

Abstract
The shift by cancer cells toward aerobic glycolysis (Warburg effect) confers selective advantages by utilizing nutrients (e.g. lipids, amino acids and nucleotides) to build biomass. Lipogenesis is generally enhanced and its inhibition diminishes proliferation and survival. Re-expression of the metastasis suppressor KISS1 in human melanoma cells results in greater mitochondrial biogenesis, inhibition of glycolysis, utilization of beta-oxidation to provide energy, elevated oxidation of exogenous fatty acids and increased expression of early-phase lipogenesis genes at both mRNA and protein levels. Correspondingly, the energy sensor AMPKβ is phosphorylated, resulting in inhibitory phosphorylation of acetyl-CoA carboxylase (ACC), which is linked to enhanced beta-oxidation. Furthermore, PGC1α is required for KISS1-mediated phosphorylation of ACC and metastasis suppression. Collectively, these data further support the linkages between macromolecular metabolism and metastasis.
Keywords: KISS1, Metastasis, Lipid Metabolism, Glycolysis
Introduction
Metabolic reprogramming has long been recognized as a biochemical change in cancer cells. Otto Warburg described how cancer cells grown under normoxic conditions utilize aerobic glycolysis, resulting in increased glucose uptake and lactic acid production [1, 2]. Herbert Crabtree described that, with the addition of glucose, neoplastic cells reversibly switch from mitochondrial respiration to aerobic glycolysis [3, 4]. Ultimately, cancer cells appear to utilize aerobic glycolysis to redirect carbons toward biomass synthesis in order to sustain proliferation. While the role(s) of metabolic reprogramming in tumor progression toward metastasis is incompletely understood, aerobic glycolysis has been thought to promote tumor invasion partly through the lactic acid-mediated acidification of the microenvironment which renders the surrounding microenvironment uninhabitable [5, 6]. Importantly, cancer cells have metabolic plasticity that allows them to shift between glycolysis and mitochondrial respiration, thereby allowing them to adapt to multiple microenvironments required for tumor progression, invasion and metastasis.
Metabolic perturbation resulting in increased glycolysis can be attributed to multiple events including alterations of tumor-associated gene expression and/or protein activities [7-9], mitochondrial structure and function [8, 10, 11], and mutations in mitochondrial DNA and enzymes [12-15]. Currently, the role of aerobic glycolysis is better defined in primary tumor growth while the role(s) of aerobic glycolysis in metastasis is not fully understood.
Metastasis is a multi-step, multi-genic process with enormous energy requirements and intense cellular and biochemical stress. During metastasis, tumor cells leave the site of origination to enter the circulation and disseminate, then leave the circulation at distant sites where they proliferate to establish secondary tumor masses [16-20]. Throughout the process, tumor cells survive multiple stresses such as hypoxia, re-oxygenation-associated redox stress, shear, immune surveillance, weak/non-adhesion, and restoration of proliferation at secondary sites [21, 22]. Metabolic reprogramming could therefore confer flexibility to meet the unique energy requirements of the metastatic cascade. However, metabolic plasticity during metastasis is neither well-described nor understood.
KISS1 is a metastasis suppressor, defined by the ability to suppress metastasis without blocking primary tumor growth [23, 24]. In the initial studies, KISS1 delayed orthotopic tumor onset, but tumors grew at a similar rate following initial establishment. Even when the time to euthanasia was delayed to compensate for the primary tumor delay, metastasis was significantly suppressed [23, 24]. Nascent KISS1 is a 145-amino acid secreted protein that is cleaved by the prohormone convertase furin into polypeptides, termed kisspeptins [25-27]. The first identified kisspeptin, KP54, binds a G protein-coupled receptor, KISS1R (formerly GRP54, AXOR12, and hOT7T175 [25, 28, 29]), and signals via phospholipase C, protein kinase C, and intracellular Ca2+ mobilization. KISS1 also activates mitogen-activated protein kinase and phosphatidylinositol-3-kinase/Akt pathways [reviewed in [30]]. Surprisingly, KISS1 anti-metastatic effects do not require KISS1R [27, 30]. Therefore, the relevance of these specific signaling cascades in mediating metastasis suppression is not fully defined.
We previously showed that KISS1, but not a mutant form of KISS1 lacking the secretion signal peptide, decreases aerobic glycolysis by enhancing mitochondrial oxidative phosphorylation and PPARγ co-activator-1 alpha (PGC1α)-mediated mitochondrial biogenesis [31]. KISS1 also appeared to have roles in regulating lipogenesis and lipid metabolism, possibly through the AMP-kinase (AMPK) pathway [31]. Therefore, we hypothesized that KISS1 could also activate alternative energy-generating pathways.
Materials and Methods
Cell Lines and Culture
These experiments were performed with a highly metastatic subclone of the human melanoma cell line C8161, named C8161.9. KFM (KISS1-Flag-Metastin) or KFMΔSS clones were described previously [27]. Briefly, KFM and KFMΔSS expression constructs contain a FLAG epitope within the proprotein convertase processing site R67 ˆ R68 allowing KP54 to be tracked. KFMΔSS was generated by deleting the 19-amino acid secretion sequence. All constructs were made using pcDNA3.1 phagemid. Lentiviral constructs were generated using Life Technologies Gateway platform (Life Technologies, Carlsbad, CA). All cell lines were cultured in a 1:1 mixture of Dulbecco’s Modified Minimum Essential Medium and Ham’s F-12 medium (Life Technologies) supplemented with 5% FBS (Life Technologies or Atlanta Biologicals, Flowery Branch, GA) and 1% nonessential amino acids (Life Technologies). Short hairpin RNA targeting KISS1 was purchased from Open Biosystems (GE Healthcare, Chicago, IL). All cells lines were tested free of Mycoplasma spp. contamination using a PCR-based assay (ATCC, Manassas, VA). All parental cell lines were validated by STR allele profiling at the University of Arizona Genetics Core.
Real-time RT-PCR and PCR array
Total RNA was isolated using Quick-RNA™ Mini prep (Zymo Research, Irvine, CA) and reverse transcribed using iScript cDNA synthesis kit (Bio Rad, Hercules, CA). cDNA then was amplified using Taqman probes (Life Technologies) and Taqman Fast Advanced Master Mix (Life Technologies). The following Taqman probes were used: ACADVL (Hs00825606_g1), ACADS (Hs00163506_m1), ACC1 (ACACA, Hs01046047_m1), ACC2 (ACACB, Hs00153715_m1), ACLY (Hs00982738_m1), FABP4 (Hs01086177_m1), FASN (Hs01005622_m1), HKII (Hs00606086_m1), LDHA (Hs00855332_g1), LDHB (Hs00929956_m1), PFKM (Hs01075411_m1), PKM1 (Hs00987255_m1), PKM2 (Hs00987261_g1), PPARα (Hs00947539_m1), SLC2A1 (Hs00892681_m1), and RNA18S5 (Hs03928985_g1).
To analyze fatty acid metabolism gene expression, RNA was extracted as previously described and reverse transcribed using RT2 cDNA synthesis kit (Qiagen, Valencia, CA). Fatty acid metabolism RT2 profiler PCR array and RT2 Real-Time SyBR Green/ROX PCR Mix were purchased from SuperArray Biosciences/Qiagen and the array was performed according to the manufacturer’s instructions using pooled samples from 3 independent experiments. qRT-PCR and PCR arrays were performed on ABI ViiA™ Real-Time PCR system (Life Technologies).
Fatty acid oxidation
To measure mitochondrial beta oxidation capacity, a Seahorse Bioscience XF24 extracellular flux analyzer and XF fatty acid oxidation assay were used according to the manufacturer’s instructions (Seahorse Bioscience, North Billerica, MA). The oxygen consumption rate (OCR) was expressed in pmol/min, and normalized by whole cell lysate. Oligomycin (ATP synthase inhibitor), carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone (FCCP) (mitochondrial uncoupling agent), rotenone (complex I inhibitor), and antimycin A (complex III inhibitor) were used to disrupt mitochondrial function and were purchased from Sigma (Sigma-Aldrich, St. Louis, MO). The calculations for various fatty acid oxidation parameters have previously been described in detail [32].
Glycolysis stress test
To measure glycolytic parameters, the Seahorse Bioscience Glycolysis Stress Test was performed according to the manufacturer’s protocol with one exception, rotenone and antimycin A were used in an additional injection step to measure the Crabtree Effect. Glycolytic [33] and Crabtree effect [34] parameters are described elsewhere.
Antibodies and immunoblotting
Cells were lysed in ice-cold RIPA buffer (ThermoFisher, Waltham, MA), supplemented with Halt Protease Inhibitor cocktail (ThermoFisher) for 30 minutes and centrifuged (12,000 x g, 10 minutes; ThermoFisher Microfuge 22R centrifuge). Mitochondrial and cytosolic fractions were isolated using Mitochondria Isolation Kit for Cultured Cells (ThermoFisher) according to manufacturer’s instructions. The mitochondrial fraction was lysed in RIPA buffer supplemented with protease inhibitor cocktails. The lysates were then diluted with 4X NuPage LDS sample buffer (ThermoFisher) with beta-mercaptoethanol and boiled at 95°C for 10 minutes. The boiled lysate was resolved using 4%-20% precast polyacrylamide gels and blotted onto PVDF membranes using the BioRad Transblot Turbo system. The blots then were blocked in either 5% milk in TBST or 5% BSA in TBST. Primary antibodies were purchased from following manufacturers and were used at the titers listed: Acc (Cell Signaling, Danvers, MA cat# 3676, 1:500), phos-Acc Ser79 (Cell Signaling cat#3661, 1:500), FASN (Cell Signaling cat#3180, 1:500), AMPK-α (Cell Signaling cat# 2603, 1:500), phos-AMPK-α Thr172 (Cell Signaling cat#2535, 1:500), AMPK-β1/2 (Cell Signaling cat#4150, 1:500), phos- AMPK-β1/2 Ser108 (Cell Signaling cat#4181, 1:500), hexokinase II (Cell Signaling cat#2867, 1:500), VDAC (Millipore cat# AB10527) , PGC1α (Millipore cat# ST1202, 1:1,000), β-actin (Sigma cat#A5441, 1:5,000), GAPDH (Santa Cruz Biotechnology, Santa Cruz, CA cat#sc-365062, 1:3000), and KISS1 (ref. [26], 1:500). Secondary antibodies were purchased from following manufacturers and were used at the titers listed: mouse HRP conjugated (GE Healthcare cat# NA931, 1:5000), rabbit HRP conjugated (GE Healthcare cat# NA934, 1:5,000), and goat HRP conjugated (Santa Cruz cat#sc-2020, 1:5,000). Antibodies were diluted in 5% milk in TBST buffer (for non-phosphorylated proteins) or 5% BSA in TBST (for phosphorylated proteins).
In vivo metastasis
Athymic mice (3-5 weeks old) were purchased from Charles River. Experimental metastasis was studied by injecting 2 × 105 (suspended in ice-cold 200 μl HBSS) viable GFP-labelled C8161.9 or its derivative cells into the lateral tail veins of athymic mice (n=10). Mice were euthanized 5 weeks post-injection. For spontaneous metastasis, 1 × 106 viable C8161.9pCDNA3.1, C8161.9KFM or C8161.9KFM/shPGC1α cells were injected intradermally into dorsolateral flank of athymic mice (n=9-10). Tumor growth was measured weekly. When mean tumor diameter reached ~1.5 cm at ~5 weeks, primary tumors were removed. Mice were followed until there was evidence of metastases before euthanasia. Organs were collected and metastatic lesions visualized by fluorescence microscopy. All protocols were approved by the Institutional Animal Care and Use Committee at the University of Kansas Medical Center.
Results
KISS1 down-regulates glycolysis
Persistent elevated glycolysis is a hallmark of the Warburg effect in cancer [35]. Neoplastic cells exhibit metabolic plasticity in terms of the short-term adaptation mechanisms depending upon the surrounding environment (Crabtree effect). We previously demonstrated that KFM expression increases extracellular pH due to decreased lactate secretion [31]. Moreover, KFM expression also increased extracellular glucose, possibly due to decreased cellular uptake of glucose [31]. Therefore, we hypothesized that KFM inhibits the glycolytic pathway. Therefore, we measured glycolytic parameters and the Crabtree effect.
KFM decreased glycolysis significantly compared to the vector control (Figures 1a +c). As with other biological functions, presence of a secretion signal sequence was required for inhibition of glycolysis. When ectopic KFM was knocked down using shRNA, KFM-mediated inhibition of glycolysis was reversed (Figure 1c). KFM-expressing cells exhibited higher glycolytic reserve after treating with 2-deoxyglucose, indicating that KFM cells are less glycolytic; whereas, vector control and KFMΔSS cells lacked similar reserve capacity (Figure 1d). In general, cells with low glycolytic reserve capacity are more dependent on glycolysis, while cells with higher glycolytic reserve are more tolerant of metabolic shift [36]. All cells presented with approximately equal glycolytic capacity (Figure 1e). Vector and KFMΔSS-expressing C8161.9 cells exhibited the Crabtree effect as mitochondrial respiration was suppressed when glucose was introduced. However, KFM-expressing C8161.9 cells were resistant to switching toward glycolysis and inhibition of mitochondrial function. This result further revealed that KFM cells have defective glycolysis and highly competent mitochondrial respiration. Therefore, increased glucose did not influence mitochondrial metabolism as it did in other cells. Altogether, these data suggested that KFM-expressing cells are capable of glycolysis, but do not utilize it as the sole or primary energy source. The same cells initiated glycolysis in response to lack of energy production by the mitochondria. Importantly, KFM cells mainly used mitochondrial metabolism and were less glycolytic and resistant to the Crabtree effect, suggesting this as a mechanism that potentially renders them less malignant.
Figure 1.
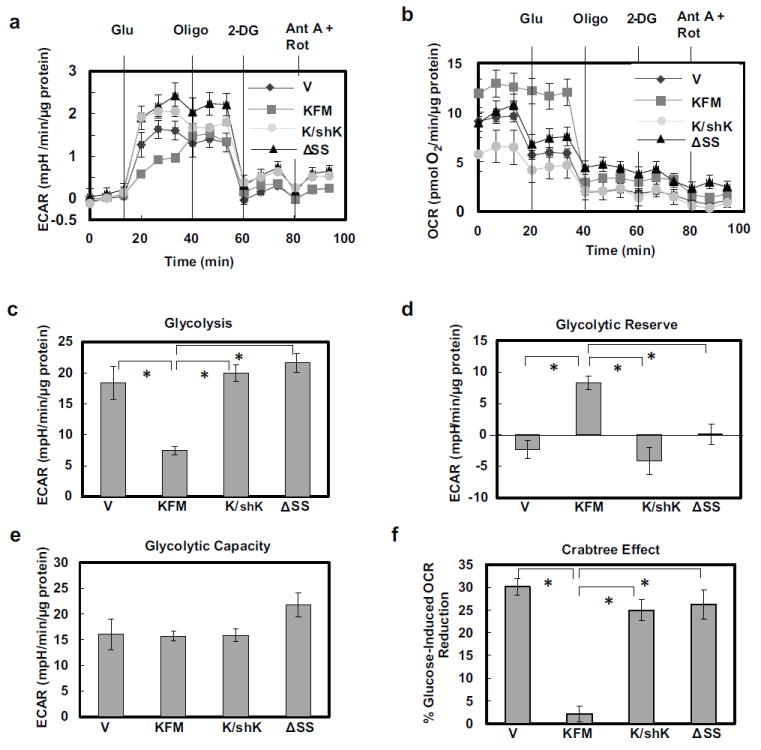
KISS1 inhibits both glycolysis and the Crabtree effect. A Seahorse XF24 bioanalyzer was used to measure extracellular acidification rate (ECAR) (a) and mitochondrial oxygen consumption rate (OCR)(b) in C1861.9Vector (V), C8161.9KFM59(KFM), C8161.9KFM59/shKISS1 (K/shK), and C8161.9KFMΔSS6 (ΔSS) cells under basal conditions followed by sequential injections of 10 mM glucose (Glu), 1 μM oligomycin (Oligo), 50 mM 2-deoxyglucose (2-DG), and 2 μM antimycin A (Ant A) and 4 μM rotenone (Rot). Basal glycolysis (c), glycolytic reserve (d), and glycolytic capacity (e) are derived from ECAR measurements in (a). Crabtree effect (f) is derived from OCR measurements in (B). N = 5; error bars, SEM; * P ,< 0.05
To delve more deeply into the glycolytic pathway, expression of selected genes and proteins were analyzed. KFM down-regulated mRNA expression of a glucose transporter gene, Slc2a1 and a key early step glycolytic enzyme, HKII (Figure 2a). The effects were specific in that knockdown of KFM partially reversed the effects. KFMΔSS-expressing cells also decreased Slc2a1 and HKII mRNA, but not significantly. mRNA expression of downstream enzymes, phosphofructokinase muscle isoform (PFKM), pyruvate kinase M1 and M2 (PKM1 and PKM2), and lactate dehydrogenase isoforms A and B (LDHA and LDHB) were unaffected by either KFM or KFMΔSS (Figure 2a). KFM did not change the protein levels of key glycolytic enzymes (SLC2A1, HKII, PKM2, and LDHA).
Figure 2.
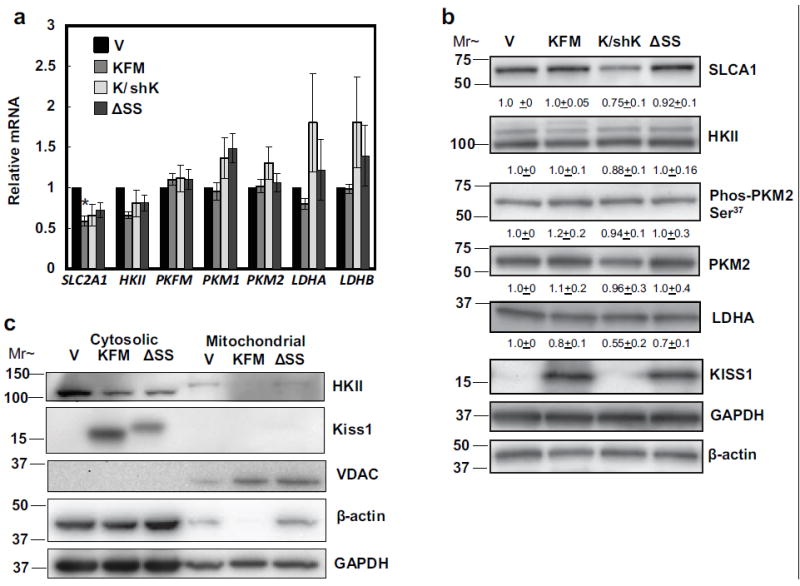
KISS1 has minimal effects on glycolytic enzyme expression, but decreases mitochondria bound hexokinase II. (a) mRNA expression of glycolytic transporter and enzymes, SLC2A1, HKII, PFKM, PKM1, PKM2, LDHA, and LDHB in C1861.9Vector (V), C8161.9KFM59 (KFM), C8161.9KFM59/shKISS1 (K/shK), and C8161.9KFMΔSS6 (ΔSS) cells was quantified by qRT-PCR. (b) SLCA1, hexokinase II, phos-PKM2 at serine 37, PKM2, LDHA, and KISS1 proteins from C1861.9Vector, C8161.9KFM59, C8161.9KFM59/shKISS1, and C8161.9KFMΔSS6 whole cell lysate were quantified using immunoblot analysis with GADPH and beta-actin as loading controls. Densitometry of proteins was normalized to beta-actin. (c) Cytosolic and mitochondrial fractions were extracted from C1861.9Vector, C8161.9KFM59, C8161.9KFM59/shKISS1, and C8161.9KFMΔSS6 cells and immunoblot analysis was performed for hexokinase II, KISS1, VDAC, beta-actin, and GAPDH. (a) N ≥ 3; error bars, SEM; * P ,< 0.05 (b) N≥ 3; mean ± SEM. Note: the KFMΔSS construct consistently runs at a higher molecular weight than full-length KISS1 [26]; however, gel density and gel size affect separation, which is why there is minor gel-gel variability between panels 2b and 2c.
PKM2 is phosphorylated at serine 37 by mitogen-activated protein kinase 1 (MAPK1/ERK2), resulting in nuclear translocation and promotion of the Warburg effect [37]. KFM did not modulate phosphorylation of PKM2 (Figure 2b). Interestingly, KISS1 attenuated expression of mitochondrial-bound hexokinase II (HKII) (Figure 2c), a rate-limiting enzyme that upregulates glycolytic pathways and suppresses apoptosis [38]. Cumulatively, these data suggest that KISS1 reverses the Warburg effect by mitigating aerobic glycolysis and boosting mitochondrial metabolism, thereby shifting energy production to rely more on oxidative phosphorylation (OXPHOS) and beta-oxidation.
KISS1 increases mitochondrial beta-oxidation
Since KISS1 promotes mitochondrial biogenesis [31], we hypothesized that KISS1 could also increase mitochondrial beta-oxidation of fatty acids. Sequential injections of oligomycin (ATP synthase inhibitor), FCCP (uncoupling agent), rotenone (Complex I inhibitor) and Antimycin A (Complex III inhibitor) allowed assessment of various mitochondrial functional parameters in the Seahorse Bioanalyzer. KFM over-expression, but not KFMΔSS, increased mitochondrial OCR compared to vector-only transduced cells (Figures 1b, 3a-c). Oligomycin inhibited ATP production and substantially decreased OCR only in KFM-expressing cells.
Figure 3.
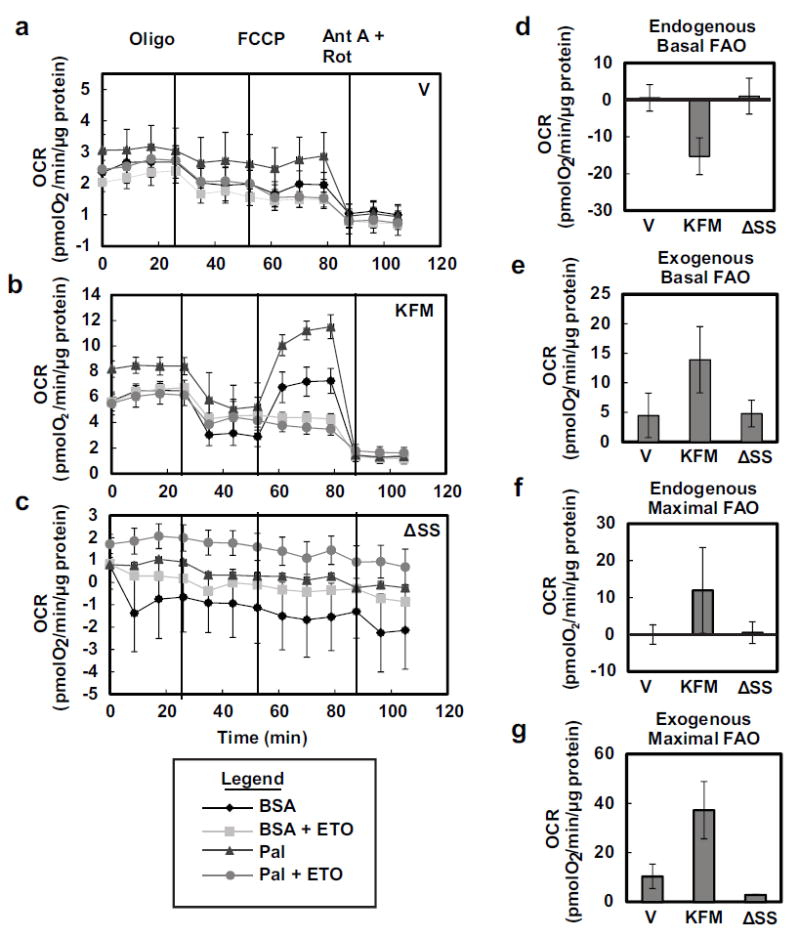
KISS1 increases mitochondrial beta-oxidation. Seahorse XF24 bioanalzyer was used to measure OCR in C1861.9Vector (V) (a), C8161.9KFM59 (KFM) (b), and C8161.9KFMΔSS6(ΔSS) (c) cells treated with BSA or palmitate-BSA (Pal) with or without 40 μM etomoxir (ETO) in low glucose (0.5 mM) conditions followed by sequential injections of oligomycin (oligo), 5 μM carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone (FCCP), and 2 μM antimycin A (Ant A) and 4 μM rotenone (Rot). Endogenous (d) and exogenous (e) basal fatty acid oxidation and endogenous (f) and exogenous (g) maximal fatty acid oxidation are derived from the OCR measurements in (a-c). N ≥ 3; error bars, SEM; * P ,< 0.05
Beta-oxidation and energy production
KFM expression in cells slightly increased the maximal oxidation of endogenous fatty acids, but there were no differences in basal oxidation (Figures 3d+f). KFM-expressing cells displayed significantly higher oxidation of exogenous fatty acids when cultured under both basal and maximal respiration conditions, suggesting that KFM-expressing cells can oxidize exogenous fatty acids more efficiently (Figures 3e+g).
To assess potential roles of KISS1 in beta oxidation, a PCR Array was performed for 84 fatty acid metabolism-related genes. KFM increased expression of 12 genes involved in fatty acid metabolism by ≥2-fold (Figures 4a+b) while down-regulating five beta-oxidation related genes (Table 1). The up-regulated genes are the key to beta oxidation and conjugation of fatty acids with acyl-CoA. These results suggest that KFM promotes conjugation of fatty acids to acyl-CoA, transport of fatty acid acyl-CoA into mitochondria, and beta-oxidation. Peroxisome proliferator-activated receptor alpha (PPARα) is a key transcriptional activator of beta-oxidation. KFM, but not KFMΔSS, down-regulated PPARα mRNA. Knockdown of KFM by shRNA reversed the KFM-induced decreases in PPARα gene expression (Figure 4c), suggesting a possible compensatory change.
Figure 4.
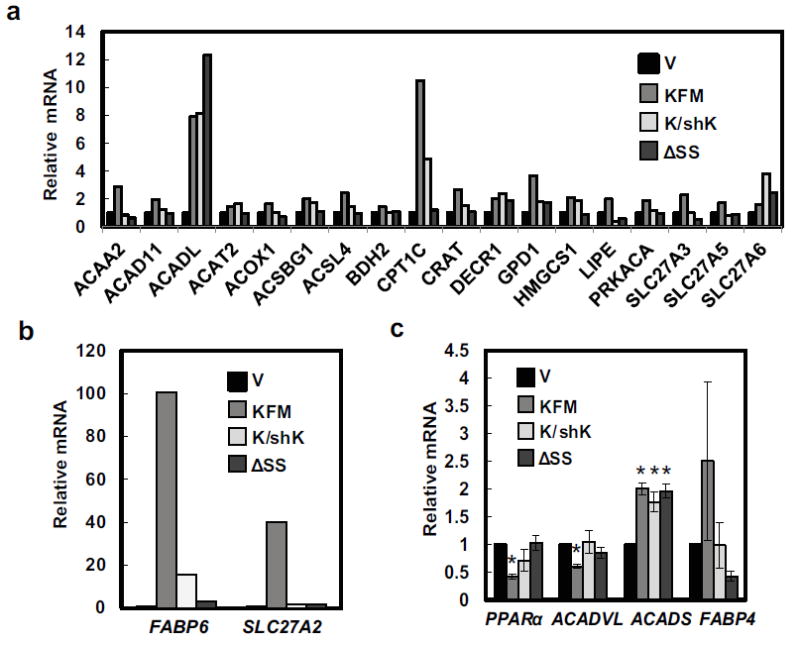
KISS1 upregulates beta-oxidation-related gene expression. A PCR Array was used to probe for various gene expressions in C1861.9Vector (V), C8161.9KFM59 (KFM), C8161.9KFM59/shKISS1 (K/shK), and C8161.9KFMΔSS6 (ΔSS) cells and selected upregulated genes are shown in (a & b). (c) mRNA expression of beta-oxidation related genes, PPARα, ACADVL, ACADS, and FABP4 in C1861.9Vector, C8161.9KFM59, C8161.9KFM59/shKISS1, and C8161.9KFMΔSS6 cells were quantified by qRT-PCR. N ≥ 3; error bars, SEM; * P , <0.05
KFM differentially regulated transcription of acyl-CoA dehydrogenase short chain (ACADS) and acyl-CoA dehydrogenase very long chain (ACADVL) (Figure 4c). Both ACADS and ACADVL are localized in mitochondrial inner matrix where they catalyze the first step of beta oxidation [39, 40]. It appears that KFM promotes catabolism of short chain fatty acids as revealed by increased ACADS gene expression; however, KFMΔSS also increased ACADS expression and knockdown of KFM did not reverse the expression. Regardless, the data suggests that KFM acts as a switch for energy generating pathways, in which KFM suppresses glycolysis and promotes beta-oxidation of fatty acids possibly to generate NAPDH and FADH2 in the citric acid cycle to promote mitochondrial oxidative phosphorylation.
KISS1 enriches key lipogenic enzymes
Lipid synthesis and metabolism pathways are mutually exclusive and reciprocally regulate each other [41]. The key function of early lipogenic enzymes is to generate building blocks (Acetyl-CoA and fatty acid Acyl-CoA) for both fatty acid synthesis and beta oxidation pathways. Acetyl-CoA carboxylase (ACC) genes are master regulators shifting Acetyl CoA toward either lipid synthesis or beta oxidation [42]. KFM increased expression of ACC at both mRNA and protein levels. Knock down of KFM reversed the increases. KFMΔSS did not affect ACC expression (Figures 5a+b). KFM did not increase ATP citrate lyase (ACLY), fatty acid synthase (FASN) expression (Figures 5a+b).
Figure 5.
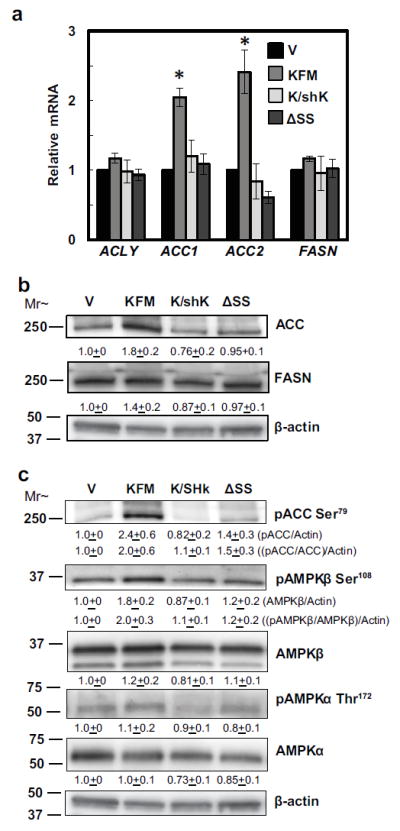
KISS1-mediated beta-oxidation induction is a result of AMPK phosphorylation of ACC at serine 79. (a) mRNA expression of early lipogenic genes, ACLY, ACC1, ACC2, and FASN in C1861.9Vector (V), C8161.9KFM59 (KFM), C8161.9KFM59/shKISS1 (K/shK), and C8161.9KFMΔSS6 (ΔSS) cells were quantified by qRT-PCR. (b & c) ACC, FASN, phos-ACC at serine 79, phos-AMPKβ at serine 108, AMPKβ, phos-AMPKα at threonine 172, and AMPKα proteins from C1861.9Vector, C8161.9KFM59, C8161.9KFM59/shKISS1, and C8161.9KFMΔSS6 whole cell lysate were quantified using immunoblot analysis with beta-actin as loading control. Densitometry of proteins was first normalized to beta-actin and then pACC:ACC ratios were determined. (a-c) N ≥ 3; error bars, SEM; * P ,< 0.05 (b & c) N≥ 3; mean ± SEM
Increased beta oxidation is a result of KISS1-induced persistent AMPK activation
To begin addressing the conundrum of why KFM expression upregulates ACC without affecting other early lipogenic enzymes, we hypothesized that KFM might promote beta-oxidation through AMPK. AMPK leads to beta oxidation as a means to generate additional ATP [43]. Increased AMP:ATP ratio induces an activating AMPK serine 108 (Ser108) autophosphorylation [44]. Additionally, AMPK is also phosphorylated at threonine 172 (Thr172) by liver kinase B1 (LKB1), resulting in activation of AMPK [45]. Activated AMPK then will phosphorylate ACC at serine 79 (Ser79) and possibly other serine sites, resulting in inhibition of ACC activity and increased beta-oxidation [46, 47]. KFM increased phosphorylation of ACC at Ser79. Upstream of ACC, KFM promotes AMPKβ phosphorylation at Ser108, but not Thr172 phosphorylation of the AMPKα subunit. Furthermore, the secretion signal sequence is required for KFM-promoted AMPK and ACC phosphorylation. Knockdown of KFM attenuated the AMPK-mediated phosphorylation of ACC (Figure 5c). Thus, KFM activates AMPK-mediated inhibitory phosphorylation of ACC, which may result in the observed increased beta-oxidation.
PGC1α is required for KISS1-induced transcription of beta-oxidation-related genes and ACC phosphorylation
We previously showed that KISS1 promotes mitochondrial biogenesis and respiration via PGC1α [31]. Loss of PGC1α ablates KISS1-mediated metastasis suppression in vitro [31]. PGC1α promotes fatty acid oxidation via the coactivators ERR and PPARδ. AMPK activates PGC1α by phosphorylation at threonine-177 and serine-538 resulting in enhanced mitochondrial respiration in skeletal muscle [48]. Thus, we assessed whether PGC1α is required for KISS1-mediated activation of beta-oxidation-related genes. Knocking down PGC1α attenuated KISS1 induction of multiple key beta-oxidation-related genes (Figures 6a+b). PGC1α knockdown also decreased Acc1, Acc2, and ACLY mRNA levels and partially restored PPARα mRNA levels (Figure 6c). At the protein level, PGC1α knockdown ablates the increases in phosphorylated and total ACC protein levels induced by KFM (Figure 6d). Altogether, the data suggest that PGC1α is indispensable for KFM-mediated broad upregulation of lipid beta oxidation genes.
Figure 6.
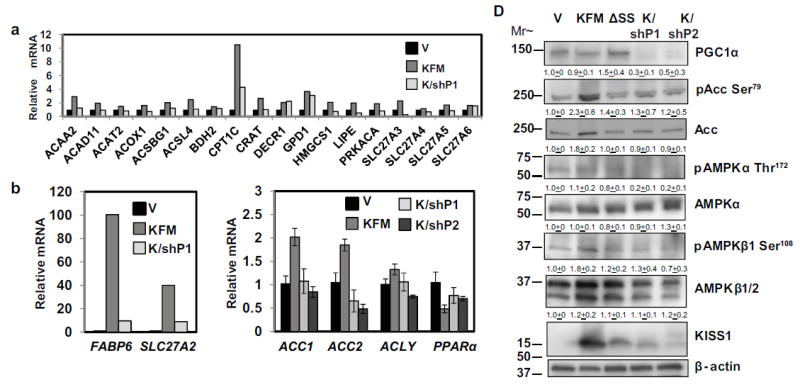
PGC-1α appears to be required for KISS1-mediated induction of beta-oxidation-related gene expression and AMPK-mediated phosphorylation of ACC. A PCR Array was used to probe expressions of various genes in C1861.9Vector (V), C8161.9KFM59 (KFM), and C8161.9KFM59/shPGC-1α (K/shP) cells. Selected upregulated genes are shown in (a & b). (c) mRNA expression of beta-oxidation-related genes, ACC1, ACC2, ACLY, and PPARα in C1861.9Vector, C8161.9KFM59, and C8161.9KFM59/shPGC-1α#1(K/shP1), and C8161.9KFM59/shPGC-1α#2 (K/shP2)cells were quantified by qRT-PCR. (d) PGC-1α, phos-ACC at serine 79, ACC, phos-AMPKβ at serine 108, AMPKβ, phos-AMPKα at threonine 172, AMPKα, and KISS1 proteins from C1861.9Vector, C8161.9KFM59, C8161.9KFMΔSS6(ΔSS), C8161.9KFM59/shPGC-1α#1 and C8161.9KFM59/shPGC-1α#2 whole cell lysates were quantified using immunoblot analysis with beta-actin as loading control. (a-c) N ≥ 3; error bars, SEM; * P , <0.05
PGC1α is required for KISS1-mediated metastasis suppression
Cumulatively our data, published here and elsewhere, suggests that PGC1α is a major molecular player in KISS1-mediated metastasis suppression. Our next question was whether PGC1α is required for the KISS1 metastasis suppressive effects in vivo. In both experimental (Figures 7a+b; C8161.9KFM vs C8161.9KFM/shP1; p = 0.0002 | C8161.9KFM vs C8161.9KFM/shP2; p = 0.0112) and spontaneous (Figures 7c+d; C8161.9Vector vs C8161.9KFM/shP1; p = 0.017 and C8161.9Vector vs C8161.9KFM/shP2; p = 0.001) metastasis assays, KISS1 suppression of metastasis was wholly or partly reversed when PGC1α was knocked down using shRNA. The proportion of mice developing metastases and the numbers of metastases were both affected and both parameters were shRNA construct-dependent. In this particular experiment, there was a greater than usual growth inhibition in the KFM-expressing population of C8161.9, which was also reversed by knockdown of PGC1α (Figure 7e).
Figure 7.
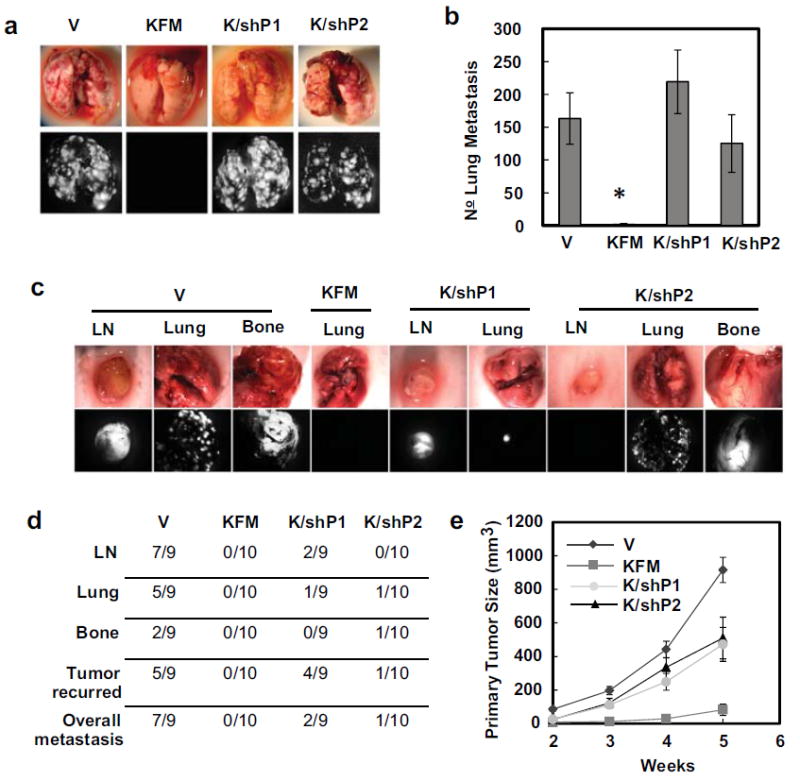
PGC-1α knockdown reverses KISS1-mediated metastasis suppression in vivo. (a) Representative images of lung metastasis after i.v. injection of C1861.9Vector (V), C8161.9KFM59 (KFM), C8161.9KFM59/shPGC-1α#1 (K/shP1), and C8161.9KFM59/shPGC-1α#2 (K/shP2) cells. The lung metastases were visualized macroscopically and using green fluorescent protein, lung metastases were quantified (b). N =10; error bars, SEM; * P , <0.05 (c) Representative images of spontaneous metastasis following orthotopic tumor growth of C1861.9Vector, C8161.9KFM59, C8161.9KFM59/shPGC-1α#1 and C8161.9KFM59/shPGC-1α#2 cells and metastases were illuminated using GFP. (d) Incidence of spontaneous metastases to distant organs. (e)Orthotopic tumor volume was measured at 2, 3, 4, and 5 weeks. n = 9-10; error bars, SEM; * P , <0.05
Discussion
Activation of glycolysis in cells growing under normoxic conditions is a hallmark of many cancers and it is believed to possibly confer a survival advantage. However, the underlying mechanisms behind the Warburg effect are not fully understood, especially as it regards metastasis. When we previously reported that KISS1 reversed the Warburg effect by enhancing mitochondrial biogenesis and PGC1α levels [31], a possible link between metabolism and metastatic potential was inferred. This study was designed to further dissect the effects of KISS1 on glycolysis and mitochondrial beta-oxidation in metastatic melanoma cells.
Most neoplastic cells can utilize either aerobic glycolysis or mitochondrial respiration; however, the types of metabolism correlate with KISS1 expression and whether the cells metastasize or not [31]. Aerobic glycolysis is not required for tumors to grow, since KISS1-expressing cells still form orthotopic tumors. However, growth rates are affected by metabolism. Importantly, the data presented here and elsewhere [49-51], suggest that metabolic shifts may contribute to metastasis.
KISS1 re-expression decreased mitochondrial hexokinase II (HKII). HKII plays a dual role in cancer cells by upregulating aerobic glycolysis and by preventing mitochondria-induced apoptosis [38, 52]. Mitochondrion-bound HKII prevents apoptosis by both preventing mitochondrial permeability transition and inhibiting Bax-mediated cytochrome c release [52]. These findings may partly explain reports of KISS1-induced apoptosis [53, 54], suggesting that reduction of HKII may increase susceptibility to apoptotic stimuli. Correspondingly, mitochondrion-bound HKII interacts with VDAC (Voltage-dependent anionic channel), suppressing respiration and increasing aerobic glycolysis [55]. The latter is consistent with our observations that KISS1 upregulates mitochondrial beta oxidation and oxidative phosphorylation.
Beta oxidation and lipid synthesis are mutually exclusive in cells. Sustained proliferation requires continuous lipid synthesis to meet requirements for membrane generation. Tumor cells reactivate de novo lipid synthesis by upregulating expression of key enzymes, including FASN, ACLY, and ACC [56, 57]. KISS1 increased ACC protein, but also simultaneously inhibit ACC activity. ACC is the rate-limiting enzyme in de novo fatty acid synthesis and is overexpressed in breast [58], prostate [59], skin [60], and liver [61] cancers. Through inhibition of ACC, KISS1 appears to suppress simultaneously de novo lipid synthesis and activate beta-oxidation, potentially to prevent membrane biogenesis and cell proliferation.
KISS1 increases mitochondrial beta-oxidation of exogenous fatty acids. Beta-oxidation and fatty acid catabolism are emerging therapeutic targets in multiple cancer types [56, 62-64] because there are higher yields of ATP from fatty acids compared to carbohydrate catabolism [65]. As a result, both are speculated to meet the high-energy requirements of tumorigenesis and metastasis [56, 65]. How might KISS1 activate beta oxidation and still suppress metastasis? For proliferating tumor cells that rely on high rates of glycolysis, it is well accepted that aerobic glycolysis generates ATP, produces reducing equivalents in the form of NADPH, and support biosynthesis, most notably in de novo fatty acid synthesis. Recent empirical studies indicate that the amount of ATP required for tumor cell growth may be less than expected. In fact, tumor cells may never reach limiting values of ATP; demand could be meet already by mitochondrial respiration. High rates of glycolysis and lactate production would serve to regenerate NAD+ that, on one hand, allows for glycolysis to continue and, on the other hand, suppresses pyruvate dehydrogenase (PDH) and mitochondrial respiration. These observations indicate that proliferating tumor cells need both high glycolysis rate and a basal level of mitochondrial respiration. From a therapeutic perspective, and both glycose and mitochondrial respiration need to be eliminated to kill tumor cells.
Our studies revealed that KISS1 inhibits glycolysis and carbon sources for endogenous fatty acid oxidation (FAO). We speculated that KISS1 increases mitochondrial respiration as a metabolic compensation for defective glycolysis so that tumor cells remain viable but do not proliferate (consistent with our published observations that KISS1-expressing cells are dormant after seeding ectopic tissues [27]). KISS1-inhibited glycolysis and activation of FAO would limit the availability of some macromolecules and fatty acid pools for membrane incorporation, storage and/or signaling. As a result, tumor cells would be less likely to actively or successfully proliferate.
AMPK is involved in lipid metabolism by inhibiting ACC, with a corresponding inhibition of fatty acid synthesis and activation of beta-oxidation [43]. KISS1, by an as-yet undetermined mechanism, appears to regulate AMPK activity via control of Ser108 phosphorylation of the AMPKβ subunit. Additionally, AMPK inhibits the mTOR pathway which is associated with diminished protein synthesis, reduced cell proliferation, and activation of autophagic pathways [66, 67]. KISS1-induced AMPK activation may therefore be an explanation for previously reported KISS1-induced autophagy [31].
Compiling the results reported here and elsewhere, KISS1 appears to be integral to the coordination of lipid and carbohydrate metabolic pathways. In the presence of KISS1, AMPK is constitutively active, resulting in inhibition of ACC and activation of beta oxidation of fatty acids. Since KISS1 has no known kinase nor phosphorylase activities, how it regulates AMPK is still only a matter of speculation. Nonetheless, since AMPK activation has been reported to suppress melanoma invasion and metastasis development [68], the findings support the notion that metabolism may be important for controlling metastasis, the most lethal attribute of cancer cells.
Moreover, the results also suggest that PGC1α is a key mediator for KISS1-induced AMPK activation and subsequent activation of beta-oxidation. PGC1α had previously been reported to be activated by AMPK via phosphorylation resulting in enhanced PGC1α expression and mitochondrial biogenesis [48, 69, 70]. Our lab reported that KISS1 promotes PGC1α-mediated mitochondrial biogenesis [31]. The results showed that AMPK activation by KISS1 was possibly responsible for the subsequent upregulation of PGC1α activity resulting in enhanced beta-oxidation. Additionally, PGC1α has been independently verified to suppress melanoma metastasis [71] and, in line with that study, our data clearly revealed that PGC1α blocks disseminated tumor cells outgrowth in lung parenchyma. PGC1α appears to be indispensable for KISS1 metastasis suppression. So far, we do not know which of the myriad metabolic aspects regulated by PGC1α (i.e. mitochondrial biogenesis, lipogenesis, β-oxidation or aerobic glycolysis) might be responsible for the KISS1 metastasis suppressor activity.
Collectively, the data presented here show the outlines a tapestry connecting cellular metabolism and cancer metastasis. A KISS1-AMPK- PGC1α axis may involve sequential components as well as synergistic ones, each affecting different steps of the metastatic cascade, including growth at the primary or secondary sites.
Figure 8.
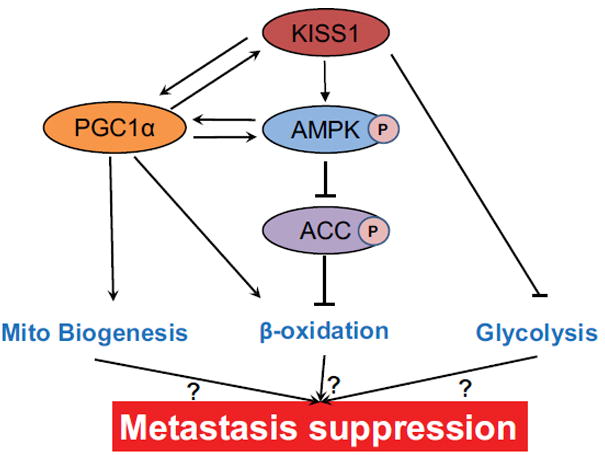
A proposed simplified model depicting how KISS1 suppresses metastasis via modulation of tumor metabolism. KISS1 boosts mitochondrial biogenesis via modulation of PGC1α expression. KISS1 also increases β-oxidation by activating AMPK and PGC1α. AMPK activation leads to inhibitory phosphorylation of ACC, thus preventing malonyl-CoA-mediated inhibition of β-oxidation. PGC1α cooperates with PPARα in transcriptional control of nuclear genes encoding β-oxidation. AMPK activates PGC1α and vice versa. KISS1 inhibits aerobic glycolysis by modulating genes expression involved in glycolysis.
Key Messages.
KISS1 alters fatty acid metabolism.
There may be connections between metastasis and metabolism.
PGC1alpha appears to be downstream mediator of KISS1 metastasis suppression.
Acknowledgments
This study was supported by grants to DRW from Susan G. Komen for the Cure [SAC11037], the National Foundation for Cancer Research-Center for Metastasis Research, U.S. National Cancer Institute RO1-CA134981, and using partial support from RO1-CA87728 and P30-CA168524. D.R. Welch is the Hall Family Professor of Molecular Medicine and a Kansas Bioscience Authority Eminent Scholar. This study also was supported by fellowship to SM from the Kansas INBRE, P20 GM103418.
Footnotes
Conflict of interest:
The authors declare no conflicts of interest.
Reference List
- 1.Warburg O. On the origin of cancer cells. Science. 1956;123:309–14. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- 2.Warburg O, Wind F, Negelein E. The metabolism of tumors in the body. J Gen Physiol. 1927;8:519–30. doi: 10.1085/jgp.8.6.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Crabtree HG. The carbohydrate metabolism of certain pathological overgrowths. Biochem J. 1928;22:1289–98. doi: 10.1042/bj0221289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Crabtree HG. Observations on the carbohydrate metabolism of tumours. Biochem J. 1929;23:536–45. doi: 10.1042/bj0230536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gatenby RA, Gillies RJ. Why do cancers have high aerobic glycolysis? Nature Rev Cancer. 2004;4:891–9. doi: 10.1038/nrc1478. [DOI] [PubMed] [Google Scholar]
- 6.Kaelin WG, Thompson CB. Q&A: Cancer: Clues from cell metabolism. Nature. 2010;465:562–4. doi: 10.1038/465562a. [DOI] [PubMed] [Google Scholar]
- 7.Cairns RA, Harris IS, Mak TW. Regulation of cancer cell metabolism. Nature Rev Cancer. 2011;11:85–95. doi: 10.1038/nrc2981. [DOI] [PubMed] [Google Scholar]
- 8.Dang CV. c-Myc target genes involved in cell growth, apoptosis, and metabolism. Mol Cell Biol. 1999;19:1–11. doi: 10.1128/mcb.19.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Semenza GL. Regulation of cancer cell metabolism by hypoxia-inducible factor 1. Semin Cancer Biol. 2009;19:12–6. doi: 10.1016/j.semcancer.2008.11.009. [DOI] [PubMed] [Google Scholar]
- 10.Lopez-Rios F, Sanchez-Arago M, García-García E, Ortega AD, Berrendero JR, Pozo-Rodríguez F, Lopez-Encuentra A, Ballestín C, Cuezva JM. Loss of the mitochondrial bioenergetic capacity underlies the glucose avidity of carcinomas. Cancer Res. 2007;67:9013–7. doi: 10.1158/0008-5472.CAN-07-1678. [DOI] [PubMed] [Google Scholar]
- 11.Chiche J, Rouleau M, Gounon P, Brahimi-Horn MC, Pouyssegur J, Mazure NM. Hypoxic enlarged mitochondria protect cancer cells from apoptotic stimuli. J Cell Physiol. 2010;222:648–57. doi: 10.1002/jcp.21984. [DOI] [PubMed] [Google Scholar]
- 12.Thompson CB. Metabolic enzymes as oncogenes or tumor suppressors. N Engl J Med. 2009;360:813–5. doi: 10.1056/NEJMe0810213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ishikawa K, Takenaga K, Akimoto M, Koshikawa N, Yamaguchi A, Imanishi H, Nakada K, Honma Y, Hayashi J. ROS-generating mitochondrial DNA mutations can regulate tumor cell metastasis. Science. 2008;320:661–4. doi: 10.1126/science.1156906. [DOI] [PubMed] [Google Scholar]
- 14.Koshikawa N, Hayashi J, Nakagawara A, Takenaga K. Reactive oxygen species-generating mitochondrial DNA mutation up-regulates hypoxia-inducible factor-1alpha gene transcription via phosphatidylinositol 3-kinase-Akt/protein kinase C/histone deacetylase pathway. J Biol Chem. 2009;284:33185–94. doi: 10.1074/jbc.M109.054221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ishikawa K, Hashizume O, Koshikawa N, Fukuda S, Nakada K, Takenaga K, Hayashi J. Enhanced glycolysis induced by mtDNA mutations does not regulate metastasis. FEBS Lett. 2008;582:3525–30. doi: 10.1016/j.febslet.2008.09.024. [DOI] [PubMed] [Google Scholar]
- 16.Pantel K, Brakenhoff RH. Dissecting the metastatic cascade. Nature Rev Cancer. 2004;4:448–56. doi: 10.1038/nrc1370. [DOI] [PubMed] [Google Scholar]
- 17.Francia G, Cruz-Munoz W, Man S, Xu P, Kerbel RS. Mouse models of advanced spontaneous metastasis for experimental therapeutics. Nature Rev Cancer. 2011;11:135–41. doi: 10.1038/nrc3001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Khanna C, Hunter K. Modeling metastasis in vivo. Carcinogenesis. 2005;26:513–23. doi: 10.1093/carcin/bgh261. [DOI] [PubMed] [Google Scholar]
- 19.Eccles SA, Welch DR. Metastasis: recent discoveries and novel treatment strategies. Lancet. 2007;369:1742–57. doi: 10.1016/S0140-6736(07)60781-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Welch DR. American Association for Cancer Research, ed AACR Education Book 2006. Philadelphia: AACR; 2006. Defining a cancer metastasis; pp. 111–115. [Google Scholar]
- 21.Payen VL, Porporato PE, Baselet B, Sonveaux P. Metabolic changes associated with tumor metastasis, part 1: tumor pH, glycolysis and the pentose phosphate pathway. Cell Mol Life Sci. 2016;73:1333–48. doi: 10.1007/s00018-015-2098-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Porporato PE, Payen VL, Baselet B, Sonveaux P. Metabolic changes associated with tumor metastasis, part 2: Mitochondria, lipid and amino acid metabolism. Cell Mol Life Sci. 2016;73:1349–63. doi: 10.1007/s00018-015-2100-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee J-H, Miele ME, Hicks DJ, Phillips KK, Trent JM, Weissman BE, Welch DR. KiSS-1, a novel human malignant melanoma metastasis-suppressor gene. J Natl Cancer Inst. 1996;88:1731–7. doi: 10.1093/jnci/88.23.1731. [DOI] [PubMed] [Google Scholar]
- 24.Lee J-H, Welch DR. Suppression of metastasis in human breast carcinoma MDA-MB-435 cells after transfection with the metastasis suppressor gene, KiSS-1. Cancer Res. 1997;57:2384–7. [PubMed] [Google Scholar]
- 25.Ohtaki T, Shintani Y, Honda S, Matsumoto H, Hori A, Kanehashi K, Torao Y, Kumano S, Takatsu Y, Matsuda Y, et al. Metastasis suppressor gene KiSS1 encodes peptide ligand of a G-protein-coupled receptor. Nature. 2001;411:613–7. doi: 10.1038/35079135. [DOI] [PubMed] [Google Scholar]
- 26.Harihar S, Pounds KM, Iwakuma T, Seidah NG, Welch DR. Furin is the major proprotein convertase required for KISS1-to-Kisspeptin processing. PLoS One. 2014;9:e84958. doi: 10.1371/journal.pone.0084958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nash KT, Phadke PA, Navenot J-M, Hurst DR, Accavitti-Loper MA, Sztul E, Vaidya KS, Frost AR, Kappes JC, Peiper SC, et al. KISS1 metastasis suppressor secretion, multiple organ metastasis suppression, and maintenance of tumor dormancy. J Natl Cancer Inst. 2007;99:309–21. doi: 10.1093/jnci/djk053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Muir AI, Chamberlain L, Elshourbagy NA, Michalovich D, Moore DJ, Calamari A, Szekeres PG, Sarau HM, Chambers JK, Murdock P, et al. AXOR12: A novel human G protein-coupled receptor, activated by the peptide KiSS-1. J Biol Chem. 2001;276:28969–75. doi: 10.1074/jbc.M102743200. [DOI] [PubMed] [Google Scholar]
- 29.Kotani M, Detheux M, Vandenbogaerde A, Communi D, Vanderwinden JM, Le Poul E, Brezillon S, Tyldesley R, Suarez-Huerta N, Vandeput F, et al. The metastasis suppressor gene KiSS-1 encodes kisspeptins, the natural ligands of the orphan G protein-coupled receptor GPR54. J Biol Chem. 2001;276:34631–6. doi: 10.1074/jbc.M104847200. [DOI] [PubMed] [Google Scholar]
- 30.Beck BH, Welch DR. The KISS1 metastasis suppressor: a good night kiss for disseminated cancer cells. Eur J Cancer. 2010;46:1283–9. doi: 10.1016/j.ejca.2010.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu W, Beck BH, Vaidya KS, Nash KT, Feeley KP, Ballinger SW, Pounds KM, Denning WL, Diers AR, Landar A, et al. Metastasis suppressor KISS1 seems to reverse the Warburg effect by enhancing mitochondrial biogenesis. Cancer Res. 2014;74:954–63. doi: 10.1158/0008-5472.CAN-13-1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rogers GW, Nadanaciva S, Swiss R, Divakaruni AS, Will Y. Assessment of fatty acid beta oxidation in cells and isolated mitochondria. Curr Protoc Toxicol. 2014;60:25–19. doi: 10.1002/0471140856.tx2503s60. [DOI] [PubMed] [Google Scholar]
- 33.TeSlaa T, Teitell MA. Techniques to monitor glycolysis. Methods Enzymol. 2014;542:91–114. doi: 10.1016/B978-0-12-416618-9.00005-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wilkins HM, Koppel S, Carl SM, Ramanujan S, Weidling I, Michaelis ML, Michaelis EK, Swerdlow RH. Oxaloacetate enhances neuronal cell bioenergetic fluxes and infrastructure. J Neurochem. 2016;137:76–87. doi: 10.1111/jnc.13545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science. 2009;324:1029–33. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cheng G, Zielonka J, Dranka BP, McAllister D, Mackinnon AC, Jr, Joseph J, Kalyanaraman B. Mitochondria-targeted drugs synergize with 2-deoxyglucose to trigger breast cancer cell death. Cancer Res. 2012;72:2634–44. doi: 10.1158/0008-5472.CAN-11-3928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yang W, Zheng Y, Xia Y, Ji H, Chen X, Guo F, Lyssiotis CA, Aldape K, Cantley LC, Lu Z. ERK1/2-dependent phosphorylation and nuclear translocation of PKM2 promotes the Warburg effect. Nature Cell Biol. 2012;14:1295–304. doi: 10.1038/ncb2629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mathupala SP, Ko YH, Pedersen PL. Hexokinase II: Cancer’s double-edged sword acting as both facilitator and gatekeeper of malignancy when bound to mitochondria. Oncogene. 2006;25:4777–86. doi: 10.1038/sj.onc.1209603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bartlett K, Eaton S. Mitochondrial beta-oxidation. Eur J Biochem. 2004;271:462–9. doi: 10.1046/j.1432-1033.2003.03947.x. [DOI] [PubMed] [Google Scholar]
- 40.Houten SM, Wanders RJ. A general introduction to the biochemistry of mitochondrial fatty acid beta-oxidation. J Inherit Metab Dis. 2010;33:469–77. doi: 10.1007/s10545-010-9061-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Santos CR, Schulze A. Lipid metabolism in cancer. FEBS J. 2012;279:2610–23. doi: 10.1111/j.1742-4658.2012.08644.x. [DOI] [PubMed] [Google Scholar]
- 42.Wang C, Rajput S, Watabe K, Liao DF, Cao D. Acetyl-CoA carboxylase-a as a novel target for cancer therapy. Front Biosci (Schol Ed) 2010;2:515–26. doi: 10.2741/s82. [DOI] [PubMed] [Google Scholar]
- 43.Hardie DG, Carling D. The AMP-activated protein kinase--fuel gauge of the mammalian cell? Eur J Biochem. 1997;246:259–73. doi: 10.1111/j.1432-1033.1997.00259.x. [DOI] [PubMed] [Google Scholar]
- 44.Warden SM, Richardson C, O’Donnell J, Jr, Stapleton D, Kemp BE, Witters LA. Post-translational modifications of the beta-1 subunit of AMP-activated protein kinase affect enzyme activity and cellular localization. Biochem J. 2001;354:275–83. doi: 10.1042/0264-6021:3540275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shaw RJ, Kosmatka M, Bardeesy N, Hurley RL, Witters LA, DePinho RA, Cantley LC. The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc Natl Acad Sci U S A. 2004;101:3329–35. doi: 10.1073/pnas.0308061100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Winder WW, Hardie DG. Inactivation of acetyl-CoA carboxylase and activation of AMP-activated protein kinase in muscle during exercise. Am J Physiol. 1996;270:E299–E304. doi: 10.1152/ajpendo.1996.270.2.E299. [DOI] [PubMed] [Google Scholar]
- 47.Hardie DG, Corton J, Ching YP, Davies SP, Hawley S. Regulation of lipid metabolism by the AMP-activated protein kinase. Biochem Soc Trans. 1997;25:1229–31. doi: 10.1042/bst0251229. [DOI] [PubMed] [Google Scholar]
- 48.Jager S, Handschin C, St-Pierre J, Spiegelman BM. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc Natl Acad Sci U S A. 2007;104:12017–22. doi: 10.1073/pnas.0705070104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tennant DA, Duran RV, Boulahbel H, Gottlieb E. Metabolic transformation in cancer. Carcinogenesis. 2009;30:1269–80. doi: 10.1093/carcin/bgp070. [DOI] [PubMed] [Google Scholar]
- 50.Simonnet H, Alazard N, Pfeiffer K, Gallou C, Beroud C, Demont J, Bouvier R, Schagger H, Godinot C. Low mitochondrial respiratory chain content correlates with tumor aggressiveness in renal cell carcinoma. Carcinogenesis. 2002;23:759–68. doi: 10.1093/carcin/23.5.759. [DOI] [PubMed] [Google Scholar]
- 51.Pavlides S, Vera I, Gandara R, Sneddon S, Pestell RG, Mercier I, Martinez-Outschoorn UE, Whitaker-Menezes D, Howell A, Sotgia F, et al. Warburg meets autophagy: cancer-associated fibroblasts accelerate tumor growth and metastasis via oxidative stress, mitophagy, and aerobic glycolysis. Antioxid Redox Signal. 2012;16:1264–84. doi: 10.1089/ars.2011.4243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pastorino JG, Shulga N, Hoek JB. Mitochondrial binding of hexokinase II inhibits Bax-induced cytochrome c release and apoptosis. J Biol Chem. 2002;277:7610–8. doi: 10.1074/jbc.M109950200. [DOI] [PubMed] [Google Scholar]
- 53.Navenot JM, Fujii N, Peiper SC. KISS1 metastasis suppressor gene product induces suppression of tyrosine kinase receptor signaling to Akt, tumor necrosis factor family ligand expression, and apoptosis. Molec Pharmacol. 2009;75:1074–83. doi: 10.1124/mol.108.054270. [DOI] [PubMed] [Google Scholar]
- 54.Navenot JM, Fujii N, Peiper SC. Activation of Rho and Rho-associated kinase by GPR54 and KiSS1 metastasis suppressor gene product induces changes of cell morphology and contributes to apoptosis. Molec Pharmacol. 2009;75:1300–6. doi: 10.1124/mol.109.055095. [DOI] [PubMed] [Google Scholar]
- 55.Lemasters JJ, Holmuhamedov E. Voltage-dependent anion channel (VDAC) as mitochondrial governator--thinking outside the box. Biochim Biophys Acta. 2006;1762:181–90. doi: 10.1016/j.bbadis.2005.10.006. [DOI] [PubMed] [Google Scholar]
- 56.Zhang F, Du G. Dysregulated lipid metabolism in cancer. World J Biol Chem. 2012;3:167–74. doi: 10.4331/wjbc.v3.i8.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Currie E, Schulze A, Zechner R, Walther TC, Farese RV., Jr Cellular fatty acid metabolism and cancer. Cell Metab. 2013;18:153–61. doi: 10.1016/j.cmet.2013.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Milgraum LZ, Witters LA, Pasternack GR, Kuhajda FP. Enzymes of the fatty acid synthesis pathway are highly expressed in in situ breast carcinoma. Clin Cancer Res. 1997;3:2115–20. [PubMed] [Google Scholar]
- 59.Swinnen JV, Vanderhoydonc F, Elgamal AA, Eelen M, Vercaeren I, Joniau S, Van PH, Baert L, Goossens K, Heyns W, et al. Selective activation of the fatty acid synthesis pathway in human prostate cancer. Int J Cancer. 2000;88:176–9. doi: 10.1002/1097-0215(20001015)88:2<176::aid-ijc5>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 60.Li W, Zhang C, Du H, Huang V, Sun B, Harris JP, Richardson Q, Shen X, Jin R, Li G, et al. Withaferin A suppresses the up-regulation of acetyl-coA carboxylase 1 and skin tumor formation in a skin carcinogenesis mouse model. Mol Carcinog. 2016;55:1739–46. doi: 10.1002/mc.22423. [DOI] [PubMed] [Google Scholar]
- 61.Yahagi N, Shimano H, Hasegawa K, Ohashi K, Matsuzaka T, Najima Y, Sekiya M, Tomita S, Okazaki H, Tamura Y, et al. Co-ordinate activation of lipogenic enzymes in hepatocellular carcinoma. Eur J Cancer. 2005;41:1316–22. doi: 10.1016/j.ejca.2004.12.037. [DOI] [PubMed] [Google Scholar]
- 62.Wu X, Daniels G, Lee P, Monaco ME. Lipid metabolism in prostate cancer. Am J Clin Exp Urol. 2014;2:111–20. [PMC free article] [PubMed] [Google Scholar]
- 63.Blum R, Kloog Y. Metabolism addiction in pancreatic cancer. Cell Death Dis. 2014;5:e1065. doi: 10.1038/cddis.2014.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rodrigues MF, Obre E, de Melo FH, Santos GC, Jr, Galina A, Jasiulionis MG, Rossignol R, Rumjanek FD, Amoedo ND. Enhanced OXPHOS, glutaminolysis and beta-oxidation constitute the metastatic phenotype of melanoma cells. Biochem J. 2016;473:703–15. doi: 10.1042/BJ20150645. [DOI] [PubMed] [Google Scholar]
- 65.Carracedo A, Cantley LC, Pandolfi PP. Cancer metabolism: fatty acid oxidation in the limelight. Nature Rev Cancer. 2013;13:227–32. doi: 10.1038/nrc3483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Shackelford DB, Shaw RJ. The LKB1-AMPK pathway: metabolism and growth control in tumour suppression. Nature Rev Cancer. 2009;9:563–75. doi: 10.1038/nrc2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13:132–41. doi: 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cerezo M, Tichet M, Abbe P, Ohanna M, Lehraiki A, Rouaud F, Allegra M, Giacchero D, Bahadoran P, Bertolotto C, et al. Metformin blocks melanoma invasion and metastasis development in AMPK/p53-dependent manner. Mol Cancer Ther. 2013;12:1605–15. doi: 10.1158/1535-7163.MCT-12-1226-T. [DOI] [PubMed] [Google Scholar]
- 69.Zong H, Ren JM, Young LH, Pypaert M, Mu J, Birnbaum MJ, Shulman GI. AMP kinase is required for mitochondrial biogenesis in skeletal muscle in response to chronic energy deprivation. Proc Natl Acad Sci U S A. 2002;99:15983–7. doi: 10.1073/pnas.252625599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rohas LM, St-Pierre J, Uldry M, Jager S, Handschin C, Spiegelman BM. A fundamental system of cellular energy homeostasis regulated by PGC-1alpha. Proc Natl Acad Sci U S A. 2007;104:7933–8. doi: 10.1073/pnas.0702683104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Luo C, Lim JH, Lee Y, Granter SR, THOMAS A, Vazquez F, Widlund HR, Puigserver P. A PGC1alpha-mediated transcriptional axis suppresses melanoma metastasis. Nature. 2016;537:422–6. doi: 10.1038/nature19347. [DOI] [PMC free article] [PubMed] [Google Scholar]