

Abstract
Ferroportin Disease (FD) is an autosomal dominant hereditary iron loading disorder associated with heterozygote mutations of the ferroportin-1 (FPN) gene. It represents one of the commonest causes of genetic hyperferritinemia, regardless of ethnicity. FPN1 transfers iron from the intestine, macrophages and placenta into the bloodstream. In FD, loss-of-function mutations of FPN1 limit but do not impair iron export in enterocytes, but they do severely affect iron transfer in macrophages. This leads to progressive and preferential iron trapping in tissue macrophages, reduced iron release to serum transferrin (i.e. inappropriately low transferrin saturation) and a tendency towards anemia at menarche or after intense bloodletting. The hallmark of FD is marked iron accumulation in hepatic Kupffer cells. Numerous FD-associated mutations have been reported worldwide, with a few occurring in different populations and some more commonly reported (e.g. Val192del, A77D, and G80S). FPN1 polymorphisms also represent the gene variants most commonly responsible for hyperferritinemia in Africans. Differential diagnosis includes mainly hereditary hemochromatosis, the syndrome commonly due to either HFE or TfR2, HJV, HAMP, and, in rare instances, FPN1 itself. Here, unlike FD, hyperferritinemia associates with high transferrin saturation, iron-spared macrophages, and progressive parenchymal cell iron load. Abdominal magnetic resonance imaging (MRI), the key non-invasive diagnostic tool for the diagnosis of FD, shows the characteristic iron loading SSL triad (spleen, spine and liver). A non-aggressive phlebotomy regimen is recommended, with careful monitoring of transferrin saturation and hemoglobin due to the risk of anemia. Family screening is mandatory since siblings and offspring have a 50% chance of carrying the pathogenic mutation.
Introduction
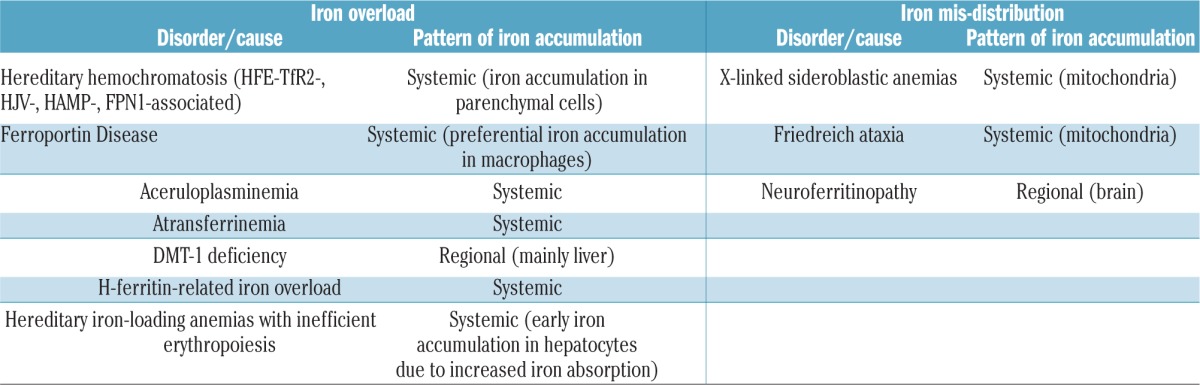
The name Ferroportin Disease (FD) refers to a clinical entity that differs from all other known forms of hereditary iron overload, including hemochromatosis (HC) [synonymous for hereditary hemochromatosis (HH)], i.e. the syndrome due to either HFE or non-HFE hemochromatosis gene mutations.1 In humans, a number of genetic disorders associate with systemic iron overload (Table 1) while others are caused by iron misdistribution and are associated with the regional accumulation of iron in subcellular compartments (e.g. mitochondria in Friedreich ataxia) or certain cell types and organs (e.g. basal ganglia in neuroferritinopathy) (Table 1). In strict terms, the latter disorders may not all qualify as true iron-overload states, as the total body iron content may not be increased. FD, which today accounts for one of the commonest forms of hereditary iron overload disorder besides HFE-hemochromatosis, is characterized by a unique pathogenic basis and clinical presentation and, unlike HC, has been reported worldwide, regardless of ethnicity.
Table 1.
Human hereditary disorders associated with iron overload and iron mis-distribution.
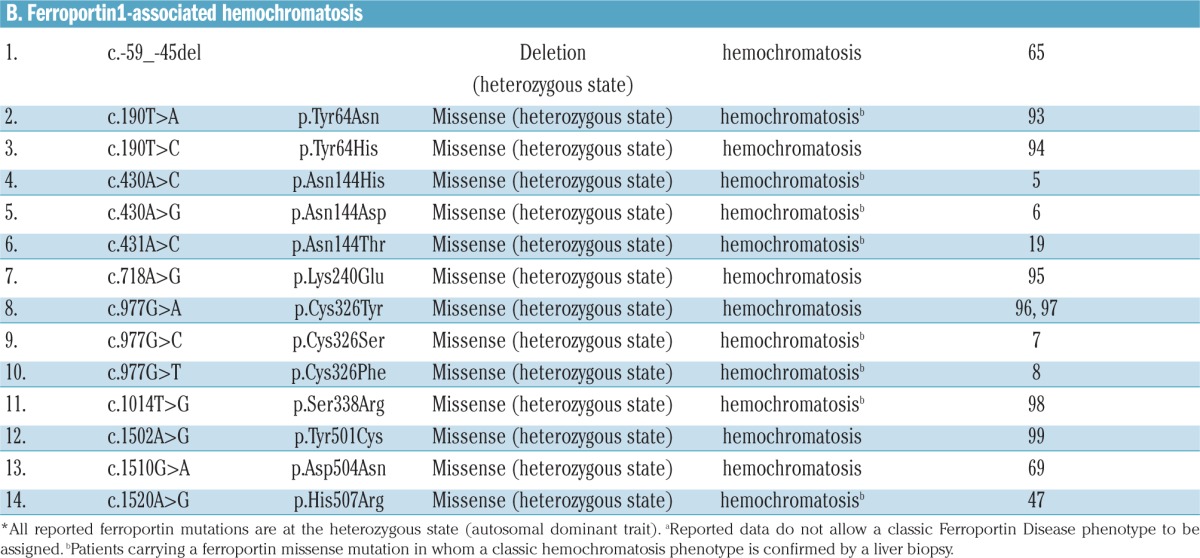
Ferroportin Disease (phenotype MIM number 606069, gene/Locus; MIM number 604653; https://www.omim.org/entry/606069?search=ferroportin%20disease&highlight=ferroportin%20disease) is due to pathogenic (usually missense) mutations of the ferroportin1 gene (FPN1; SLC40A1) which encodes the only iron exporter so far identified in mammals;2–4 lack-of-function mutations impair the iron-export capability of FPN1, particularly in cells with high iron turnover, such as tissue macrophages. Unlike the mutations causing FD, other rare mutations of FPN1 (such as N144H, C326Y, C326S and C326F),5–8 do not impair protein expression at the cell membrane or its iron export capability, but make FPN1 resistant to the inhibitory effect of hepcidin, the physiological FPN1 inhibitor (see below under Pathogenesis). This causes unchecked iron export activity of FPN1; the resulting clinical disorder is different from FD and indistinguishable from other forms of hereditary HC (Tables 1 and 2).
Table 2.
Main features of Ferroportin Disease and other hereditary iron overload disorders in humans.

Definition and classification
The OMIM database classifies the two forms of hereditary iron overload due to FPN1 mutations within the same taxonomic category as “hemochromatosis type 4” (https://www.omim.org/entry/606069?search=ferroportin%20disease&highlight=ferroportin%20disease). Similar terminology has then been adopted by Orphanet with the inclusion of two subcategories: hemochromatosis type 4A (referring to classic FD due to lack-of-function FPN1 mutations) and hemochromatosis type 4B (referring to FD due to gain-of-function FPN1 mutations) (http://www.orpha.net/consor/cgibin/OC_Exp.php?Lng=EN&Expert=139491). These classifications have been incorporated into recent publications, with some variants.9,10 Disease naming and classification (taxonomy) can vary depending on different criteria, such as pathogenic genes, mechanisms, clinical manifestations, etc. Ideally, disease taxonomy (and names) should also help clinicians to recognize, diagnose, and cure diseases. In this context, the taxonomy adopted by OMIM and Orphanet, by embracing two pathogenically and clinically different disorders caused by mutations in the same gene under the term “hemochromatosis”, may fail to reach those objectives. Over the past decades, the term hemochromatosis has been inconsistently used in the literature and in clinical practice to imprecisely refer to: i) any form of body iron overload; ii) tissue iron overload causing organ damage and disease; iii) genetically determined iron overload; and, recently, iv) HFE-related iron overload.11 Recent discoveries in the field have shown that, regardless of the underlying genetic defect, a number of hereditary iron loading disorders (i.e. those due to loss-of-function mutations of HFE, TfR2, HJV, HAMP and gain-of-function mutations of FPN1) belong to the same syndromic entity as they share the pathogenic basis (lack of hepcidin function-activity), biochemical expressivity (high transferrin saturation and high serum ferritin), liver pathology features (iron accumulation in parenchymal cells with iron-spared Kupffer cells until late stage), damage and disease of distinct target organs (liver, heart, endocrine glands, joints), and the therapeutic approach with optimal response to phlebotomy.11 As discussed in the following sections, each individual feature reported above is different in classic FD.1 Therefore, using the term “hemochromatosis” for the classic FD or the term “Ferroportin Disease” for FPN1-associated HC, is misleading, particularly for clinicians, since clinical suspicion, diagnostic strategy and management differ profoundly. Based on these considerations, and on our present understanding of the pathogenesis and clinical manifestations of these disorders, it is proposed that the disorder due to lack-of-function mutations of FPN1 is termed “Ferroportin Disease”, as originally described,1 and the disorder due to gain-of-function FPN1 mutations is termed “FPN-1 associated hemochromatosis” (Table 1). Instead, in analogy with other protein-related classifications (e.g. ferritinopathies; hemoglobinopathies), both disorders due to lack- and gain-of-function mutations of FPN1 may well be included in a broader taxonomic category named “ferroportinopathies”.
Historical aspects
In 1996, the HFE hemochromatosis gene, whose C282Y homozygote mutation is responsible for most cases of HH in Caucasians, was identified.12 Soon after, it became apparent that not all hereditary iron overload disorders could be explained by HFE mutations, particularly in Southern Europe, where an active search for other genes linked to genetic iron overload flourished. From 2000 to 2004, all known non-HFE genes associated so far with HC, namely transferrin receptor 2 (TfR2),13 FPN1,5 hepcidin (HAMP),14 and hemojuvelin (HJV)15 were identified.
A few years earlier, in 1999, a distinct and somehow unusual phenotype had been reported in a large family with hereditary iron overload from Italy. Selective iron loading of liver macrophages, hyperferritinemia co-existing with normal-low transferrin saturation, and tendency to anemia after intense phlebotomy were the hallmarks of the disorder.16 In 2001, all affected family members were reported to be heterozygous for a c. 230 C→A substitution resulting in the replacement of alanine 77 with aspartate in FPN1.17 This entity was subsequently named FD.1
On the other hand, FPN1-related HC, due to a gain-of-function mutation of FPN1 (p.N144H), was first reported by Njajou et al. in 2001.5 Yet, it is worth mentioning that the first clinical description of an “autosomal dominant” form of classic HC had been already reported by Eason et al. in a Melanesian kindred in 1990.18 In this same population, Arden et al.19 have later linked the HC phenotype to the NI44T gain-of-function mutation of FPN1.
Ferroportin biology and physiology and FD
FPN1, the product of the FPN1 (SLC40A1) gene, transfers iron from the external milieu (i.e. maternal blood or intestinal lumen) and from internal sites of iron storage and recycles it into the bloodstream. In fact, it is highly expressed in liver and spleen macrophages, the luminal site of enterocytes and placental syncytiotrophoblasts.2–4
FPN1 is regulated at different levels by a number of factors, including transcriptionally by heme,20 translationally by the iron-regulatory proteins (IRPs),21 and posttranslationally mainly by hepcidin, the iron hormone. Hepcidin is produced by the liver in response to iron, inflammation, and a variety of stressors.22–25 Hepcidin binds to the extracellular loop of FPN1 and triggers its ubiquitinylation on lysine residues located in the intracellular domain leading to internalization and degradation in lysosomes.26–28 This mechanism allows a finely-tuned control of iron efflux from enterocytes and macrophages toward the bloodstream when more iron is needed during active erythropoiesis (in this case, hepcidin synthesis is inhibited by erythroid signals), or blood iron must be controlled due to pathogen proliferation/growth or incipient iron overload (here hepcidin synthesis is induced by inflammatory or iron mediators, respectively) (reviewed by Drakesmith, Nemeth and Ganz29).
FPN1 topology and membrane organization have long been addressed with controversial results concerning localization of the N- and C-terminal extremities and number of transmembrane segments.30–39 Recently, an inward-open conformation of the transporter has been predicted,34,37 with a cluster of residues lying in the central core of the protein important for iron traffic and consistent with an iron-binding site37 and residues involved in hepcidin binding fully accessible in the outward-open model.37 The inward-open form may represent the resting state of the protein, and the outward-open state as a conformation attainable only in the presence of intracellular iron, i.e. when FPN1 shuttles between the two conformations (Figure 1A). The selective binding of hepcidin to the outer-facing conformation would, therefore, guarantee that FPN1 degradation can occur only when intracellular iron is abundant37 and actively pumped through the channel38 (Figure 1B). Recently, the crystal structures of a bacterial homolog of FPN1, BbFPN, has been resolved in both the outward- and inward-facing states, and a homology model with human FPN1 has been developed.39 According to the Au, FPN1 has 12 TM helices, as previously predicted,32 and is divided into two halves, one forming the N lobe, the other the C lobe connected by a long cytosolic loop, with a central cavity between the lobes that is open towards the extracellular side and not accessible from the intracellular side (Figure 1A). FPN1 undergoes an intra-domain conformational rearrangement during the transport cycle. When hepcidin enters the central cavity between the N and C lobes, and interacts with the hepcidin-binding site located on the C lobe, it elicits two effects: a) it increases the accessibility of the intracellular loops that harbor the ubiquitination sites to the ubiquitin ligases; and b) it arrests the conformational transition of FPN1 from the outward-facing state to the inward-facing state, inhibiting the access of iron from the cytoplasm to the substrate-binding site within the intracellular gate (Figure 1B).39
Figure 1.

Biology of ferroportin and postulated pathobiology of Ferroportin Disease (FD). (A) Structure-function relationship of iron-export ferroportin activity.39 (B) Putative mechanisms of hepcidin binding to FPN and its degradation.39 (C) Postulated basis for FD. (Upper panel) In cells undergoing relatively low iron flux, such as enterocytes, the product of the FPN wild-type allele is able to reach the plasma membrane and export iron. For clarity, mutated FPN1 was not depicted at the cell surface: based on previous in vitro work, it has been postulated that some mutant FPN1 can still reach the cell surface and preserve some iron-transport competence, but this is still controversial. (Lower panel) In cells undergoing high iron turnover, such as macrophages, increased requests for iron export impose high demands on FPN traffic leading to a ‘traffic jam’ within the endocytic/plasmamembrane and degradation compartments and inappropriately low wild-type allele product targeting to the cell membrane.54 (D) Postulated effect of FPN mutations that affect formation of the intracellular gate and access to the iron binding site.39
Molecular pathogenesis
The general pathophysiological basis of FD is well defined and relies on the impaired iron export from the iron storage/recycling site (particularly macrophages) towards the bloodstream. Figure 2 shows the basic iron transport defect of the FD as opposed to FPN1-associated HC. In the latter cases, as discussed above, mutations that affect the hepcidin binding site and or FPN1 ubiquitination result in reduced FPN ‘sensitivity’ to hepcidin, leading to the FPN1-related HC phenotype. This has been nicely exemplified by an informative murine model corresponding at the mutation of the hepcidin binding site.40
Figure 2.

The basis for abnormal iron transfer into the bloodstream in Ferroportin Disease as opposed to FPN-associated hereditary hemochromatosis.
In spite of these advances, the molecular pathogenesis of FD has long been elusive. A number of in vitro studies, mostly using over-expressed exogenous wild-type and mutant FPN1 in a variety of cell lines, have investigated FPN1 biology and function and the effect of different FPN1 mutants on protein traffic and iron-transfer capability, although with conflicting results, depending on the cell line or methodology used.30–32,34,35,38,41–47
In this context, it has been actively debated whether FPN1 haplo-insufficiency would explain FD or whether the disease results from a dominant-negative effect. It has been argued that if haplo-insufficiency was the explanation for FD, then nonsense mutations should also result in the disorder; however, so far, the vast majority of reported mutations in FD are missense mutations.48 In addition, a targeted gene deletion in the murine Fpn1 gene has little effect in heterozygous animals,49 whereas the flatiron (ffe) mouse with a missense mutation in Fpn1 that affects its localization and iron export activity when over-expressed in vitro, present a phenotype similar to human patients.50 In studies using exogenous tagged protein in vitro, Fpn1 forms multimers and mutant Fpn1 prevents cell membrane localization of wild-type Fpn1.33,42,50 A multimeric protein, through a dominant-negative effect, would better explain the autosomal dominant trait of FD. However, other studies from different groups have provided experimental evidence to support the opposite conclusion and showed that Fpn1 is a monomer in cultured cells35,51,52 and in vivo.53 More recently, Sabelli et al.,54 using for the first-time cultured macrophages from FD patients, found that endogenous FPN1 shows a similar localization to that in donor macrophages, except for greater accumulation in lysosomes, suggesting a higher degradation rate of mutant FPN1. Unexpectedly, and contrary to previous studies using over-expressed mutant protein in cell lines, FPN1 in FD macrophage circulates in the early endocytic compartment, does not multimerize, it reaches the plasma membrane, is iron-transport competent (although to a lesser extent than normal macrophages), and is promptly internalized and degraded upon exposure to hepcidin. However, when FD macrophages are exposed to large amounts of heme iron, in contrast to donor macrophages, FPN1 can no longer reach the cell surface, leading to marked intracellular iron retention. Based on these observations, a model of FD has been proposed in which FPN1 monomers, in spite of the fact that half proteins are mutated, can still reach the cell surface and export iron in cells that are exposed in vivo to a relatively low flux of iron, such as enterocytes (Figure 1C).54 On the contrary, in cells undergoing high iron turnover in vivo, such as tissue macrophages, sufficient FPN1 is prevented from reaching the plasma membrane, possibly due to a ‘traffic jam’ in the degradation and/or endocytic cycling pathways. This model is consistent with the clinical manifestation of FD characterized by early iron accumulation in hepatic Kupffer cells and normal transferrin saturation, indicating that mutant FPN1 activity does not limit intestinal iron transfer; the latter becomes critically low in young females at menarche or after aggressive phlebotomy, when high iron demands for erythropoiesis likely impose increased FPN1 traffic/cycling within tissue macrophages.1,16,17 (See below under Clinical Manifestations and Diagnosis and Treatment). This study did not address the question as to whether mutant or only wild-type FPN1 reaches the plasma membrane and whether mutant-FPN1 is transport competent. Previous studies have not been conclusive. Exogenously expressed p.A77D and p.Val162del FPN1 mutants have been found to be iron-transport incompetent in all studies, but able to reach the cell membrane in some,34,35,38,41,43 and not in others.30,31 The p.G80S FPN1 mutant has been localized at the cell surface in two published studies,43,55 and found iron transport competent in one55 and incompetent in the other.43
According to Taniguchi et al.,39 the mutation sites associated with FD are mainly mapped onto the inter-lobe interface, mostly on the intracellular side, and form the intracellular gate. These mutations would, therefore, destabilize the inter-lobe interactions, thereby affecting the stable formation of the intracellular gate and reducing the iron transport activity of FPN1 (Figure 1D). It is possible that different mutants differently affect iron transport capability of FPN1; while this may be better overcome by the normal allele product in cells with low iron turnover such as enterocytes or hepatocytes, it may be further hampered in cells like macrophages where the additional ‘traffic jam’ in the endocytic-plasmamembrane compartment will aggravate the basic defect (see above).
Genetics and epidemiology
A list of published mutations associated with FD and FPN1-related HC is reported in Table 3.5–8,17,43,56–102 Numerous mutations of the FPN1 gene have been identified so far in probands with the classic FD phenotype of French-Canadian, Melanesian, Thai, Japanese and European heritages.
Table 3.
Disease-associated mutations of the FPN1 gene.
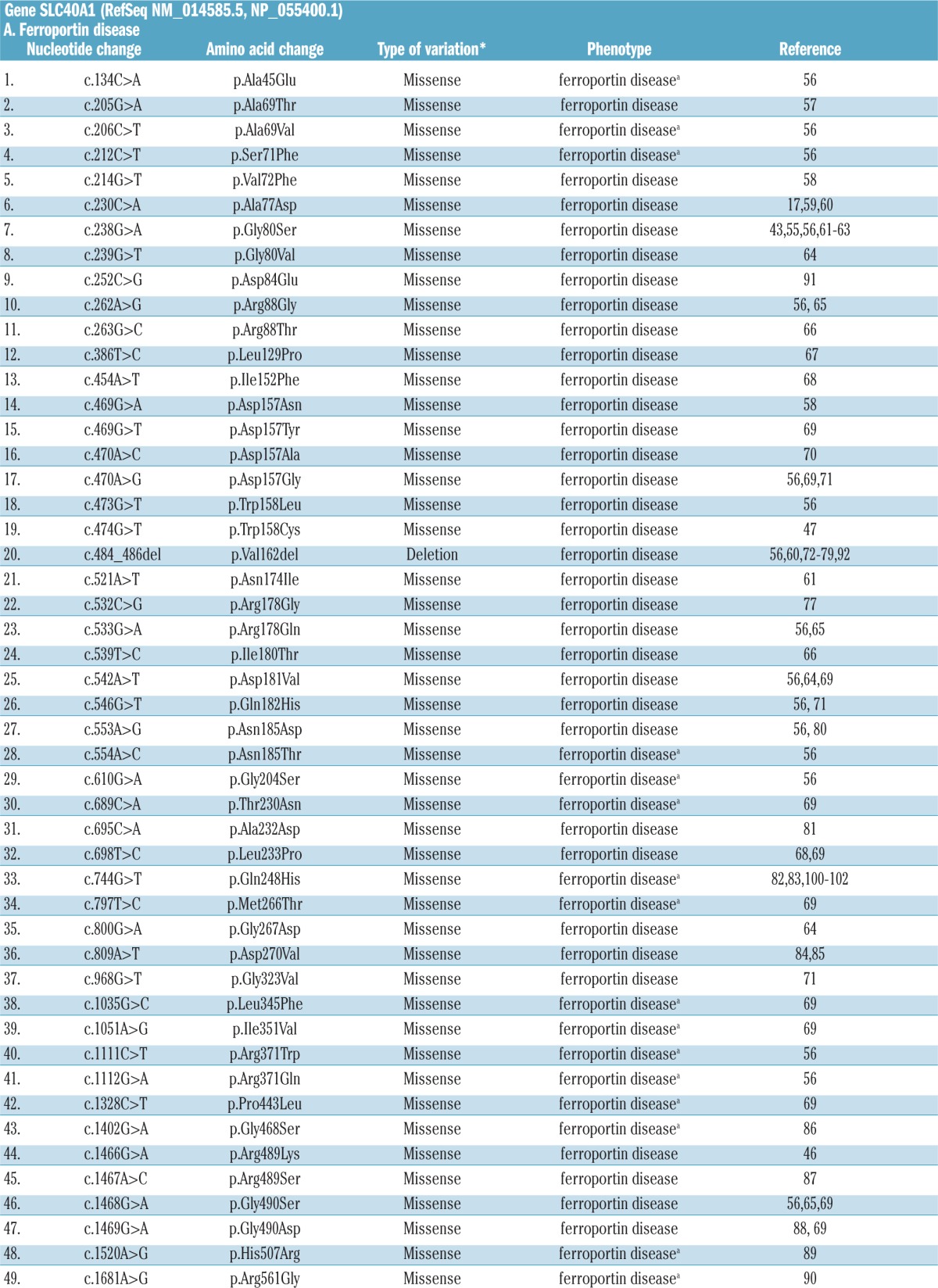
A few common FPN1 mutations have been reported in independent pedigrees, in different countries (e.g. Val192del;56,60,72–79,92 A77D,17,59,60 G80S.43,55,56,61–63 It is now believed that the most frequently reported FPN1 mutations, such as the p.Val162del, are more frequently identified than other SLC40A1 mutations because they have occurred multiple times in isolated populations rather than occurring once and spreading to different populations, as indicated by the identification of a de novo p.Val162del variant in an isolated case of FD.79
FPN1 variants are highly prevalent in African populations. The first prevalent FPN1 variant reported in Africans and Black Americans was the common Q248H polymorphism (p.Gln248His).82,83,100–102 Interestingly, global analysis of variants in the SLC40A1 gene (which includes mutations associated with both the FD and FPN1-associated HH) revealed an allele frequency of 0.0364%, giving a predicted pathogenic genotype carrier rate of 1 in 1373, a figure that approaches the frequency of HFE-HC.103 This was largely due to the relatively high allele frequencies for two SLC40A1 variants (p.Asp270Val84,85 and p.Arg371Trp56) in the African populations; the predicted SLC40A1 pathogenic genotype carrier rate of these two variants is 1 in 197 among the African population.103 The Q248H,101,102 the p.Asp270Val and the p.Arg371Trp and other FPN1 polymorphic variants84 may also predispose to iron overload; but no clear evidence for this has been provided (e.g. lack of functional studies), while the possibility remains that, because of the small sample size, these observations could be attributable to chance or that the polymorphisms identified may be in linkage disequilibrium with other disease-causing loci. Yet, taken together, the collected data make FPN1 the gene most frequently associated with hereditary hyperferritinemia in Africans.
Clinical manifestations and diagnosis
As discussed, FD is caused by loss-of-function mutations in FPN1. These mutations impair iron export, particularly from reticuloendothelial macrophages. The result is iron accumulation in macrophages of the spleen, liver, and bone (reflected by high levels of SF) (Figure 3). At liver histology, parenchymal cells of these organs are largely spared (Figure 3), but discrete hepatocytic iron deposits are also appreciable, due to defective FPN1 activity in hepatocytes, even at early stages.16 Clinical presentation appears heterogeneous, but overall expressivity is milder than classic HC, and the associated liver disease is usually not as severe (Table 2 and Figure 3).1,16,17,56 As occurs in classic forms of HFE HC, also in the FD host factors (menses, blood loss, etc.), co-inheritance of mutations of other iron-genes or variants in genes associated to antioxidant defense and organ fibrosis, and associated pathological conditions (metabolic syndrome, viral hepatitis, etc.) may all affect the phenotype. Hypochromic anemia is not uncommon in young menstruating females.
Figure 3.

The different stages and outcomes of “iron retention” in Ferroportin Disease versus “iron accumulation” in FPN1-associated hemochromatosis (HC). Liver histology pictures are reproduced with the permission of Sabelli et al.54
Owing to the mild clinical expressivity reported in the literature, doubts have been raised on the penetrance of the genetic defect and the rationale for iron-removal therapy. However, there is limited and usually not detailed clinical information in the published reports; this, and the lack of prospective studies, still hamper our understanding of the actual clinical impact of the disorder. In the only report published so far, 6 members of the pedigree in which FD was first described16 were followed for 11–24 years.104 The proband, aged 83, who had carried an occult HBV infection since the age of 56, developed a liver cancer in a non-cirrhotic liver after discontinuation of a 20-year long phlebotomy program; 2 siblings, who had also interrupted treatment, showed a fibrosis progression. These clinical data, while of interest, do not allow definite conclusions to be drawn as to a pathogenic link between iron accumulation in FD and liver damage and disease.
The hallmark of classic FD is marked iron accumulation in Kupffer cells (Figure 3). Kupffer cells are vital to the production of fibrogenic mediators, to immunological tumor surveillance, and disposal of transformed hepatocytes.105 Selective and massive iron overload may impair these activities and favor fibrogenesis and carcinogenesis. Moreover, as discussed above, hepatocytic iron accumulation also takes place in FD, although to a much lesser extent than in HFE- and non-HFE HC, and the established pro-oxidant damaging activity of iron in parenchymal cells may also contribute to disease progression.
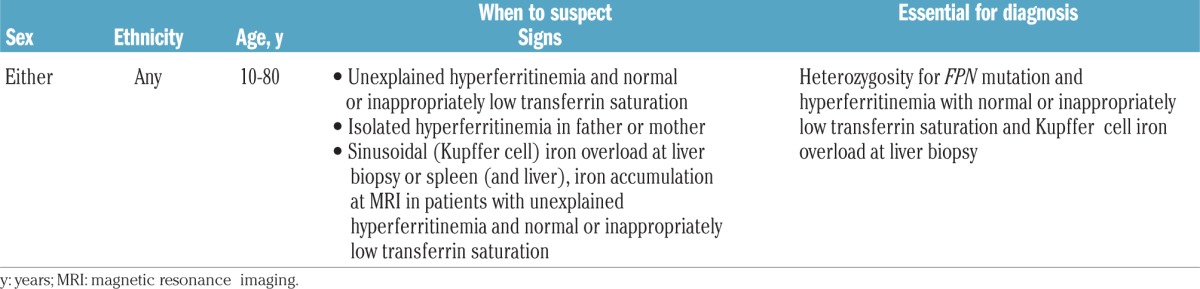
Unlike HFE-HC, the pattern of inheritance of FD is autosomal dominant. Therefore, either parent carries the pathogenic mutation of FPN1 and presents with unexplained hyperferritinemia. In addition, the proband carries a 50% risk of having an affected child. The disease must be suspected in any individual with unexplained hyperferritinemia and low-normal transferrin saturation (TS), or non-parenchymal cell siderosis at liver biopsy or liver and spleen iron accumulation at MRI (Table 4).
Table 4.
Suspecting and diagnosing Ferroportin Disease.
Hyperferritinemia in FD appears very early in life, and unexplained hyperferritinemia with normal TS in a child should prompt MRI evaluation to evaluate iron accumulation in liver, spleen and bone marrow106 (see below).
Figure 4 shows a proposed algorithm for the diagnosis of FD. If hyperferritinemia associates with high TS (as confirmed in at least two sequential determinations) but in the absence of anemia, a typical picture of HFE- and non HFE-HC (including FPN1-associated HC due to gain-of-function FPN1 mutations) is ruled out a priori. If hyperferritinemia associates with high TS and anemia, a typical picture of hereditary hemoglobinopathies and red cell defects or atransferrinemia, FD is again ruled out (Figure 4).
Figure 4.

Diagnostic algorithm for Ferroportin Disease and hereditary hyperferritinemia. ACD/AI: anemia of chronic disease/anemia of inflammation. *Gaucher disease may present with or without siderosis depending on the disease stage. **Advanced ACD/AI may also present with siderosis at MRI (usually spleen and bone marrow).
On the contrary, in subjects with increased serum ferritin and low or normal TS, the workup should focus on common causes of secondary hyperferritinemia and other rare causes of hereditary hyperferritinemia to confirm the diagnosis of FD (Figure 4). First, common causes of hyperferritinemia, such as metabolic disorders, inflammation, cancer, etc., should be considered. If they are not found, or if the hyperferritinemia persists after their treatment, the next step depends on whether or not anemia is present. In the absence of overt anemia, if liver and spleen iron content are increased at MRI or liver biopsy shows prominent Kupffer cell iron load, FD disease should be considered and genetic testing performed for confirmation of diagnosis (Figure 4). Another common cause of hereditary hyperferritinemia with normal TS associated with iron accumulation and anemia is Gaucher disease, usually associated with hepatosplenomegaly, cytopenia, abnormal coagulation, bone disease, and neuropathic manifestations.107
In the absence of body iron accumulation, but in the presence of elevated SF levels and normal TS, autosomal dominant hyperferritinemia with cataract (due to mutations of the iron responsive element in the 5′ untranslated region of the L ferritin mRNA108) or without cataract,109 should be considered. If overt anemia is present, but TS is normal/low, aceruloplasminemia should be suspected, a rare autosomal recessive disease due to loss of function mutations in ceruloplasmin (CP) and resulting in iron overload in the liver and pancreas, and progressive neurodegeneration, diabetes and retinal degeneration.110
Brain MRI with typical iron accumulation in basal ganglia and thalamus may help confirm the diagnosis. As mentioned above, another rare genetic disease presenting with hyperferritinemia and anemia is atransferrinemia/hypotransferrinemia111 which, however, is characterized by increased transferrin saturation due to extremely low serum transferrin levels.
Differential diagnosis mainly includes the classic (HFE) and nonclassic (TfR2, HAMP, HJV and FPN1) forms of HH, all characterized by early and progressive increase of TS followed by elevation of serum ferritin as iron accumulation increases in parenchymal cells of the liver, pancreas, heart and other organs (Table 2). As discussed, unlike HH, in FD clinical expressivity is milder.
Abdominal MRI is a useful non-invasive tool to categorize and diagnose the disorder, as it can differentiate patients with FD, characterized by the SSL triad (spleen, spine, liver) iron retention (Figure 5B), from all other forms of HH, including FPN1-HC, associated with liver iron overload but normal spleen and bone marrow iron content (Figure 5D).112
Figure 5.

Abdominal magnetic resonance imaging (MRI) pattern of Ferroportin Disease (FD). MRI scans. T2*-weighted gradient-echo sequences were used to detect iron accumulation. (A) Normal subject. (B) FD. (C) FD after completion of phlebotomy program (note that excess iron is still detectable in the liver and spine in spite of normal serum ferritin and transferrin saturation levels). (D) Ferroportin-associated hereditary hemochromatosis: iron accumulation involves only the liver and spares the spleen and spine (arrows).
Treatment
Venesection is the cornerstone of therapy also in FD, but it may not be tolerated equally in all patients, and low TS with anemia may be rapidly established despite SF still being elevated.1 Macrophage iron overload is very resistant to iron withdrawal in this disorder, even in patients who are apparently well-treated (Figure 5C). Therefore, unlike HH, not only serum ferritin, but especially TS should be carefully monitored during therapy. In addition, therapy should not aim at reaching the usual HH targets for iron depletion (TS below 20%, SF 50 ng/L or slight anemia) but be more conservative. There are no studies on the optimal phlebotomy schedule in FD. In practical terms, a monthly/bi-monthly phlebotomy session for 1–2 years, depending on the underlying mutation, allows an acceptable state of iron depletion to be reached, while maintenance therapy (usually a phlebotomy session every 4–6 months) should be continued for life. A reasonable target for therapy is an SF level of 100–200 ng/mL. In certain cases, such Ft values may still reflect some iron loading of tissue macrophages (Figure 5C), but the associated clinical risk is negligible. Ideally, the optimal target is the lowest acceptable ferritin level for TS and hemoglobin levels not below the lower limit of normal. The (controversial) dietary restrictions sometimes recommended for patients with HH (avoiding vitamin C or iron-rich or enriched foods) do not apply to FD due to the different pathogenic basis as compared to HH: normal/sufficient enterocyte iron absorption and normal/marginally increased iron accumulation in parenchymal cells in the FD versus increased iron absorption and marked iron accumulation in parenchymal cells in HH.
Iron chelation may be an option in selected cases.79
Siblings of patients with FD, like their offspring, must undergo screening since they have a 50% chance of being susceptible.
Conclusions
FPN1 is a multipass membrane iron-exporter that has evolved in mammals to assure sufficient iron delivery from the external milieu and internal sites of iron storage and recycling to the bloodstream, mainly to support the erythron activity. Overall, the FPN1/SLC40A1 gene is essential for humans and total loss (homozygote mutation) of its product is incompatible with life.49 Loss-of-function of one FPN1 allele in humans results in FD, characterized by a preserved intestinal iron export activity but compromised iron export from tissue macrophages. This leads to progressive iron retention in liver, spleen and bone marrow macrophages, resulting in inappropriately low iron delivery to circulating transferrin and marginal iron-restricted erythropoiesis that may result in overt anemia when bone marrow demands are increased (e.g. menarche, aggressive phlebotomy regimen). Gain-of-function mutations of FPN1 preclude the inhibitory activity of hepcidin, thereby leading to unrestricted iron transfer to the bloodstream and causing a rare form of HH.
The pathogenic, biochemical and clinical signatures of FD are symmetrical and opposite to HFE and non HFE-HH: normal/sufficient enterocyte iron absorption, marked iron accumulation in non parenchymal cells in the FD versus increased iron absorption and marked iron accumulation in parenchymal cells in HH; hyperferritinemia with normal/low transferrin saturation in FD versus hyperferritinemia and high transferrin saturation in HH; intolerance to aggressive phlebotomy regimens in FD versus optimal response to intense phlebotomy in HH; mild and benign clinical course in FD versus potentially severe clinical expressivity in HH; vertical hereditary transmission and presentation at each generation of FD versus recessive transmission of most forms of HH (except FPN1-HH).
While the molecular pathogenesis of FD is becoming more and more defined, the long-term effect of massive iron retention in tissue macrophages in the setting of chronic inflammatory/infectious or degenerative disorders is still unclear.
Today, isolated or unexplained hyperferritinemia represents one of the commonest reasons for referral. Knowing that FD is one of the most frequent genetic causes of hyperferritinemia, regardless of ethnicity, it is important to maintain a high diagnostic suspicion for this disorder.
Supplementary Material
Footnotes
Check the online version for the most updated information on this article, online supplements, and information on authorship & disclosures: www.haematologica.org/content/102/12/1972
References
- 1.Pietrangelo A. The ferroportin disease. Blood Cells Mol Dis. 2004;32(1):131–138. [DOI] [PubMed] [Google Scholar]
- 2.Abboud S, Haile DJ. A novel mammalian iron-regulated protein involved in intracellular iron metabolism. J Biol Chem. 2000;275(26):19906–19912. [DOI] [PubMed] [Google Scholar]
- 3.Donovan A, Brownlie A, Zhou Y, et al. Positional cloning of zebrafish ferroportin1 identifies a conserved vertebrate iron exporter. Nature. 2000;403(6771):776–781. [DOI] [PubMed] [Google Scholar]
- 4.McKie AT, Marciani P, Rolfs A, et al. A novel duodenal iron-regulated transporter, IREG1, implicated in the basolateral transfer of iron to the circulation. Mol Cell. 2000;5(2):299–309. [DOI] [PubMed] [Google Scholar]
- 5.Njajou OT, Vaessen N, Joosse M, et al. A mutation in SLC11A3 is associated with autosomal dominant hemochromatosis. Nat Genet. 2001;28(3):213–214. [DOI] [PubMed] [Google Scholar]
- 6.Wallace DF, Clark RM, Harley HA, Subramaniam VN. Autosomal dominant iron overload due to a novel mutation of ferroportin1 associated with parenchymal iron loading and cirrhosis. J Hepatol. 2004;40(4):710–713. [DOI] [PubMed] [Google Scholar]
- 7.Sham RL, Phatak PD, West C, Lee P, Andrews C, Beutler E. Autosomal dominant hereditary hemochromatosis associated with a novel ferroportin mutation and unique clinical features. Blood Cells Mol Dis. 2005;34(2):157–161. [DOI] [PubMed] [Google Scholar]
- 8.Chen SR, Yang LQ, Chong YT, et al. Novel gain of function mutation in the SLC40A1 gene associated with hereditary haemochromatosis type 4. Intern Med J. 2015;45(6):672–676. [DOI] [PubMed] [Google Scholar]
- 9.Brissot P, Loreal O. Iron metabolism and related genetic diseases: A cleared land, keeping mysteries. J Hepatol. 2016;64(2): 505–515. [DOI] [PubMed] [Google Scholar]
- 10.Hollerer I, Bachmann A, Muckenthaler MU. Pathophysiological consequences and benefits of HFE mutations: 20 years of research. Haematologica. 2017;102(5):809–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pietrangelo A. Hemochromatosis: an endocrine liver disease. Hepatology. 2007;46(4):1291–1301. [DOI] [PubMed] [Google Scholar]
- 12.Feder JN, Gnirke A, Thomas W, et al. A novel MHC class I-like gene is mutated in patients with hereditary haemochromatosis. Nat Genet. 1996;13(4):399–408. [DOI] [PubMed] [Google Scholar]
- 13.Camaschella C, Roetto A, Cali A, et al. The gene TFR2 is mutated in a new type of haemochromatosis mapping to 7q22. Nat Genet. 2000;25(1):14–15. [DOI] [PubMed] [Google Scholar]
- 14.Rivard SR, Lanzara C, Grimard D, et al. Juvenile hemochromatosis locus maps to chromosome 1q in a French Canadian population. Eur J Hum Genet. 2003;11(8):585–589. [DOI] [PubMed] [Google Scholar]
- 15.Papanikolaou G, Samuels ME, Ludwig EH, et al. Mutations in HFE2 cause iron overload in chromosome 1q-linked juvenile hemochromatosis. Nat Genet. 2004;36(1): 77–82. [DOI] [PubMed] [Google Scholar]
- 16.Pietrangelo A, Montosi G, Totaro A, et al. Hereditary hemochromatosis in adults without pathogenic mutations in the hemochromatosis gene [see comments]. N Engl J Med. 1999;341(10):725–732. [DOI] [PubMed] [Google Scholar]
- 17.Montosi G, Donovan A, Totaro A, et al. Autosomal-dominant hemochromatosis is associated with a mutation in the ferroportin (SLC11A3) gene. J Clin Invest. 2001;108(4):619–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Eason RJ, Adams PC, Aston CE, Searle J. Familial iron overload with possible autosomal dominant inheritance. Aust NZ J Med. 1990;20:226–230. [DOI] [PubMed] [Google Scholar]
- 19.Arden KE, Wallace DF, Dixon JL, et al. A novel mutation in ferroportin1 is associated with haemochromatosis in a Solomon Islands patient. Gut. 2003;52(8):1215–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Marro S, Chiabrando D, Messana E, et al. Heme controls ferroportin1 (FPN1) transcription involving Bach1, Nrf2 and a MARE/ARE sequence motif at position -7007 of the FPN1 promoter. Haematologica. 2010;95(8):1261–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lymboussaki A, Pignatti E, Montosi G, et al. The role of the iron responsive element in the control of ferroportin1/IREG1/MTP1 gene expression. J Hepatol. 2003;39(5):710–715. [DOI] [PubMed] [Google Scholar]
- 22.Krause A, Neitz S, Magert HJ, et al. LEAP-1, a novel highly disulfide-bonded human peptide, exhibits antimicrobial activity. FEBS Lett. 2000;480(2–3):147–150. [DOI] [PubMed] [Google Scholar]
- 23.Pigeon C, Ilyin G, Courselaud B, et al. A new mouse liver-specific gene, encoding a protein homologous to human antimicrobial peptide hepcidin, is overexpressed during iron overload. J Biol Chem. 2001;276(11): 7811–7819. [DOI] [PubMed] [Google Scholar]
- 24.Park CH, Valore EV, Waring AJ, Ganz T. Hepcidin, a urinary antimicrobial peptide synthesized in the liver. J Biol Chem. 2001;276(11):7806–7810. [DOI] [PubMed] [Google Scholar]
- 25.Vecchi C, Montosi G, Zhang K, et al. ER stress controls iron metabolism through induction of hepcidin. Science. 2009;325(5942):877–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nemeth E, Tuttle MS, Powelson J, et al. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science. 2004;306(5704):2090–2093. [DOI] [PubMed] [Google Scholar]
- 27.Qiao B, Sugianto P, Fung E, et al. Hepcidin-induced endocytosis of ferroportin is dependent on ferroportin ubiquitination. Cell Metab. 2012;15(6):918–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ross SL, Tran L, Winters A, et al. Molecular Mechanism of Hepcidin-Mediated Ferroportin Internalization Requires Ferroportin Lysines, Not Tyrosines or JAK-STAT. Cell Metab. 2012;15(6):905–917. [DOI] [PubMed] [Google Scholar]
- 29.Drakesmith H, Nemeth E, Ganz T. Ironing out Ferroportin. Cell Metab. 2015;22(5):777–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schimanski LM, Drakesmith H, Merryweather-Clarke AT, et al. In vitro functional analysis of human ferroportin (FPN) and hemochromatosis-associated FPN mutations. Blood. 2005;105(10):4096–4102. [DOI] [PubMed] [Google Scholar]
- 31.Drakesmith H, Schimanski LM, Ormerod E, et al. Resistance to hepcidin is conferred by hemochromatosis-associated mutations of ferroportin. Blood. 2005;106(3):1092–1097. [DOI] [PubMed] [Google Scholar]
- 32.Liu XB, Yang F, Haile DJ. Functional consequences of ferroportin 1 mutations. Blood Cells Mol Dis. 2005;35(1):33–46. [DOI] [PubMed] [Google Scholar]
- 33.De Domenico I, Ward DM, Musci G, Kaplan J. Evidence for the multimeric structure of ferroportin. Blood. 2007;109(5):2205–2209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wallace DF, Harris JM, Subramaniam VN. Functional analysis and theoretical modeling of ferroportin reveals clustering of mutations according to phenotype. Am J Physiol Cell Physiol. 2010;298(1):C75–84. [DOI] [PubMed] [Google Scholar]
- 35.Rice AE, Mendez MJ, Hokanson CA, Rees DC, Bjorkman PJ. Investigation of the biophysical and cell biological properties of ferroportin, a multipass integral membrane protein iron exporter. J Mol Biol. 2009;386(3):717–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Le Gac G, Ka C, Joubrel R, et al. Structure-function analysis of the human ferroportin iron exporter (SLC40A1): effect of hemochromatosis type 4 disease mutations and identification of critical residues. Hum Mutat. 2013;34(10):1371–1380. [DOI] [PubMed] [Google Scholar]
- 37.Bonaccorsi di Patti MC, Polticelli F, Cece G, et al. A structural model of human ferroportin and of its iron binding site. FEBS J. 2014;281(12):2851–2860. [DOI] [PubMed] [Google Scholar]
- 38.Praschberger R, Schranz M, Griffiths WJ, et al. Impact of D181V and A69T on the function of ferroportin as an iron export pump and hepcidin receptor. Biochim Biophys Acta. 2014;1842(9):1406–1412. [DOI] [PubMed] [Google Scholar]
- 39.Taniguchi R, Kato HE, Font J, et al. Outward- and inward-facing structures of a putative bacterial transition-metal transporter with homology to ferroportin. Nat Commun. 2015;6:8545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Altamura S, Kessler R, Grone HJ, et al. Resistance of ferroportin to hepcidin binding causes exocrine pancreatic failure and fatal iron overload. Cell Metab. 2014;20(2):359–367. [DOI] [PubMed] [Google Scholar]
- 41.McGregor JA, Shayeghi M, Vulpe CD, et al. Impaired iron transport activity of ferroportin 1 in hereditary iron overload. J Membr Biol. 2005;206(1):3–7. [DOI] [PubMed] [Google Scholar]
- 42.De Domenico I, Ward DM, Nemeth E, et al. The molecular basis of ferroportin-linked hemochromatosis. Proc Natl Acad Sci USA. 2005;102(25):8955–8960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.De Domenico I, McVey Ward D, Nemeth E, et al. Molecular and clinical correlates in iron overload associated with mutations in ferroportin. Haematologica. 2006;91(8):1092–1095. [PMC free article] [PubMed] [Google Scholar]
- 44.De Domenico I, Ward DM, Langelier C, et al. The molecular mechanism of hepcidin-mediated ferroportin down-regulation. Mol Biol Cell. 2007;18(7):2569–2578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mayr R, Janecke AR, Schranz M, et al. Ferroportin disease: A systematic meta-analysis of clinical and molecular findings. J Hepatol. 2010;53(5):941–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Griffiths WJ, Mayr R, McFarlane I, et al. Clinical presentation and molecular pathophysiology of autosomal dominant hemochromatosis caused by a novel ferroportin mutation. Hepatology. 2010;51(3): 788–795. [DOI] [PubMed] [Google Scholar]
- 47.Mayr R, Griffiths WJ, Hermann M, et al. Identification of Mutations in SLC40A1 That Affect Ferroportin Function and Phenotype of Human Ferroportin Iron Overload. Gastroenterology. 2011;140(7): 2056–2063 e2051. [DOI] [PubMed] [Google Scholar]
- 48.Pietrangelo A, Caleffi A, Corradini E. Non-HFE hepatic iron overload. Sem Liv Dis. 2011;31(3):302–318. [DOI] [PubMed] [Google Scholar]
- 49.Donovan A, Lima CA, Pinkus JL, et al. The iron exporter ferroportin/Slc40a1 is essential for iron homeostasis. Cell Metab. 2005;1(3):191–200. [DOI] [PubMed] [Google Scholar]
- 50.Zohn IE, De Domenico I, Pollock A, et al. The flatiron mutation in mouse ferroportin acts as a dominant negative to cause ferroportin disease. Blood. 2007;109(10):4174–4180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Goncalves AS, Muzeau F, Blaybel R, et al. Wild-type and mutant ferroportins do not form oligomers in transfected cells. Biochem J. 2006;396(2):265–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schimanski LM, Drakesmith H, Talbott C, et al. Ferroportin: lack of evidence for multimers. Blood Cells Mol Dis. 2008;40(3):360–369. [DOI] [PubMed] [Google Scholar]
- 53.Pignatti E, Mascheroni L, Sabelli M, Barelli S, Biffo S, Pietrangelo A. Ferroportin is a monomer in vivo in mice. Blood Cells Mol Dis. 2006;36(1):26–32. [DOI] [PubMed] [Google Scholar]
- 54.Sabelli M, Montosi G, Garuti C, et al. Human macrophage ferroportin biology and the basis for the ferroportin disease. Hepatology. 2017;65(5):1512–1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.McDonald CJ, Wallace DF, Ostini L, Bell SJ, Demediuk B, Subramaniam VN. G80S-linked ferroportin disease: classical ferroportin disease in an Asian family and reclassification of the mutant as iron transport defective. J Hepatol. 2011;54(3):538–544. [DOI] [PubMed] [Google Scholar]
- 56.Le Lan C, Mosser A, Ropert M, et al. Sex and acquired cofactors determine phenotypes of ferroportin disease. Gastroenterology. 2011;140(4):1199–1207.e1–2. [DOI] [PubMed] [Google Scholar]
- 57.Ferbo L, Manzini PM, Badar S, et al. Detection of a rare mutation in the ferroportin gene through targeted next generation sequencing. Blood Transfus. 2016;14(6):531–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pelucchi S, Mariani R, Salvioni A, et al. Novel mutations of the ferroportin gene (SLC40A1): analysis of 56 consecutive patients with unexplained iron overload. Clin Genet. 2008;73(2):171–178. [DOI] [PubMed] [Google Scholar]
- 59.Subramaniam VN, Wallace DF, Dixon JL, Fletcher LM, Crawford DH. Ferroportin disease due to the A77D mutation in Australia. Gut. 2005;54(7):1048–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lim FL, Dooley JS, Roques AW, Grellier L, Dhillon AP, Walker AP. Hepatic iron concentration, fibrosis and response to venesection associated with the A77D and V162del “loss of function” mutations in ferroportin disease. Blood Cells Mol Dis. 2008;40(3):328–333. [DOI] [PubMed] [Google Scholar]
- 61.Corradini E, Montosi G, Ferrara F, et al. Lack of enterocyte iron accumulation in the ferroportin disease. Blood Cells Mol Dis. 2005;35(3):315–318. [DOI] [PubMed] [Google Scholar]
- 62.Mougiou A, Pietrangelo A, Caleffi A, Kourakli A, Karakantza M, Zoumbos N. G80S-linked ferroportin disease: the first clinical description in a Greek family. Blood Cells Mol Dis. 2008;41(1):138–139. [DOI] [PubMed] [Google Scholar]
- 63.Wolff F, Bailly B, Gulbis B, Cotton F. Monitoring of hepcidin levels in a patient with G80S-linked ferroportin disease undergoing iron depletion by phlebotomy. Clin Chim Acta. 2014;430:20–21. [DOI] [PubMed] [Google Scholar]
- 64.Cremonesi L, Forni GL, Soriani N, et al. Genetic and clinical heterogeneity of ferroportin disease. Br J Haematol. 2005;131(5): 663–670. [DOI] [PubMed] [Google Scholar]
- 65.Cunat S, Giansily-Blaizot M, Bismuth M, et al. Global sequencing approach for characterizing the molecular background of hereditary iron disorders. Clin Chem. 2007;53(12):2060–2069. [DOI] [PubMed] [Google Scholar]
- 66.Bach V, Remacha A, Altes A, Barcelo MJ, Molina MA, Baiget M. Autosomal dominant hereditary hemochromatosis associated with two novel Ferroportin 1 mutations in Spain. Blood Cells Mol Dis. 2006;36(1):41–45. [DOI] [PubMed] [Google Scholar]
- 67.Moreno-Carralero MI, Munoz-Munoz JA, Cuadrado-Grande N, et al. A novel mutation in the SLC40A1 gene associated with reduced iron export in vitro. Am J Hematol. 2014;89(7):689–694. [DOI] [PubMed] [Google Scholar]
- 68.Girelli D, De Domenico I, Bozzini C, et al. Clinical, pathological, and molecular correlates in ferroportin disease: A study of two novel mutations. J Hepatol. 2008;49(4):664–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Callebaut I, Joubrel R, Pissard S, et al. Comprehensive functional annotation of 18 missense mutations found in suspected hemochromatosis type 4 patients. Hum Mol Genet. 2014;23(17):4479–4490. [DOI] [PubMed] [Google Scholar]
- 70.Saja K, Bignell P, Robson K, Provan D. A novel missense mutation c.470 A>C (p.D157A) in the SLC40A1 gene as a cause of ferroportin disease in a family with hyperferritinaemia. Br J Haematol. 2010;149(6):914–916. [DOI] [PubMed] [Google Scholar]
- 71.Hetet G, Devaux I, Soufir N, Grandchamp B, Beaumont C. Molecular analyses of patients with hyperferritinemia and normal serum iron values reveal both L ferritin IRE and 3 new ferroportin (slc11A3) mutations. Blood. 2003;102(5):1904–1910. [DOI] [PubMed] [Google Scholar]
- 72.Devalia V, Carter K, Walker AP, et al. Autosomal dominant reticuloendothelial iron overload associated with a 3-base pair deletion in the ferroportin 1 gene (SLC11A3). Blood. 2002;100(2):695–697. [DOI] [PubMed] [Google Scholar]
- 73.Wallace DF, Pedersen P, Dixon JL, et al. Novel mutation in ferroportin1 is associated with autosomal dominant hemochromatosis. Blood. 2002;100(2):692–694. [DOI] [PubMed] [Google Scholar]
- 74.Cazzola M, Cremonesi L, Papaioannou M, et al. Genetic hyperferritinaemia and reticuloendothelial iron overload associated with a three base pair deletion in the coding region of the ferroportin gene (SLC11A3). Br J Haematol. 2002;119(2):539–546. [DOI] [PubMed] [Google Scholar]
- 75.Melis MA, Cau M, Congiu R, et al. A mutation in the TMPRSS6 gene, encoding a transmembrane serine protease that suppresses hepcidin production, in familial iron deficiency anemia refractory to oral iron. Haematologica. 2008;93(10):1473–1479. [DOI] [PubMed] [Google Scholar]
- 76.Roetto A, Merryweather-Clarke AT, Daraio F, et al. A valine deletion of ferroportin 1: a common mutation in hemochromastosis type 4. Blood. 2002;100(2):733–734. [DOI] [PubMed] [Google Scholar]
- 77.Speletas M, Kioumi A, Loules G, et al. Analysis of SLC40A1 gene at the mRNA level reveals rapidly the causative mutations in patients with hereditary hemochromatosis type IV. Blood Cells Mol Dis. 2008;40(3):353–359. [DOI] [PubMed] [Google Scholar]
- 78.Zoller H, McFarlane I, Theurl I, et al. Primary iron overload with inappropriate hepcidin expression in V162del ferroportin disease. Hepatology. 2005;42(2):466–472. [DOI] [PubMed] [Google Scholar]
- 79.Unal S, Piperno A, Gumruk F. Iron chelation with deferasirox in a patient with de-novo ferroportin mutation. J Trace Elem Med Biol. 2015;30:1–3. [DOI] [PubMed] [Google Scholar]
- 80.Morris TJ, Litvinova MM, Ralston D, Mattman A, Holmes D, Lockitch G. A novel ferroportin mutation in a Canadian family with autosomal dominant hemochromatosis. Blood Cells Mol Dis. 2005;35(3):309–314. [DOI] [PubMed] [Google Scholar]
- 81.Relvas L, Claro MT, Bento MC, Ribeiro ML. Novel human pathological mutations. Gene symbol: SLC40A1. Disease: haemochromatosis, type 4. Hum Genet. 2009;125(3):338. [PubMed] [Google Scholar]
- 82.Gordeuk VR, Caleffi A, Corradini E, et al. Iron overload in Africans and African-Americans and a common mutation in the SCL40A1 (ferroportin 1) gene small star, filled. Blood Cells Mol Dis. 2003;31(3):299–304. [DOI] [PubMed] [Google Scholar]
- 83.Beutler E, Barton JC, Felitti VJ, et al. Ferroportin 1 (SCL40A1) variant associated with iron overload in African-Americans. Blood Cells Mol Dis. 2003;31(3):305–309. [DOI] [PubMed] [Google Scholar]
- 84.Zaahl MG, Merryweather-Clarke AT, Kotze MJ, van der Merwe S, Warnich L, Robson KJ. Analysis of genes implicated in iron regulation in individuals presenting with primary iron overload. Hum Genet. 2004;115(5):409–417. [DOI] [PubMed] [Google Scholar]
- 85.Lee PL, Gaasterland T, Barton JC. Mild iron overload in an African American man with SLC40A1 D270V. Acta Haematol. 2012;128(1):28–32. [DOI] [PubMed] [Google Scholar]
- 86.Lee PL, Gelbart T, West C, Barton JC. SLC40A1 c.1402G–>a results in aberrant splicing, ferroportin truncation after glycine 330, and an autosomal dominant hemochromatosis phenotype. Acta Haematol. 2007;118(4):237–241. [DOI] [PubMed] [Google Scholar]
- 87.Koyama C, Wakusawa S, Hayashi H, et al. A Japanese family with ferroportin disease caused by a novel mutation of SLC40A1 gene: hyperferritinemia associated with a relatively low transferrin saturation of iron. Intern Med. 2005;44(9):990–993. [DOI] [PubMed] [Google Scholar]
- 88.Jouanolle AM, Douabin-Gicquel V, Halimi C, et al. Novel mutation in ferroportin 1 gene is associated with autosomal dominant iron overload. J Hepatol. 2003;39(2):286–289. [DOI] [PubMed] [Google Scholar]
- 89.Yamakawa N, Oe K, Yukawa N, et al. A Novel Phenotype of a Hereditary Hemochromatosis Type 4 with Ferroportin-1 Mutation, Presenting with Juvenile Cataracts. Intern Med. 2016;55(18):2697–2701. [DOI] [PubMed] [Google Scholar]
- 90.Sussman NL, Lee PL, Dries AM, Schwartz MR, Barton JC. Multi-organ iron overload in an African-American man with ALAS2 R452S and SLC40A1 R561G. Acta Haematol. 2008;120(3):168–173. [DOI] [PubMed] [Google Scholar]
- 91.Wallace DF, McDonald CJ, Ostini L, Iser D, Tuckfield A, Subramaniam VN. The dynamics of hepcidin-ferroportin internalization and consequences of a novel ferroportin disease mutation. Am J Hematol. 2017;92(10):1052–1061. [DOI] [PubMed] [Google Scholar]
- 92.Wallace DF, Browett P, Wong P, Kua H, Ameratunga R, Subramaniam VN. Identification of ferroportin disease in the Indian subcontinent. Gut. 2005;54(4):567–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Rivard SR, Lanzara C, Grimard D, et al. Autosomal dominant reticuloendothelial iron overload (HFE type 4) due to a new missense mutation in the FERROPORTIN 1 gene (SLC11A3) in a large French-Canadian family. Haematologica. 2003;88(7):824–826. [PubMed] [Google Scholar]
- 94.Raszeja-Wyszomirska J, Caleffi A, Milkiewicz P, Pietrangelo A. Ferroportin-related haemochromatosis associated with novel Y64H mutation of the SCL40A1 gene. Prz Gastroenterol. 2014;9(5):307–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Del-Castillo-Rueda A, Moreno-Carralero MI, Alvarez-Sala-Walther LA, et al. Two novel mutations in the SLC40A1 and HFE genes implicated in iron overload in a Spanish man. Eur J Haematol. 2011;86(3): 260–264. [DOI] [PubMed] [Google Scholar]
- 96.Viprakasit V, Merryweather-Clarke AT, Chinthammitr Y, et al. Molecular Diagnosis of the First Ferroportin Mutation (C326Y) in the Far East Causing a Dominant Form of Inherited Iron Overload. Blood. 2004;104(11):3204. [Google Scholar]
- 97.Lok CY, Merryweather-Clarke AT, Viprakasit V, et al. Iron overload in the Asian community. Blood. 2009;114(1):20–25. [DOI] [PubMed] [Google Scholar]
- 98.Wallace DF, Dixon JL, Ramm GA, Anderson GJ, Powell LW, Subramaniam VN. A novel mutation in ferroportin implicated in iron overload. J Hepatol. 2007;46(5):921–926. [DOI] [PubMed] [Google Scholar]
- 99.Letocart E, Le Gac G, Majore S, et al. A novel missense mutation in SLC40A1 results in resistance to hepcidin and confirms the existence of two ferroportin-associated iron overload diseases. Br J Haematol. 2009;147(3):379–385. [DOI] [PubMed] [Google Scholar]
- 100.Barton JC, Acton RT, Rivers CA, et al. Genotypic and phenotypic heterogeneity of African Americans with primary iron overload. Blood Cells Mol Dis. 2003;31(3):310–319. [DOI] [PubMed] [Google Scholar]
- 101.Gordeuk VR, Diaz SF, Onojobi GO, et al. Ferroportin Q248H, dietary iron, and serum ferritin in community African-Americans with low to high alcohol consumption. Alcohol Clin Exp Res. 2008;32(11):1947–1953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Barton JC, Acton RT, Lee PL, West C. SLC40A1 Q248H allele frequencies and Q248H-associated risk of non-HFE iron overload in persons of sub-Saharan African descent. Blood Cells Mol Dis. 2007;39(2): 206–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Wallace DF, Subramaniam VN. The global prevalence of HFE and non-HFE hemochromatosis estimated from analysis of next-generation sequencing data. Genet Med. 2016;18(6):618–626. [DOI] [PubMed] [Google Scholar]
- 104.Corradini E, Ferrara F, Pollicino T, et al. Disease progression and liver cancer in the ferroportin disease. Gut. 2007;56(7):1030–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Manifold IH, Triger DR, Underwood JC. Kupffer-cell depletion in chronic liver disease: implications for hepatic carcinogenesis. Lancet. 1983;2(8347):431–433. [DOI] [PubMed] [Google Scholar]
- 106.Galicia-Poblet G, Cid-Paris E, Lopez-Andres N, et al. A Pediatric Case Report of Ferroportin Disease. J Pediatr Gastroenterol Nutr. 2015;63(6):e205–e207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Mistry PK, Lopez G, Schiffmann R, Barton NW, Weinreb NJ, Sidransky E. Gaucher disease: Progress and ongoing challenges. Mol Genet Metab. 2017;120(1–2):8–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Beaumont C, Leneuve P, Devaux I, et al. Mutation in the iron responsive element of the L ferritin mRNA in a family with dominant hyperferritinaemia and cataract. Nat Genet. 1995;11(4):444–446. [DOI] [PubMed] [Google Scholar]
- 109.Kannengiesser C, Jouanolle AM, Hetet G, et al. A new missense mutation in the L ferritin coding sequence associated with elevated levels of glycosylated ferritin in serum and absence of iron overload. Haematologica. 2009;94(3):335–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Miyajima H, Nishimura Y, Mizoguchi K, Sakamoto M, Shimizu T, Honda N. Familial apoceruloplasmin deficiency associated with blepharospasm and retinal degeneration. Neurology. 1987;37(5):761–767. [DOI] [PubMed] [Google Scholar]
- 111.Heilmeyer L, Keller W, Vivell O, Betke K, Woehler F, Keiderling W. [Congenital atransferrinemia]. Schweiz Med Wochenschr. 1961;91:1203. [PubMed] [Google Scholar]
- 112.Pietrangelo A, Corradini E, Ferrara F, et al. Magnetic resonance imaging to identify classic and nonclassic forms of ferroportin disease. Blood Cells Mol Dis. 2006;37(3):192–196. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.