

Abstract
A recombinant ribonuclease, cusativin, was characterized for its cytidine-specific cleavage ability of RNA to map chemical modifications. Following purification of native cusativin protein as described before [Rojo et al. (1994) Planta 194:328], partial amino acid sequencing was carried out to identify the corresponding protein coding gene in cucumber genome. Cloning and heterologous expression of the identified gene in Escherichia coli resulted in successful production of active protein as a C-terminal His-tag fusion protein. The ribonuclease activity and cleavage specificity of the fusion protein were confirmed with a variety of tRNA isoacceptors and total tRNA. Characterization of cusativin digestion products by ion-pairing reverse phase liquid-chromatography coupled with mass spectrometry (IP-RP-LC-MS) analysis revealed cleavage of CpA, CpG, and CpU phosphodiester bonds at the 3′-terminus of cytidine under optimal digestion conditions. Ribose methylation or acetylation of cytosine inhibited RNA cleavage. The CpC phosphodiester bond was also resistant to cusativin-mediated RNA cleavage; a feature to our knowledge has not been reported for other nucleobase-specific ribonucleases. Here, we demonstrate the analytical utility of such a novel feature for obtaining high sequence coverage and accurate mapping of modified residues in substrate RNAs.
Keywords: Cusativin, cytidine-specific, modification mapping, acetylation, LC-MS
Graphical Abstract
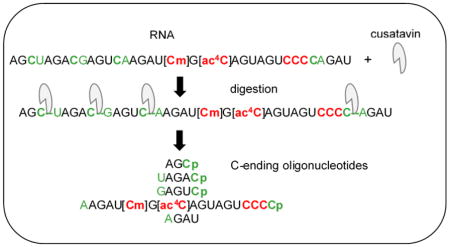
INTRODUCTION
In RNA modification mapping analysis, nucleobase-specific ribonucleases are a critical requirement for locating chemical modifications on the nucleotide sequence (1, 2). The post-transcriptional modifications that decorate RNA sequences (3) have been implicated in a number of cellular events ranging from regulation of gene expression (4–6), immunomodulation (7) to diseases (8). Assigning a definitive role to the targeted chemical modification requires precise identification and mapping of its location on the overall RNA sequence. Reverse transcription-dependent DNA-based sequencing technologies require a very small sample amount (9), however, the analytical capability is limited to one modification at a time in a given RNA. Other shortcomings of such high-throughput protocols include reproducibility related analytical characteristics such as resolution, precision, sensitivity, and quantification (6, 10).
Mass spectrometry is a highly sensitive and powerful analytical technique that can measure the mass shift associated with additional chemical group/s attached to RNA nucleosides accurately (11) in a single experiment. Initially pioneered by the McCloskey laboratory (12), the mass spectrometry-based approach identifies and locates modifications by two separate analyses. First, a census of modified nucleosides is obtained through complete hydrolysis of RNA. Subsequently, the RNA is subjected to nucleobase-specific digestion to determine the location of modifications in the oligonucleotides by tandem mass spectrometry (MS/MS) analysis (2). Their studies emphasized that the mass spectral analysis is simplified when the compositional value of the one of four nucleotides is known and restricted to a single residue. Such information imposes a constraint on the number of allowable base compositions within the specified mass measurement errors (12). In practice, the oligoribonucleotides of digested RNA are progressively separated based on their size and hydrophobicity on a reverse phase column with an ion-pairing agent before being subjected to tandem mass spectrometry (referred to as ion pairing-reverse phase-liquid chromatography-mass spectrometry (IP-RP-LC-MS)). The oligonucleotides, further resolved based on their mass-to-charge (m/z) values, are mass selected for collision-induced dissociation (CID) to yield sequence-informative product (also referred to as fragment) ions. The sequence location of the modified nucleoside is subsequently deciphered from the exhibited mass shift of the dissociated product ions corresponding to the mass of the attached chemical group. Ribonucleases cleave phosphodiester bonds in RNA through a 2′, 3′-cyclic phosphate intermediate producing mono- or oligonucleotides with a terminal 3′ phosphate. These enzymes are organized into RNase A, T1 and T2 families based on their characteristic features (13–14). T2 enzymes generally cleave at all 4 bases compared to pyrimidine (RNase A) and guanosine (T1) specificity of the respective family members. However, RNase MC1 (uridine-specific), a member of the T2 family (15, 16), is an exception to this general rule with its high specificity towards uridine. Addition of a methyl group or even substitution of oxygen with sulfur at position 4 (S4U) can make uridine a non-substrate for this enzyme. Similarly, cusativin protein initially purified from cucumber seed, presumably belonging to T2 family, is reported to exhibit high preference towards cytidine-specific RNA cleavage (17). Commercially available RNase T1 (guanosine specific) and RNase A (pyrimidine specific) have been extensively used for RNA modification mapping (18). However, employing C-specific or U-specific enzyme in RNA modification mapping analysis could potentially help in resolving ambiguities associated with the interference of M+1 isotope from the 1 Da mass difference observed between the nitrogen bases C and U (NH vs O). Thus, to expand the base specificity of enzymes further, we recently described overexpression systems for RNase U2 (purine specific) (19) and RNase MC1 (uridine specific) (16) enzymes.
RNase cusativin isolated from cucumber seed seems to be a promising cytidine-specific enzyme suitable for RNA modification mapping (17) compared to poly(C)-preferential ribonuclease purified from chicken liver (with significant levels of cleavage at uridine) (20). MALDI–MS based analysis of cusativin digestion products revealed predominant cleavage at cytidine and significant cleavage at uridine (21). Here, we report identification of the amino acid sequence of cusativin and its heterologous expression for easy purification and use as a bioanalytical tool. A systematic LC-MS based analysis of the RNA cleavage properties of purified cusativin has identified conditions for cleavage of CpA, CpG, and CpU phosphodiester bonds, with no cleavage of phosphodiester linkage between consecutive cytidines (CpC). We demonstrate the analytical utility of such behavior, not observed with other ribonucleases, for RNA modification mapping of single tRNA species or a total tRNA mixture.
MATERIALS AND METHODS
Cusativin purification from cucumber seed
Cucumber seed (Cucumis sativus, var. Pickling) was purchased from Eden brothers (http://www.edenbrothers.com/). All chemicals used in purification, unless otherwise specified, were procured from Fisher Scientific. The protein purification protocol initially described by Rojo et al. (17) was followed which included protein extraction from seed, clarification, gel filtration, CM-cellulose based ion exchange chromatography and size exclusion chromatography. The final size-exclusion chromatography fractions (1.5 mL) exhibiting the 23–25 kDa polypeptide were pooled, buffer exchanged (with 100 mM ammonium acetate (pH 5.5)) and concentrated with an Amicon Ultra-5 Centrifugal filter device (size=3k). The purified protein was stored in aliquots at 4 °C. Serial dilutions of purified protein (356 μg/mL) were made to test the nucleobase-specific ribonuclease activity.
Partial sequencing of cusativin by trypsin digest and LC-MS/MS
Purified protein (~1600 ng) was digested with trypsin (150 ng) as described before (22). The tryptic peptides were subsequently extracted from the digestion mixture using Pierce C18 spin columns (#89870) as per manufacturer’s instructions, dried on a speedvac and stored at −20 °C. The tryptic peptides were resolved on a Titan-C18 column (Supelco, 2.1 × 20 mm) at 35 °C using mobile phase A (95% water, 5% acetonitrile, 0.1% formic acid) and B (95% acetonitrile, 5% water, 0.1% formic acid) using a Thermo Vanquish UHPLC system. The MS/MS analysis was performed on an Orbitrap Fusion Lumos™ mass spectrometer equipped with an electrospray ionization (ESI) source. The chromatographic gradient composed of linear increase of mobile phase B from 2 to 5% in 3 min, 10% in 5 min, 50% in 55 min, and 95% in 4 min was applied before equilibration at 98% A, 2% B at a 120 μl/min flow rate.
Mass spectra were recorded in positive polarity under the optimal electrospray conditions (capillary temperature (299 °C), spray voltage (3.5 kV), and settings of 20, 7, and 0 arbitrary units for sheath, auxiliary, and sweep gas, respectively). Collision-induced dissociation (CID, 35% normalized collision energy) tandem mass spectrometry was used in data-dependent mode to switch automatically between MS (scan 1), and four CID scans to obtain sequence information of the digestion products. The full mass spectra in the Orbitrap mode (m/z 300–1500) was acquired at 120 000 mass resolution to determine the accurate mass (<5 ppm) and charge states of the precursor ions. The raw mass spectral data files were analyzed and matched by Thermo Proteome Discoverer (version 2.1) featuring the SEQUEST™ protein search algorithm and annotated cucumber proteome database for protein identification.
Cusativin gene cloning
By utilizing the identified amino acid sequence of the polypeptide as a template, a synthetic gene based on the observed codon usage in E. coli (23) was designed using Integrated DNA Technologies (IDT) web tool, (http://www.idtdna.com/CodonOpt) for heterologous expression. The custom designed gene was cloned into pET22b vector and the inducible C-terminal His-tag (His)6 fusion protein was purified as described before (16).
Enzyme activity assay
Enzyme activity was tested by incubating either an RNA oligonucleotide (200 pmol), AUCACCUCCUUUCU, or E. coli tRNA (Tyr or Phe) (0.2 μg) with serial dilutions of purified protein and 1 μL of 220 mM ammonium acetate at 37 °C or 50 °C in a volume of 10 μL for 60–90 min. The change in absorbance at 260 nm was compared against the protein omitted control while keeping the volume constant. One unit of enzyme activity is defined as the amount of protein required to bring ~10-fold increase in absorbance at 260 nm.
Characterization of cusativin for cytidine-specific ribonuclease activity
Purified protein was tested for its cytidine-specific cleavage using E. coli tRNA (Tyr or Phe) or total tRNA from E. coli. Serial dilutions of purified protein ranging from 35 ng to 1 μg of protein were mixed with 50 pmol (1.5 μg) of tRNA and incubated initially at 37 °C or 50 °C in 120 mM ammonium acetate (pH 6–6.5) for 1 h. The digests were dried in a speedvac and stored at 4 °C until analysis by IP-RP-LC-MS/MS using LTQ-XL mass spectrometer as described before (24). Mass-to-charge (m/z) values of theoretically expected precursor ions and corresponding product ions obtained by CID were computed using the Mongo Oligo Mass Calculator (http://mods.rna.albany.edu/masspec/Mongo-Oligo). The LC-MS/MS data was also probed for the appearance of products that indicated non-specific digestion of RNA using in-house developed software.
RESULTS
Purification of cusativin protein
Nucleobase-specific ribonuclease availability is a critical requirement for RNA modification mapping. To complement the RNA modification mapping mediated by RNase T1 (guanosine-specific) and more recently by RNase MC1 (16), we have identified cusativin (17) as an appropriate candidate enzyme for its highly preferential cytidine specific RNA cleavage. To enable its easy expression and purification from a heterologous bacterial host, we embarked on identification of the amino acid sequence of the 23–25 kDa polypeptide. We purified the protein from locally available Pickling variety of cucumber seed as described by Rojo et al. (17) to ensure its cytidine-specific RNA cleavage and identify its amino acid sequence.
The presence of cusativin in the purification fractions was initially ascertained by the observation of a 23–25 kDa polypeptide on a denaturing gel. As shown in Figure 1, such a polypeptide was seen only after cation-exchange chromatography on the CM-cellulose column (lane 3), while it was not detectable in the fractions of seed extract and initial gel filtration steps (lanes 1 and 2). After evaluating the non-specific ribonuclease activity of putative cusativin polypeptide (23–25 kDa) found in 200 mM NaCl eluent of CM-cellulose column, the pooled fractions were subjected to size exclusion chromatography. This step succeeded in eliminating the abundant low molecular weight (5–10 kDa) polypeptides, which had co-purified until cation-exchange chromatography, from the enriched putative cusativin protein fraction (Figure 1, lane 4).
Figure 1.
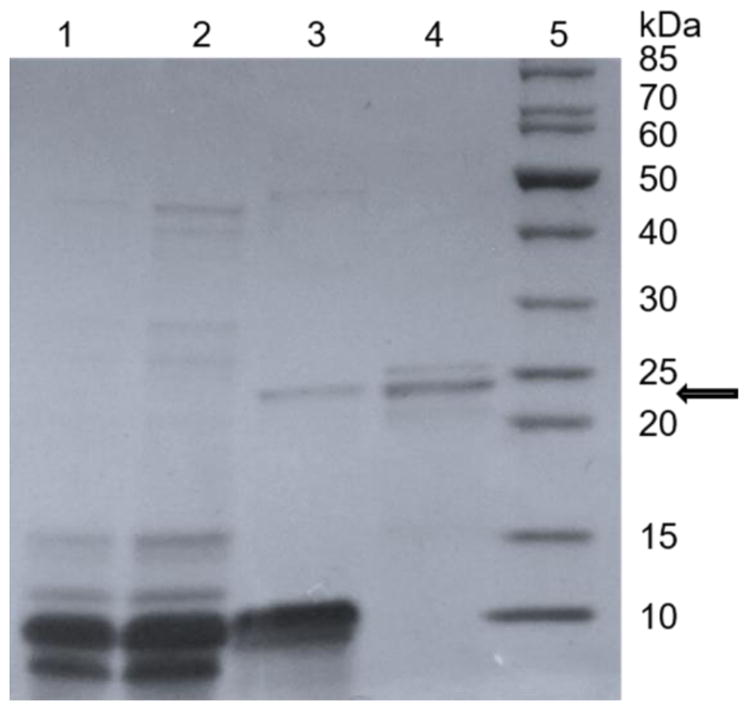
SDS-PAGE analysis of cusativin enriched at different stages of purification from seed extracts.
Lanes 1, seed extract; 2, flow-through of Sephadex G-25; 3, CM-Cellulose column fraction eluted with 200 mM NaCl; 4, CM-cellulose fraction purified by Sephadex G-75 column; and 5, polypeptide size standards. About 20–25 μL of protein sample from each step of purification was concentrated to 10 μL, boiled with equal volume of Laemmli buffer and analyzed on a 4–21% polyacrylamide gradient gel. Position of ~23 kDa polypeptide is indicated by an arrow.
Characterization of ribonuclease activity of purified cusativin
An increase in absorbance (at 260 nm-A260) of the oligonucleotide, AUCACCUCCUUUCU, following incubation with the purified protein at 37 °C or 50 °C was attributed to the ribonuclease activity. Both CM-cellulose and Sephadex G-75 purified protein fractions exhibited an increase in A260 following incubation at 37 °C and 50 °C for 1 h. No appreciable increase in A260 was observed beyond 1 h incubation, presumably due to no further cleavage of the oligonucleotide substrate.
Cleavage preferences of cusativin
To illustrate the cleavage properties of ribonuclease cusativin and its nucleobase specificity, the purified protein was incubated with E. coli tRNA(Tyr) at 37 °C and 50 °C and the digestion products were independently subjected to LC-MS/MS. As described with the studies of RNase MC1 (16), systematic examination of the MS/MS spectra of the resulting digestion products revealed the consistent presence of a 3′-end specific fragment ion corresponding to a cytidine residue (y1, m/z 322.2), which became part of the sequence informative product ions (exclusively yn ion series) scored for confirmation of the sequence. Two such representative oligonucleotides, GAGCp and GUCp, are shown in Figure 2 and Fig. S1 in the Electronic Supplementary Material (ESM), respectively. However, the 5′-termini exhibited wide variability in nucleotide identity with the exception of cytidine, which was conspicuously absent, indicating that the putative cusativin protein cleaves RNA at the 3′-phosphodiester bond of cytidine.
Figure 2.
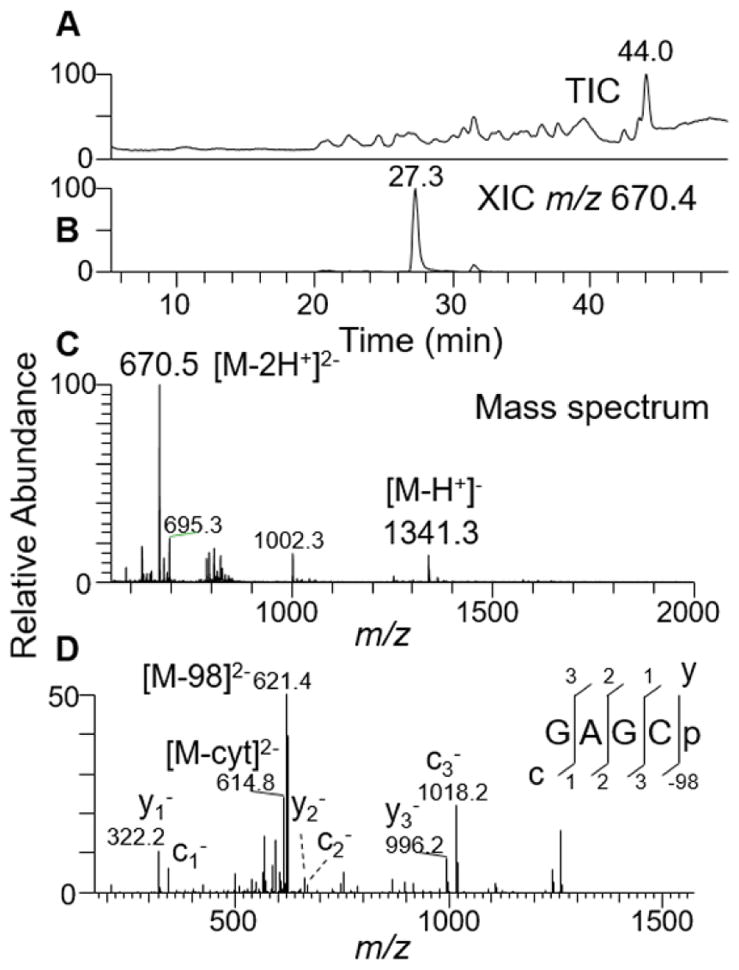
LC-MS/MS analysis of RNase cusativin digestion product GAGCp from E. coli tRNA(Tyr I).
(A) Total ion chromatogram (TIC) of all the ions eluted from reverse phase column. (B) Extracted ion chromatogram (XIC) for m/z 670.1, corresponding to a digestion product GAGCp (position 13–16). (C) Mass spectrum of the XIC at 27.3 min depicting the presence of single and doubly deprotonated oligonucleotide anions. (D) Tandem mass spectrum (MS/MS) of the precursor ion m/z 670.1 following collision-induced dissociation (CID) is depicted. The observed sequence informative product ion series, cn (sharing common 5′ end) and yn (with common 3′ end) with a subscript denoting the position of cleavage on phosphodiester backbone or the loss of phosphate (−98 Da), are labeled and plotted.
We also tested the previously reported observation (21) about the incomplete cleavage of RNA in cytidine rich regions by scoring the presence of digestion products that have multiple cytidine residues in the sequence. We noticed high levels of a number of digestion products in the LC-MS data, which include UUCCCCCACCACCA-OH, ACCAp, ACCp, UUCCCCCp, GAAUCCp, UGCCp, [Gm]GCCp, and pGGUGGGG[s4U]UCCCp. Of these, ACCA-OH and ACCp (ESM Figs. S2 and S3) result from the cleavage of the phosphodiester bond between C and A from any of the three potential sites in the oligomer UUCCCCCACCACCA-OH (ESM Fig. S4). Interestingly, no digestion product that would arise from the cleavage of the bond between consecutive cytidines could be detected. Absence of such digestion products indicates that the phosphodiester bond located between two consecutive cytidine residues is not a substrate under the specified digestion conditions of the enzyme. In other words, mononucleotide phosphates (Cp) are not expected to be present in this digest, a behavior unique to this enzyme. These observations might also explain the incomplete cleavage of RNA at the location of multiple cytidines observed in the previous report (21).
A further examination of the LC-MS data revealed the presence of digestion products bearing 3′-linear and 2′, 3′-cyclic phosphates. One such representative digestion product, [Gm]GCCp and [Gm]GCC>p, is shown in ESM Fig. S5. The presence of cyclic phosphates is consistent with an RNase T2 mechanism, which proceeds via the 2′, 3′-cyclic phosphate intermediate before forming the 3′ linear phosphate as the final product (25, 26). This feature was not enzyme concentration dependent, as excess enzyme (up to 5X enzyme) did not affect cyclic phosphate levels. Concentration-independent formation of the cyclic phosphate is more consistent with a slow rate of phosphodiester bond hydrolysis. Based on these initial observations, a list of m/z values of expected E. coli tRNA (Tyr) digestion products and their CID fragment ions were computed using Mongo Oligo and subsequently confirmed for their presence in LC-MS/MS data (Table 1). Positive scoring of all of the expected digestion products spanning the whole sequence of E. coli tRNA(Tyr) in the digested sample demonstrate the utility of cusativin in obtaining high sequence coverage of modified RNA.
Table 1.
Comparison of the observed cusativin digestion product peak areas of tRNA(Tyr) bearing 3′-PO4 and 2′,3′-cyclic phosphate at two different protein amounts
Identical amount of RNA was treated with different amounts of protein as described in methods and the resulting digestion product ion abundances for 3′-linear and 2′, 3′-cyclic phosphates were compared between the optimal and 10X of optimal protein amount. RNA to protein ratio is critical for cytidine specific cleavage of cusativin based on the observed ion abundances. ND-not detected
| Nucleotide sequence (position) | 3′-PO4 | 2′,3′-cyclic PO4 | ||
|---|---|---|---|---|
| 35.6 ng | 356 ng | 35.6 ng | 356 ng | |
| pGGUGGGG[s4U]UCCC (1–12) | 1.5E5 | 2.5E2 | 1.9E5 | 5.9E2 |
| GAGC (13–14) | 5.6E5 | 4.5E3 | 1.4E6 | ND |
| [Gm]GCC (15–18) | 2.7E5 | ND | 7.7E5 | 2.1E4 |
| AAAGGGAGC (19–27) | 2.2E5 | ND | 3.1E5 | ND |
| AGAC (28–31 & 49–52) | 4.3E5 | ND | 5.1E5 | ND |
| U[Q][Ψ]A[ms2i6A]A[Ψ]C (32–39) | 2.7E6 | 3.1E3 | 8.3E5 | ND |
| UGCC (40–43) | 8.4E5 | 1.1E4 | 1.5E5 | ND |
| GUC (44–46) | 6.7E4 | ND | 1.3E4 | ND |
| AC (47–48) | ND | 6.2E5 | 1.9E4 | 3.1E5 |
| UUC (53–55) | 1.2E5 | 6.8E5 | 7.0E5 | ND |
| GAAGG[m5U][Ψ]C (56–63) | 8.5E4 | ND | 2.7E5 | ND |
| GAAUCC (64–69) | 7.2E5 | ND | 1.7E6 | ND |
| UUCCCCCACCACCA-OH (70–83) | 8.5E5 | 8.7E2 | 8.5E5 | 8.7E2 |
| ACCA-OH (80–83) | 6.5E4 | ND | 6.5E4 | ND |
| ACC (77–79 or 80–82) | 4.6E4 | 4.1E5 | 5.9E4 | 3.4E4 |
Cusativin is a cytidine-specific ribonuclease
The LC-MS data was examined for the presence of digestion products that are rich in uridine but ending with cytidine (indication of cytidine specific cleavage) or nonspecific digestion products ending with uridine (21) or other nucleotides at the 3′-terminus. One digestion product with cytidine-specific cleavage arising from the anticodon region of E. coli tRNA(Tyr), U[Q][Ψ]A[ms2i6A]A[Ψ]Cp, has two pseudouridines and one uridine. This digestion product exhibited an abundant MS signal (ESM Fig. S6) and its tandem mass spectrum of the precursor ion registered the presence of all the sequence informative product ions (both c- and y-type ion series’), thus confirming the nucleotide sequence. If cusativin is to hydrolyze phosphodiester bonds at uridine, one would not expect this anticodon digestion product to be present at such high abundance. Similarly, other digestion products with multiple uridine residues, such as pGGUGGGG[s4U]UCCC, were consistently observed at high abundance in the same LC-MS data (Table 1). These results strongly suggest that the uridine rich regions of an RNA sequence do not significantly alter the cytidine-specific RNA cleavage ability of cusativin under the described digestion conditions.
Impact of higher amount of native protein in RNA digests
We next investigated whether the RNA cleavage properties of cusativin are affected by protein amount and incubation temperature. To accomplish this, the relative abundance levels of 5-mer and longer digestion products were examined following incubation of E. coli tRNA (Tyr) (1.5 μg) at different protein amounts (35 to 350 ng) and two different temperatures 37 °C and 50 °C. While the abundance levels of the longer digestion products did not vary significantly between 35 to 178 ng of added enzyme, they exhibited a sharp decrease in abundance when the enzyme amount was increased to 356 ng (10X the lowest protein amount tested). This dramatic decrease in abundance is noticeable for both linear and cyclic phosphate digestion products (Table 1). Only five out of 14 oligomers with a 3′-linear phosphate and two out of 14 digestion products with a cyclic phosphate were detected when higher protein amounts of ribonuclease (10X) were used. Changes in abundance of a representative digestion product, U[Q]ΨA[ms2i6A]A[Ψ]Cp corresponding to each tested conditions are shown in ESM Fig. S6E; note the dramatic decrease in abundance of this oligomer at the highest protein amount used. The LC-MS data of the digest, where the highest protein amount was used, exhibited fewer digestion products and no bias towards any specific nucleoside, indicating the increased non-specific activity of the protein preparation. Digestion of RNA at two different temperatures did not make a significant impact on the digestion product pattern as per the changes in observed abundances (ESM Fig. S6E). These observations suggest that either the enzyme exhibits non-specific cleavage at higher amounts or a co-purifying non-specific ribonuclease becomes significant at higher protein amounts used, thereby, resulting in loss of signal for multiple digestion products.
LC-MS/MS-based sequencing of cusativin tryptic peptides
Heterologous expression of cusativin requires identification of the nucleotide or amino acid sequence of the coding gene. Therefore, the purified protein was subjected to amino acid sequencing by LC-MS following trypsin-mediated digestion. The LC-MS behavior of one of the tryptic peptides is shown in Figure 3. Proteome Discoverer was used to process the LC-MS/MS data and subsequent identification of the amino acid sequence of an associated protein following database search analysis of the cucumber proteome (GCF_0000040752_ASM407V2_protein.faa). This exercise identified a protein under Gene Bank Accession number 449459530 and an entry code of XP_004147499.1. The corresponding entry for mRNA (XM_004147451.1) was annotated as predicted mRNA for ribonuclease MC1-like (LOC101213213) protein. The observed tryptic peptides of purified cusativin mapped to ~38% of the amino acid sequence of this protein (Figure 3E). Incidentally, a couple of tryptic peptides, STIHGLWPNK, YFQTAINMR, reported in the previous study (17) matched well with those observed in the current study suggesting that the protein preparation purified in both studies could be identical.
Figure 3.

LC-MS/MS analysis of trypsin digest of RNase cusativin.
(A) Total ion chromatogram (TIC) of all the observed peptide ions. (B) Extracted ion chromatogram (XIC) for m/z 561.8331 (3.5 ppm), corresponding to tryptic peptide HGIDLLSVLR. (C) Mass spectrum associated with the XIC at 21.3 min. (D) Tandem mass spectrum (MS/MS) of the precursor ion, m/z 561.8, following collision-induced dissociation (CID) in the ion trap. The observed sequence informative product ion series, bn (sharing common N-terminal end) and yn (with common C-terminal end), with a subscript denoting the position of cleavage on peptide backbone, are labeled and plotted. (E) Predicted amino acid sequence of the polypeptide following cucumber proteome database search analysis with the tandem mass spectrometry data. The observed tryptic peptides are shown bold type and underlined.
Amino acid sequence alignment of Ribonuclease MC1 (26) and cusativin indicated extensive conservation (~55%) of amino acid identities (ESM Fig. S7). For example, the key catalytic residues of His34, His83, Glu84, Lys87 and His88 of MC1 (26) are conserved at corresponding positions (60, 109, 110, and 113, respectively) in cusativin. Similarly, highly conserved Gln9 and Phe80 residues in RNases of T2 family and MC1 (26) are also found in cusativin at positions 36 and 106, respectively. Of the two other key residues of MC1, Asn71 and Leu73, only Asn (at position 97) is conserved while the leucine was replaced by valine (position 99).
Cloning and heterologous expression of cusativin
After confirming the sequence and reading frame of the recombinant cusativin-(His)6 gene fusion in pET22b vector, analysis of various protein expression conditions revealed higher protein yield when cells were induced around ~0.6 OD600 with 0.4 mM IPTG for 2–3 h (data not shown). SDS-PAGE analysis of the affinity purified protein from a nickel column yielded a major polypeptide band at Mr ~ 24 kDa with a few minor polypeptides. The expected molecular mass of the cusativin-(His)6 fusion protein (24.1 kDa) is similar to that observed in SDS-PAGE (data not shown) suggesting the production of the anticipated polypeptide in the bacterial host.
Cleavage preferences of recombinant cusativin
To investigate the nucleobase specificity and cleavage properties of the recombinant ribonuclease cusativin-(His)6 fusion protein, yeast tRNA(Phe), E. coli tRNA(Tyr) and total tRNA were digested and analyzed by LC-MS/MS. One clear difference between native cusativin and recombinant cusativin is that higher protein amounts in the digestion mixture did not affect the cleavage specificity. For optimal detection of digestion products, at least 1 μg of purified protein (higher amounts, up to 5 μg, did not significantly influence the specificity) is required for 1 μg of RNA in contrast to the 35 ng of native protein. However, evaluation of the tandem mass spectral data of E. coli tRNA(Tyr) revealed that the observed oligonucleotide digestion products were identical to those shown in Table 1 indicating that the recombinant protein retained the enzyme characteristics of the native protein.
Analysis of digestion products from yeast tRNA (Phe) revealed similar behavior of cytidine specific RNA cleavage. Examination of these digestion products indicated that the cleavage was inhibited at ribose methylated cytidines, however, 5-methylcytosine was still recognized as a substrate. This feature was indicated by the abundant presence of a digestion product corresponding to AGA[Cm]U[Gm]AA[yW]A[Ψ] [m5C]>p (Figure 4) in the yeast tRNA(phe) digest. The CID-based MS/MS spectrum exhibited a dominant product ion corresponding to base loss of wybutosine (yW). The remainder of the tandem mass spectrum, although complex, did account for a number of predicted sequence informative c- and y-type fragment ions (Figure 4). Such fragmentation is not entirely unexpected because of the presence of ribose methylated residues [Cm] and [Gm] apart from the wybutosine in the sequence. Appearance of a digestion product due to base loss of wybutosine in the gas phase is also seen in the MS1 scan, which is not subjected to CID (Figure 4, panel C), suggesting a higher level of this digestion product in the treated sample. To confirm that no partial cleavages at 2′-O-methylcytidine occurred, the predicted digestion products with linear and 2′, 3′-cyclic phosphates consistent with such cleavage, AGA[Cm] at m/z 1339.2 and m/z 1321.2, were also searched. No ions for these m/z values were detected, indicating absence of cleavage at 2′-O-methylcytidine. These data strongly support that the RNA cleavage is inhibited by ribose methylation of cytidine, but not by cytosine base methylation.
Figure 4.

LC-MS/MS analysis of recombinant cusativin digestion product, AGA[Cm]U[Gm]AA[yW]A[Ψ][m5C]>p from yeast tRNA(Phe).
(A) Total ion chromatogram (TIC). (B) Extracted ion chromatogram (XIC) for m/z 1389.7, corresponding to AGA[Cm]U[Gm]AA[yW]A[Ψ][m5C]>p (position 29–40). (C) Mass spectrum of the XIC at 40.5 min. Note the presence of triply deprotonated anions in the spectrum The spectrum also shows the presence of oligonucleotide anion corresponding to the loss of [yW] base (m/z 1264.1). (D) Tandem mass spectrum (MS/MS) of the precursor ion, m/z 1389.7, following collision-induced dissociation (CID). The observed sequence informative product ion series, cn (sharing common 5′ endare indicated by *) and yn (sharing common 3′ end and indicated by ◆) with a subscript denoting the position of cleavage on phosphodiester backbone are labeled and plotted.
To test the effect of other cytidine modifications such as acetylation, E. coli total tRNA was digested with purified cusativin and the status of digestion products from tRNA(Met) that harbors [ac4C] was investigated. Analysis of LC-MS/MS data revealed inhibition of RNA cleavage at acetylated cytidine following the detection of abundant amount of U[ac4C]AU[t6A]A[Ψ]GAUGGG[m7G][acp3U]C>p in the mass spectrum (Figure 5). Further, the CID-based MS/MS spectrum exhibited a majority of sequence-informative c- and y-type fragment ion series’ supporting the sequence prediction. More importantly, the MS/MS spectrum exhibited product ions that correspond to loss of [m7G] base or side chain fragmentation of [t6A] (18) confirming the predicted sequence. This experimental data clearly identifies the specific instances (consecutive cytidine residues or specific modifications), where cusativin-mediated RNA cleavage is inhibited.
Figure 5.

LC-MS/MS analysis of recombinant cusativin digestion product, U[ac4C]AU[t6A]A[Ψ]GAUGGG[m7G][acp3U]C>p of tRNA(Met) from E.coli total tRNA.
(A) Total ion chromatogram (TIC). (B) Extracted ion chromatogram (XIC) for m/z 1371.0, corresponding to U[ac4C]AU[t6A]A[Ψ]GAUGGG[m7G][acp3U]C>p, (position 33–48). (C) Mass spectrum of the XIC at 35.9 min. Note the presence of triply and quadruple deprotonated anions in the spectrum. D) Tandem mass spectrum (MS/MS) of the precursor ion, m/z 1370.1, following collision-induced dissociation (CID). The MSMS spectrum is complex with a number of base losses typical to the specific modifications reported before. The spectrum also shows the presence of oligonucleotide anion corresponding to the loss of [m7G] base (m/z 1329.2), and potentially side chain cleavage of [t6A] corresponding to the mass of 147 Da (m/z 1332.9). (The observed sequence informative product ion series, cn (sharing common 5′ end and indicated by *) and yn (sharing common 3′ end and indicated by ◆) with a subscript denoting the position of cleavage on phosphodiester backbone are labeled and plotted.
Discussion
Nucleobase-specific RNases provide critical base composition constraints to simplify MS data interpretation (12), thereby allowing the placement of modified residues on a sequence in RNA modification mapping procedures (2). Employment of T1 alone does not result in high sequence coverage of modified RNA, especially if there are sequence redundancies or G-rich regions in RNA. The other commercially available enzyme, RNase A, or the recently overexpressed RNase U2 (19) will generate shorter RNase digestion products that are not particularly useful for modification placement. In this context, we recently described uridine-specific ribonuclease MC1 (16) that can enhance the sequence coverage in combination with RNase T1 (16). The availability of additional nucleobase-specific ribonucleases can further enhance sequence coverage especially in G-rich or U-rich regions of RNA.
Cusativin, initially purified and characterized from cucumber seed (17), was identified as a potential enzyme for C-preferential/specific cleavage based modification mapping of RNA. Although further studies revealed incomplete cleavage and occasional cleavage of substrate RNA at uridine residues (21), we reasoned that this enzyme is more promising than chicken liver enzyme (27) as cusativin exhibited less preference towards uridine. As the cucumber genome was not sequenced at the time of initiation of this study, the cusativin protein was purified by basic biochemical methods as described previously (17). The strategy of detecting a ~23–25 kDa polypeptide by SDS-PAGE with ribonuclease activity of substrate RNA as requirements for ensuring the purification of cusativin was successful as revealed by biochemical and mass spectrometry analysis.
The observation of both 3′-linear and 2′,3′-cyclic digestion products in the cusativin treated RNA is consistent with the two-stage endonucleolytic cleavage process operating through the 2′,3′-cyclicphosphate intermediate (25). One striking feature of cusativin-mediated enzymatic digestion is that the cleavage of RNA is inhibited when cytidines are present in tandem in the sequence at optimal RNA/protein ratio. Such a characteristic was not observed with RNases T1, A, U2 or even MC1. Nevertheless, such a characteristic analytical feature is helpful in RNA modification mapping where one could obtain near complete sequence coverage of substrate RNA (for example, the E. coli tRNA (Tyr) as used in the current study). The LC-MS based analytical studies revealed the requirement of optimal RNA/protein ratio to facilitate cytidine-specific cleavage. A higher RNA/protein ratio (1500 ng RNA/36 ng protein or 40:1) was ideal for effecting cytidine-specific cleavage of RNA by native cusativin. A lower RNA/protein ratio (<8:1) could result in loss of cleavage specificity as revealed by the loss of signal for various digestion products in LC-MS. Since it was not clear whether this behavior was due to loss of specificity at higher protein amount or co-purifying ribonuclease, we embarked on making recombinant cusativin to eliminate the co-purifying nucleases.
Partial sequencing of the purified protein had successfully identified the cucumber gene responsible for encoding the cusativin protein. Alignment of the amino acid sequences of RNase MC1 and cusativin by homology modeling (Fig. S7) indicated extensive conservation of residues throughout the sequence. The conserved residues include the catalytic residues and a majority of base-recognizing amino acids. However, the amino acid residue corresponding to leucine at position 73 of RNase MC1 is replaced by valine (corresponding position 99) in cusativin. In spite of high similarity and identity of amino acid residues including the catalytic site, the digestion behavior of the two enzymes is quite different. RNase MC1 catalyzes RNA cleavage at the 5′-end of uridine (16). In contrast, cusativin cleaves RNA at 3′-end of cytidine, a feature similar to RNase T1, A and U2 enzymes. This difference suggests the existence of other determinants in the amino acid sequences of RNase MC1 and cusativin that might be dictating their cleavage behavior.
Higher amounts of recombinant cusativin protein purified from BL21 cells were required to effect similar RNA cleavage as that of native protein (1500 ng vs 36 ng for 1.5 μg RNA). However, nonspecific cleavage of RNA was not noticed at higher concentrations of recombinant protein when purified from BL21 cells. This suggests that the non-specific activity observed with native protein might be due to the co-purifying non-specific ribonuclease and that can be eliminated with the recombinant protein. Nevertheless, the cytidine-specific cleavage behavior of recombinant protein was identical to that of native protein at higher protein amounts than native protein. This also suggests that either a portion of expressed protein is inactive or not folded in an identical fashion to that of native protein, thereby exhibiting lower enzyme activity.
The RNA cleavage preferences of both native and recombinant cusativin indicate that the substrate binding site of the enzyme does not distinguish 5-methylated cytidine from cytidine. This feature is in contrast to RNase MC1 where methylated uridine is a non-substrate. Nevertheless, certain modifications of cytidine did inhibit RNA cleavage including ribose sugar methylation [Cm] and acetylation of cytidine [ac4C] (Figure 5). Loss of cleavage at 2′-O methylated cytidines is consistent with a T2-mechanism of enzyme activity. Inhibition of cleavage at acetylation suggests that certain chemical groups at this position may not be accommodated in the enzyme’s binding site. One surprising behavior of cusativin is the lack of phosphodiester bond cleavage between consecutive cytidine residues. Such behavior, to our knowledge, has not been reported for any other RNase before. However, it could offer an explanation for the previously reported incomplete/missed cleavage of RNA by the native cusativin protein purified from cucumber seed (21).
This kind of analytical characterization of the cleavage preferences of cusativin demonstrates a number of features that warrant using this enzyme in RNA modification mapping. Its nucleobase specificity complements well with RNase T1 (28) or MC1 (16) through generation of overlapping digestion products, thereby improving sequence coverage of target RNA. Use of more than one nucleobase-specific enzyme can also help validate the modification placement data of one enzyme with the other enzyme thereby providing high confidence and accuracy of modified nucleoside sequence assignments. In cases, where a single smaller RNA species is being characterized, the use of cusativin alone can provide near complete sequence coverage due to its inability to cleave the bond between the two cytidine residues, as illustrated with modification mapping of E. coli tRNA(Tyr).
Cusativin can also minimize the challenges associated with the differentiation of cytidine from uridine (1 Da, NH versus O). As digestion of RNA by cusativin results in cytidine/s at the 3′-terminus of the oligonucleotide, uridine/s can only be found at the 5′-terminus or internal position within the sequence; conversely, cytidine is not expected to be observed at the 5′-end of oligomer, thus, providing additional constraints to improve MS data interpretation (12, 29). Inhibition of RNA cleavage by the presence of specific cytidine modifications could be used to identify the modification location in both LC-MS/MS, shown in this work, as well as RNA-seq analysis. For the latter, a comparison of RNA-seq transcripts generated using this nuclease against the predicted sequence of genome can reveal the presence of post-transcriptional modifications of cytidine in the C-rich regions of UCCUC repeats (30) on a genome-wide basis. Similarly, biochemical approaches such as RNA foot-printing or those that elucidate sequence preferences for protein binding could benefit from the employment of this additional nucleobase-specific ribonuclease.
Supplementary Material
Fig. S1 LC-MS/MS analysis of Cusativin digestion product GUCp from E. coli tRNATyr I.
(A) Total ion chromatogram (TIC). (B) Extracted ion chromatogram (XIC) for m/z 973.4, a peak with retention time of 21.4 min corresponding to a digestion product GUCp (position 44–46) is shown. (C) Mass spectrum associated with the XIC at 21.4 min depicting the presence of deprotonated singly charged anion. (D) Tandem mass spectrum (MS/MS) of the precursor ion m/z 973.4 following collision-induced dissociation (CID). The observed sequence informative product ion series, cn (sharing common 5′ end) and yn (with common 3′ end) with a subscript denoting the position of cleavage on phosphodiester backbone, or loss of phosphate (−98 D) are labeled and plotted
Fig. S2 LC-MS/MS analysis of RNase Cusativin digestion product ACCA from E. coli tRNATyr I.
(A) Total ion chromatogram (TIC). (B) Extracted ion chromatogram (XIC) for m/z 602.4, corresponding to a digestion product ACCA (position 80–83). (C) Mass spectrum of the XIC at 15.3 min. Note the presence of double and singly deprotonated oligonucleotide anions in the spectrum. (D) Tandem mass spectrum (MS/MS) of the precursor ion, m/z 602.4, following collision-induced dissociation (CID) is depicted. The observed sequence informative product ion series, cn and yn with a subscript denoting the position of cleavage on phosphodiester backbone are labeled and plotted
Fig. S3 LC-MS/MS analysis of RNase Cusativin digestion product ACCp from E. coli tRNATyr I.
(A) Total ion chromatogram (TIC). (B) Extracted ion chromatogram (XIC) for m/z 956.3, corresponding to a digestion product ACCp (position 77–79). (C) Mass spectrum of the XIC at 24.1 min. Note the presence of singly deprotonated oligonucleotide anions in the spectrum, (D) tandem mass spectrum (MS/MS) of the precursor ion, m/z 956.3 following collision-induced dissociation (CID) is depicted. The observed sequence informative product ion series, cn and yn with a subscript denoting the position of cleavage on phosphodiester backbone are labeled and plotted
Fig. S4 LC-MS/MS analysis of RNase Cusativin digestion product, UUCCCCCACCACCA from E. coli tRNATyr I.
(A) Total ion chromatogram (TIC). (B) Extracted ion chromatogram (XIC) for m/z 1427.2, corresponding to a digestion product UUCCCCCACCACCA (position 70–83) with a 3′-OH. (C) Mass spectrum associated with the XIC at 39.1 min. Note the presence of triply deprotonated oligonucleotide anion in the spectrum. (D) Tandem mass spectrum (MS/MS) of the precursor ion, m/z 1427.2, following collision-induced dissociation (CID). The observed sequence informative product ion series, cn (sharing common 5′ end indicated by *) and yn (with common 3′ end indicated by ◆) with a subscript denoting the position of cleavage on phosphodiester backbone are labeled and plotted
Fig. S5 LC-MS/MS analysis of cusativin digestion product [Gm]GCC>p (position 15–18) from E. coli tRNA(Tyr I).
(A) Extracted ion chromatogram (XIC) for m/z 665.4, corresponding to [Gm]GCC>p with linear phosphate. (B) Extracted ion chromatogram (XIC) for m/z 656.4, corresponding to same digestion product with 2′, 3′-cyclic phosphate. (C) Mass spectrum associated with the XIC at 24.8 min depicting the singly and doubly charged molecular ions. (D) Tandem mass spectrum (MS/MS) of the precursor ion m/z 656.4 following collision-induced dissociation (CID) is depicted. The observed sequence informative product ion series, cn (sharing common 5′ end and indicated by *) and yn (with common 3′ end and indicated by ◆) with a subscript denoting the position of cleavage on phosphodiester backbone, are labeled and plotted
Fig. S6 LC-MS/MS analysis of RNase cusativin digestion product U[Q][Ψ]A[ms2i6A]A[Ψ]Cp from E. coli tRNA(Tyr I).
(A) Total ion chromatogram (TIC). (B) Extracted ion chromatogram (XIC) for m/z 1406.5, corresponding to U[Q][Ψ]A[ms2i6A]A[Ψ]Cp (position 32–39). (C) Mass spectrum associated with the XIC at 44 min. Note the presence of double and triple deprotonated oligonucleotide anions in the spectrum. (D) Tandem mass spectrum (MS/MS) of the precursor ion, m/z 1406.5, following collision-induced dissociation (CID). The observed sequence informative product ion series, cn (sharing common 5′ end and indicated by *) and yn (with common 3′ end and indicated by ◆) with a subscript denoting the position of cleavage on phosphodiester backbone, are labeled and plotted. (E) Changes in abundance levels of oligonucleotide, U[Q][Ψ]A[ms2i6A]A[Ψ]Cp, at different protein concentrations and incubation temperature is shown
Fig. S7 Homology based alignment of amino acid sequences of Rnase MC1 and cusativin. Identical amino acid residues at respective positions are indicated by *, Substitutions with residues of similar polarity are indicated by “:” or “.” symbols. Identical catalytic residues in both sequences are depicted in bold type, whereas the identical nucleobase recognizing residues are represented in bold italics. The nucleobase recognizing residue that is different in cusativin is underlined
Acknowledgments
Financial support was provided by the National Institute of Health (GM058843 and OD018485 to P.A.L.) and the University of Cincinnati.
Footnotes
Conflict of Interest
The authors declare no conflict of interest.
References
- 1.Giessing AMB, Kirpekar F. Mass spectrometry in the biology of RNA and its modifications. J Proteomics. 2012;75(12):3434–49. doi: 10.1016/j.jprot.2012.01.032. [DOI] [PubMed] [Google Scholar]
- 2.Kowalak JA, Pomerantz SC, Crain PF, McCloskey JA. A novel method for the determination of post-transcriptional modification in RNA by mass spectrometry. Nucleic Acids Res. 1993;21(19):4577–85. doi: 10.1093/nar/21.19.4577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Machnicka MA, Milanowska K, Osman Oglou O, Purta E, Kurkowska M, Olchowik A, et al. MODOMICS: a database of RNA modification pathways--2013 update. Nucleic Acids Res. 2013;41(Database issue):D262–7. doi: 10.1093/nar/gks1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yi C, Pan T. Cellular dynamics of RNA modification. Accounts of Chem Res. 2011;44(12):1380–8. doi: 10.1021/ar200057m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang X, He C. Dynamic RNA Modifications in Posttranscriptional Regulation. Mol Cell. 2014;56(1):5–12. doi: 10.1016/j.molcel.2014.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Song J, Yi C. Chemical Modifications to RNA: A New Layer of Gene Expression Regulation. ACS Chem Biol. 2017 doi: 10.1021/acschembio.6b00960. [DOI] [PubMed] [Google Scholar]
- 7.Karikó K, Buckstein M, Ni H, Weissman D. Suppression of RNA recognition by Toll-like receptors: The impact of nucleoside modification and the evolutionary origin of RNA. Immunity. 2005;23(2):165–75. doi: 10.1016/j.immuni.2005.06.008. [DOI] [PubMed] [Google Scholar]
- 8.Torres AG, Batlle E, Ribas de Pouplana L. Role of tRNA modifications in human diseases. Trends Mol Med. 2014;20(6):306–14. doi: 10.1016/j.molmed.2014.01.008. [DOI] [PubMed] [Google Scholar]
- 9.Van Dijk EL, Jaszczyszyn Y, Thermes C. Library preparation methods for next-generation sequencing: Tone down the bias. Exp Cell Res. 2014;322(1):12–20. doi: 10.1016/j.yexcr.2014.01.008. [DOI] [PubMed] [Google Scholar]
- 10.Dominissini D, Moshitch-Moshkovitz S, Schwartz S, Salmon-Divon M, Ungar L, Osenberg S, et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature. 2012;485(7397):201–6. doi: 10.1038/nature11112. [DOI] [PubMed] [Google Scholar]
- 11.Gaston KW, Limbach PA. The identification and characterization of non-coding and coding RNAs and their modified nucleosides by mass spectrometry. RNA biol. 2014;11(12):1568–85. doi: 10.4161/15476286.2014.992280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pomerantz SC, Kowalak JA, McCloskey JA. Determination of oligonucleotide composition from mass spectrometrically measured molecular weight. J Am Soc Mass Spectr. 1993;4(3):204–9. doi: 10.1016/1044-0305(93)85082-9. [DOI] [PubMed] [Google Scholar]
- 13.Luhtala N, Parker R. T2 Family ribonucleases: ancient enzymes with diverse roles. Trends Biochem Sci. 2010;35(5):253–9. doi: 10.1016/j.tibs.2010.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Deshpande RA, Shankar V. Ribonucleases from T2 family. Crit Rev Microbiol. 2002;28(2):79–122. doi: 10.1080/1040-840291046704. [DOI] [PubMed] [Google Scholar]
- 15.Numata T, Kashiba T, Hino M, Funatsu G, Ishiguro M, Yamasaki N, et al. Expression and Mutational Analysis of Amino Acid Residues Involved in Catalytic Activity in a Ribonuclease MC1 from the Seeds of Bitter Gourd. Biosci Biotech Bioch. 2000;64(3):603–5. doi: 10.1271/bbb.64.603. [DOI] [PubMed] [Google Scholar]
- 16.Addepalli B, Lesner NP, Limbach PA. Detection of RNA nucleoside modifications with the uridinespecific ribonuclease MC1 from Momordica charantia. RNA. 2015;21(10):1746–56. doi: 10.1261/rna.052472.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rojo MA, Arias FJ, Iglesias R, Ferreras JM, Muñoz R, Escarmís C, et al. Cusativin, a new cytidine-specific ribonuclease accumulated in seeds of Cucumis sativus L. Planta. 1994;194(3):328–38. doi: 10.1007/BF00197532. [DOI] [PubMed] [Google Scholar]
- 18.Ross R, Cao X, Yu N, Limbach PA. Sequence mapping of transfer RNA chemical modifications by liquid chromatography tandem mass spectrometry. Methods. 2016;107:73–8. doi: 10.1016/j.ymeth.2016.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Houser WM, Butterer A, Addepalli B, Limbach PA. Combining recombinant ribonuclease U2 and protein phosphatase for RNA modification mapping by liquid chromatography-mass spectrometry. Anal biochem. 2015;478:52–8. doi: 10.1016/j.ab.2015.03.016. [DOI] [PubMed] [Google Scholar]
- 20.Hayano K, Iwama M, Sakamoto H, Watanabe H, Sanda A, Ohgi K, et al. Characterization of poly C preferential ribonuclease from chicken liver. J Biochem. 1993;114(1):156–62. doi: 10.1093/oxfordjournals.jbchem.a124132. [DOI] [PubMed] [Google Scholar]
- 21.Hahner S, Lüdemann HC, Kirpekar F, Nordhoff E, Roepstorff P, Galla HJ, et al. Matrix-assisted laser desorption/Ionization mass spectrometry (MALDI) of endonuclease digests of RNA. Nucleic Acids Res. 1997;25(10):1957–64. doi: 10.1093/nar/25.10.1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Addepalli B, Limbach PA, Hunt AG. A disulfide linkage in a CCCH zinc finger motif of an Arabidopsis CPSF30 ortholog. FEBS Lett. 2010;584(21):4408–12. doi: 10.1016/j.febslet.2010.09.043. [DOI] [PubMed] [Google Scholar]
- 23.Fuglsang A. Codon optimizer: a freeware tool for codon optimization. Protein expres and purif. 2003;31(2):247–9. doi: 10.1016/s1046-5928(03)00213-4. [DOI] [PubMed] [Google Scholar]
- 24.Wong SY, Javid B, Addepalli B, Piszczek G, Strader MB, Limbach PA, et al. Functional role of methylation of G518 of the 16S rRNA 530 loop by GidB in Mycobacterium tuberculosis. Antimicrob Agents Ch. 2013;57(12):6311–8. doi: 10.1128/AAC.00905-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kaiser PM, Bonacker L, Witzel H, Holy A. Studies on the reaction mechanism of a ribonuclease II from Aspergillus oryzae (author’s transl) H-S Z Physiol Chem. 1975;356(2):143–55. [PubMed] [Google Scholar]
- 26.Suzuki A, Yao M, Tanaka I, Numata T, Kikukawa S, Yamasaki N, et al. Crystal structures of the ribonuclease MC1 from bitter gourd seeds, complexed with 2′-UMP or 3′-UMP, reveal structural basis for uridine specificity. Biochem Bioph Res Co. 2000;275(2):572–6. doi: 10.1006/bbrc.2000.3318. [DOI] [PubMed] [Google Scholar]
- 27.Boguski MS, Hieter PA, Levy CC. Identification of a cytidine-specific ribonuclease from chicken liver. J Biol Chem. 1980;255(5):2160–3. [PubMed] [Google Scholar]
- 28.Durairaj A, Limbach PA. Improving CMC-derivatization of pseudouridine in RNA for mass spectrometric detection. Anal Chim Acta. 2008;612(2):173–81. doi: 10.1016/j.aca.2008.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Meng Z, Limbach PA. RNase mapping of intact nucleic acids by electrospray ionization Fourier transform ion cyclotron resonance mass spectrometry (ESI-FTICRMS) and 18O labeling. Int J Mass Spectrom. 2004;234(1–3):37–44. [Google Scholar]
- 30.Delatte B, Wang F, Ngoc LV, Collignon E, Bonvin E, Deplus R, et al. RNA biochemistry. Transcriptome-wide distribution and function of RNA hydroxymethylcytosine. Science. 2016;351(6270):282–5. doi: 10.1126/science.aac5253. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 LC-MS/MS analysis of Cusativin digestion product GUCp from E. coli tRNATyr I.
(A) Total ion chromatogram (TIC). (B) Extracted ion chromatogram (XIC) for m/z 973.4, a peak with retention time of 21.4 min corresponding to a digestion product GUCp (position 44–46) is shown. (C) Mass spectrum associated with the XIC at 21.4 min depicting the presence of deprotonated singly charged anion. (D) Tandem mass spectrum (MS/MS) of the precursor ion m/z 973.4 following collision-induced dissociation (CID). The observed sequence informative product ion series, cn (sharing common 5′ end) and yn (with common 3′ end) with a subscript denoting the position of cleavage on phosphodiester backbone, or loss of phosphate (−98 D) are labeled and plotted
Fig. S2 LC-MS/MS analysis of RNase Cusativin digestion product ACCA from E. coli tRNATyr I.
(A) Total ion chromatogram (TIC). (B) Extracted ion chromatogram (XIC) for m/z 602.4, corresponding to a digestion product ACCA (position 80–83). (C) Mass spectrum of the XIC at 15.3 min. Note the presence of double and singly deprotonated oligonucleotide anions in the spectrum. (D) Tandem mass spectrum (MS/MS) of the precursor ion, m/z 602.4, following collision-induced dissociation (CID) is depicted. The observed sequence informative product ion series, cn and yn with a subscript denoting the position of cleavage on phosphodiester backbone are labeled and plotted
Fig. S3 LC-MS/MS analysis of RNase Cusativin digestion product ACCp from E. coli tRNATyr I.
(A) Total ion chromatogram (TIC). (B) Extracted ion chromatogram (XIC) for m/z 956.3, corresponding to a digestion product ACCp (position 77–79). (C) Mass spectrum of the XIC at 24.1 min. Note the presence of singly deprotonated oligonucleotide anions in the spectrum, (D) tandem mass spectrum (MS/MS) of the precursor ion, m/z 956.3 following collision-induced dissociation (CID) is depicted. The observed sequence informative product ion series, cn and yn with a subscript denoting the position of cleavage on phosphodiester backbone are labeled and plotted
Fig. S4 LC-MS/MS analysis of RNase Cusativin digestion product, UUCCCCCACCACCA from E. coli tRNATyr I.
(A) Total ion chromatogram (TIC). (B) Extracted ion chromatogram (XIC) for m/z 1427.2, corresponding to a digestion product UUCCCCCACCACCA (position 70–83) with a 3′-OH. (C) Mass spectrum associated with the XIC at 39.1 min. Note the presence of triply deprotonated oligonucleotide anion in the spectrum. (D) Tandem mass spectrum (MS/MS) of the precursor ion, m/z 1427.2, following collision-induced dissociation (CID). The observed sequence informative product ion series, cn (sharing common 5′ end indicated by *) and yn (with common 3′ end indicated by ◆) with a subscript denoting the position of cleavage on phosphodiester backbone are labeled and plotted
Fig. S5 LC-MS/MS analysis of cusativin digestion product [Gm]GCC>p (position 15–18) from E. coli tRNA(Tyr I).
(A) Extracted ion chromatogram (XIC) for m/z 665.4, corresponding to [Gm]GCC>p with linear phosphate. (B) Extracted ion chromatogram (XIC) for m/z 656.4, corresponding to same digestion product with 2′, 3′-cyclic phosphate. (C) Mass spectrum associated with the XIC at 24.8 min depicting the singly and doubly charged molecular ions. (D) Tandem mass spectrum (MS/MS) of the precursor ion m/z 656.4 following collision-induced dissociation (CID) is depicted. The observed sequence informative product ion series, cn (sharing common 5′ end and indicated by *) and yn (with common 3′ end and indicated by ◆) with a subscript denoting the position of cleavage on phosphodiester backbone, are labeled and plotted
Fig. S6 LC-MS/MS analysis of RNase cusativin digestion product U[Q][Ψ]A[ms2i6A]A[Ψ]Cp from E. coli tRNA(Tyr I).
(A) Total ion chromatogram (TIC). (B) Extracted ion chromatogram (XIC) for m/z 1406.5, corresponding to U[Q][Ψ]A[ms2i6A]A[Ψ]Cp (position 32–39). (C) Mass spectrum associated with the XIC at 44 min. Note the presence of double and triple deprotonated oligonucleotide anions in the spectrum. (D) Tandem mass spectrum (MS/MS) of the precursor ion, m/z 1406.5, following collision-induced dissociation (CID). The observed sequence informative product ion series, cn (sharing common 5′ end and indicated by *) and yn (with common 3′ end and indicated by ◆) with a subscript denoting the position of cleavage on phosphodiester backbone, are labeled and plotted. (E) Changes in abundance levels of oligonucleotide, U[Q][Ψ]A[ms2i6A]A[Ψ]Cp, at different protein concentrations and incubation temperature is shown
Fig. S7 Homology based alignment of amino acid sequences of Rnase MC1 and cusativin. Identical amino acid residues at respective positions are indicated by *, Substitutions with residues of similar polarity are indicated by “:” or “.” symbols. Identical catalytic residues in both sequences are depicted in bold type, whereas the identical nucleobase recognizing residues are represented in bold italics. The nucleobase recognizing residue that is different in cusativin is underlined