

Abstract
Rationale
Strategies to reduce the misuse of mu opioid agonists are critically needed. Previous work has shown that kappa opioid agonists can diminish the abuse-related effects and augment the antinociceptive effects of mu agonists. However, use of traditional kappa agonists is limited by their dysphoric side effects.
Objectives
The current study examined the effects of nalfurafine, a clinically available atypical kappa agonist, on the reinforcing, thermal antinociceptive, and respiratory depressant effects of oxycodone in male rats.
Methods
To determine oxycodone:nalfurafine mixture proportions to be examined intravenously across procedures, a progressive-ratio (PR) self-administration procedure compared the reinforcing effects of oxycodone (56 μg/kg/inj) available alone or as a mixture with co-administered nalfurafine (0.32, 1, or 3.2 μg/kg/inj), corresponding to oxycodone:nalfurafine proportions of 175:1, 56:1 and 18:1, respectively. Next, PR and thermal antinociception dose-effect functions were each determined for oxycodone, nalfurafine, and the same oxycodone:nalfurafine mixture proportions. Finally, the respiratory depressant effects of equi-antinociceptive doses of oxycodone, nalfurafine, and the mixtures were compared.
Results
Nalfurafine decreased the reinforcing effects of oxycodone, and the 18:1 mixture did not function as a reinforcer. Oxycodone and nalfurafine each produced dose-dependent antinociception, and the mixtures produced additive antinociception. In addition, antinociceptive doses of the 56:1 and 18:1 mixtures did not produce respiratory depression.
Conclusions
These results suggest that nalfurafine may augment the thermal antinociceptive effects while reducing the reinforcing and respiratory depressant effects of oxycodone.
Introduction
Mu opioid receptor agonists possess high clinical utility for the treatment of pain. However, their use has become a major public health issue in the United States, exemplified by the 4.2-fold increase in the number of fatal prescription-opioid overdoses from 1999–2014 (Centers for Disease Control and Prevention 2015). This surge in overdose deaths correlates with increased prescribing rates (Frenk et al.,2015), which expose a greater number of individuals to medications with high abuse liability and potentially-fatal effects (Duthie and Nimmo 1987). In response to the scope of the current opioid overdose epidemic, interest in the development of safer opioids with reduced abuse liability has recently intensified (e.g., United States Food and Drug Administration 2015).
One strategy for developing safer analgesic medications is to combine a mu opioid agonist with an agent that diminishes its reinforcing effects. Kappa opioid receptor agonists have emerged as a class of drugs that may be useful for this application, as they have been reported to reduce the abuse-related effects of drugs of abuse, including mu opioids (see Bruijnzeel 2009 for review). For example, kappa agonists (e.g., salvinorin A; U50,488) decrease the reinforcing effects of mu agonists in both rhesus monkeys (Freeman et al. 2014; Negus et al. 2008) and rats (Glick et al. 1995; Kuzmin et al. 1997). Similar findings have been reported in the rodent place-conditioning literature, with kappa agonists decreasing place preferences induced by morphine at doses that are not aversive when administered alone (Bolanos et al. 1996; Funada et al. 1993; Hasebe et al. 2004). Taken together, these results raise the possibility that a kappa agonist could be combined with a mu agonist to produce an analgesic medication with reduced abuse liability.
Although abuse liability is a major concern when developing opioid analgesics, the ability to reduce pain is paramount. Like mu agonists, kappa agonists produce antinociceptive effects in animal models of pain (see Jones et al. 2016 for review). Additionally, kappa agonists are reported to have fewer respiratory depressant effects than mu agonists (Shook et al. 1990), suggesting that these drugs will have a lower risk of fatal overdose. However, the development of kappa agonists as standalone analgesics has been limited by their dysphoric and psychotomimetic effects in humans (Millan 1990). Although these aversive effects have limited the clinical utility of kappa agonists, previous work has shown that mixtures of mu and kappa agonists can produce additive thermal antinociception in rhesus monkeys (Ko and Husbands 2009; Negus et al. 2008). These findings suggest that a mu/kappa agonist mixture could produce antinociception with a relatively low dose of each of the constituent drugs, which may reduce the side effects associated with higher doses of each drug in isolation (i.e., mu agonist-mediated respiratory depression and kappa agonist-mediated aversion) as well as reduce abuse liability.
The atypical kappa agonist, nalfurafine, is the only selective kappa agonist currently approved for clinical use in humans (Inui 2015). Since 2009, nalfurafine has been used in Japan for the treatment of uremic pruritus (i.e., Remitch®: 2.5 μg nalfurafine HCl/tablet; P.O.), with no reports of dysphoric mood disturbances or abuse liability (Kumagai et al. 2010; Kumagai et al. 2012; Ueno et al. 2013). Similar to prototypical kappa agonists, nalfurafine produces antinociception and diminishes the abuse-related effects of morphine in rodents and primates (Hasebe et al. 2004; Ko and Husbands 2009). Additionally, nalfurafine has recently been shown to function as a G-protein-biased agonist at human and, to a lesser degree, rat kappa opioid receptors (Schattauer et al. 2017), suggesting that the drug may produce fewer of the p38 MAPK-associated dysphoric effects than the relatively unbiased, traditional kappa agonists (see Dogra and Yadav 2015 for review). Based on these findings, nalfurafine may reduce the reinforcing effects of prescription opioids without causing untoward effects typical of traditional kappa agonists.
The purpose of the current study was to evaluate the effects of a clinically relevant mu opioid agonist (i.e., oxycodone) and nalfurafine in rodent models that are applicable to the development of abuse-deterrent opioid analgesics. To model a compounded medication (i.e., two drugs incorporated into a single formulation), drug combinations were tested as fixed-proportion mixtures across dose determinations in tests of drug self-administration, thermal antinociception, and respiratory depression in male rats.
2. Methods
2.1. Subjects
Forty-one male Sprague-Dawley rats were acquired at 10 weeks of age (Envigo Laboratories, New Jersey, U.S.A.) and acclimated to the laboratory for at least one week before surgery. Initial weights ranged from 300–324g. Rats were pair housed with ad-libitum access to food and water. In Experiments 1–3, rats were maintained on a reversed 12-h light/dark cycle (lights off at 0800) and testing occurred in the dark phase. To minimize the likelihood of nocturnal hyper-locomotion (e.g., Honma and Hiroshige, 1978) interfering with plethysmography measurements, the rats of Experiment 4 were maintained on a non-reversed 12-h light/dark cycle (lights off at 1900) and testing occurred in the light phase. Procedures were conducted in compliance with the National Research Council’s Guide for Care and Use of Laboratory Animals (2011) and approved by the University of Mississippi Medical Center’s Institutional Animal Care and Use Committee.
2.2. Catheter Implantation and Maintenance
Intravenous catheters were implanted and maintained as described previously (Huskinson et al. 2017). Rats were allowed to recover for 5–7 days following surgery. In cases where rats were untested for extended periods of time, catheters were flushed approximately once per week with heparinized saline (30 U/ml) to prevent clots from forming. Patency was verified by intravenous injection of 5 mg/kg methohexital after the final test of each animal. Catheters were considered patent if ataxia was apparent within 3 s.
2.3. Apparatus
2.3.1. Operant Chambers
Eight operant test chambers (Med-Associates, St. Albans, VT, USA), which have been described previously (Townsend et al. 2015), were used in self-administration procedures. Testing occurred 7 days a week, beginning at ~9:00. A PC equipped with Med-Associates software (Med-PC for Windows; St. Albans, VT, USA) controlled experimental conditions and recorded data.
2.3.2. Hot Plate
A hot plate (Omnitech, Hot Plate Analgesiometer) with a plastic enclosure (11″ L, 11″ W, 8″ H) was used for thermal antinociceptive testing. The hot plate was maintained at 52.5°C (±1°C) on test days. Latencies were recorded by an experimentally blinded observer with a digital timer. Testing occurred on Tuesdays and Fridays, separating tests by at least 72 h.
2.3.4. Plethysmography Recording Chamber
Pulmonary ventilation in the presence of 21% oxygen and a 79% nitrogen balance was determined by assessing pressure fluctuations within a plastic plethysmography chamber (~5 L) using a spirometer (model ML141, AD Instruments, Colorado Springs, USA) and a PC equipped with analyzing software (PowerLab data acquisition, AD Instruments, Colorado Springs, USA). Testing occurred Monday through Friday.
2.4. Drugs
Oxycodone HCl was generously provided by the National Institute on Drug Abuse (NIDA) Drug Supply Program (Rockville, MD, USA). Nalfurafine HCl was synthesized and provided by Dr. Christopher McCurdy at the University of Mississippi (University, MS, USA). All drugs were prepared in 0.9% sterile saline and doses are expressed as the salt.
2.5. Procedure
2.5.1. Oxycodone self-administration training and saline determination
Animals were initially trained to self-administer oxycodone (56 μg/kg/inj) on a fixed-ratio one (FR1) schedule of reinforcement. These sessions would end after 2-h had elapsed or if the animal earned 20 injections. The response requirement was increased to FR5 if a rat earned 15 more more injections over 3 consecutive sessions or 20 injections over 2 consecutive sessions. The FR5 schedule of reinforcement was tested until the animal met the FR1 criteria.
Next, animals self-administered oxycodone (56 μg/kg/inj) on a progressive-ratio (PR) schedule of reinforcement. The ratios were determined by a logit equation described by Thomsen et al. (2005): ratio = 19 X [1 + log(step/7-0.3 X step))], rounded to the nearest integer up to step 23, after which the ratio was increased linearly by 12, resulting in ratios of 3, 9, 13, 16, 18, 20, 22, 24, 25, 27, 28, 29, 31, 32, 34, 35, 37, 39, 41, 44, 47, 52, 64, 76, 88, and so forth. This condition was tested for at least 3-d and until the number of earned injections was within 20% of a 3 session mean with no systematic trends. However, if the animal earned fewer than 20 injections per session, the condition was tested for 5 days before assessing stability criteria.
In the final component of training, saline was substituted for oxycodone for three consecutive sessions. The number of saline injections earned on the third day of the substitution represented saline responding in subsequent analyses.
2.5.2. Experiment 1: Potency of nalfurafine to decrease oxycodone self-administration (Ratio determination)
To select the proportions of oxycodone and nalfurafine to be compared in subsequent experiments, the potency of nalfurafine to decrease the reinforcing effects of a dose of oxycodone was determined using three-day substitution tests. Rats (n=6) were given access to oxycodone (56 μg/kg/inj) until the number of earned injections was greater than the number of saline injections. Next, rats were given access to the same dose of oxycodone mixed with either 0.32, 1, or 3.2 μg/kg/inj nalfurafine, thus constituting oxycodone:nalfurafine mixture proportions of 175:1, 56:1, and 18:1, respectively. Each mixture condition was tested for 3 days in a counterbalanced order. The number of injections earned on the third day of the substitution test was the primary dependent variable. The data collected on the first two days were not counted, as we have observed that responding for saline or low doses of drugs tends to be relatively high on the initial days of a substitution test, likely attributable to extinction of lever-pressing behavior maintained by the training dose of oxycodone. Between test conditions, each rat was given access to the training dose of oxycodone until the number of earned injections was greater than the number it had earned for saline.
2.5.3. Experiment 2: Progressive-ratio dose-response for oxycodone, nalfurafine, and oxycodone:nalfurafine mixtures
Experiment 2 determined progressive-ratio dose-effect functions for oxycodone alone, nalfurafine alone, and oxycodone:nalfurafine mixtures with the same fixed proportions of oxycodone:nalfurafine as in Experiment 1 (i.e., 175:1, 56:1, and 18:1). Drug doses were counterbalanced and tested using three-day substitution tests as described in Experiment 1. Group 1 (n=8) self-administered 4 oxycodone doses: 32, 56, 100, 180 μg/kg/inj. Groups 2 (n=8), 3 (n=8), and 4 (n=8) were given access to the same doses of oxycodone with the addition of nalfurafine as a mixture at oxycodone:nalfurafine ratios of 175:1. 56:1, and 18:1, respectively. Additionally, the rats of Group 2 were used to determine a dose-effect function for nalfurafine alone (0.32–10 μg/kg/inj) following completion of the 175:1 dose-effect function. Saline values were re-determined in these rats prior to nalfurafine testing.
2.5.4. Experiment 3: Thermal antinociceptive potency of oxycodone, nalfurafine, and oxycodone:nalfurafine mixtures
Seven rats with an oxycodone self-administration history (i.e., 7 rats from Experiment 2, Group 1) were used in an intravenous hot-plate experiment following at least 1 month of drug abstinence. On the day preceding the first test session, rats were administered i.v. saline through their self-administration ports and placed onto an unheated hot plate for 60-s.
During test sessions, the hot plate was heated to 52.5°C and each rat completed a cumulative dose-effect function for either oxycodone (0.1–5.6 mg/kg), nalfurafine (0.0032–0.32 mg/kg), or the oxycodone:nalfurafine mixture proportions tested in Experiments 1 and 2 (175:1, 56:1, 18:1). The session began by each animal receiving an i.v. saline injection. Following a 15-minute pretreatment period, rats were placed onto the hotplate and a latency to emit a nociceptive response (i.e., paw lift, paw lick, jumping) was recorded. The rat was removed from the hotplate and administered a drug injection. Doses increased in ½ or ¼ logarithmic units until a maximum latency (60-s) was achieved. Rats experienced oxycodone as the first drug condition, followed by nalfurafine as the second drug condition. Rats were exposed to the mixtures in a counterbalanced order.
2.5.5. Experiment 4: Respiratory-depressant effects of an antinociceptive dose of oxycodone, nalfurafine, and oxycodone:nalfurafine mixtures
Experiment 4 compared the effects of doses that were experimentally-determined to be antinociceptive in Experiment 3 (i.e., ED80 values derived from the hotplate results for oxycodone, nalfurafine and the mixtures) on pulmonary ventilation. These equi-antinociceptive doses were compared in 9 rats. To screen for a possible effect of drug exposure on the effects of these drugs on pulmonary ventilation, 3 experimentally naïve rats and 6 rats from Experiment 2, Group 4 were tested. Rats from Experiment 2 experienced at least one month of drug abstinence before testing in Experiment 4. Using a method adapted from Bassi et al. (2015), rats were allowed ~30-minutes to acclimate to the recording chamber before each baseline test. Subsequently, the chamber was filled with 21% oxygen, sealed, and pressure fluctuations were measured for 1-minute. Next, the chamber was opened, the animal was administered a drug and completed a 15-minute pretreatment time within the chamber. The chamber was subsequently refilled with 21% oxygen, sealed, and a 1-minute measurement was taken. The order of drug conditions was counterbalanced across subjects.
2.6. Data analysis
In Experiment 1, injections earned across drug conditions and saline were compared using a repeated-measures one-way analysis of variance (ANOVA) with a Bonferroni multiple-comparisons test.
In Experiment 2, the numbers of injections earned across dose conditions were compared to the saline determination within each group using repeated measures one-way ANOVA and a Bonferroni multiple-comparison test. Additionally, a repeated measures two-way ANOVA compared injections between groups, with the within-subjects factor of oxycodone dose and between-subjects factor of drug condition (oxycodone alone or in a fixed-proportion mixture with nalfurafine). A Bonferroni multiple-comparisons test was used to assess differences in responding for the drug mixtures relative to oxycodone.
In Experiment 3, hotplate latencies were expressed as %Maximum Possible Effect (%MPE), which were calculated using the following equation:
where the test latency was the latency for the rat to emit a nociceptive response after administration of a dose of a drug, and saline latency was the latency to emit a response following the initial saline injection. Individual %MPE for each drug condition was plotted as a function of logarithmic dose. %MPE dose-effect functions were organized for each condition such that the lowest dose corresponded to ≤20% responding and the highest dose was corresponded to ≥80% responding.
The relationship between oxycodone and nalfurafine within the hotplate assay was presented graphically with an isobologram (see Tallarida 2012) and assessed statistically with dose-addition analysis, which compares predicted additive ED50 values (Zadd) with experimentally determined ED50 values (Zmix) of drug mixtures. Specifically, Linear regression was used to determine the dose and 95% confidence limits at which %MPE scores were increased to 50% of control (i.e., ED50). For mixtures, ED50 values were determined in terms of both ox codone and nalfurafine. Additionall, “Zmix” values were calculated for mixtures, which were defined as the total drug dose (i.e., dose oxycodone + dose nalfurafine) that increased %MPE scores to 50%.
Drug relationships were displayed graphically using an isobologram. Here, mean ED50 values for nalfurafine and oxycodone were plotted on the x- and y-axes, respectively. These ED50 values were connected by a line, which represented the coordinates of predicted additive ED50 values of mixtures (i.e., line of additivity). If the ED50 coordinates of a mixture fell above the line of additivity, a sub-additive interaction was suggested. Conversely, if the ED50 coordinates of a mixture fell below the line of additivity, a supra-additive interaction was suggested.
Drug relationships were statistically assessed by comparing Zmix with predicted additive ED50 values (i.e., Zadd), as described by Tallarida, (2000). Individual Zadd values was determined using the equation:
where A was the ED50 value of oxycodone alone, B was the ED50 value of nalfurafine alone, and f was a mixture-specific fractional multiplier. The value of f was determined using the equation:
where MR was the mixture ratio of oxycodone:nalfurafine in a particular mixture, and RPA was the relative potency ratio of oxycodone:nalfurafine when the drugs were tested alone. Mean Zmix and Zadd values were considered to be significantly different if 95% confidence limits did not overlap. Identical calculations were performed with ED25 and ED75 values.
Due to differences in the slope of the oxycodone and nalfurafine dose-effect functions (data not shown), an identical dose-addition analysis was performed for the ED25 and ED75 effect estimations to determine if the nonlinearity of two drugs rendered different results.
The doses of oxycodone, nalfurafine, and the mixtures administered in Experiment 4 were derived from ED80 values calculated using linear regression from %MPE scores from Experiment 3. Changes in pulmonary ventilation (i.e., daily baseline versus treatment) were compared across treatments using a repeated measures one-way ANOVA, with a Bonferroni multiple comparisons test assessing for differences from saline. Pulmonary ventilation was calculated using a plethysmography method adapted from Malan (1973). Respiratory frequency (f) and tidal volume (VT) were determined for each baseline and drug condition by selecting data within the 1-minute sampling period that contained rhythmic breathing (i.e., signals that included oscillations caused by gross body movement were excluded from analysis) using PowerLab data acquisition software. All statistical tests were performed with Prism 6 (GraphPad Software, San Diego, CA).
3. Results
As depicted in Figure 1, nalfurafine produced a proportion-dependent decrease in the reinforcing effects of 56 μg/kg/inj oxycodone [F(4,20)=24.40; p<0.0001]. In comparison to saline, significant differences were observed in the number of injections earned for oxycodone (p<0.0001), the 175:1 mixture (p<0.0001), the 56:1 mixture (p=0.006), but not the 18:1 mixture (p>0.99), indicating all drug conditions functioned as reinforcers with the exception of the 18:1 mixture. In comparison to oxycodone, the number of injections earned for the 175:1 mixture was not different (p=0.18), but rats earned fewer injections of the 56:1 and 18:1 mixtures (56:1; p=0.02, 18:1; p<0.0001).
Figure 1.

Potency of nalfurafine to decrease oxycodone self-administration on a progressive-ratio schedule of reinforcement (Experiment 1). Mean ±SEM intake across drug conditions, which include saline, oxycodone 56 μg/kg/inj and the same dose of oxycodone combined with 0.32, 1, or 3.2 μg/kg/inj nalfurafine. Numbers within the bars correspond to relative proportions of oxycodone:nalfurafine. Filled bars indicate that the condition functioned as a reinforcer (i.e., greater then saline). “ns” indicates that no significant difference between a mixture condition and ox codone alone was observed. “**” and “***” indicate that a significant difference between oxycodone alone and a mixture condition of either p<0.01 or p<0.001 was observed, respectively.
Figure 2A shows progressive-ratio dose-effect curves for oxycodone alone and the three mixtures plotted as a function of oxycodone dose. Although oxycodone produced a characteristic ascending limb between the 32 and 56 μg/kg/inj doses followed by an asymptote between the 56 and 180 μg/kg/inj doses, dose-effect functions for the mixtures were relatively flat. Nevertheless, oxycodone, the 175:1 mixture, and the 56:1 mixture functioned as reinforcers at all doses tested (oxycodone: [F(2.16,15.13)=18.03; p<0.0001]; 175:1: [F(1.8,12.9)=8.9; p=0.004]; 56:1: [F(2.2,13)=9.3; p=0.003]). However, the 18:1 mixture did not function as a reinforcer at any dose tested [F(1.3,9.4)=1.7; p=0.22]. In addition, responding differed across groups [F(3,28)=5.4; p=0.005], with animals in the 18:1 group earning significantly fewer drug injections than the oxycodone group when the 100 and 180 μg/kg/inj oxycodone doses were available (0.1; p=0.02, 0.18; p=0.001). Figure 2B shows the dose-effect series for nalfurafine. Nalfurafine did not function as a reinforcer. Rather, the injections earned for all doses were significantly lower than saline [F(2.1,14.5)=16.3; p=0.0002].
Figure 2.

Progressive-ratio dose-response functions oxycodone, nalfurafine, and oxycodone:nalfurafine mixtures (Experiment 2). Figure 2A depicts mean ±SEM intake across drug conditions in terms of oxycodone dose (32-180 μg/kg/inj) relative to saline responding for each group. Filled symbols indicate that the condition functioned as a reinforcer (i.e., greater then saline). “#” indicates a significant difference between oxycodone and the 18:1 mixture at a dose (p<0.05). Figure 2B depicts mean ±SEM intake of nalfurafine across dose (0.32-10 μg/kg/inj). ignificant decreases from saline are depicted with “*” (p<0.05), “**” (p<0.01), or “***” (p<0.001).
Oxycodone and nalfurafine independently produced full thermal antinociception (oxycodone ED50=2.75 mg/kg, nalfurafine ED50=0.048), yielding a relative potency ratio of 57:1 oxycodone:nalfurafine at the ED50 effect level. %MPE scores are plotted in separate graphs as a function of oxycodone or nalfurafine dose in Figures 3A and 3B, respectively. The relationship of oxycodone:nalfurafine is illustrated with an isobologram in Figure 3C. The 175:1, 56:1, and 18:1 mixtures were all found to produce additive thermal antinociception, as evidenced by overlapping 95% confidence limits between the experimentally determined Zmix and the predicted Zadd values at the ED50 effect level (see Table 1). Similar results were observed at the ED25 and ED75 effect levels, although the 18:1 mixture was found to be supra-additive at the ED75 effect level (Table 1).
Figure 3.

Thermal antinociceptive potency of oxycodone, nalfurafine, and three mixture proportions (Experiment 3). Effects of cumulative oxycodone and nalfurafine (mg/kg, i.v.) are depicted as percent maximum possible effect (%MPE) ±SEM across dose of either oxycodone (Figure 3A) or nalfurafine (Figure 3B). Thermal antinociceptive relationship of oxycodone and nalfurafine at the ED50 effect level are depicted as an isobologram in Figure 3C. The y-axis represents the ED50 value and 95% confidence limits in terms of oxycodone dose (mg/kg) and the x-axis represents the ED50 value and 95% confidence limits in terms of nalfurafine dose. The line connecting the ED50 value for oxycodone and nalfurafine alone represents the coordinates of predicted additive values. The dashed line represents values within the 95% confidence limits of additivity.
Table 1.
Predicted additive ED50, ED25, and ED75 values (Zadd) and experimentally determined ED50, ED25, and ED75 values (Zmix) for oxycodone and nalfurafine mixtures from a measure of thermal antinociception (Experiment 3).
| Effect Level | Zadd (95% CL) | Zmix (95% CL) | Relationship |
|---|---|---|---|
| ED50 | |||
|
| |||
| 175:1 | 2.3 (1.3–3.4) | 1.8 (1.3–2.2) | Additive |
| 56:1 | 1.6 (0.9–2.3) | 1.2 (0.4–2.1) | Additive |
| 18:1 | 0.8 (0.5–1.1) | 0.5 (0.3–0.6) | Additive |
| ED25 | |||
|
| |||
| 175:1 | 1.1 (0.5–1.8) | 1.2 (0.9–1.5) | Additive |
| 56:1 | 0.6 (0.3–0.9) | 0.8 (0.2–1.4) | Additive |
| 18:1 | 0.2 (0.1–0.3) | 0.3 (0.2–0.4) | Additive |
| ED75 | |||
|
| |||
| 175:1 | 4.3 (2.5–6.0) | 2.6 (1.9–3.3) | Additive |
| 56:1 | 3.5 (2.1–4.9) | 2.1 (1.0–3.2) | Additive |
| 18:1 | 2.2 (1.3–3.1)* | 0.7 (0.5–0.8)* | Synergistic* |
indicates a significant difference between Zadd and Zmix (non-overlapping 95% confidence limits). If Zadd > Zmix, a supra-additive interaction is suggested. Conversely, if Zadd < Zmix, a sub-additive interaction is suggested.
The drug dose conditions of Experiment 4 are expressed as oxycodone+nalfurafine and are as follows (mg/kg): oxycodone: 5.9+0; nalfurafine: 0+0.317; 175:1: 2.74+0.015; 56:1: 2.5+0.044; 18:1: 0.73+0.041. The one-way ANOVA found a significant effect of drug condition [F(2.3, 18.3)=4.4; p=0.02]. As depicted in Figure 4, relative to saline, both an antinociceptive dose of oxycodone alone and the 175:1 mixture decreased pulmonary ventilation (oxycodone; p=0.012, 175:1; p=0.012). In contrast, nalfurafine, the 56:1, and the 18:1 mixtures did not significantly affect pulmonary ventilation at the doses tested (nalfurafine; p=0.5, 56:1; p=0.07, 18:1; p=0.94). Differences in the rank order of the mean respiratory depressant effects of the drugs were not observed between the 3 naïve rats and the 6 rats from Experiment 2, Group 4.
Figure 4.
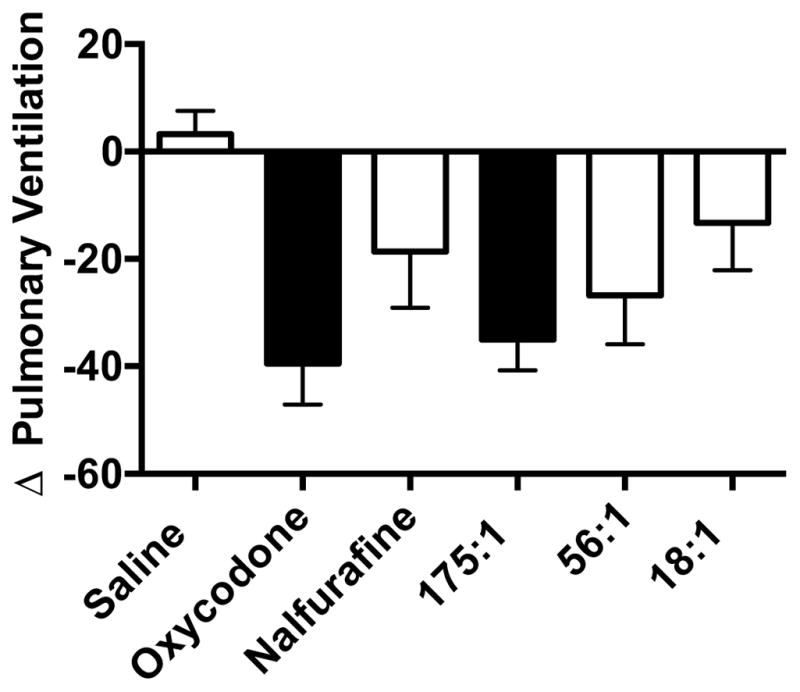
Effects of a thermal antinociceptive dose of oxycodone, nalfurafine, and three mixtures on pulmonary ventilation (Experiment 4). Mean ±SEM percent change in pulmonary ventilation following treatment with a thermal antinociceptive dose (ED80 value from Experiment 3) of the drug mixtures. Doses (oxycodone+nalfurafine) are as follows (mg/kg): oxycodone: 5.9+0; nalfurafine: 0+0.317; 175:1: 2.74+0.015; 56:1: 2.5+0.044; 18:1: 0.73+0.041. Filled bars indicate a significant difference between saline and a drug condition (p<0.05).
4. Discussion
The current study was designed to model a candidate abuse-deterrent opioid analgesic by combining oxycodone and nalfurafine and assessing the effectiveness of the drug mixtures across three experimental endpoints: self-administration, thermal antinociception, and respiration. By matching the mixture proportions and route of administration (intravenous) across studies, we were able to identify an optimal delivery strategy that concurrently enhanced antinociception and mitigated abuse-related and respiratory-depressant effects. The main finding of the current report was that the 18:1 mixture did not function as a reinforcer, produced additive antinociception, and did not produce respiratory depression at an antinociceptive dose. These results suggest that nalfurafine, or kappa agonists with similarly modest side-effect profiles, could be combined with oxycodone to produce a medication with decreased abuse liability and a decreased likelihood of producing respiratory depression when administered at an antinociceptive dose.
The key finding of the self-administration portion of the current report was that nalfurafine decreased the reinforcing effectiveness of oxycodone in a proportion-dependent manner. This finding agrees with previous reports that assessed the effects of mu and kappa agonists in drug self-administration (Freeman et al. 2014; Glick et al. 1995; Kuzmin et al. 1997; Negus et al. 2008). However, the current report is the first to demonstrate that a clinically available kappa agonist, nalfurafine, can diminish the reinforcing effects of a mu agonist. Given that nalfurafine appears to be well tolerated in humans (Inui 2015), these results may be useful in establishing a rationale for examining the abuse liability of oxycodone and nalfurafine mixtures in human volunteers.
When interpreting results of the self-administration portion of this report, it is important to consider the mechanisms that ma have contributed to nalfurafine’s effects on ox codone reinforcement. One possibility is that nalfurafine attenuated the abuse-related neurochemical effects of oxycodone. Indeed, kappa agonism has been shown to decrease drug-induced extracellular dopamine accumulation within mesocorticolimbic structures (see Bruijnzeel 2009 for review), which would be expected to diminish drug reinforcement. Although not mutually exclusive from dopamine modulation, another possibility is that nalfurafine, in agreement with the place-conditioning literature (e.g., Mori et al. 2002), produced an unpleasant or aversive effect that punished oxycodone self-administration. In support of a punishment mechanism, the current report found nalfurafine to maintain significantly fewer injections than saline, which could be interpreted as nalfurafine punishing lever-pressing behavior maintained by saline. Alternatively, the observed effects could have been influenced by non-specific, rate-decreasing effects, preventing the rats from maintaining drug injections at high response requirements. Although this caveat is difficult to circumvent when using single-lever self-administration procedures, we have previously demonstrated that the highly selective kappa opioid agonist, salvinorin A, can dose-dependently decrease the reinforcing effects of remifentanil in a concurrent schedule of reinforcement (i.e., a drug-choice procedure; Freeman et al., 2014), indicating that a kappa opioid agonist can decrease the reinforcing effects of a mu opioid agonist independent of effects on response rate (see Freeman et al. 2014 for a discussion of this issue). In addition, nalfurafine has been shown to decrease the place-conditioning effects of morphine (Mori et al., 2002), an effect that was observed when the rats were in a drug-free state. Finally, given that mixtures of mu and kappa agonists, including oxycodone and nalfurafine, produce sub-additive rate-decreasing effects on food-maintained responding (Negus et al. 2008; our unpublished observations), it seems unlikely that the observed effects are solely attributable to non-specific effects on response rate. Nevertheless, to better address the topic of rate suppression, drug-choice studies are currently underway in our laboratory to determine if nalfurafine can decrease the reinforcing effects of oxycodone independent of effects on response rate.
The current report found oxycodone and nalfurafine to produce additive thermal antinociceptive effects, which agrees with the observations of Ko and Husbands (2009) with nalfurafine, although the mu agonist (i.e., oxycodone versus morphine) and species tested (i.e., rat versus rhesus monkey) were different. Taken together, these results suggest that a nalfurafine and mu opioid agonist mixture can produce antinociceptive effects with a relatively low dose of each of the constituent drugs, which may produce fewer of the side effects associated with higher doses of each drug in isolation. However, it should be noted that both studies assessed the interaction of oxycodone and nalfurafine using a single noxious thermal stimulus (current report: 52.5°C; Ko and Husbands: 50°C), leaving the possibility that differential interactions could occur at other temperatures. In addition, oxycodone:nalfurafine mixtures will need to be tested in complementary preclinical models of pain (e.g., inflammatory pain, neuropathic pain, amelioration of pain-suppressed behaviors) to determine their generality of effect across different pain modalities (see Negus et al. 2006 for review). Finally, the effect of sex was not assessed in any measure of the current report, which is a highly relevant variable to consider, as sex-dependent differences in the effects of mu and kappa opioid have been widely reported (e.g., Barrett 2006). With the rationale of combining oxycodone and nalfurafine established with the current data, we are currently determining if the abuse-related and antinociceptive effects of these mixtures differ between male and female rats.
With the aim of comparing the respiratory depressant effects of equi-antinociceptive doses of oxycodone, nalfurafine, and the mixtures, the current report assessed the effects of a thermal antinociceptive dose of each drug or drug combination (ED80 values from Experiment 3) on pulmonary ventilation. Given the relatively weak respiratory depressant effects of kappa opioid agonists (Shook et al. 1990), we hypothesized that oxycodone would decrease respiration, and the mixtures would produce fewer respiratory depressant effects as a function of decreasing oxycodone concentration in the mixture. Indeed, we observed that oxycodone and the 175:1 mixture proportion each produced a respiratory depressant effect. In contrast, a thermal antinociceptive dose of nalfurafine, as well as the 56:1 and 18:1 mixture proportions, did not significantly affect pulmonary ventilation. These data suggest that an oxycodone and nalfurafine mixture could produce analgesia with fewer respiratory-depressant effects than oxycodone alone. In addition, the diminished reinforcing effects of the mixtures may decrease the likelihood of medication overconsumption, further decreasing the chance of fatal respiratory depression. However, additional studies are needed to formally characterize the relationship between oxycodone and nalfurafine on respiration (e.g., dose-addition analysis).
The misuse of prescription opioid analgesics is an increasingly prevalent and complex health care issue in the United States. With a current lack of high efficacy, non-opioid analgesics available clinically, interest in optimizing the therapeutic effects while minimizing the side effects of existing mu opioid agonists has garnered increasing attention. The current study found the 18:1 oxycodone:nalfurafine mixture to have the most favorable effect profile, as it produced fewer abuse-related and respiratory depressant effects than oxycodone while also preserving its antinociceptive effect. However, when interpreting these results, one should consider that nalfurafine appears to have less functional selectivity for the G-protein pathway in rats compared to humans (Schattauer et al. 2017), which would be expected to affect the potency of nalfurafine to produce antinociceptive and anti-reinforcing effects. In addition, the relatively long half life of nalfurafine (14h: Inui 2015) may lead to its accumulation if administered at oxycodone-appropriate dosing intervals (e.g., OxyContin®: every 12h: Purdue Pharmaceuticals 2016). Future studies could attempt to model this issue of poly-drug pharmacokinetics by assessing the oral bioavailability of these drugs when administered as a mixture. Nevertheless, the demonstrated tolerabilit of nalfurafine in humans encourages further investigation of this drug’s ability to increase the safety of mu opioid analgesics, which may lead to the development of atypical kappa opioid agonists that are tailored for this application.
Acknowledgments
Funding: The study was supported by R01-DA039167 to KBF from the National Institute on Drug Abuse.
Funding: The preparation of this manuscript was supported by grant R01-DA039167 to KBF from the National Institute on Drug Abuse.
The authors would like to thank Hunter Bruce, Georganna Nutt, Yvonne Zuchowski, and Josh Woods for their expert technical assistance.
References
- Barrett AC. Low efficacy opioids: implications for sex differences in opioid antinociception. Exp Clin Psychopharmacol. 2006;14:1–11. doi: 10.1037/1064-1297.14.1.1. [DOI] [PubMed] [Google Scholar]
- Bassi M, Nakamura NB, Furuya WI, Colombari DS, Menani JV, do Carmo JM, da Silva AA, Hall JE, Colombari E. Activation of the brain melanocortin system is required for leptin-induced modulation of chemorespiratory function. Acta Physiol. 2015;213:893–901. doi: 10.1111/apha.12394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolanos CA, Garmsen GM, Clair MA, McDougall SA. Effects of the kappa-opioid receptor agonist U-50,488 on morphine-induced place preference conditioning in the developing rat. Eur J Pharmacol. 1996;317:1–8. doi: 10.1016/s0014-2999(96)00698-x. [DOI] [PubMed] [Google Scholar]
- Bruijnzeel AW. Kappa-opioid receptor signaling and brain reward function. Brain Res Rev. 2009;62:127–46. doi: 10.1016/j.brainresrev.2009.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Centers for disease control and prevention: National center for health statistics. Number of age-adjusted rates of drug-poisoning deaths involving opioid analgesics and heroin: United States, 1999–2014. 2015 https://www.cdc.gov/nchs/data/health_policy/AADR_drug_poisoning_involving_OA_Heroin_US_2000-2014.pdf.
- Dogra S, Yadav PN. Biased agonism at kappa opioid receptors: Implication in pain and mood disorders. Eur J Pharmacol. 2015;763:184, 90. doi: 10.1016/j.ejphar.2015.07.018. [DOI] [PubMed] [Google Scholar]
- Duthie DJ, Nimmo WS. Adverse effects of opioid analgesic drugs. Br J Anaesth. 1987;59:61–77. doi: 10.1093/bja/59.1.61. [DOI] [PubMed] [Google Scholar]
- Freeman KB, Naylor JE, Prisinzano TE, Woolverton WL. Assessment of the kappa opioid agonist, salvinorin A, as a punisher of drug self-administration in monkeys. Psychopharmacology. 2014;231:2751–8. doi: 10.1007/s00213-014-3436-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frenk SM, Porter KS, Paulozzi LJ Centers for disease control and prevention: National center for health statistics. Prescription Opioid Analgesic Use Among Adults: United States, 1999 – 2012. 2015 https://www.cdc.gov/nchs/products/databriefs/db189.htm.
- Funada M, Suzuki T, Narita M, Misawa M, Nagase H. Blockade of morphine reward through the activation of kappa-opioid receptors in mice. Neuropharmacology. 1993;32:1315–23. doi: 10.1016/0028-3908(93)90026-y. [DOI] [PubMed] [Google Scholar]
- Glick SD, Maisonneuve IM, Raucci, Archer S. Kappa opioid inhibition of morphine and cocaine self-administration in rats. Brain Res. 1995;681:147–52. doi: 10.1016/0006-8993(95)00306-b. [DOI] [PubMed] [Google Scholar]
- Hasabe K, Kawai K, Suzuki T, Kawamura K, Tanaka T, Narita M, et al. Possible pharmacotherapy of the opioid kappa receptor agonist for drug dependence. Ann N Y Acad Sci. 2004;1025:404–13. doi: 10.1196/annals.1316.050. [DOI] [PubMed] [Google Scholar]
- Honma K, Hiroshige T. Simultaneous determination of circadian rhythms of locomotor activity and body temperature in the rat. Jpn J Physiol. 1978;28:159–69. doi: 10.2170/jjphysiol.28.159. [DOI] [PubMed] [Google Scholar]
- Huskinson SL, Naylor JE, Townsend EA, Rowlett JK, Blough BE, Freeman KB. Self-administration and behavioral economics of second-generation synthetic cathinones in male rats. Psychopharmacology. 2017;234:589–598. doi: 10.1007/s00213-016-4492-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inui S. Nalfurafine hydrochloride to treat pruritus: a review. Clin Cosmet investing Dermatol. 2015;8:249–55. doi: 10.2147/CCID.S55942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones MR, Kaye AD, Kaye AJ, Urman RD. The emerging therapeutic roles of kappa-opioid agonists. J Opioid Manag. 2016;12:101–7. doi: 10.5055/jom.2016.0321. [DOI] [PubMed] [Google Scholar]
- Ko MC, Husbands SM. Effects of atypical kappa-opioid receptor agonists on Intrathecal morphine-induced itch and analgesia in primates. J Pharmacol Exp Ther. 2009;328:193–200. doi: 10.1124/jpet.108.143925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumagai H, Ebata T, Takamori K, Miyasato K, Muramatsu T, Nakamoto H, et al. Efficacy and safety of a novel κ-agonist for managing intractable pruritus in dialysis patients. Am J Nephrol. 2012;36:175–83. doi: 10.1159/000341268. [DOI] [PubMed] [Google Scholar]
- Kumagai H, Ebata T, Takamori K, Muramatsu T, Nakamoto H, Suzuki H. Effect of a novel kappa-receptor agonist, nalfurafine hydrochloride, on severe itch in 337 haemodialysis patients: a phase III, randomized, double-blind, placebo-controlled study. Nephrol Dial Transplant. 2010;25:1251–7. doi: 10.1093/ndt/gfp588. [DOI] [PubMed] [Google Scholar]
- Kuzmin AV, Semenova S, Gerrits MA, Zvartau EE, Van Ree JM. Kappa-opioid receptor agonist U50,488H modulates cocaine and morphine self-administration in drug naïve rats and mice. Eur J Pharmacol. 1997;321:265–71. doi: 10.1016/s0014-2999(96)00961-2. [DOI] [PubMed] [Google Scholar]
- Milan A. Ventilation measured by body plethysmography in hibernating mammals and in poikitotherms. Resp Physiol. 1973;17:32–44. doi: 10.1016/0034-5687(73)90108-4. [DOI] [PubMed] [Google Scholar]
- Millan MJ. Kappa-opioid receptors and analgesia. Trends Pharmacol Sci. 1990;11:70–6. doi: 10.1016/0165-6147(90)90321-x. [DOI] [PubMed] [Google Scholar]
- Mori T, Nomura M, Nagase H, Narita M, Suzuki T. Effects of a newly synthesized kappa opioid receptor agonist, TRK-820, on the discriminative stimulus and rewarding effects of cocaine in rats. Psychopharmacology. 2002;161:17–22. doi: 10.1007/s00213-002-1028-z. [DOI] [PubMed] [Google Scholar]
- Negus SS, Schrode K, Stevenson GW. Mu/kappa opioid interactions in rhesus monkeys: implications for analgesia and abuse liability. Exp Clin Psychopharmacol. 2008;16:386–99. doi: 10.1037/a0013088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negus SS, Vanderah TW, Brandt MR, Bilsky EJ, Becerra L, Borsook D. Preclinical assessment of candidate analgesic drugs: recent advancements and future challenges. J Pharmacol Exp Ther. 2006;319:507–14. doi: 10.1124/jpet.106.106377. [DOI] [PubMed] [Google Scholar]
- OxyContin® [package insert] Purdue Pharma LP; Stamford, CT, USA: Dec, 2016. [Google Scholar]
- Schattaurer SS, Kuhar JR, Song A, Chavkin C. Nalfurafine is a G-protein biased agonist having significantly greater bias at the human than rodent form of the kappa opioid receptor. Cell Signal. 2017;32:59–65. doi: 10.1016/j.cellsig.2017.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shook JE, Watkins WD, Camporesi EM. Differential roles of opioid receptors in respiration, respiratory disease, and opiate-induced respiratory depression. Am Rev Respir Dis. 1990;142:895–909. doi: 10.1164/ajrccm/142.4.895. [DOI] [PubMed] [Google Scholar]
- Tallarida RJ. Drug synergism and dose-effect data analysis. Boca Raton, FL: Chapman and Hall/CRC; 2000. [Google Scholar]
- Tallarida RJ. Revisiting the isobole and related quantitative methods for assessing drug synergism. J Pharmacol Exp Ther. 2012;342:2–8. doi: 10.1124/jpet.112.193474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomsen M, Woldbye Dp, Wortwein G, Fink-Jensen A, Wess J, Caine SB. Reduced cocaine self-administration in muscarinic M5 acetylcholine receptor-deficient mice. J Neurosci. 2005;25:8141–9. doi: 10.1523/JNEUROSCI.2077-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townsend EA, Beloate LN, Huskinson SL, Roma PG, Freeman KB. Corn oil, but not cocaine, is a more effective reinforcer in obese than in lean Zucker rats. Physiol Behav. 2015;143:136–41. doi: 10.1016/j.physbeh.2015.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueno Y, Mori A, Yanagita T. One year long-term study on the abuse-liability of nalfurafine in hemodialysis patients. Int J Clin Pharmacol Ther. 2013;51:823–31. doi: 10.5414/CP201852. [DOI] [PubMed] [Google Scholar]
- United States food and drug administration center for drug evaluation and research. Abuse-deterrent opioids evaluation and labeling guidance for industry. 2015 https://www.fda.gov/downloads/Drugs/Guidances/UCM334743.pdf.