microRNA biomarkers for detection of gentamicin-induced acute kidney injury in rats. Regul Toxicol Pharmacol. 2016;78:78–84. doi: 10.1016/j.yrtph.2016.04.001. [DOI] [PubMed] [Google Scholar]
- 125.Sun SQ, et al. Circulating microRNA-188, -30a, and -30e as early biomarkers for contrast-induced acute kidney injury. J Am Heart Assoc. 2016;5:e004138. doi: 10.1161/JAHA.116.004138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Wang N, et al. Urinary microRNA-10a and microRNA-30d serve as novel, sensitive and specific biomarkers for kidney injury. PLoS One. 2012;7:e51140. doi: 10.1371/journal.pone.0051140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Szeto CC, et al. Micro-RNA expression in the urinary sediment of patients with chronic kidney diseases. Dis Markers. 2012;33:137–144. doi: 10.3233/DMA-2012-0914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Wang G, et al. Elevated levels of miR-146a and miR-155 in kidney biopsy and urine from patients with IgA nephropathy. Dis Markers. 2011;30:171–179. doi: 10.3233/DMA-2011-0766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Barutta F, et al. Urinary exosomal microRNAs in incipient diabetic nephropathy. PLoS One. 2013;8:e73798. doi: 10.1371/journal.pone.0073798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Delic D, et al. Urinary exosomal miRNA signature in type II diabetic nephropathy patients. PLoS One. 2016;11:e0150154. doi: 10.1371/journal.pone.0150154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Te JL, et al. Identification of unique microRNA signature associated with lupus nephritis. PLoS One. 2010;5:e10344. doi: 10.1371/journal.pone.0010344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Sui W, et al. Microarray analysis of microRNA expression in acute rejection after renal transplantation. Transpl Immunol. 2008;19:81–85. doi: 10.1016/j.trim.2008.01.007. [DOI] [PubMed] [Google Scholar]
- 133.Scian MJ, et al. MicroRNA profiles in allograft tissues and paired urines associate with chronic allograft dysfunction with IF/TA. Am J Transp. 2011;11:2110–2122. doi: 10.1111/j.1600-6143.2011.03666.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Maluf DG, et al. The urine microRNA profile may help monitor post-transplant renal graft function. Kidney Int. 2014;85:439–449. doi: 10.1038/ki.2013.338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Ben-Dov IZ, et al. Urine microRNA as potential biomarkers of autosomal dominant polycystic kidney disease progression: description of miRNA profiles at baseline. PLoS One. 2014;9:e86856. doi: 10.1371/journal.pone.0086856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Chen W, et al. Microarray analysis of long non-coding RNA expression in human acute rejection biopsy samples following renal transplantation. Mol Med Rep. 2014;10:2210–2216. doi: 10.3892/mmr.2014.2420. [DOI] [PubMed] [Google Scholar]
- 137.Kriegel AJ, et al. MiR-382 targeting of kallikrein 5 contributes to renal inner medullary interstitial fibrosis. Physiol Genomics. 2012;44:259–267. doi: 10.1152/physiolgenomics.00173.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Putta S, et al. Inhibiting microRNA-192 ameliorates renal fibrosis in diabetic nephropathy. J Am Soc Nephrol. 2012;23:458–469. doi: 10.1681/ASN.2011050485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Zhao B, et al. MicroRNA-23b targets Ras GTPase-activating protein SH3 domain-binding protein 2 to alleviate fibrosis and albuminuria in diabetic nephropathy. J Am Soc Nephrol. 2016;27:2597–2608. doi: 10.1681/ASN.2015030300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Patel V, et al. miR-17∼92 miRNA cluster promotes kidney cyst growth in polycystic kidney disease. Proc Natl Acad Sci U S A. 2013;110:10765–10770. doi: 10.1073/pnas.1301693110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Lakhia R, et al. MicroRNA-21 aggravates cyst growth in a model of polycystic kidney disease. J Am Soc Nephrol. 2016;27:2319–2330. doi: 10.1681/ASN.2015060634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Serino G, et al. Abnormal miR-148b expression promotes aberrant glycosylation of IgA1 in IgA nephropathy. J Am Soc Nephrol. 2012;23:814–824. doi: 10.1681/ASN.2011060567. [DOI] [PMC free article] [PubMed] [Google Scholar]