

Abstract
NHP iPSCs provide a unique opportunity to test safety and efficacy of iPSC-derived therapies in clinically relevant NHP models. To monitor these cells in vivo, there is a need for safe and efficient labeling methods. Gene insertion into genomic safe harbors (GSHs) supports reliable transgene expression while minimizing the risk the modification poses to the host genome or target cell. Specifically, this protocol demonstrates targeting of the adeno-associated virus site 1 (AAVS1), one of the most widely used GSH loci in the human genome, with CRISPR/Cas9, allowing targeted marker or therapeutic gene insertion in rhesus macaque induced pluripotent stem cells (RhiPSCs). Furthermore, detailed instructions for screening targeted clones and a tool for assessing potential off-target nuclease activity are provided.
Keywords: Rhesus macaque, induced pluripotent stem cells, CRISPR/Cas9, safe-harbor, non-human primate
INTRODUCTION
Cell therapies using iPSC-derived cells possess tremendous potential to treat human diseases and are expected to enter early phase clinical trials in the near future. The similarity of rhesus macaque anatomy and physiology to that of humans makes it an ideal animal model for development of novel cell therapies. NHP iPSCs are a powerful pre-clinical tool to further study the safety and efficacy of proposed therapies. Methods to track transplanted iPSC derived therapies are necessary to allow evaluation of their integration into recipient tissue architecture, assess potential safety issues, and enable long-term monitoring of the persistence and localization of iPSC-derived transplanted cells. GSHs have been utilized as targets for integration of transgenes to maintain stable and robust expression of the transgene in iPSC and their progeny cells, without detrimental genotoxicity. This unit includes protocols for targeting the AAVS1-like safe harbor loci in RhiPSCs with CRISPR/Cas9 for transgene insertion (Basic Protocol 1), characterization of the modified RhiPSCs, and Cre-mediated excision of puromycin resistance gene (Basic protocol 2), and an approach to assess potential off-target effects in the rhesus genome (Basic Protocol 3). Figure 1 illustrates the entire protocol to generate and screen AAVS1 targeted RhiPSCs.
Figure 1.

Timeline and workflow for CRISPR/Cas9-based safe-harbor gene editing in RhiPSCs.
RhiPSCs in feeder-free condition are harvested. A donor plasmid and the CRISPR/Cas9 “all-in-one” vector are delivered to RhiPSCs via nucleofection. The donor plasmid described in this protocol allows puromycin-mediated selection to enrich cells that have targeted insertion at the desired AAVS1 locus. Optionally, live-cell staining with the antibody against the transgene in the donor plasmid can be performed to make the colony selection process easier. After targeting, colonies need to be screened by PCR, Southern blot, and off-target analyses. Examples of a correct clone (clone #1) and a non-desirable clone (clone #2) for each assay is shown. Clones that have targeted insertion in one or both alleles (monoallelic or biallelic) with no additional random integration or off-target mutation should be selected for further steps. Right panel depicts additional subcloning step to remove the puromycin resistance gene. In this case, RhiPSCs are transfected with the GFP-Cre plasmid and positive cells are purified by FACS. Of note, GFP reporter in sorted cells is transient, hence will be lost by the time of colony growth and picking as illustrated on Day 48. Excised clones are confirmed by Cre PCR/qPCR. An example of a correctly excised clone (Clone A) and partial/non-excised clone (Clone B) is shown.
STRATEGIC PLANNING
A guide to establish optimal culture conditions for RhiPSCs can be found in Unit 4A.11. The plasmids used during transfection reactions of this protocol are adaptable to experimental requirements. The donor plasmid used in this protocol has the original human AAVS1 homology arms replaced with the homologous rhesus sequences, and can be found in Addgene (#84209). Of note, this vector contains copGFP as the transgene, but can be readily replaced with any transgene of interest (Hong et al., 2017). The “all-in-one” CRISPR/Cas9 plasmid (Addgene #79145) used in this protocol has a CAG promoter, which is known to have robust expression in human pluripotent stem cells (Ran et al., 2013; Zou et al., 2011) and has eSpCas9, which has been shown to be more specific than previous Cas9 nucleases (Slaymaker et al., 2016). The gRNA selected for this protocol (ggggccactagggacaggac (chr19:61115594–61115613)) targets the AAVS1-like loci in the rhesus genome. This gRNA has a one nucleotide difference compared to the gRNA used for targeting the AAVS1 T2 site in the human genome (Mali et al., 2013). The cutting efficiency of this gRNA and other rhesus macaque targeted guides can be assessed by the T7E1 assay using the rhesus FRhK-4 cell line, available from ATCC (CRL-1688) (see Unit 5B.6.14 for further information on gRNA screening and cutting efficiency assessment). The steps necessary to clone the selected gRNA into the backbones provided can be found on the Zhang lab website CRISPR resources (http://zlab.mit.edu/resources.html).
BASIC PROTOCOL 1. Gene Targeting and Colony Isolation
The CRISPR/Cas9 “all-in-one” and donor plasmids will be delivered into RhiPSCs via nucleofection. Cells will undergo homology directed repair (HDR) and gain puromycin resistance since this gene is included in the donor plasmid. Following puromycin selection for targeted clones, screening of selected clones with PCR and Southern Blot identifies desirable RhiPSC clones to be used for further studies. Removing the puromycin resistance gene via Cre-mediated excision is important to prevent an immunogenic response if these cells are to be transplanted back into an immunocompetent donor rhesus macaque.
Materials
4D-Nucleofector™ Core Unit (Lonza cat. no. AAF-1002B)
P3 Primary Cell 4D-Nucleofector® X Kit L (12 RCT) (Lonza cat. no. V4XP-3012)
Accutase (EMD Millipore cat. no. SCR005)
DMEM, high glucose, pyruvate (Gibco cat. no. 11995)
DMEM (Lonza cat. no. 12-614F)
Fetal Bovine Serum (FBS) (Hyclone cat. no. SV30014.03)
KnockOut DMEM (KO/DMEM) (Gibco cat. no. 10829)
Knockout Serum Replacement (KSR) (Gibco cat. no. 10828)
Human Fibroblast like Growth Factor-basic (bFGF) (PeproTech cat. no. 100-18B)
MEM nonessential amino acids (NEAA) (Gibco cat. no. 11140)
Penicillin-Streptomycin-Glutamine (PSG) (Gibco cat. no. 10378)
2-mercaptoethanol (Sigma-Aldrich cat. no. M7522)
Matrigel™ (BD Bioscience cat. no. 354277)
Puromycin (Sigma cat. no. P8833)
ROCK inhibitor (Y-27632) (Stemgent cat. no. 04-0012)
Mouse embryonic fibroblasts (MEF), 2 million cells/vial (GlobalStem cat. no. GSC-6201G)
Puro MEF IRR, 2 million cells/vial (GlobalStem cat no. GSC-6220G)
Ultrapure Water with 0.1% Gelatin (Millipore cat. no. ES-006-B)
Hypoxia Chamber (BioSpherix cat. no. C-274)
O2 Controller (BioSpherix cat. no. P110)
CO2 Controller (BioSpherix cat. no. P120)
MultiGrip™ 200μl Tips (Optional) (Denville cat. no. P3133-F)
Falcon™ 15mL Conical Centrifuge Tube (Fisher Scientific cat. no. 14-959-53A)
PBS-1X, w/o Ca++, Mg++ (Lonza cat. no. 17-516F)
Gene Targeting
Day 1
-
1
Passage RhiPSCs from culture on MEF to feeder free conditions (Matrigel plates)
RhiPSCs should be cultured in hypoxia chambers (5% CO2 and 5% O2). Cells in feeder free conditions require conditioned media (CM) (see Reagents and Solutions). ROCK inhibitor (10 μM) needs to be supplemented in the CM when passaging RhiPSCs from MEF to feeder free conditions. Aim to passage cells on Matrigel at least once before nucleofection to enable proper adaptation to feeder-free conditions.
Day 2–3
-
2
Change CM daily.
Day 4
-
3
Passage RhiPSCs onto at least 6 Matrigel-coated wells in a 6 well plate.
Each transfection condition requires 1 million cells. Typically, each well of RhiPSCs (on Matrigel) yields 0.5–2 million cells. Prepare 3 million cells (3 conditions) for each RhiPSC cell line – positive control (lonza pmaxGFP), negative control (gRNA only), and experimental wells.
Day 5–6
-
4
Change CM daily.
-
5
Thaw puromycin resistant MEFs.
Follow the same thawing and plating protocol as normal MEFs (Unit 4A.11.2). For each experimental condition prepare 6 MEF wells, and 3 MEF wells for positive and negative controls.
Day 7
-
6
Warm Accutase, CM + ROCK inhibitor to 37°C.
See reagents and solutions for ROCK inhibitor preparation. -
7
Take out nucleofection reagents, donor plasmid, CRISPR/Cas9 “all-in-one” plasmid from refrigerator/freezer to equilibrate to room temperature (10–15 minutes).
-
8
Aspirate CM from RhiPSCs wells.
-
9
Add 1 mL Accutase per well.
-
10
Place in incubator for 3–4 minutes.
Remove cells from incubator once RhiPSCs have clearly detached from plate after gentle shaking. -
11
Add 4 mL CM + Rock Inhibitor to each well and combine all wells into one 50ml conical tube.
Pipet cells thoroughly to break up any clumps to make single cells. -
12
Count cells.
-
13
Centrifuge the conical tube at 250 × g for 5 minutes.
-
14
Aspirate supernatant off the cell pellet.
-
15
Resuspend the pellet with PBS to reach a concentration of 1 million/mL.
-
16
Transfer 1 million cell aliquots to new 15 ml conical tubes and add 2 mL PBS.
-
17
Centrifuge conical tubes at 250 × g for 5 minutes.
-
18
During centrifugation step, change the medium of Puro-MEFs to CM + ROCK inhibitor (2.5ml per well).
-
19
Aspirate supernatant off RhiPSC cell pellets.
-
20
Resuspend pellet with 100 μL of room temperature nucleofection buffer.
-
21
Add appropriate plasmids to their respective cell suspension.
The amount of plasmid added can be titered to reach optimal transfection efficiency. Here, we suggest 10 μg of both donor and all-in-one plasmids. 5 μg of GFP plasmid is sufficient for the positive control. We have seen transfection efficiencies in the range of 5–20% with these concentrations, which in turn yielded a reasonable number of targeted cells. -
22
Transfer the cell + plasmid suspensions to nucleofection vessels.
-
23
Place nucleofection vessels in the 4D Nucleofector Core Unit and run the desired program.
Select the appropriate vessel format and the number of conditions. Select the “hES-H9” condition under the protocol list. Refer to equipment manual for basic operation instructions. -
24
Immediately add 750 μL of CM + ROCK inhibitor to each nucleofection vessel.
-
25
Transfer the solution in the nucleofection vessels to puro-MEF plates using the eye-droppers provided in the kit.
Each of the vessel contents (1 million) can be transferred to 6 puro-MEF wells since this has been shown to yield a good density (minimal overlap of colonies) when colonies grow up after selection. For GFP and negative control conditions, 3 wells are sufficient, so transfer half the volume of the vessel to the wells. -
26
Gently shake the plate to evenly distribute cells throughout.
-
27
Move plates to hypoxia incubator.
Day 8
-
28
Observe wells under fluorescent microscope to confirm whether the nucleofection protocol was completed successfully.
Both the GFP plasmid and all-in-one plasmid wells should be positive for GFP (to varying degrees). The lack of GFP expressing cells in the positive control cells is indicative that the nucleofection protocol was not successful. -
29
Change RhiPSC media daily.
Clone Selection
Day 9 – Day 13
-
30
Change media to RhiPSC media + puromycin (0.5 μg/mL).
See Reagents and Solutions for puromycin preparation. Positive control (GFP) cells can be discarded at this stage since they will not be puromycin resistant. The puromycin concentration needs to be titrated if colonies grow up from negative control wells. Only RhiPSCs that underwent HDR with the donor plasmid should grow up through the selection process.
Day 14 – until colonies appear
-
31
Change media to RhiPSC media daily (no puromycin).
Once colonies are visible, live staining (optional) (see Support Protocol 1) works to make the colony selection process more efficient, since colonies with transgene expression suggests integration of the transgene at the intended AAVS1 locus. (Note: If the transgene is a fluorescent marker (GFP), live staining is unnecessary and colonies can be visualized under a fluorescent microscope). After live staining, selecting 5–6 transgene positive colonies should be sufficient. If the proposed transgene is not a fluorescent marker or does not enable live staining, random colonies (at least 10–15 in our experience) need to be selected. Following colony selection, PCR and Southern Blot analysis of selected clones is necessary to confirm targeted integration and the absence of random integrations (see Basic Protocol 2). -
32
Manually select and transfer colonies to individual MEF plate wells.
RhiPSCs no longer require puromycin resistant MEFs. -
33
Expand colonies to at least 2 wells.
-
34
Harvest one well for genomic DNA extraction using the standard protocols from a kit and maintain a well for sustained culture.
BASIC PROTOCOL 2. Screening and Cre-Excision
After selection of colonies following live staining for transgene integration, or random picking, selected RhiPSCs clones need to be assessed for targeted versus random integration via PCR and Southern Blot analysis. The primers used in this protocol can be found in Table 2. After screening clones, the puromycin resistance gene can be removed via Cre-mediated excision. This is necessary if the modified RhiPSCs will be transplanted into an immunocompetent animal. In our experience, we have not seen a noticeable difference in transgene expression after the Cre-excision process. A Cre-plasmid with a GFP reporter is transfected into the selected RhiPSCs followed by FACS to select for GFP positive cells. If the transgene of interest is GFP, then Cre-excision by this method will not work. Alternatively, using a Cre-neomycin or hygromycin resistance plasmid can be used to confer resistance to the drugs and select for clones like the selection strategy utilized above for puromycin selection after targeting (see Addgene #34565 or #34568). Drug concentration and length of selection needs to optimized for your respective RhiPSC line(s). After the gene editing and Cre-excision, it is recommended to perform cytogenetic analysis (Research Cytogenetics Lab, Oregon Health & Science University) on RhiPSC clones intended to be used for further studies to confirm the genetic integrity of the clones.
Table 2.
Primers and probes used in this protocol.
| Assay | Site | Forward Primer (5′-3′) | Reverse Primer (5′-3′) |
|---|---|---|---|
| Southern Blot | Rhesus AAVS1 5′ homology arm | tatgaccatgattacgccgccacctccttcaggttccagcttcct | tatgctatacgaagttatgcctgtccctagtggccccacggtggg |
| qPCR | Puromucin N-acetyl-transferase | gcaacctccccttctacgag | cgggcttgcgggtcatg |
| NHEJ PCR | Rhesus AAVS1 WT Allele | tgctttctttgcctggacac | tgatgcacagaggaacagtac |
| Targeted integration PCR | tcctgctttcactgacctgc | ggcttgtactcggtcatcctag | |
| Off-target analysis | OT-1 | agagagtgtgtaggtgggtatt | gcctcttggaatggtgctataa |
| OT-2 | aaagttcaaagtggagacagga | aagagcaagcagggtgttta | |
| OT-3 | ccaggttagaggaggcataaac | ggaactgaagagatgcccaa | |
| OT-4 | tttgaaccgtgctcctagc | gctggacttcaggcctattt | |
| OT-5 | gctcatcttgagagagagaagaaa | gctgtagcgggttatttgaatg | |
| OT-6 | atgtcacaagcgtgcctt | atccatgaagcctccaagatg | |
| OT-7 | ttctggtggttacacttcattca | agggctctttgacagatcattac | |
| OT-8 | ggttactgttcttagccactgt | gactcagggaatgcgtttct | |
| OT-9 | tgtgaggtactgtgcctgga | gaaaggccattgcattagga | |
| OT-10 | tccacctctcaggttcaagc | acccaaggaacctttgcttt |
Materials
REDTaq® ReadyMix™ PCR Reaction Mix (Sigma cat. no. R2523-20RXN)
Platinum PCR Supermix (ThermoFisher cat. no. 11306016)
MultiGrip™ 200μl Tips (Optional) (Denville cat. no. P3133-F)
Opti-MEM Reduced Serum Medium (ThermoFisher cat. no. 31985062)
EditPro™ Stem Transfection Reagent (MTI-GlobalStem cat. no. GST-2174)
KnockOut DMEM (KO/DMEM) (Gibco cat. no. 10829)
Knockout Serum Replacement (KSR) (Gibco cat. no. 10828)
Human Fibroblast like Growth Factor-basic (bFGF) (PeproTech cat. no. 100-18B)
MEM nonessential amino acids (NEAA) (Gibco cat. no. 11140)
Penicillin-Streptomycin-Glutamine (PSG) (Gibco cat. no. 10378)
2-mercaptoethanol (Sigma-Aldrich cat. no. M7522)
Mouse embryonic fibroblasts (MEF), 2 million cells/vial (GlobalStem cat. no. GSC-6201G)
Ultrapure Water with 0.1% Gelatin (Millipore cat. no. ES-006-B)
Hypoxia Chamber (BioSpherix cat. no. C-274)
O2 Controller (BioSpherix cat. no. P110)
CO2 Controller (BioSpherix cat. no. P120)
MultiGrip™ 200μl Tips (Optional) (Denville cat. no. P3133-F)
Falcon® 5mL Round Bottom Polystyrene Test Tube, with Cell Strainer Snap Cap (Corning cat. no. 352235)
Micro tube 1.5ml (SARSTEDT cat. no. 72.692.005)
Screening
Day ~25 (continuing from the end of Basic Protocol 1)
-
1
PCR amplify the RhAAVS1 target site with the validated primers and thermal cycler conditions.
See Table 2 for primer information. The forward primer is found in the genomic DNA flanking the AAVS1 locus near the gRNA target site while the reverse primer is located within the donor plasmid (puromycin resistance gene); therefore, a PCR reaction yielding a ~964 base pair (bp) product (as seen on a 1% gel) indicates targeted integration of the transgene.-
Red Taq polymerase thermal cycler settings:
1 cycle: 94°C for 3 min 35 cycles: 94°C for 30 sec 66°C for 30 sec 72°C for 25 sec 1 cycle: 72°C for 10 min
-
-
2
Run the PCR products on a 1% agarose gel.
-
3
Visualize the bands using a spectrophotometer.
Select clones with a single band of the appropriate length (~964 bp) for Southern Blot.
Day ~26–36
-
4
Perform Southern Blot analysis.
The probe sequence we use hybridizes with a sequence in the left homology arm (see Table 2 and Figure 2). Following a standard Southern Blot protocol (see Unit. 8.2 in Essential Laboratory Techniques) with an initial DNA digestion with SphI restriction enzyme will yield bands specific to the WT allele and targeted integration. Clones on the Southern Blot can have 2 bands (wild type allele (6.5 kb) and targeted integration (3.8 kb)), 1 band (biallelic integration if 3.8 kb or no integration if 6.5 kb), or more than two bands which would indicate random integration of the donor plasmid.
Figure 2.
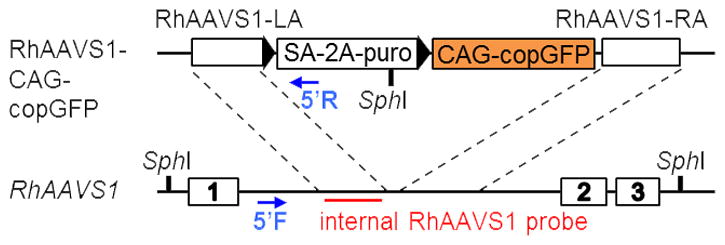
The RhAAVS1 locus (bottom) is located in the first intron of the PPP1R12C gene. The rhesus AAVS1-CAG-human truncated copGFP donor plasmid (top) was modified from the human AAVS1-CAG-copGFP vector by replacing the human AAVS1 homology arms with orthologous rhesus AAVS1. The Southern Blot probe is found in the left homology arm (red bar) while the forward and reverse PCR primers used for detecting targeted integration of the donor plasmid in RhiPSCs are located outside of the donor plasmid and within the SA-2A-puro gene of the plasmid respectively (blue arrows).
Cre-excision
Day ~37
-
5
Passage selected RhiPSCs from MEF to feeder free conditions.
Clones with only monoallelic or biallelic integration and without random integration of the transgene should be selected to undergo Cre-excision. Typically, it will require one additional round of passaging after RhiPSCs are transferred to feeder free conditions to reach the required number of wells (6) for Cre-excision. Transfection of RhiPSCs with the Cre-plasmid should be completed one day after passaging RhiPSCs to 6 wells (small colonies).
Day ~41
-
6
Change CM.
-
7
Prepare transfection mixture in a 1.5ml micro centrifuge tube.
-
For each well undergoing transfection:
62.5 μL warmed Opti-MEM medium
1 μg Cre plasmid (we use the Puro-T2A-Cre-GFP plasmid (Merling et al., 2013)
6 μL EditPro™ Stem Transfection reagent
-
-
8
Incubate transfection mixture at room temperature for 10 minutes.
-
9
Add equal volumes of transfection mixture to each well.
Gently shake plate. -
10
Transfer plate(s) to incubator.
-
11
Visualize cells with fluorescent microscope (GFP) to verify transfection occurred.
GFP expression can be detected as early as 24 hours after transfection and fluorescence is detected at the edges of colonies.
Day ~42
-
12
Change CM.
-
13
Prepare MEF plates.
Make 3 wells in a 6-well plate for each RhiPSC clone undergoing Cre-excision.
Day ~43
-
14
Dissociate colonies with warmed accutase.
Incubate cells with accutase for 3–4 minutes at 37°C. -
15
Spin down cells at 250 × g for 5 minutes.
-
16
Discard supernatant.
-
17
Resuspend cells in 500 μL of RhiPSC medium supplemented with ROCK inhibitor.
-
18
Transfer cells to round-bottom polystyrene tubes.
Keep tubes on ice while transporting samples to FACS machine. -
19
Change medium on MEF plates to RhiPSC medium supplemented with ROCK inhibitor.
-
20
Sort 103 GFP positive RhiPSCs directly into each MEF well.
Seek guidance from the FACS machine coordinator about how to make the sorting process gentler on RhiPSCs to increase viability. Gentle shake wells to distribute cells after sorting. -
21
Transfer plates to hypoxia incubator.
-
22
Change RhiPSC medium daily.
Day ~48–51
-
23
Mechanically transfer clones to new MEF plates after colonies arise.
Colonies appear roughly 5–8 days after sorting. Select 10–15 clones.
Day ~56–59
-
24
Expand selected colonies to at least two wells.
Maintain at least one well for sustained culture. Harvest one well for DNA extraction. -
25
Perform qPCR to detect the presence or loss of the puromycin resistance gene.
See Table 2 for primer information.
SUPPORT PROTOCOL 1
Live-cell staining of RhiPSCs after targeting can help to make the colony selection process easier, if the transgene of interest can be detected by commercial antibodies. All steps should be performed in a sterile working environment.
Materials
KnockOut DMEM (KO-DMEM) (Gibco cat. no. 10829)
Knockout Serum Replacement (KSR) (Gibco cat. no. 10828)
Human Fibroblast like Growth Factor-basic (bFGF) (PeproTech cat. no. 100-18B)
MEM nonessential amino acids (NEAA) (Gibco cat. no. 11140)
Penicillin-Streptomycin-Glutamine (PSG) (Gibco cat. no. 10378)
2-mercaptoethanol (Sigma-Aldrich cat. no. M7522)
Human Fibroblast like Growth Factor-basic (bFGF) (PeproTech cat. no. 100-18B)
-
Dilute RhiPSC media 1:5 with KO- DMEM.
This 1:5 dilution reduces the KSR from 20% to 4% which helps to minimize the potential interference of the media components on the fluorescent signal. -
Add conjugated antibody for the transgene of interest at 1:50 dilution to 4% RhiPSC media.
Each well for live staining needs 1 mL of 4% RhiPSC media. All steps should be performed in the biological safety cabinet.Note: Live staining can be performed with an unconjugated antibody. The primary and secondary antibodies need to be added simultaneously. -
Add 1 mL of 4% RhiPSC media + antibody solution to each well and place in hypoxia incubator for 1–2 hours.
Confirm specificity of antibody binding with side-by-side staining with parental RhiPSCs. Observe wells under a fluorescent microscope to identify and mark positively stained RhiPSCs.
Change to regular RhiPSC media.
BASIC PROTOCOL 3. Off-target Analysis
While CRISPR/Cas9 is highly specific, off-target mutations have been reported in various cell types (Cho et al., 2014; Cradick, Fine, Antico, & Bao, 2013; Fu et al., 2013; Hsu et al., 2013; Slaymaker et al., 2016). To investigate off-target effects of CRISPR/Cas9 in RhiPSCs, we developed an in-house Python package that lists potential off-target sites associated with the given gRNA sequence up to 4 mismatches in the rhesus macaque genome (Hong et al., 2017). The scores for each site were calculated based on the previous published algorithm (Hsu et al., 2013) as well as the updated formula posted by the same group (http://crispr.mit.edu/about). This tool is available online with detailed guidelines (https://bcbcore.nhlbi.nih.gov/tunc/simpCrispr/). Using this pipeline, one can computationally identify candidate off-target sites for their gRNA of interest in the rhesus macaque genome, rank them, and sequence top loci using Sanger-sequencing. Each sequencing chromatogram can be analyzed with the Tracking of In/dels by Decomposition (TIDE) software (http://tide.nki.nl). In clones which have off-target mutations, a precise sequence of indels can be confirmed by subcloning of PCR fragments using the Zero Blunt® TOPO® cloning kit (Invitrogen). Here, we provide a list of top 10 potential off-target sites for the rhesus AAVS1 gRNA used in this protocol (Table 1) as well as primers set information for each site (Table 2). Conditions for OT PCR with Platinum PCR Supermix (ThermoFisher) were as follows:
Table 1.
Locations of the Top 10 Potential Off-Target Sites
| Off-target Sequence | PAM | Chr | Position | Strand | MM Locations | # of MMs | Score | Overlapping Genea | |
|---|---|---|---|---|---|---|---|---|---|
| 1 | AGGGCCACTTGGGACAGGAC | TAG | 4 | 77699284 | − | 1,10 | 2 | 7.414830508 | None |
| 2 | GGCCCCACTAGGGACAGGAC | AAG | 3 | 160355267 | + | 3,4 | 2 | 5.146703297 | CADPS2b |
| 3 | GGGGCCAGTGGGGACAGGAC | AGG | 13 | 127282774 | − | 8,10 | 2 | 5.028448276 | None |
| 4 | GGCCCCATTAGGGACAGGAC | CAG | 1 | 18536016 | + | 3,4,8 | 3 | 2.448888889 | None |
| 5 | GGGACCATGAGGGACAGGAC | TGG | 19 | 19145046 | − | 4,8,9 | 3 | 1.51751634 | ENSMMUG00000003164b |
| 6 | GGGGGCAGCAGGGACAGGAC | TGG | 17 | 93563191 | − | 5,8,9 | 3 | 1.482630907 | None |
| 7 | GGAGCTAATAGGGACAGGAC | TGG | 14 | 30064062 | − | 3,6,8 | 3 | 1.481577778 | None |
| 8 | GGGAGCACTAAGGACAGGAC | CGG | 7 | 46978018 | − | 4,5,11 | 3 | 1.446502058 | ITGA11b |
| 9 | GGGAGCAGTGGGGACAGGAC | TGG | 11 | 3181345 | − | 4,5,8,10 | 4 | 1.257112069 | None |
| 10 | GGGGCCAGTGGGGACAGGAA | GGG | 12 | 95806523 | − | 8,10,20 | 3 | 1.141953521 | None |
MM stands for Mismatch.
Gene information was found using BLAST online analysis (www.ensembl.org).
Off-target sequence is located in the intron of the corresponding gene.
| 1 cycle: | 94°C for 5 min |
| 35 cycles: | 94°C for 30 sec |
| 57°C for 30 sec | |
| 68°C for 1 min | |
| 1 cycle: | 68°C for 5 min |
Note: OT10 PCR reaction requires an annealing temperature of 61°C.
REAGENTS AND SOLUTIONS
RhiPSC media
KnockOut DMEM (Gibco) supplemented with:
20% Knockout Serum Replacement
0.1mM NEAA
1% PSG
0.1 mM 2-mercaptoethanol
20 ng/ml bFGF
Store for up to 2 weeks at 4°C
Conditioned Media (RhiPSC media w/o bFGF)
See Unit 4A.11 for CM preparation.
Add 20 ng/mL bFGF after thawing CM.
Store for up to 2 weeks at 4°C
MEF Culture Media
DMEM (Gibco cat. no. 11995) supplemented with:
10% FBS
1% NEAA
ROCK inhibitor
To make a 10 mM stock solution of Y27632, reconstitute 2 mg of the compound by adding 624.4 μl of DMSO. Make aliquots of 50 μl per tube and store at −20°C. To reach working concentration, stock can be used as 1000× – use 50 μl per 50 mL of medium to reach 10 μM. Store for up to 6 months at −20°C.
Puromycin
Puromycin powder should be reconstituted with sterile water to reach a stock solution of 50 μg/ml. Store for up to 4 years at −20°C.
COMMENTARY
Background Information
Traditional genome modification tools for gene insertion, such as gamma-retroviral or lentiviral vectors have widely been studied; however, transgene insertion into unpredictable loci of the host genome can lead to transgene silencing or even insertional mutagenesis (Bestor, 2000; Chen & Goncalves, 2016). Specificity and toxicity have plagued editing techniques; however, new programmable nuclease based methods like CRISPR/Cas9 have allowed for robust and reliable targeted genomic modifications (Chen & Goncalves, 2016). This system enables both knockout of specific genes or knock-in of a desired transgene. Specifically, knock-in of marker genes or potentially therapeutic genes holds tremendous promise if the transgene can be stably expressed.
Genomic safe harbors (GSHs) are regions that are amenable to genomic modification without deleterious effects to the host cell while still supporting robust and stable expression. While stringent criteria for putative GSHs, namely – CCR5, Rosa26, CLYBL and AASV1 have been proposed, these theoretical GSHs need to be fully validated. NHP iPSC targeted with marker genes at the GSH site will provide a great opportunity for safety and functional validation. Here we have detailed how the rhesus ortholog of the human AAVS1 locus can be used for transgene insertion and stable transgene expression. GFP or human, truncated CD19 have been used as transgenes with this approach and have been shown to be stably expressed when RhiPSCs are differentiated to cells of lineages from all three germ layers (Hong et al., 2017). Furthermore, this approach can be used to introduce reporter genes that are suitable for in vivo imaging, such as the sodium idodide symporter (NIS) (Ahn, 2012; Penheiter, Russell, & Carlson, 2012) to enable non-invasive monitoring of transplanted cells in NHPs.
Current CRISPR/Cas9 nuclease systems are powerful tools for efficient disruption of specific endogenous genes or introduction of transgenes at desired loci; however, the potential for off-target effects has been widely documented (Pattanayak et al., 2013; Trounson & DeWitt, 2016; Tsai et al., 2015). Tools for identifying non-specific nuclease activity have been useful for screening edited cells before translation to therapeutic applications (Tsai et al., 2017; Tsai et al., 2015). The Zhang lab offers an online software tool for gRNA target selection and off-target screening (http://crispr.mit.edu), but the rhesus macaque genome is not available. As described above (Protocol 3), the tool provided here is useful for research groups working in the rhesus model for identifying potential CRISPR/Cas9 off-target sites in the rhesus genome. Going forward, methods allowing genome-wide identification of off-target effects, such as CIRCLE-seq (Tsai et al., 2017) will be useful for more accurate and sensitive Cas9 off-target determination before in vivo use of these edited cells.
Critical Parameters and Trouble Shooting
Successful targeting of the AAVS1 locus with CRISPR/Cas9 in RhiPSCs is reliant on the quality of the RhiPSCs in culture, the concentrations of the donor and the CRISPR/Cas9/gRNA plasmids, and the establishment of a live staining system for colony selection. Proper maintenance of RhiPSCs in hypoxic conditions and mechanically passaging colonies to appropriate sizes is essential for generating edited clones that possess appropriate morphology and pluripotency potential. For issues with maintenance of RhiPSCs, see Unit 4A.11for further guidelines. It is important to use endotoxin-free plasmids for transfection. As discussed above, the concentrations of the donor or all-in-one plasmid can vary the extent of HDR or the level of cleavage at the desired site versus off-target loci. If less than 10% of the clones after puromycin selection have targeted integration, then consider optimizing DNA concentrations of the donor and CRISPR/Cas9 plasmids. When trouble shooting, you can pool the DNA from all the clones picked after puromycin selection and do NHEJ PCR to detect the cutting efficiency on the target site. If the NHEJ efficiency is significant (>50%) but you have <10% clones with targeted integration, then increasing the donor plasmid concentration will probably help to increase the targeting efficiency. If NHEJ is less than 50%, consider increasing the CRISPR/Cas9 vector concentration. Also, consider checking the integrity of the donor plasmid to survey for signs of degradation or rearrangement by gel electrophoresis of undigested and digested plasmids using restriction enzymes. The choice of gRNA can affect cutting efficiency and potential off-target nuclease activity. Therefore, if selecting a gRNA other than the one suggested above see Unit 5B.6.2 for troubleshooting gRNA design and assessment of its activity.
Anticipated Results
The measureable parameters to look for with these protocols include targeting efficiency, Cre-excision efficiency, and off-target propensity. After puromycin selection, roughly three-fourths of the RhiPSCs clones tested in our laboratory have had targeted integration of the donor plasmid at the intended AAVS1 site. Excision of the puromycin resistance gene was successful in almost half of the RhiPSC clones screened. Off-target mutations after CRISPR/Cas9-mediated gene editing have been reported with varying efficiency, depending on the choice of gRNA and type of Cas9. Using the eSpCas9 (Slaymaker et al., 2016), we could effectively eliminate off-target editing caused by wild-type SpCas9.
Time Considerations
The editing process timeline typically ranges from 4–6 weeks from initial nucleofection to performing Southern Blot analysis of selected clones. Since managing many clones after the puromycin selection process can be both reagent and time intensive, it is essential to verify properly targeted clones through PCR and Southern Blot early on and either freeze down or discard non-targeted clones. Cre-excision of the puromycin resistance gene takes about 4–5 weeks to complete.
Significance Statement.
Non-human primate (NHP) induced pluripotent stem cells (iPSCs) are useful for the preclinical development of safe and effective cell therapies. Use of the CRISPR/Cas9 nuclease system for safe harbor targeting in NHP iPSCs enables stable transgene expression and allows for tracking of iPSC progeny. This platform also provides the foundation for future therapeutic gene delivery in a clinically relevant NHP model.
Acknowledgments
This research was supported by funding from the Division of Intramural Research at the National Heart, Lung and Blood Institute (NHLBI), National Institutes of Health.
LITERATURE CITED
- Ahn BC. Sodium iodide symporter for nuclear molecular imaging and gene therapy: from bedside to bench and back. Theranostics. 2012;2(4):392–402. doi: 10.7150/thno.3722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bestor TH. Gene silencing as a threat to the success of gene therapy. J Clin Invest. 2000;105(4):409–411. doi: 10.1172/JCI9459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Goncalves MA. Engineered Viruses as Genome Editing Devices. Mol Ther. 2016;24(3):447–457. doi: 10.1038/mt.2015.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho SW, Kim S, Kim Y, Kweon J, Kim HS, Bae S, Kim JS. Analysis of off-target effects of CRISPR/Cas-derived RNA-guided endonucleases and nickases. Genome Res. 2014;24(1):132–141. doi: 10.1101/gr.162339.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cradick TJ, Fine EJ, Antico CJ, Bao G. CRISPR/Cas9 systems targeting beta-globin and CCR5 genes have substantial off-target activity. Nucleic Acids Res. 2013;41(20):9584–9592. doi: 10.1093/nar/gkt714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y, Foden JA, Khayter C, Maeder ML, Reyon D, Joung JK, Sander JD. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat Biotechnol. 2013;31(9):822–826. doi: 10.1038/nbt.2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong SG, Yada RC, Choi K, Carpentier A, Liang TJ, Merling RK, … Dunbar CE. Rhesus iPSC Safe Harbor Gene-Editing Platform for Stable Expression of Transgenes in Differentiated Cells of All Germ Layers. Molecular Therapy. 2017;25(1):44–53. doi: 10.1016/j.ymthe.2016.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu PD, Scott DA, Weinstein JA, Ran FA, Konermann S, Agarwala V, … Zhang F. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol. 2013;31(9):827–832. doi: 10.1038/nbt.2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, … Church GM. RNA-guided human genome engineering via Cas9. Science. 2013;339(6121):823–826. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merling RK, Sweeney CL, Choi U, De Ravin SS, Myers TG, Otaizo-Carrasquero F, … Malech HL. Transgene-free iPSCs generated from small volume peripheral blood nonmobilized CD34+ cells. Blood. 2013;121(14):e98–e107. doi: 10.1182/blood-2012-03-420273. blood-2012-03-420273 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pattanayak V, Lin S, Guilinger JP, Ma E, Doudna JA, Liu DR. High-throughput profiling of off-target DNA cleavage reveals RNA-programmed Cas9 nuclease specificity. Nat Biotechnol. 2013;31(9):839–843. doi: 10.1038/nbt.2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penheiter AR, Russell SJ, Carlson SK. The sodium iodide symporter (NIS) as an imaging reporter for gene, viral, and cell-based therapies. Curr Gene Ther. 2012;12(1):33–47. doi: 10.2174/156652312799789235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat Protoc. 2013;8(11):2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slaymaker IM, Gao L, Zetsche B, Scott DA, Yan WX, Zhang F. Rationally engineered Cas9 nucleases with improved specificity. Science. 2016;351(6268):84–88. doi: 10.1126/science.aad5227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trounson A, DeWitt ND. Pluripotent stem cells progressing to the clinic. Nat Rev Mol Cell Biol. 2016;17(3):194–200. doi: 10.1038/nrm.2016.10. [DOI] [PubMed] [Google Scholar]
- Tsai SQ, Nguyen NT, Malagon-Lopez J, Topkar VV, Aryee MJ, Joung JK. CIRCLE-seq: a highly sensitive in vitro screen for genome-wide CRISPR-Cas9 nuclease off-targets. Nat Methods. 2017;14(6):607–614. doi: 10.1038/nmeth.4278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai SQ, Zheng Z, Nguyen NT, Liebers M, Topkar VV, Thapar V, … Joung JK. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat Biotechnol. 2015;33(2):187–197. doi: 10.1038/nbt.3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou J, Sweeney CL, Chou BK, Choi U, Pan J, Wang H, … Malech HL. Oxidase-deficient neutrophils from X-linked chronic granulomatous disease iPS cells: functional correction by zinc finger nuclease-mediated safe harbor targeting. Blood. 2011;117(21):5561–5572. doi: 10.1182/blood-2010-12-328161. [DOI] [PMC free article] [PubMed] [Google Scholar]