

Abstract
Molecular diagnosis of patients with von Willebrand disease is pending in most populations due to the complexity and high cost of conventional molecular analyses. The need for molecular and clinical characterization of von Willebrand disease in Spain prompted the creation of a multicenter project (PCM-EVW-ES) that resulted in the largest prospective cohort study of patients with all types of von Willebrand disease. Molecular analysis of relevant regions of the VWF, including intronic and promoter regions, was achieved in the 556 individuals recruited via the development of a simple, innovative, relatively low-cost protocol based on microfluidic technology and next-generation sequencing. A total of 704 variants (237 different) were identified along VWF, 155 of which had not been previously recorded in the international mutation database. The potential pathogenic effect of these variants was assessed by in silico analysis. Furthermore, four short tandem repeats were analyzed in order to evaluate the ancestral origin of recurrent mutations. The outcome of genetic analysis allowed for the reclassification of 110 patients, identification of 37 asymptomatic carriers (important for genetic counseling) and re-inclusion of 43 patients previously excluded by phenotyping results. In total, 480 patients were definitively diagnosed. Candidate mutations were identified in all patients except 13 type 1 von Willebrand disease, yielding a high genotype-phenotype correlation. Our data reinforce the capital importance and usefulness of genetics in von Willebrand disease diagnostics. The progressive implementation of molecular study as the first-line test for routine diagnosis of this condition will lead to increasingly more personalized and effective care for this patient population.
Introduction
Von Willebrand disease (VWD) is the most common bleeding disorder, with a reported incidence of 0.01% to 1%.1,2 The condition is caused by abnormalities related to the von Willebrand factor protein (VWF), which has an important role in primary hemostasis.3,4 A diagnostic algorithm based on the patient’s clinical history and the results of laboratory testing to assess VWF levels and functionality has enabled the classification of VWD into quantitative (type 1 and type 3) and qualitative (type 2) abnormalities.1 The first-line tests used to classify VWD include the combination of VWF: antigen (Ag), VWF: ristocetin cofactor (RCo), factor VIII:C (FVIII:C), and the VWF:RCo/VWF:Ag ratio, whereas second-line tests include ristocetin-induced platelet aggregation (RIPA), VWF:FVIIIB, VWF: collagen binding (CB), VWF propeptide (pp), and multimer analysis, which are especially useful to differentiate between the type 2 VWD subtypes (2A, 2B, 2M, and 2N). It is essential to correctly identify these subtypes because their treatment differs. Nonetheless, this can be a challenging task as clinical and laboratory phenotypes are very heterogeneous,5 and some tests, such as RIPA and multimeric analysis, are not available in all laboratories.6 Furthermore, there is often a lack of clinical information, and the available laboratory tests are known to have intrinsic limitations.1,7,8 In addition, sequential application of these analyses makes phenotypic VWD diagnosis increasingly more costly.
The VWF glycoprotein is encoded by VWF gene, a large (178 kb comprising 52 exons) and very polymorphic gene (>100 coding single-nucleotide polymorphisms (cSNPs)) with a highly homologous partial pseudogene (VWFP; exons 23–34),9 characteristics that make specific amplification and sequencing of VWF difficult. Thus, molecular analysis of VWF is a challenge. Although genetic analysis is considered a valuable tool to support the diagnosis of VWD,10,11 classic Sanger sequencing of the entire VWF is too costly for general use in all patients. Hence, this method has been applied, with slight variations, only to specific exons of VWF, depending on the VWD subtype. Exon 28 is sequenced for 2A, 2B, and 2M patients, and in type 2A, if a mutation is not found in exon 28, gene study is extended to exons 11–17 and 50–52. In 2N cases, exons 18 to 20 are sequenced, corresponding to the FVIII-binging site. Finally, analysis of the complete coding sequence is required in type 1 and type 3 patients, as potential mutations may be spread all along VWF.12 Nonetheless, the advent of next-generation sequencing (NGS) technology is making molecular diagnosis of VWD by complete VWF sequencing faster and progressively less costly,11 in particular when the new “desktop instruments” and simplified, optimized technologies for library preparation are used.13
Based on the results of a previous Spanish survey elucidating the difficulties of diagnosing VWD,7 a multicenter, prospective project (PCM-EVW-ES, Molecular and Clinical Profile of von Willebrand Disease in Spain) was designed to centrally characterize a large multicenter cohort of VWD patients, with inclusion of NGS molecular analysis. As described in our previous report on this project,14 this new technology enabled us to undertake a nationwide molecular epidemiologic study that supported a new scenario for VWD diagnostics, with genetic analysis being an indispensable first-line tool.
Herein, we provide an in-depth description of the technical aspects of NGS-based molecular characterization of VWF in the 556 individuals studied, complete details of the identified variants, and a description of the results of a genotypic-phenotypic correlation study in patients diagnosed with all types of VWD.
Methods
Patients
Molecular studies were performed on samples from 556 individuals with locally diagnosed VWD recruited in 38 Spanish hospitals participating in the PCM-EVW-ES project.7,14 The inclusion criteria were one or more of the following: 1) VWF:Ag, VWF:RCo and/or VWF:CB ≤30% on at least two occasions regardless of the blood group, 2) detection of multimeric abnormalities, 3) evidence of decreased VWF:FVIIIB level in isolated FVIII deficiency, 4) presence of VWF candidate mutations, and 5) RIPA at a low ristocetin concentration.14 Bleeding score (BS) and central laboratory phenotypic assessment are available in Online Supplementary Methods. The study was performed according to the Declaration of Helsinki, was approved by the local Research Ethics Committee, and all participants provided written informed consent.
VWF Access Array amplification and sequencing
We designed 61 pairs of oligonucleotides (Accession NG_029217) capable of amplifying exons 1 to 52, intronic flanking regions, and approximately 1300 bp of the promoter region that were described in our previous report.14 The 48.48 Access Array integrated fluidic circuit (IFC; Fluidigm, San Francisco, CA, USA) is a nanofluidic chip that allows 2304 different polymerase chain reactions (PCRs) in a final volume of 30 nL by combining 48 samples with 48 primer pairs. As 61 primer pairs are needed for total VWF amplification, 12 multiplex PCRs were created, which allowed 2928 reactions per chip. The Access Array was processed following the manufacturers’ recommendations (Online Supplementary Methods and Online Supplementary Table S1). The outcome was a pool of all VWF amplicons from the same sample, plus a distinctive short sequence identification label that acted as a barcode, incorporated into each set of PCRs during the amplification step. The final pools of up to 192 samples (four Access Array fusion libraries) were then combined and sequenced in a MiSeq platform (Illumina, San Diego, CA, USA; Online Supplementary Figure S1).
Data analysis and identification of genetic variants
Barcoded sequences were demultiplexed and analyzed individually. The NGS pipeline output, paired sequence files (FASTQ format), was used as input for analysis with the CLC Genomic Workbench software 8.0.2 (Qiagen, Hilden, Germany). After variant calling, the resulting files (VCF) were used as input for VariantStudio 2.2.1 (Illumina). The result of this step is the identification of potential pathogenic variants and filtering of the polymorphisms described to date in the SNP (dbSNP) and 1000 Genomes databases (Figure 1). The specific analytical parameters used are described in Online Supplementary Methods. Selected mutations detected were validated/confirmed by Sanger sequencing.10 The nomenclature and criteria used to establish variant pathogenicity is also provided in Online Supplementary Methods.
Figure 1.

Flowchart depicting the molecular analysis and identification of potential mutations in individuals enrolled in the study. VWF from 48 patients were simultaneously amplified in a 48.48 Access-Array. Alignment, variant calling and annotation of each variant identified was performed by the CLC Genomic Workbench software. This analysis allowed selection of mutations/variants aligned against VWF (shown in gray) and elimination of those aligned against VWFP (shown in black). Variant filtering was performed by the Variant Studio software. *MAF<0.01 for all variants except three (p.Arg854Gln, p.Arg924Gln, p.Pro2063Ser). VWF: von Willebrand factor gene; VWFP: von Willebrand factor pseudogene; dbSNP: dbSNP database; 1000G: 1000 Genomes Project; ExAC: Exome Aggregation Consortium; MAF: minor allele frequency; NGS: next-generation sequencing.
In silico analysis
In silico prediction to evaluate the functional effects of putative pathogenic variants was performed using the Alamut Visual v.2.6.1 software (Interactive Biosoftware, Rouen, France). Missense prediction tools included PolyPhen-2, the Sorting Intolerant From Tolerant (SIFT), Mutation Taster, Mutation Assessor and Provean, whereas predictive tools for synonymous and splice site candidate mutations included GeneSplicer, Splicing Sequences Finder (SSF), Human Splicing Finder (HSF), Neural Network (NN) Splice and Maximum Entropy Modeling (MaxEnt). Upstream VWF variants were localized, visualized by the Integrative Genomics Viewer software, and compared in parallel with datasets from the Encyclopedia of DNA Elements (ENCODE) project to identify potential regulatory regions.15
Multiplex ligation-dependent probe amplification (MLPA)
VWF deletions/duplications were detected by MLPA using the SALSA MLPA P011 and P012 VWF kits (version B2; MRC–Holland, Amsterdam, The Netherlands), as previously described.14,16 This method was applied to 5 patients whose genotype did not correlate with the phenotype
Microsatellite analysis
A multiplex fluorescent PCR described by our group,17 comprising 3 VWF intragenic tetranucleotide short tandem repeats (STR-1, STR-2, and STR-3) and 1 dinucleotide repeat in the promoter region (WPA) was applied to genomic DNA samples.
Genotype-phenotype correlation
The correlation between genotype and phenotype was assessed by experts from the central laboratories of the PCM-EVW-ES who contrasted the results of the phenotypic test panel and the genetic analysis on the basis of the effect and localization of mutations and previous descriptions in the literature and/or databases.
Results
Nanofluidic VWF amplification and NGS output
The selected VWF regions were amplified with the Fluidigm Access Array. As each library is distinctively labeled, up to 384 samples can be pooled and sequenced in a single run. Fifteen Access Array and 5 MiSeq 500-cycle runs were needed to process the 556 samples (Figure 1, Online Supplementary Results). The output quality parameters obtained for each run and the resulting mean values are shown in Online Supplementary Table S2. In total, 25620 bp of VWF were sequenced in all samples, with homogeneous coverage for nearly all 61 amplicons (Online Supplementary Figure S2). Regions with low or no coverage were completed by Sanger sequencing.10
Genotype-phenotype correlation and classification
Of the 556 individuals recruited in the PCM-EVW-ES database, 442 had confirmed VWD based on central phenotype characterization. Mutation analysis by NGS was performed in all 556 individuals, and MLPA was additionally used in five (03012, 05011, 10011, 23010, 25005). Two large deletions were identified: c.1157_5620del in patient 03012 (type 2A) due to homologous recombination between short sequences of introns 10 and 32, and c.(1945+1_1946–1)_(7437+1_7438–1)del in the type 3 patient, 25005 (exact breakpoints pending characterization). Following mutation analysis, 105 were reclassified to a different subtype, five were excluded and 43 patients were reincluded (Figure 2) due to the presence of a candidate mutation in VWF: 19 type 3 VWD carriers, one type 2N VWD carrier, four type 1 VWD with borderline levels, nine type 1H VWD, three type 2M VWD with collagen binding mutations, and seven patients with uncertain classification. All these data, based on phenotyping and genotyping results, leads to a final diagnosis of VWD in 480 patients from 280 families (mean 1.7 members per family; range 1–21) who met the criteria for inclusion in the VWF registry (Figure 3). A total of 704 variants were identified, 237 were different, and 155 had not been described in the VWF European Association for Haemophilia and Allied Disorders (EAHAD) Coagulation Factor Variant Database compiled in the Leiden Open Variation Database (LOVD; EAHAD-VWD-LOVD).18 The patients’ phenotypic and molecular data were categorized according to the VWD type and used to determine genotype-phenotype correlations, which were established in 94.6% of subjects (Figure 4). Distribution of the families by VWD subtypes is depicted in Figure 5.
Figure 2.

Classification of VWD patients based on central phenotypic diagnosis and final assignment according to the genotype-phenotype correlation. The dataset highlights the number of patients reincluded or reclassified after molecular study, from the initial classification based on central phenotypic results (shown in black) to a definite, refined classification based on molecular data analysis together with phenotypic results (shown in gray). *Patients diagnosed initially as type 1 VWD and reclassified to type 2M VWD due to the presence of collagen binding mutations.† Patient with uncertain classification reclassified to type 3 VWD due to the presence of nonsense mutation in homozygous state. This is a discrepant case since laboratory levels do not correlate and it is pending a new analysis from a freshly obtained sample. Out: number of patients removed from this subtype; To: final definite classification; In: number of patients reclassified to this subtype; From: previous phenotypic classification; UC: uncertain classification; AVWS: acquired von Willebrand Syndrome.
Figure 3.

Summary of the final diagnosis of patients in terms VWD type. On the basis of phenotype data, 98 of the 556 recruited individuals did not meet any inclusion criteria and were initially excluded. Following the molecular analysis, 43 of these patients were reincluded due to the presence of a candidate mutation in VWF. The remaining 55 patients did not meet any inclusion criteria. Moreover, molecular analysis prompted the exclusion of 21 additional individuals: eight HA patients, five HA carriers, and eight patients finally diagnosed as AVWS and confirmed by the absence of mutations in VWF or GP1BA. Of note, 280 families were finally included (shown in black) and nine of them had members with different VWD types. In those particular cases, the family may be counted more than once; that is, within each VWD type where a family member was classified. VWD: von Willebrand disease.
Figure 4.

Schematic representation of phenotype/genotype correlations by VWD type. The correlation between genotype and phenotype in each patient was based on the concordance between the results of the phenotypic test panel and the results of the genetic analysis.
Figure 5.

Classification of the 280 families included according to VWD type. Numbers in parentheses refer to the total of families included in each VWD type. Of note, nine of the 280 families included in the PCM-EVW-ES presented a mixed phenotype (families in which some members had different VWD subtypes). UC: patients with an uncertain classification.
Molecular epidemiology per type of VWD
VWD type 1 was diagnosed in 159 patients. In total, 216 variants were identified in 146 patients (91.8%; Online Supplementary Table S3). Eighty-five different variants (54 undescribed in EAHAD-VWD-LOVD)18 were scattered over VWF, being more frequent in the D3 and D4 domains (45.9%). The five most recurrent mutations (Table 1) had been previously reported in type 1 VWD, except for p.Leu1733Pro, with a deleterious in silico score of 4. Patients with the p.Arg1205His, p.Pro1824His, or p.Leu1733Pro mutations showed the lowest levels of VWF:Ag and VWF:RCo (mean 9.1 and 7, respectively). In 30 patients subclassified as VWD 1H,14,19 the most frequent mutation was p.Arg924Gln (six patients), previously described as a mutation and a polymorphism (Online Supplementary Results). With the exception of 13 patients with no detected variant (mean VWF:Ag=31%; range 16–48; 78% blood group O) and patient 01030 who carried the classic p.Arg854Gln 2N mutation, a good phenotype-genotype correlation was established in 145 patients.
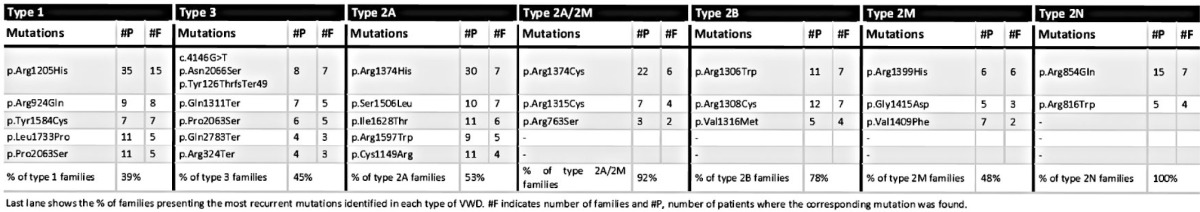
Table 1.
VWF recurrent mutations from 480 patients included in PCM-EVW-ES, classified by VWD types.
Forty-two patients classified as type 3 VWD (mean VWF:Ag=1.2%; mean BS=12.6) and 26 as carriers (mean VWF:Ag=55%; mean BS=1.7) were present in 51 different families (Online Supplementary Table S4). In total, 60 different potential mutations were identified throughout VWF, and 34 had not been previously described in EAHAD-VWD-LOVD,18 of which 12 were mutations causing null alleles. Genotyping was clear and correlated with the clinical and laboratory data in all patients except three: patients 05011 and 10011 with a mutation (p.Cys1946Phe and c.5455+1G>A, respectively) in heterozygous state, and patient 10006 with a homozygous nonsense mutation (p.Gln2470Ter) and normal laboratory levels (pending new analysis from a freshly obtained sample). Excluding these three cases, the 74.4% of type 3 patients had mutations leading to incontrovertible null alleles in both chromosomes (Online Supplementary Figure S3A).
Type 2A VWD was finally diagnosed in 111 patients (detailed classification is disclosed in Online Supplementary Results). In total, 158 variants were identified (Online Supplementary Table S5) and most were clustered in the A1 and A2 domains (38 and 57 potential mutations, respectively) encoded by exon 28. Ninety-eight patients (88%) showed a mutation classified as 2A in EAHAD-VWD- LOVD,18 and the remaining 13 patients had novel candidate mutations in one of the 2A-associated domains (D2, D3, A1, A2, and CK). Hence, an unflawed phenotype-genotype correlation was found in all except four patients: patient 42010, who showed a previously described 2A mutation (p.Arg1527Gln)20 with healthy compatible VWF:Ag and VWF:RCo levels (pending new analysis of a freshly obtained sample), patient 03012, who had a large in-frame deletion affecting residue 386–1873 (domains D2-A3), although this type of mutation seldom explains a 2A phenotype21 and additional studies are needed to investigate the molecular origin, and patients 02071 and 02072, who had a nearly normal multimeric pattern and VWF:RCo/VWF:Ag>0.7 due to the heterozygous missense mutations, p.Arg976Cys (described as 2A/IIE)22 and p.Pro2063Ser.
Thirty-four patients were classified as type 2A/2M VWD (Figure 3 and Online Supplementary Table S6). Remarkably, all families save three presented one of the widely described p.Arg1315Cys and p.Arg1374Cys mutations, located in the A1 domain.23,24 These two controversial mutations were difficult to classify in the light of previous reports and were assigned to 2A/2M, mainly because of their abnormal VWF multimers in medium-resolution gels.25 The new p.Arg763Ser potential mutation, involved in proteolytic processing of the VWF precursor, was found in heterozygous state in two families with symptomatic patients (BS >4), a smeary multimeric pattern, and VWF:FVIIIB <0.7. Of note, mutations in the same amino acid (p.Arg763Gly) have been described in type 2N, but heterozygous patients for this mutation were asymptomatic and classified as 2N carriers.26,27 The remaining family showed p.Cys2491Arg, described as type 3, which involves loss of a cysteine essential for VWF secretion, but these patients were classified as 2A/2M based on their multimeric pattern. Taking these data together, a complete genotype-phenotype correlation could be established for all 2A/2M patients.
Thirty-five patients were diagnosed as having type 2B VWD (detailed classification is disclosed in Online Supplementary Results and Online Supplementary Table S7). Interestingly, among the 16 residues associated with type 2B VWD, one out of five of them—p.Pro1266, p.Arg1306, p.Arg1308, p.Val1316 or p.Pro1337—was found to be affected. A good phenotype-genotype correlation could be established for all patients, as most showed a loss of high-molecular-weight multimers and discordance between VWF:Ag and VWF:RCo levels (mean ratio=0.51; range 0.19–1.1) and a classical type 2B mutation. A normal multimeric pattern and VWF:RCo/VWF:Ag>0.7 were observed in only two families. As expected, the classical p.Pro1266Leu and p.Pro1266Gln molecular defects were found, both being responsible for type 2B Malmö/New York VWD.28,29
Thirty-nine patients were classified as type 2M VWD (Online Supplementary Table S8). Twenty-one different missense variants were found, ten undescribed in EAHAD-VWD-LOVD.18 Of interest, in eight patients with a high VWF:RCo/VWF:Ag ratio, collagen-binding mutations were identified: p.Arg1399His, p.Ser1731Thr, and the novel p.Arg1395Trp.30 All patients showed mutations in the 2M-associated domains A1 and A3, with the exception of three patients who had mutations in other domains, and patient 27022 (carrying p.Ser1731Thr, a collagen I/III-binding mutation, but with VWF:CB=41), considered to have a discrepant genotype-phenotype correlation.
A perfect phenotype-genotype correlation was established in all type 2N VWD patients (Online Supplementary Table S9). Two classical 2N mutations were present in all patients: p.Arg816Trp in exon 19, and p.Arg854Gln in exon 20, the latter seen in 75% of cases. Different combinations of mutation types were found (Online Supplementary Figure S3B): five patients homozygous for a 2N mutation, and four patients compound heterozygous in trans for p.Arg854Gln and a nonsense mutation or p.Gln895His, reported to cause low VWF expression at the messenger ribonucleic acid (mRNA) level.31
In 14 patients the classification was uncertain (Online Supplementary Table S10) because phenotyping or genotyping data were insufficient or inconclusive to indubitably establish the classification. This occurred in eight patients with novel variants of unknown significance (VUS) who had a normal or nearly normal phenotype, and six patients with an abnormal multimeric pattern, VWF:RCo/VWF:Ag <0.7, and a novel candidate mutation. In these cases, it was difficult to distinguish between 2A or 2B VWD since RIPA assay was not assessed.
Molecular epidemiology per type of mutation
A total of 237 different variants were identified in our cohort (Online Supplementary Figure S4A) and a potential ancestral origin was established in 19 of them. All variants, classified by type, are listed in Online Supplementary Tables S11-S17.
In total, 119 different missense candidate mutations were identified, scattered over all domains of VWF (33 of 52 exons). Sixty were not included in EAHAD-VWD-LOVD18 (Online Supplementary Figure S4B) and the mean in silico score in this group was 3.50 (standard deviation (SD) 1.60). The remaining 59 mutations, previously found in VWD patients, had a similar score of 3.54 (SD 1.62). Twenty-three different nonsense mutations were found, 12 unrecorded in EAHAD-VWD-LOVD.18 As expected, nonsense mutations predominantly occurred in type 3 VWD (41 patients). However, we also found this type of mutation in nine type 1 VWD patients, two type 2N, and one type 2A. Of particular note, several mutations were detected in different VWD types. For example, p.Arg324Ter was found in types 3 and 2N, and p.Gln840Ter, p.Gln1311Ter and p.Gln2470Ter were found in types 1 and 3. Five new inframe candidate mutations (two insertions and three deletions) causing different types of VWD were identified. Thirteen potential splicing mutations were found, eight undescribed in EAHAD-VWD-LOVD.18 Their deleterious effect was assessed by five in silico tools: 11 had a predicted effect on splicing, but two of them, those farthest from the exon to be precise, were predicted to have no significant impact on splicing. Ten different upstream variants were found in 20 families with all types of VWD. Only c.-1896C>T and c.-1873A>G have been previously described,32,33 and these were found in type 2A/2M and 2M, respectively. According to their genomic localization, all but the three most upstream (c.-3151T>G, c.-2692C>T, c.-2627C>T) were mapped in regions with well-defined or potential regulatory elements. Sixteen synonymous and 51 intronic variants were identified, but only 5 and 3, respectively, were recorded in EAHAD-VWD-LOVD.18 Although it is difficult to demonstrate, any nucleotide change can be considered a potential candidate to alter splicing, especially when an intronic or synonymous variant appears next to an intron/exon junction.34,35
Discussion
This report describes the molecular study of VWF in the largest prospective multicenter cohort of VWD patients to date, made possible by the development and successful application of NGS technology. The method presented herein has also been used in a smaller Portuguese cohort, with a similar diagnostic yield.16 Heretofore, VWF genetic analysis in large cohorts has been performed by Sanger sequencing.32,33,36,37 In the study herein, considerable time and cost savings were attained by combining two high-throughput technologies, nanofluidics and NGS. The hands-on time and instrument run times for sequencing the 556 patients initially enrolled can be measured in days or weeks rather than months, whereas the cost per sample in our setting was less than $70, 10-fold lower than Sanger sequencing.
The method developed for VWF sequencing is fast and cost-effective for large patient groups; hence, it would seem inappropriate for routine diagnostic laboratories that accumulate small numbers of patients. Nonetheless, NGS can also be applied to individual samples by using the gene panel approach.38 In our opinion, the debate about whether genetic analysis is appropriate for all types of VWD12 will become obsolete in the light of this new technological scenario. It is reasonable to believe that NGS will be progressively incorporated in routine VWD diagnostics.
Centralized comprehensive population studies, although logistically complex, would be the best way to obtain a clear picture of this disease and investigate connections between the multifactorial parameters that influence the diagnosis and etiology.12 In this study, 704 variants (237 unique) were compiled (Online Supplementary Tables S11-S17), whereas the total number of entries in EAHAD-VWD-LOVD18 is about 1,200 (708 unique), an indication of the effectiveness of this approach for the genetic study of large cohorts. In contrast to other studies, the present project included patients with all VWD types and used an identical protocol (whole VWF sequencing) for their genetic analysis. The mutation detection rate was high compared with that of previous studies, being close to 100% in some VWD types (Figure 3). Candidate mutations were identified in 91.8% of type 1 individuals versus the 65–82% reported in previous studies,32,33,36,39 a result that may be related to the limiting recruitment of patients with VWF level <30%.
With regard to the most recurrent mutations identified (Table 1), the Vicenza mutation (p.Arg1205His) proved to be the most common in our type 1 patients, in contrast to previous studies where p.Tyr1584Cys was prevalent.32,33,40 In type 3 VWD, p.Gln1311Ter41 was one of the most frequent mutations detected, as was also seen in the Portuguese population study,16 whereas in patients from northern Europe, p.Arg2535Ter is predominant. As to type 2, the most common mutations found were similar to those reported in previous cohort studies.
Perhaps more important than elucidation of the molecular epidemiology of VWD were the data obtained by NGS, that can resolve many of the drawbacks and limitations of phenotyping. First, molecular characterization of VWD patients enables accurate classification: 27.5% of our patients (153/556) were re-diagnosed after the initial phenotypic study, 43 reincluded, five patients excluded, and 105 reclassified. Of note, 68 patients with uncertain classification were definitively classified by NGS, as in the case of 25 type 2A patients and ten type 2B missing previous RIPA data. Furthermore, four patients whose phenotype pointed to VWD type 1 were found to have 2B Malmö or collagen-binding mutations (Figure 2), the latter being undetectable by standard laboratory tests unless specific binding capacity for collagen types III, IV and VI is performed. Accurate classification is particularly relevant for genetic counseling and detection of carriers in VWD types with autosomal recessive inheritance. We identified 11 asymptomatic carriers 2N and 26 type 3 with borderline plasmatic levels, who would likely have been excluded if the diagnosis had been based only on clinical and laboratory data. Knowledge of the molecular defect also led to diagnostic reassessment of some type 1 patients who could be considered carriers of type 3 mutations (e.g., patient 06024 with the p.Arg2434Ter mutation).
Second, genetic study showed that 37.5% of patients in our cohort had more than one variation, a finding essential to unravelling the potential contribution of different variants to the final phenotype. The situation of two 2M patients is paradigmatic in this regard: 44003, with the p.Gly1415Asp mutation and very low FVIII levels (11%) due to a 2N mutation in heterozygous state, and 03024, who had the p.Val1409Phe plus the p.Arg1399His mutation in trans, which explains the reduction in collagen-binding affinity in this patient.
Third, genetic analysis in large cohorts could shed light on differentiating a variant previously described as a mutation or polymorphism, such as p.Pro1162Leu caused by the c.3485C>T substitution, described as a polymorphism in African Americans.42 Conversely, in our cohort p.Pro1162Leu was caused by c.3485_3486delinsTG (predicted by in silico analysis with effect on splicing), leading to type 3 phenotype in homozygous state.16 These data suggest that different variants resulting in identical amino acid changes may have different consequences at the transcriptional level. Additionally, regarding the controversial p.Pro2063Ser variant, a recent report by Kasatkar et al.43 has provided evidence that p.Pro2063Ser in homozygous state causes type 3 VWD. Hence, five patients in our cohort heterozygous for p.Pro2063Ser were classified as type 3 carriers.
Fourth, genetic studies can offer valuable data regarding the molecular pathophysiological mechanisms responsible for the symptoms observed, such as defects in the structure, intracellular transport, or secretion of VWF. The location of new mutations in certain domains or specific amino acids helps to predict their potential effect, particularly if previous in vitro studies are performed. Examples of this are the new mutations p.Arg273Pro, located in the propeptide and related with formation of disulphide-linked multimers,44 p.Arg763Ser, located in the VWF propeptide cleavage site,26 and p.Arg1395Trp, which affects an essential amino acid for collagen IV binding.30
In addition, although this approach cannot resolve some discrepant cases and those in which no candidate mutation is found (3.1% of our cohort), exome and genome studies by NGS are useful to identify modifying mutations in other genes45 and reveal structural variations undetectable by MLPA.46 It is also important to point out that the technique has provided molecular data for more than 100 cSNPs described in VWF, which will be further analyzed to determine their potential influence on VWF:Ag, FVIII:C and bleeding in our cohort, as has been done in some healthy populations.42,47 If some of these SNPs are found to contribute to the disease, they could be redefined as hemorrhagic risk polymorphisms (similar to the thrombotic risk polymorphisms).
The final obstacle to an accurate diagnosis is the uncertain pathogenicity of novel variations. In silico analysis is considered a suitable supporting tool for genetic diagnosis, and we analyzed all variants using algorithms recommended in specialized guidelines.48 However, the predictive capacity of these programs remains modest: some well-established deleterious mutations have a lower overall score (e.g., p.Leu1580Pro, score=2) than some polymorphisms (e.g., p.Cys325Phe, predicted to be deleterious by SIFT and PolyPhen).49 Therefore, in order to unequivocally determine the potential deleterious effect of new variants, in vitro functional studies remain essential.50
Herein, we present the largest prospective study to date of a cohort of VWD including an exhaustive description of their molecular epidemiology. This data will improve knowledge about the molecular mechanisms that contribute to the complexity of disease diagnosis and will help to elucidate the relationship between bleeding parameters and the patients’ laboratory and genetic profiles. The approach used implies a change in the diagnostic paradigm of VWD, which is also occurring in other genetic diseases. The cost reduction and simplification of genetic analysis by NGS will likely lead to the use of molecular study as an additional first-line routine diagnostic test, with the ultimate aim of providing more personalized and effective care for this patient population.
Supplementary Material
Footnotes
Check the online version for the most updated information on this article, online supplements, and information on authorship & disclosures: www.haematologica.org/content/102/12/2005
Funding
We are indebted to Baxalta US Inc., now a part of Shire, for its support of the PCM-EVW-ES (H13-000845 Grant). This work was also supported by the Spanish Ministerio de Economía y Competitividad (MINECO)-Instituto de Salud Carlos III (ISCIII) (PI12/01494, PI15/01643 and RD12/0042/0053). We thank Technoclone GmbH, Austria, Labclinics, SA, Spain for donating the VWF:CB (type VI collagen) kit. We are very grateful for the kind collaboration of the participating patients and their families. CIBERCV is an initiative of ISCIII co-financed by Fondo Europeo de Desarrollo Regional (FEDER) a way to build Europe.
References
- 1.Sadler JE, Budde U, Eikenboom JC, et al. Update on the pathophysiology and classification of von Willebrand disease: a report of the Subcommittee on von Willebrand Factor. J Thromb Haemost. 2006;4(10): 2103–2114. [DOI] [PubMed] [Google Scholar]
- 2.Rodeghiero F, Castaman G, Dini E. Epidemiological investigation of the prevalence of von Willebrand’s disease. Blood. 1987;69(2):454–459. [PubMed] [Google Scholar]
- 3.Ruggeri ZM. Von Willebrand factor, platelets and endothelial cell interactions. J Thromb Haemost. 2003;1(7):13351–1342. [DOI] [PubMed] [Google Scholar]
- 4.Vlot AJ, Koppelman SJ, Meijers JC, et al. Kinetics of factor VIII-von Willebrand factor association. Blood. 1996;87(5):1809–1816. [PubMed] [Google Scholar]
- 5.Castaman G, Hillarp A, Goodeve A. Laboratory aspects of von Willebrand disease: test repertoire and options for activity assays and genetic analysis. Haemophilia. 2014;20 Suppl 4:65–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hamilton A, Ozelo M, Leggo J, et al. Frequency of platelet type versus type 2B von Willebrand disease. An international registry-based study. Thromb Haemost. 2011;105(3):501–508. [DOI] [PubMed] [Google Scholar]
- 7.Batlle J, Pérez-Rodríguez A, Costa-Pinto J, Lourés E, Rodriguez-Trillo A, López-Fernández MF. Diagnosis and management of von Willebrand disease in Spain. Semin Haemost Thromb 2011; 37(5):503–510. Semin Haemost Thromb. 2011;37(5):503–510. [DOI] [PubMed] [Google Scholar]
- 8.Flood VH. New insights into genotype and phenotype of VWD. Hematology Am Soc Hematol Educ Program. 2014;2014(1):531–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mancuso DJ, Tuley EA, Westfield LA, et al. Human von Willebrand factor gene and pseudogene: structural analysis and differentiation by polymerase chain reaction. Biochemistry. 1991;30(1):253–269. [DOI] [PubMed] [Google Scholar]
- 10.Corrales I, Ramirez L, Altisent C, Parra R, Vidal F. Rapid molecular diagnosis of von Willebrand disease by direct sequencing. Detection of 12 novel putative mutations in VWF gene. Thromb Haemost. 2009;101(3):570–576. [DOI] [PubMed] [Google Scholar]
- 11.Corrales I, Catarino S, Ayats J, et al. High-throughput molecular diagnosis of von Willebrand disease by next generation sequencing methods. Haematologica. 2012; 97(7):1003–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Goodeve AC. The genetic basis of von Willebrand disease. Blood Rev. 2010;24(3): 123–134. [DOI] [PubMed] [Google Scholar]
- 13.Desai A, Jere A. Next-generation sequencing: ready for the clinics? Clin Genet. 2012;81(6):503–510. [DOI] [PubMed] [Google Scholar]
- 14.Batlle J, Perez-Rodriguez A, Corrales I, et al. Molecular and clinical profile of von Willebrand disease in Spain (PCM-EVW-ES): Proposal for a new diagnostic paradigm. Thromb Haemost. 2016;115(1):40–50. [DOI] [PubMed] [Google Scholar]
- 15.Consortium EP, Bernstein BE, Birney E, et al. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489(7414):57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fidalgo T, Salvado R, Corrales I, et al. Genotype-phenotype correlation in a cohort of Portuguese patients comprising the entire spectrum of VWD types: impact of NGS. Thromb Haemost. 2016;116(1):17–31. [DOI] [PubMed] [Google Scholar]
- 17.Vidal F, Julia A, Altisent C, Puig L, Gallardo D. Von Willebrand gene tracking by single-tube automated fluorescent analysis of four short tandem repeat polymorphisms. Thromb Haemost. 2005;93(5):976–981. [DOI] [PubMed] [Google Scholar]
- 18.European Association for Haemophilia and Allied Disorders (EAHAD). Coagulation Factor Variant Databases. October 01, 2010. ed. [DOI] [PubMed] [Google Scholar]
- 19.Montgomery RR, Christopherson P, Bellissimo DB, et al. The complete type I VWD cohort of the Zimmerman Program for the molecular and clinical biology of VWD - phenotypic assignment, mutation frequency, and bleeding assessment. Blood. 2013;122(21):332. [Google Scholar]
- 20.Ahmad F, Jan R, Kannan M, et al. Characterisation of mutations and molecular studies of type 2 von Willebrand disease. Thromb Haemost. 2013;109(1):39–46. [DOI] [PubMed] [Google Scholar]
- 21.Haberichter SL, Allmann AM, Jozwiak MA, Montgomery RR, Gill JC. Genetic alteration of the D2 domain abolishes von Willebrand factor multimerization and trafficking into storage. J Thromb Haemost. 2009;7(4):641–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schneppenheim R, Michiels JJ, Obser T, et al. A cluster of mutations in the D3 domain of von Willebrand factor correlates with a distinct subgroup of von Willebrand disease: type 2A/IIE. Blood. 2010;115(23): 4894–4901. [DOI] [PubMed] [Google Scholar]
- 23.Penas N, Perez-Rodriguez A, Torea JH, et al. von Willebrand disease R1374C: type 2A or 2M? A challenge to the revised classification. High frequency in the northwest of Spain (Galicia). Am J Hematol. 2005;80(3):188–196. [DOI] [PubMed] [Google Scholar]
- 24.Batlle J, Perez-Rodriguez A, Franqueira MD, Lopez-Fernandez MF. Type 2M von Willebrand disease: a variant of type 2A? J Thromb Haemost. 2008;6(2):388–390. [DOI] [PubMed] [Google Scholar]
- 25.Gadisseur A, van der Planken M, Schroyens W, Berneman Z, Michiels JJ. Dominant von Willebrand disease type 2M and 2U are variable expressions of one distinct disease entity caused by loss-of-function mutations in the A1 domain of the von Willebrand factor gene. Acta Haematol. 2009;121(2–3):145–153. [DOI] [PubMed] [Google Scholar]
- 26.Hilbert L, Nurden P, Caron C, et al. Type 2N von Willebrand disease due to compound heterozygosity for R854Q and a novel R763G mutation at the cleavage site of von Willebrand factor propeptide. Thromb Haemost. 2006;96(3):290–294. [DOI] [PubMed] [Google Scholar]
- 27.van den Biggelaar M, Meijer AB, Voorberg J, Mertens K. Intracellular cotrafficking of factor VIII and von Willebrand factor type 2N variants to storage organelles. Blood. 2009;113(13):3102–3109. [DOI] [PubMed] [Google Scholar]
- 28.Weiss HJ, Sussman II. A new von Willebrand variant (type I, New York): increased ristocetin-induced platelet aggregation and plasma von Willebrand factor containing the full range of multimers. Blood. 1986;68(1):149–156. [PubMed] [Google Scholar]
- 29.Weiss HJ. Type 2B von Willebrand disease and related disorders of patients with increased ristocetin-induced platelet aggregation: what they tell us about the role of von Willebrand factor in hemostasis. J Thromb Haemost. 2004;2(11):2055–2056. [DOI] [PubMed] [Google Scholar]
- 30.Flood VH, Schlauderaff AC, Haberichter SL, et al. Crucial role for the VWF A1 domain in binding to type IV collagen. Blood. 2015;125(14):2297–2304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cabrera N, Casana P, Cid AR, Haya S, Moret A, Aznar JA. Novel missense mutation c.2685G>C (p.Q895H) in VWF gene associated with very low levels of VWF mRNA. Ann Hematol. 2009;88(3):245–247. [DOI] [PubMed] [Google Scholar]
- 32.Goodeve AC, Eikenboom J, Castaman G, et al. Phenotype and genotype of a cohort of families historically diagnosed with type 1 von Willebrand disease in the European study, Molecular and Clinical Markers for the Diagnosis and Management of Type 1 von Willebrand Disease (MCMDM-1VWD). Blood. 2007; 109(1):112–121. [DOI] [PubMed] [Google Scholar]
- 33.James PD, Notley C, Hegadorn C, et al. The mutational spectrum of type 1 von Willebrand disease: Results from a Canadian cohort study. Blood. 2007; 109(1):145–154. [DOI] [PubMed] [Google Scholar]
- 34.Yadegari H, Biswas A, Akhter MS, et al. Intron retention resulting from a silent mutation in the VWF gene that structurally influences the 5′ splice site. Blood. 2016; 128(17):2144–2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sauna ZE, Kimchi-Sarfaty C. Understanding the contribution of synonymous mutations to human disease. Nat Rev Genet. 2011;12(10):683–691. [DOI] [PubMed] [Google Scholar]
- 36.Cumming A, Grundy P, Keeney S, et al. An investigation of the von Willebrand factor genotype in UK patients diagnosed to have type 1 von Willebrand disease. Thromb Haemost. 2006;96(5):630–641. [PubMed] [Google Scholar]
- 37.Veyradier A, Boisseau P, Fressinaud E, et al. A laboratory phenotype/genotype correlation of 1167 French patients from 670 families with von Willebrand disease: a new epidemiologic picture. Medicine (Baltimore). 2016;95(11):e3038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Borràs N, Corrales I, Ramírez L, Altisent C, Parra R, Vidal F. Diseño, optimitzación y validación de un panel de secuenciación de 23 genes como herramienta de diagnóstico e investigación de las coagulopatías congénitas. XXXI Congreso Nacional de la Sociedad Española de Trombosis y Hemostasia; 2015 October; Valencia, Spain: Thrombosis and Haemostasis; 2015. p. 95. [Google Scholar]
- 39.Flood VH, Christopherson PA, Gill JC, et al. Clinical and laboratory variability in a cohort of patients diagnosed with type 1 VWD in the United States. Blood. 2016; 127(20):2481–2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bowen DJ, Collins PW, Lester W, et al. The prevalence of the cysteine1584 variant of von Willebrand factor is increased in type 1 von Willebrand disease: co-segregation with increased susceptibility to ADAMTS13 proteolysis but not clinical phenotype. Br J Haematol. 2005; 128(6):830–836. [DOI] [PubMed] [Google Scholar]
- 41.Casana P, Martinez F, Haya S, Lorenzo JI, Espinos C, Aznar JA. Q1311X: a novel nonsense mutation of putative ancient origin in the von Willebrand factor gene. Br J Haematol. 2000;111(2):552–555. [DOI] [PubMed] [Google Scholar]
- 42.Bellissimo DB, Christopherson PA, Flood VH, et al. VWF mutations and new sequence variations identified in healthy controls are more frequent in the African-American population. Blood. 2012; 119(9):2135–2140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kasatkar P, Ghosh K, Shetty S. A common founder mutation p.P2063S in exon 36 of VWF in 11 unrelated Indian von Willebrand disease (VWD) families. Ann Hematol. 2013;92(8):1147–1148. [DOI] [PubMed] [Google Scholar]
- 44.Allen S, Abuzenadah AM, Hinks J, et al. A novel von Willebrand disease-causing mutation (Arg273Trp) in the von Willebrand factor propeptide that results in defective multimerization and secretion. Blood. 2000;96(2):560–568. [PubMed] [Google Scholar]
- 45.Souto JC, Almasy L, Soria JM, et al. Genome-wide linkage analysis of von Willebrand factor plasma levels: results from the GAIT project. Thromb Haemost. 2003;89(3):468–474. [PubMed] [Google Scholar]
- 46.Suzuki T, Tsurusaki Y, Nakashima M, et al. Precise detection of chromosomal translocation or inversion breakpoints by whole-genome sequencing. J Hum Genet. 2014; 59(12):649–654. [DOI] [PubMed] [Google Scholar]
- 47.Johnsen JM, Auer PL, Morrison AC, et al. Common and rare von Willebrand factor (VWF) coding variants, VWF levels, and factor VIII levels in African Americans: the NHLBI Exome Sequencing Project. Blood. 2013;122(4):590–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang QY, Song J, Gibbs RA, Boerwinkle E, Dong JF, Yu FL. Characterizing polymorphisms and allelic diversity of von Willebrand factor gene in the 1000 Genomes. J Thromb Haemost. 2013; 11(2):261–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Corrales I, Ramirez L, Altisent C, Parra R, Vidal F. The study of the effect of splicing mutations in von Willebrand factor using RNA isolated from patients’ platelets and leukocytes. J Thromb Haemost. 2011; 9(4):679–688. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.