

Abstract
Ovarian cancer is the most deadly gynecological cancer and unlike most other neoplasms, survival rates for ovarian cancer have not significantly improved in recent decades. We show that RAD6, an ubiquitin conjugating enzyme is significantly overexpressed in ovarian tumors and its expression increases in response to carboplatin chemotherapy. RAD6 expression correlated strongly with acquired chemoresistance and malignant behavior of OC cells, expression of stem cell genes and poor prognosis of OC patients suggesting an important role for RAD6 in ovarian tumor progression. Upregulated RAD6 enhances DNA damage tolerance and repair efficiency of OC cells and promotes their survival. Increased RAD6 levels cause H2B ubiquitination-mediated epigenetic changes that stimulate transcription of stem cell genes, including ALDH1A1 and SOX2, leading to a cancer stem cell phenotype, which is implicated in disease recurrence and metastasis. Downregulation of RAD6 or its inhibition using a small molecule inhibitor attenuated DNA repair signaling and expression of CSC markers and sensitized chemoresistant OC cells to carboplatin. Together, these results suggest that RAD6 could be a therapeutic target to prevent and treat acquired chemoresistance and disease recurrence in OC and enhance the efficacy of standard chemotherapy.
Keywords: RAD6, ovarian cancer, DNA repair, cancer stem cells, chemoresistance
Introduction
Ovarian cancer (OC) is often diagnosed at advanced stages with widespread intra-abdominal disease or distant metastasis due to its insidious development. Although most OC patients respond well to initial surgical debulking and platinum-based chemotherapy, a significant percent of patients present with recurrent disease (reported rates ranging from 65–80%)1, and these recurrent tumors are frequently resistant to front line chemotherapeutic drugs thus making it the most deadly gynecological cancer2,3. It is believed that recurrent tumors are largely due to a small population of cancer cells that dedifferentiated as cancer stem cells (CSC) or tumor initiating cells. CSCs have properties, such as quiescence, enhanced DNA damage tolerance, increased multidrug efflux pumps, and anti-apoptotic signaling to evade treatment4–6. Moreover, CSCs are capable of proliferating, differentiating back into tumor cells and reestablishing tumors once treatment is concluded4. Thus, signaling mechanisms that promote acquired chemoresistance and CSC phenotype have been investigated as therapeutic targets to prevent and treat disease recurrence.
RAD6 is an ubiquitin conjugating enzyme that was originally identified as a DNA repair protein in yeast and is highly conserved in all eukaryotes7. Humans have two RAD6 genes (RAD6A and RAD6B also known as UBE2A and UBE2B) that have many overlapping functions, and both complement mutant RAD6 of Saccharomyces cerevisiae in DNA repair8. RAD6, in association with RAD18, regulates mutagenic DNA damage tolerance (DDT; translesion synthesis or TLS) and Fanconi anemia (FA) DNA repair pathways in response to various genomic insults, including chemo and radiation therapy9. Additionally, RAD6 regulates gene transcription in association with RNF20/40 ubiquitin ligase by histone 2B (H2B) ubiquitination-mediated chromatin modifications10–12. RAD6 is overexpressed in melanoma and breast cancer where it correlates with tumor development, progression and metastasis13–15. Our recent studies showed increased expression of RAD6 in ovarian tumors and its levels correlated with progressive disease16. In OC cell lines, RAD6 expression promoted development of a stem cell-like phenotype and resistance to carboplatin16. In this report we show that RAD6 promotes acquired chemoresistance in OC by stimulating monoubiquitination of FANCD2 and PCNA, proteins that are important for platinum drugs-induced DNA crosslink repair and DDT mechanisms, respectively17–21. Similarly, RAD6 is upregulated in response to chemotherapy and significantly correlated with expression of OC stem cell signaling genes ALDH1A1 and SOX2 and poor prognosis of OC patients. Additionally, RAD6 downregulation or inhibition using a small molecule inhibitor attenuated DNA repair signaling, expression of CSC markers and sensitized chemoresistant OC cells to carboplatin. Collectively, these results demonstrate that RAD6 could be a therapeutic target to prevent and treat acquired chemoresistance and disease recurrence in OC and enhance the efficacy of standard chemotherapeutic drugs in OC patients.
Results
RAD6 promotes CSC gene expression and is necessary for proper DNA damage response following carboplatin treatment
We previously showed upregulation of both RAD6A and RAD6B genes and RAD6 protein levels in ovarian tumors and tumor cell lines compared to normal ovarian tissues and cells16. In isogenic chemoresistant and sensitive OC cells, RAD6 levels correlated with chemoresistance and ability to form spheroids (a stemness trait). Therefore, we hypothesized that upregulated RAD6 promotes survival of ovarian tumor cells through increased DNA repair and acquisition of a cancer stem cell (CSC) phenotype. To examine whether RAD6 status affects expression of stem cell genes and characteristics, OV90 cells were transfected with control or RAD6-specific siRNAs, treated with carboplatin and CSC and DNA damage response (DDR) protein levels were analyzed. Since both the proteins known to have overlapping functions in DNA repair, we transfected with siRNAs that target both the RAD6 genes (siRAD6A and siRAD6B) and designated as siRAD6. Consistent with previous studies, carboplatin treatment increased expression and monoubiquitination of DDR proteins FANCD2, PCNA, RAD18 and γH2AX (Fig 1A). However, RAD6 downregulation attenuated monoubiquitination of these proteins, both basally and in carboplatin-treated cells (Fig 1A). Carboplatin treatment increased levels of pro-stemness transcription factor β-catenin as well as ALDH1A1 and SOX2, and RAD6 depletion significantly diminished expression of these proteins, both basally and in response to carboplatin (Fig 1B,C). RAD6 has previously been shown to promote stability of β-catenin14 and work with RNF20/40 to regulate gene transcription by ubiquitination of H2B10,11,22. Consistent with these findings, the levels of ubiquitinated H2B increased along with RAD6 in carboplatin-treated cells and decreased in RAD6-downregulated cells (Fig 1B and C). The decrease in expression of stemness factors in RAD6-depleted cells was accompanied by decreased anchorage-independent growth, as measured by number of stem cell spheroids (Fig 1D). To rule out any off-target effects of siRNAs we evaluated two siRNAs targeting RAD6B and one siRNA targeting RAD6A and all caused decrease in ALDHI1A1 and SOX2 protein levels (Fig 1E). These results suggest that upregulated RAD6 activates DDR by monoubiquitination of FANCD2, PCNA and γH2AX and regulates stability of β-catenin and expression of CSC genes by ubiquitination of H2B. Combined this data suggest RAD6-driven increases in DNA repair and CSC signaling promotes chemoresistance and stemness phenotype, two factors that contribute to treatment relapse and disease recurrence in ovarian cancer patients1,2.
Figure 1.
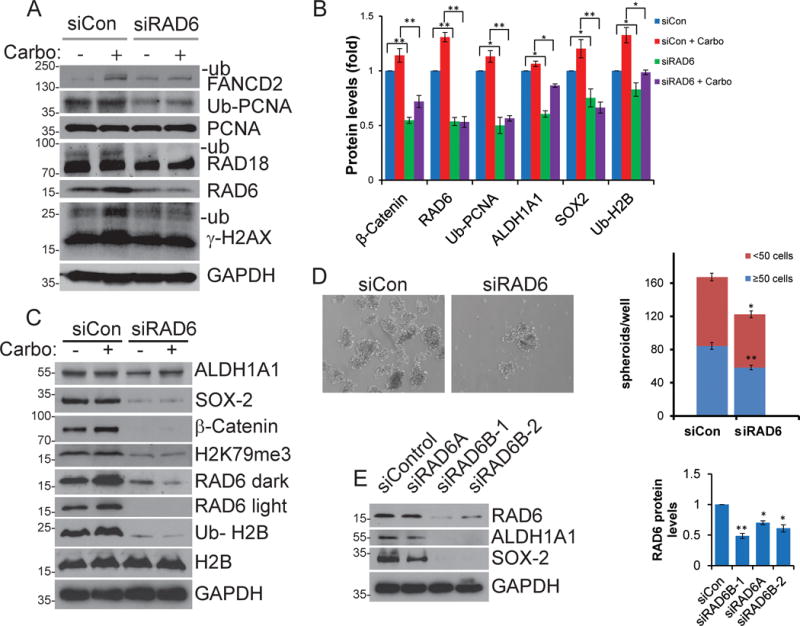
RAD6 promotes expression of CSC proteins and acquisition of stemness phenotype and is important for efficient DDR. Control and siRAD6 transfected OV90 cells were treated with 20 μM carboplatin and DDR and CSC proteins were assessed by Western blot. RAD6 knockdown impairs carboplatin induced DDR (A & B) and decreases pro-transcription histone modifications H2B-Ub and H3K79me3 and expression of CSC signaling proteins (B & C). The results presented are representative blots and the average values obtained from three independent experiments with each band adjusted to the corresponding GAPDH control. (D) Consistent with the expression studies, knocking down RAD6 impaired the ability of OV90 cells to form stem cell spheres by anchorage-independent growth, as both number and size of number of spheroids decreased. Left panel shows 1 view field of a representative stem cell sphere culture and the right panel is total spheroid count from multiple experiments. (E) Multiple siRNAs targeting either RAD6A or RAD6B were used to ensure the impact was not siRNA specific and to confirm that regulation occurs through both RAD6 isoforms. Left panel is a representative blot and the right is a compilation of 3 experiments. *p<0.05 and **p<0.01.
RAD6 promotes CSC gene expression by H2B monoubiquitination-mediated epigenetic changes
Similar to the protein levels, analysis of SOX2 and ALDH1A1 transcript levels showed over 50% decrease in RAD6-depleted OV90 cells compared to RAD6 proficient cells, in both untreated and in response to carboplatin exposure (Fig 2A and B), indicating upregulated RAD6 stimulates CSC gene expression. RAD6 is known to stimulate gene transcription through the monoubiquitination of histone H2B at lysine 120 (H2B-Ub), which promotes downstream H3K4 and H3K79 methylation (forming H3K4me3 and H3K79me3)23–25. These modified histones promote and maintain open chromatin conformation allowing enhanced gene transcription within that region26–28. In order to confirm that increased RAD6 stimulates expression of these genes by chromatin modification, chromatin immunoprecipitation (ChIP) assays were performed in SKOV3 cells to detect changes in levels of H2B-Ub within the promoter region of ALDH1A1 and SOX2. RAD6B depletion by siRNA substantially reduced levels of H2B-Ub in both promoters, while there was no change within the β-actin promoter, which is not regulated by RAD6 and served as the negative control (Fig 2C). As β-catenin stability and nuclear localization are regulated by RAD6B (Fig S1)14,29, the promoters for ALDH1A1, and SOX2 were analyzed for putative β-catenin/TCF binding sites. Two consensus sites [CTTTG(A/T)(A/T)]30 were identified in the ALDH1A1 promoter (on coding strand) and one in SOX2 (on non-coding strand). SKOV3 cells (transfected with control or RAD6B siRNA) were treated with carboplatin (2.5 μM, 8 hours) to stimulate expression of these genes and ChIP was performed on the regions containing these potential β-catenin binding sites. The putative β-catenin/TCF binding site nearest the ALDH1A1 start codon (284–290 bp upstream of the ATG) was pulled down and the signal intensity decreased in siRAD6B-treated cells (Figure 2D) while the distal site in the ALDH1A1 promoter (833–839 bp upstream of start codon) and the putative site in the SOX2 promoter (651–657 bp upstream of the ATG) had no detectable β-catenin ChIP signal (data not shown). As expected, there was no detectable β-catenin bound to the β-actin promoter (negative control). Interestingly, the β-catenin binding site seen to be actively regulated in this study was previously shown to bind β-catenin in ovarian cancer mammospheres31. To further evaluate the possibility that chemotherapy-induced stress promotes CSC phenotype in OC by upregulated RAD6-mediated ubiquitin signaling, SKOV3 cells were passaged in the presence of low levels of carboplatin (2.5 μM). Continuous exposure to sub-lethal concentration of carboplatin progressively increased the level of RAD6 with each passage (Fig 2E). As predicted, H2B-Ub levels increased, as did expression of CSC proteins SOX2, ALDH1A1 and β-catenin in response to carboplatin treatment. RAD6B has previously been shown to stabilize β-catenin and increase its nuclear localization in breast epithelial cells14. Consistently, RAD6 knockdown also affected β-catenin protein levels and its nuclear localization in OC cells (Fig S1). SKOV3 cells were systematically exposed to increasing concentrations of carboplatin to generate carboplatin resistant cells. We previously showed that RAD6 protein levels correlated with increased resistance to carboplatin (from 0 to 20μM)16 and this correlates with ability to form stem cell spheres (Fig 2F) indicating a link between chemoresistance, stemness and expression of RAD6.
Figure 2.
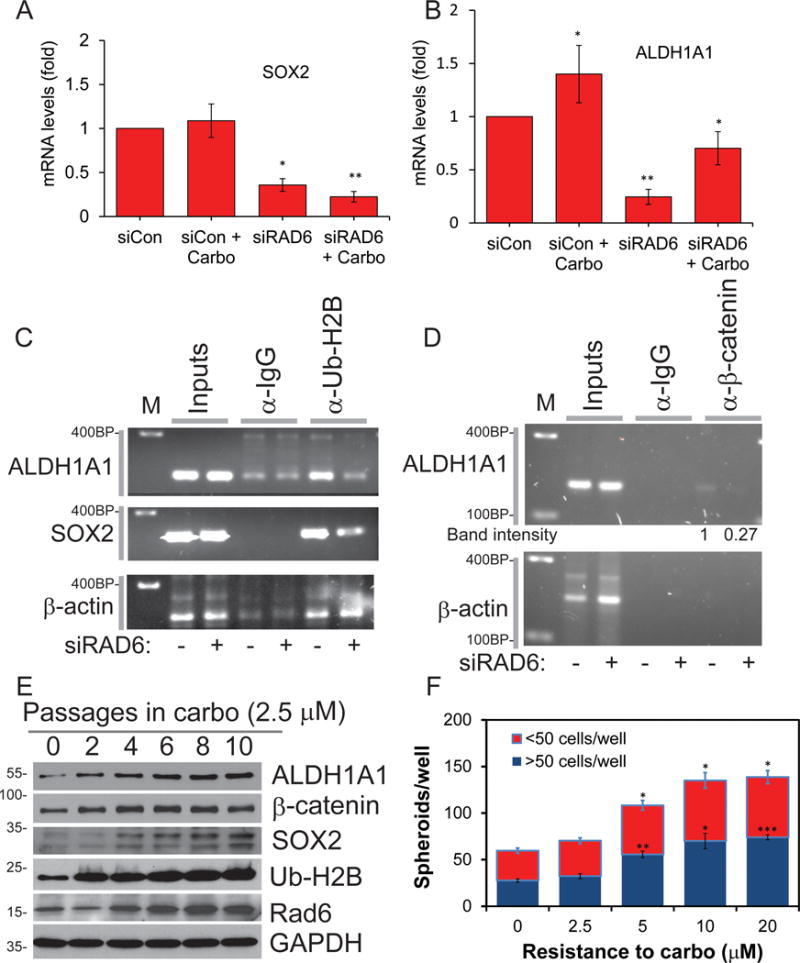
Carboplatin treatment induces expression of RAD6 and RAD6 subsequently regulates ALDH1A1 and SOX2 through pro-transcription histone modifications. (A) To determine if RAD6 regulates CSC gene expression, OV90 cells transfected with control siRNA or siRAD6B-1 and treated with carboplatin or vehicle (DMSO) were used for qRT-PCR. Knockdown of RAD6B decreased both basal and carboplatin-induced expression of (A) SOX2 and (B) ALDH1A1. (C) ChIP was used in SKOV3 cells to determine if depletion of RAD6 (siRAD6B-1) altered the level of pro-transcription H2B-Ub in the promoters of ALDH1A1 (243 to 418 bp upstream of ATG) and SOX2 (542 TO 778 bp before ATG) genes. The β-actin is not regulated by RAD6 and was used as a negative control. (D) ChIP for β-catenin binding to putative binding sites ([CTTTG(A/T)(A/T)] located in same regions analyzed for H2B-Ub in C). RAD6B depletion (siRNA) reduces β-catenin interaction at a putative β-catenin/TCF binding site located at 284–290 bp upstream of the ALDH1A1 ATG. There are no putative β-catenin binding sites in the β-actin promoter thus it was used as a negative control. (E) Systematic exposure of SKOV3 cells to sub-lethal carboplatin (2.5μM) caused progressive increase in RAD6 and associated H2B-Ub levels as well as expression of RAD18 and CSC proteins. (F) Ability to grow as stem cell spheres increased with acquired carboplatin resistance indicating correlation between stemness and chemoresistance. *p<0.05, **p<0.01 and ***p<0.001.
Chemotherapy induces RAD6 expression and stem cell signaling genes in ovarian tumors and poor prognosis in OC patients
The previous results indicate a connection between chemotherapy-induced upregulation of RAD6 and acquired chemoresistance and stemness in OC cells through RAD6-mediated ubiquitin signaling, resulting in increased DDR and expression of CSC proteins (Fig 1A–C and 2). In order to determine if these findings translate to the clinical setting, a small pilot study was conducted with 26 OC patients. The expression of RAD6B was measured in matched tumor samples collected before and after the patients received standard carboplatin/taxane chemotherapy. RAD6B expression increased by an average of 4.9 fold (p<0.01) in tumor specimens collected after chemotherapy compared to their matched pre-chemo samples (Fig 3A and B). Moreover, increased RAD6B expression correlated with poor progression free survival (PFS) in these patients (Fig 3C). Statistically there was no difference in RAD6 levels based on the debulking status (p= 0.23). Interestingly, when stratifying patients by mean cut point of 5.0 (<= 5.0 vs. >= 5.1), higher levels of RAD6 correlated to the recurrence rates 37.5 vs. 70% (p=0.01), respectively. Consistent with the in vitro data, expression of CSC markers ALDH1A1 and SOX2 were also increased in response to carboplatin chemotherapy by 2.7 (p<0.05) and 2.6 (p<0.05) fold, respectively (Fig 3A and B). The changes in RAD6B, ALDH1A1 and SOX2 transcript levels from individual patients are given in figure S2. To further confirm the mRNA results, pre- and post-chemotherapy tumor samples were analyzed by immunohistochemistry (IHC) for RAD6 and SOX2 protein levels. Due to the limited tumor samples collected, especially post-chemotherapy samples, the number of proteins that can be analyzed was restricted. Consistent with the mRNA levels, IHC staining showed significant increase in the expression of RAD6 and SOX2 in tumor samples collected after chemotherapy compared to the tumors collected before chemotherapy (Fig 3D, E and S3). These results combined with DDR and CSC expression data strongly implicate RAD6 in chemoresistance and poor patient outcomes. The findings are in agreement with previous studies showing that RAD6B levels are increased in breast cancer and correlate with chemoresistance and decreased survival12,13,32.
Figure 3.
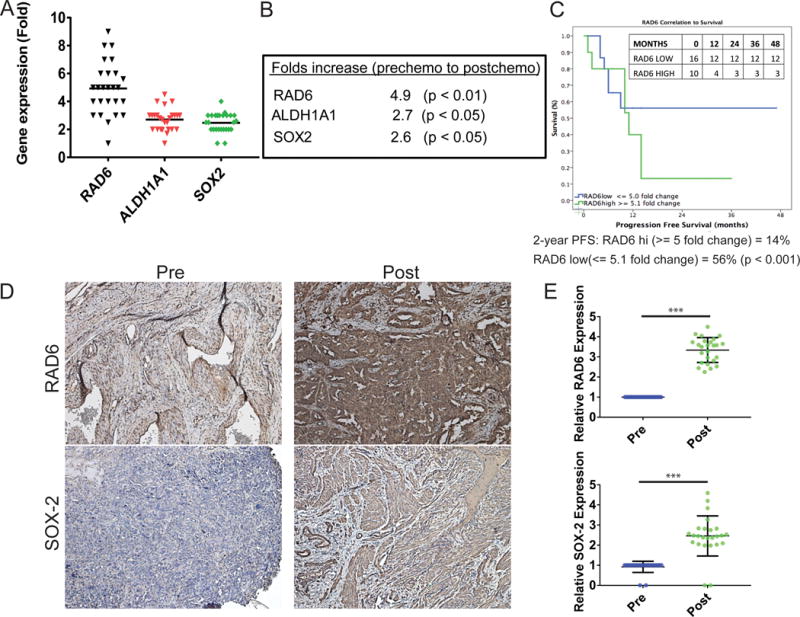
Expression of RAD6, ALDH1A1 and SOX2 increase in OC patient tumors in response to chemotherapy and RAD6 expression correlates with progression free survival. A small pilot study was conducted with 26 advanced stage III/IV epithelial OC patients. Tumor samples were taken from OC patients (n=26) prior to chemotherapy (carboplatin/Taxane; designated pre-chemo) and following chemotherapy (post-chemo) and gene expression analyzed and compared. (A) The post-chemo samples showed increased expression of RAD6B, ALDH1A1 and SOX2 compared to the matched pre-chemo sample from the same patient. (B) The mean fold increase for the 3 genes in all matched patient samples. (C) RAD6 expression correlated with poor PFS in OC patients. High RAD6 was considered anything over 5 fold and PFS study was conducted for 2 years. Inset table shows the number of cases present at different time points in the two groups. (D and E) IHC staining of post-chemo samples showed increased expression of RAD6 and SOX2 compared to the matched pre-chemo sample form the same patient. ***p<0.001.
Inhibition of RAD6 using a small molecule inhibitor (SMI) TZ9 induces DNA replication stress, decreased DNA repair and a CSC phenotype
To test whether pharmacological inhibition of RAD6 can attenuate its ubiquitination signaling mediated acquired chemoresistance and stemness in OC cells, we examined TZ9 [4-Amino-6-(phenylamino)-[1,3,5]triazin-2-yl)methyl-4-nitrobenzoate], a previously identified RAD6-specific inhibitor (Fig 4A)33,34. Exposure of OV90 cells to TZ9 induced γH2AX foci, a marker of DNA double strand breaks (DSB) and stalled replication forks (Fig 4B). Consistent with RAD6 downregulation by siRNA, TZ9 treatment reversed activation of DDR in response to carboplatin as evidenced by decreased ubiquitination of FANCD2, PCNA and γH2AX (Fig 4C). Additionally, TZ9 treatment in OV90 cells for 24 or 48 hours results in accumulation of cells in S and G2 phases compared to DMSO treated cells (Fig 4D and E). These proteins represent activation of multiple pathways as PCNA ubiquitination indicates translesion synthesis and FANCD2 is a member of the Fanconi anemia tumor suppressor and homologous recombination repair pathways, suggesting that RAD6 inhibition could sensitize cells to DNA damaging chemotherapeutic agents such as carboplatin. Furthermore, TZ9 treatment significantly decreased invasiveness of both isogenic platinum-sensitive (A2780) and resistant (A2780/CP70) cells in matrigel (Fig 4F) suggesting that RAD6 inhibition could reduce aggressiveness of ovarian tumors.
Figure 4.
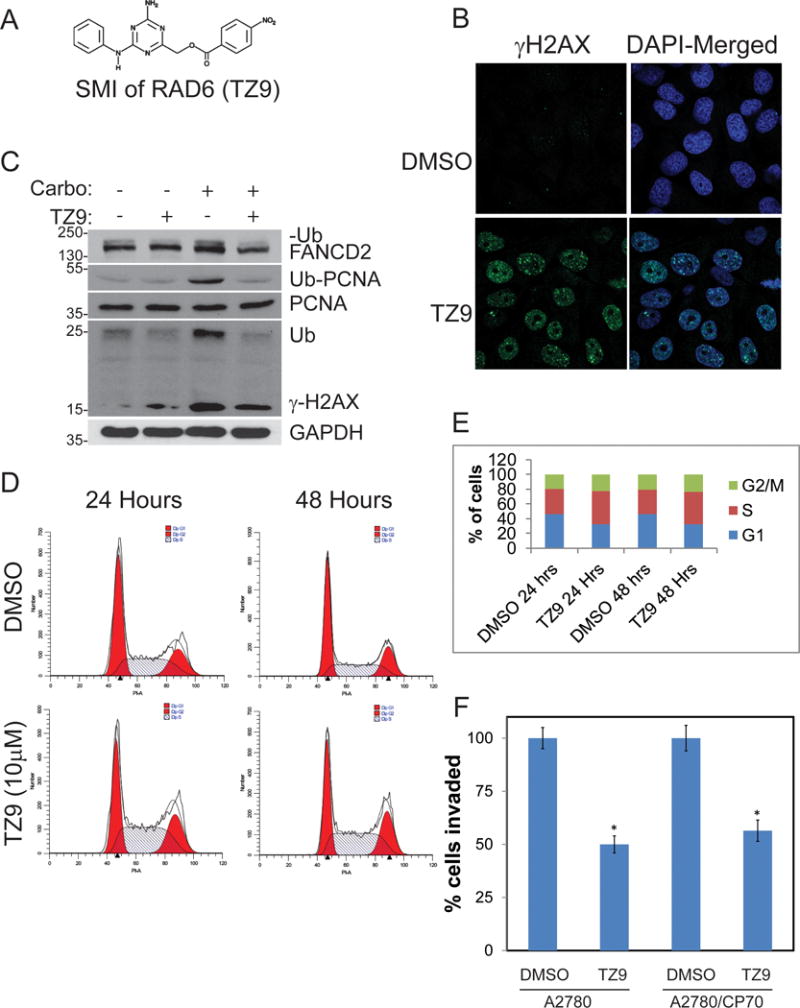
RAD6 SMI TZ9 impairs activation of DNA damage response to carboplatin and induces replication stress in OV90 cells. (A) Structure of the RAD6 SMI TZ9. (B) TZ9 causes γH2AX foci formation indicating that it causes DNA double strand breaks and (C) blocks carboplatin-induced monoubiquitination of FANCD2, PCNA, and γH2AX. (D and E) OV90 cells treated with TZ9 (10μM) for 24 or 48 hours were tested for cell cycle profile. TZ9 causes G2/M cell cycle arrest indicating the presence of DNA damage and activation of damage checkpoint. (F) Matrigel invasion assay in isogenic sensitive (A2780) and chemoresistant (A2780/CP70) cells showed that the RAD6 inhibitor decreased their ability for invasion, suggesting RAD6 may be a valid target for treatment of OC. *p<0.05.
The increase in γH2AX in RAD6 knockdown or inhibited cells suggests replication stress. This is supported by the substantial co-localization of γH2AX foci with regions of active DNA replication, as determined by EdU incorporation (Fig 5A). Consistently, depletion (siRNA) or inhibition (TZ9) of RAD6 significantly diminished clonogenic survival in OC cells (Fig 5B). Moreover, treatment of RAD6 downregulated cells with TZ9 did not show any additional effects over siRNA, suggesting TZ9-mediated effects are specific for RAD6. To further examine replication dynamics in RAD6-deficient cells, single cell DNA fiber analysis was conducted in SKOV3 cells as outlined in figure 5C. Treating cells with either TZ9 or knocking down RAD6 significantly attenuated replication fork velocities compared to control siRNA treated cells (Fig 5E). As expected, carboplatin treated cells showed increased stalled/collapsed/terminal forks compared to control cells (Fig 5D, F and S4). Similarly, TZ9 or RAD6 knockdown cells also exhibited increased stalled/terminal forks. Consistent with the attenuated PCNA and FANCD2 ubiquitination, combination of carboplatin and RAD6 deficiency (siRNAs or TZ9) resulted in several fold increase in stalled/terminal forks (Fig 5F). These findings indicate that increased RAD6 levels protect OC cells from replication stress, likely through promoting expression of DDR and DDT proteins.
Figure 5.
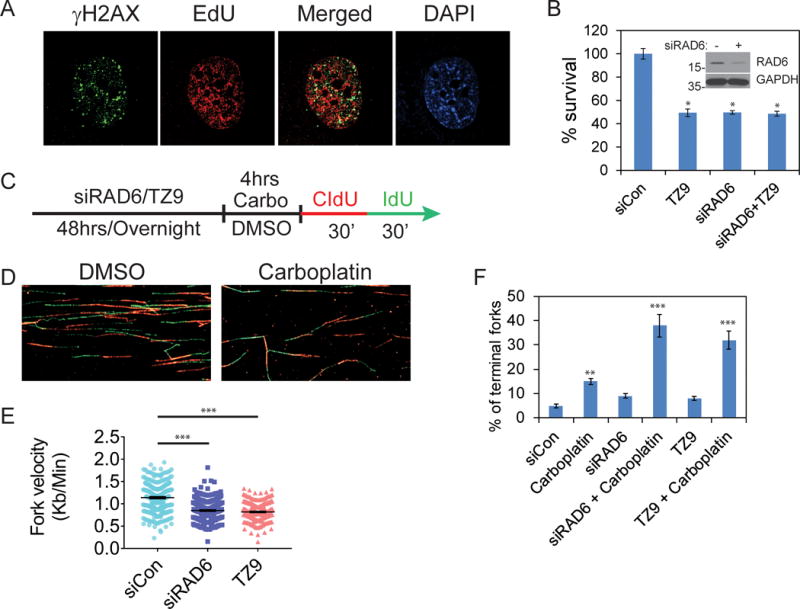
Knocking down or inhibiting RAD6 causes replication stress and decreases viability in OC cells. (A). Sites of active DNA replication was labeled using EdU and sites of DSB identified by visualizing γH2AX foci in RAD6-depleted SKOV3 cells. Many of the γH2AX foci co-localize with active DNA replication (EdU foci) indicating loss of RAD6 in tumor cells leads to replication stress. (B) Small molecule inhibitor (TZ9) of RAD6 specifically inhibits clonogenic potential of SKOV3 cells expressing RAD6. Downregulation of RAD6 does not show any additional toxic effects by TZ9. (C) Single cell DNA fiber analysis was used to assess DNA replication fork dynamics. To measure fork velocity, SKOV3 cells were treated with control siRNA or siRAD6B-1 for 48 hours or TZ9 (60μM) for overnight and labeled for 30 min with 25 μM CldU followed by 30 min labeling with 250 μM IdU as indicated. (D) Representative DNA fiber images for the control siRNA or carbo treated samples. (E) The length of fibers was measured in 300 individual fiber structures from three independent experiments and their fork velocity is represented with S.E. (F) To measure stalled forks, SKOV3 cells were treated with control siRNA or siRAD6B-1 for 48 hours or TZ9 overnight and then treated with DMSO or carboplatin for 4 hours. RAD6 inhibited (siRAD6B-1 or TZ9) cells that are exposed to carboplatin showed increased stalled replication fork (red only) compared to the control cells. *p<0.05, **p<0.01 and ***p<0.001.
Similar to RAD6 deficiency, TZ9 treatment caused loss of pro-transcription histone modifications H2B-Ub and H3K79me3 as well as expression of CSC genes ALDH1A1 and SOX2 both basally and in response to carboplatin (Fig 6A–C). ALDH1A1 has been strongly linked to stem cell characteristics in multiple normal and cancer cell lines including ovarian35–37. This loss of CSC gene expression was accompanied by a substantial decrease in their ability to form tumor spheres. TZ9 inhibited both number and size of the spheroids in a dose-dependent manner in OV90 (Fig 6D and E) and SKOV3 (Fig S5A and B) cells.
Figure 6.
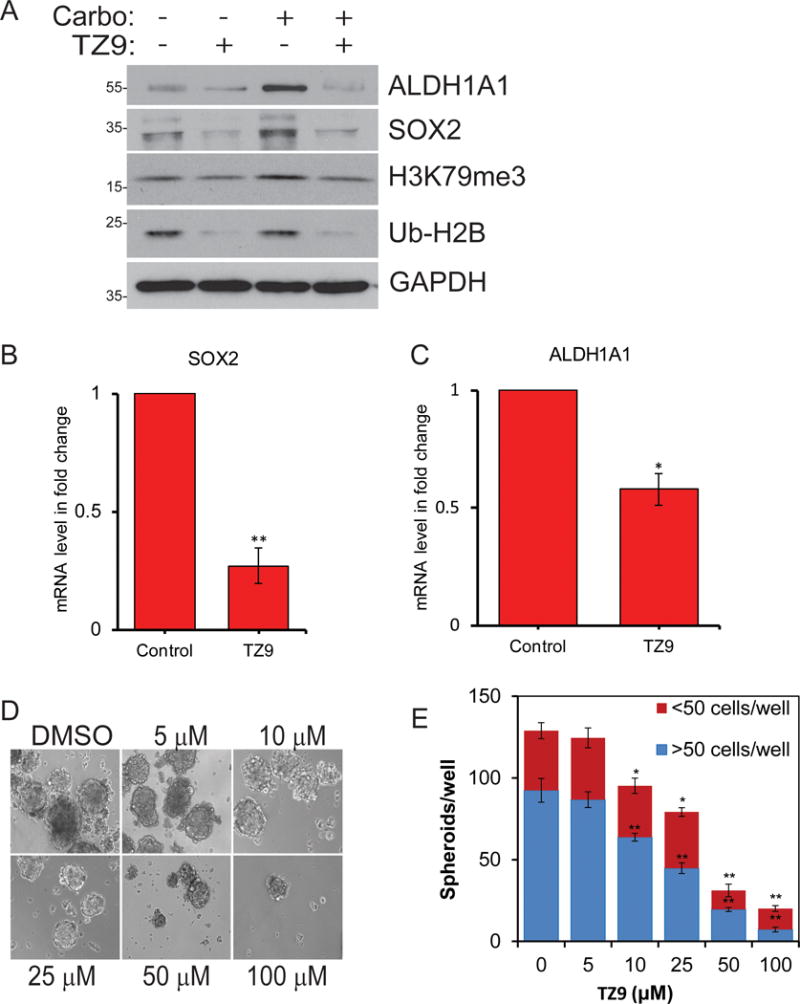
RAD6 inhibition with TZ9 prevents RAD6-induced expression of ALDH1A1 and SOX2 and stemness. (A) OV90 cells were treated with vehicle control or carboplatin to induce expression of RAD6 and with the RAD6 inhibitor TZ9. TZ9 prevented RAD6-induced histone modification and CSC gene expression both basally and in carboplatin treated cells. To determine if TZ9 blocks transcription of (B) ALDH1A1 and (C) SOX2, qRT-PCR was done on mRNA isolated TZ9 treated and control OV90 cells. The levels of both mRNAs were reduced. RAD6 inhibitor TZ9 decreased stem cell sphere-forming ability in OV90 cells in a dose-dependent manner. (D) Representative image and (E) compiled data from 3 experiments. *p<0.05 and **p<0.01.
RAD6 SMI sensitizes OC cells to carboplatin treatment
Collectively, these studies indicate RAD6 inhibition impairs DDR as well as CSC signaling, suggesting that RAD6 inhibition may sensitize cancer cells to DNA damaging chemotherapeutic agents, like carboplatin. To evaluate this, multiple OC cell lines were tested in clonogenic survival assays using various concentrations of TZ9 and carboplatin. First, we determined the concentration-dependent responses for both TZ9 and carboplatin in all cell lines used (Fig 7, S5C–D and data not shown)16,33,35. Both TZ9 and carboplatin alone cause varying degrees of OC cell death; however, the combination with TZ9 caused significantly increased sensitivity to carboplatin in all OC cell lines tested (Fig 7 and S5C and D). Analysis of different concentrations of the drug combinations revealed 4μM carboplatin and 50μM TZ9 as the most effective conditions in all the OC cell lines tested (Fig 7C). These findings are consistent with a study in breast cancer cells, where a RAD6 inhibitor induced apoptosis and enhanced lethality of the platinum drug cisplatin38. Together these data conclusively demonstrate that RAD6 SMI significantly sensitizes OC cells to carboplatin, a drug that is typically used to treat OC patients. Furthermore, our studies suggest RAD6 could be an effective therapeutic target to prevent and treat acquired chemoresistance and disease recurrence in OC, the main factors contributing to the low survival rate of these patients. Moreover, normal tissues including ovarian express low levels of RAD6; therefore, it is likely that RAD6 inhibitors would selectively target the RAD6 overexpressing cancer cells, as was seen in breast cancer16,38. To test this, normal immortalized fallopian tube epithelial and ovarian surface epithelial cell lines (FTEC and IOSE80, respectively) and OC cell lines (SKOV3, OV90 and A2780) were evaluated in growth inhibition assays using different concentrations of TZ9 (Fig S6). The OC cell lines were significantly more sensitive to RAD6 inhibition than the normal ovarian cells, suggesting RAD6 inhibitors may have less toxic effects to normal cells and tissues.
Figure 7.
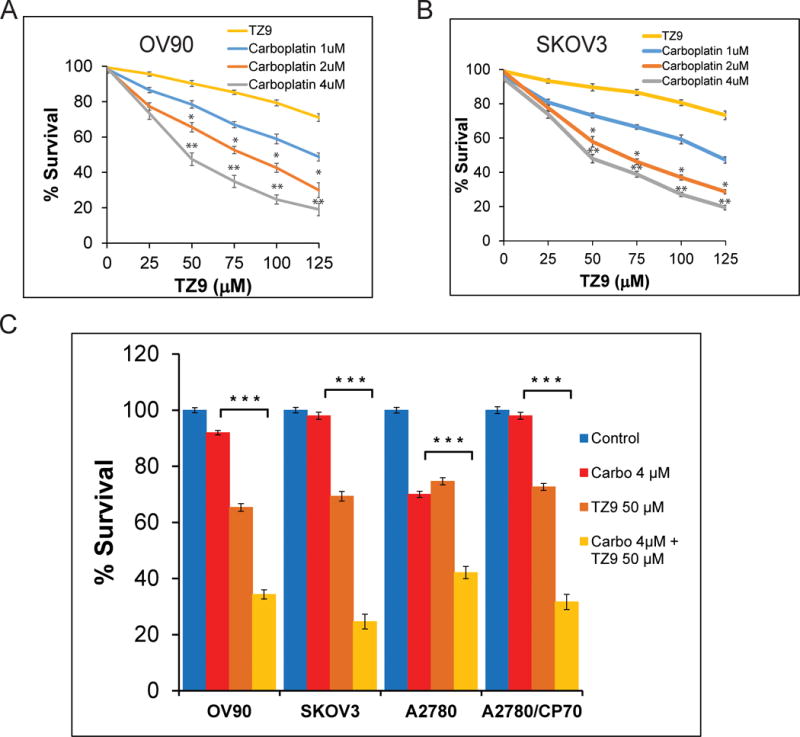
TZ9 sensitizes OC cells to carboplatin treatment. Clonogenic survival assays were conducted using a range of concentrations of the RAD6 SMI TZ9 (0–125μM) and carboplatin (0–4 μM) in multiple OC cells lines. Both OV90 (A) and SKOV3 (B) showed significant sensitization to carboplatin treatment at multiple concentrations of TZ9. Isogenic A2780 and A2780/CP70 cell lines are shown in figure S1. (C) Histogram showing compiled data from multiple experiments at the best combination of drugs (50μM TZ9 and 4μM carboplatin). *p<0.05, **p<0.01 and ***p<0.001.
Discussion
RAD6 levels are increased in melanoma, breast and ovarian cancers13,15,16,39. Both RAD6 proteins promoted cellular proliferation and invasion in normal human liver cells and RAD6B is associated with tumorigenesis and platinum resistance in breast12,13,32 and ovarian16 cancer. Though initial treatment is typically quite successful in OC patients, tumor recurrence is extremely high in this disease and frequently accompanied by acquired chemoresistance causing poor survival among these patients1,2. One mechanism cancer cells use to evade chemotherapy is altered regulation of DDR and repair mechanisms4–6,40. RAD6 is well known as a DNA repair protein and was initially identified in yeast as a key component of DNA repair pathways, where it functions with RAD18 in allowing replication to bypass bulky DNA lesions and in DSB repair7. RAD6 and RAD18 regulate DNA polymerase switching during TLS through PCNA monoubiquitination41–43. RAD6-mediated histone modification is vital for efficient DNA repair and damage tolerance44,45. Furthermore, increased levels of these proteins provide resistance to chemotherapeutic drugs that impact DNA replication, such as platinum drugs like carboplatin and cisplatin, as higher levels of RAD18 enhance TLS and DSB repair by monoubiquitination of PCNA and FANCD2 respectively46–48. Consistent with this we have shown that knocking down or inhibiting RAD6 in OC cells triggered replication stress (Fig 4B–E and 5) and decreased activation of DNA repair and damage tolerance pathways in response to carboplatin treatment (Fig 1A and 4C). Recent work with RAD6 SMI in breast cancer cells showed that they cause PARP-1 activation and apoptosis supporting RAD6 as an effective target for treating multiple neoplasms38. Therefore, treating OC tumors with a RAD6 inhibitor could be an effective new treatment strategy to counter acquired chemoresistance.
The CSC hypothesis states that recurrent tumors are largely due to a small population of cancer cells that dedifferentiated as CSCs. CSCs have properties which allow them to evade treatment and proliferating, differentiating back into tumor cells and reestablishing tumors once treatment is concluded4–6. Many chemotherapeutic strategies currently being developed target CSCs that evade approved treatments. We have shown that RAD6B is upregulated in response to carboplatin treatment in OC cells and patient tumors where it correlates with poor PFS (Fig 2E and 3) and that increased RAD6B levels promote expression of CSC proteins and acquisition of stem cell phenotype in OC cell lines (Fig 1B–C and 2)16. Knocking down RAD6 decreased expression of CSC proteins and impaired the ability of OC cells to form stem cell spheres (Fig 1C and D). These results were recapitulated using RAD6 SMI TZ9 (Fig 6) indicating that RAD6 inhibitors may be effective for preventing OC disease recurrence in patients. TZ9 alone induced cell death in OC cells and sensitized these cells to carboplatin (Fig 5B, 7, and S5) which is frequently utilized in treating OC patients indicating that combination therapy may be warranted. There are several RAD6 inhibitors currently being investigated, though an ideal clinical RAD6 SMI has yet to be discovered34,38,49. Thus we have designed a family of RAD6 inhibitors based upon the SMI TZ9 and these compounds show promise as they have performed well in in vitro RAD6 ubiquitin conjugation activity assays and in blocking RAD6 activity in an array of cancer cell lines, including OC33. The LC50 of several of these new SMI are superior to that of TZ9 suggesting that they may be more effective and more extensive evaluation is being undertaken to determine their efficacy.
RAD6 is also known to regulate gene transcription through histone modification. RAD6 partners with RNF20/40 to monoubiquitinate histone H2B at lysine residue 12010,50,51. This triggers further histone modification, including H3K4me3 and H3K79me3, and together these modified histones promote an open chromatin structure that allows gene transcription26,52,53. H2B monoubiquitination is required for efficient differentiation of embryonic stem cells54, and this mechanism is likely to be active in CSCs as well. There are increased H2B-Ub and H3K79me3 levels in response to carboplatin treatment (Fig 1B and C) or overexpression of RAD6B16 and knocking down or inhibiting RAD6B decreases these modifications (Fig 1B, 1C and 6A). Western blot and ChIP analysis confirm that H2B-Ub levels increase in response to carboplatin and decrease within the promoters of two CSC genes, ALDH1A1 and SOX2, when the cells are depleted of RAD6B (Fig 2C). Loss of RAD6B also resulted in decreased β-catenin binding to the ALDH1A1 gene promoter (Fig 2D). Therefore, it appears that RAD6 is also promoting CSC protein levels by both the WNT/β-catenin pathway and histone modification and chromatin rearrangement, suggesting that RAD6 inhibition would be more effective than chemotherapeutic agents that target WNT signaling alone.
RAD6B increases the levels of multiple proteins that have been proposed as potential targets for treating various types of cancer, including β-catenin, SOX2 and ALDH1A1 making it a very attractive target as multiple cancer-associated proteins will be simultaneously impacted. β-catenin is a transcription factor and effector of the WNT signaling pathway which is highly conserved and essential for regulating developmental processes and stemness55–58. It is likely that RAD6B promotes CSC gene expression both by H2B ubiquitination and stabilization of β-catenin14,54,59. ALDH1A1 regulates normal stem cell proliferation and is frequently overexpressed in cancers and is a prominent member of ALDH protein family that is associated with resistance to radiation and chemotherapy and stemness. We have previously shown that ALDH1A1 is an important marker for OC stem cell phenotype and chemoresistance and cells expressing high levels of ALDH1A1 display increased migration and invasion potential35. Furthermore, ALDH1A1 has been reported as a direct target of β-catenin in ovarian cancer and β-catenin regulated ALDH1A1 is critical for the maintenance of ovarian tumorspheres31. SOX2 is a transcription factor regulating expression of genes involved in chromatin remodeling, RNA scaffolding, pluripotency, proliferation, cell migration, cell invasion, transcription, and maintenance of stemness60. Importantly, reciprocal regulation of ALDH1A1 and SOX2 has been demonstrated in ovarian cancer and impacts CSC proliferation and tumor progression61. Our findings suggest that novel treatment regimens utilizing RAD6 SMI in combination with existing frontline chemotherapeutic drugs such as carboplatin would be an effective therapeutic strategy. This report shows that inhibiting RAD6B decreases DNA repair and damage tolerance mechanisms, causing replication stress and DNA damage. The RAD6 SMI TZ9 increased sensitivity to carboplatin, strongly suggesting that this combination could be effective at reducing acquired chemoresistance and protecting against disease recurrence (Fig 8). This would represent a significant improvement in treating patients with this deadly disease. Furthermore, RAD6B expression is relatively low in normal cells and thus RAD6 inhibitors would likely cause little damage to surrounding normal tissue and combination therapy typically allows reduced levels of each chemotherapeutic agent further reducing side effects associated with high doses of these drugs.
Figure 8.
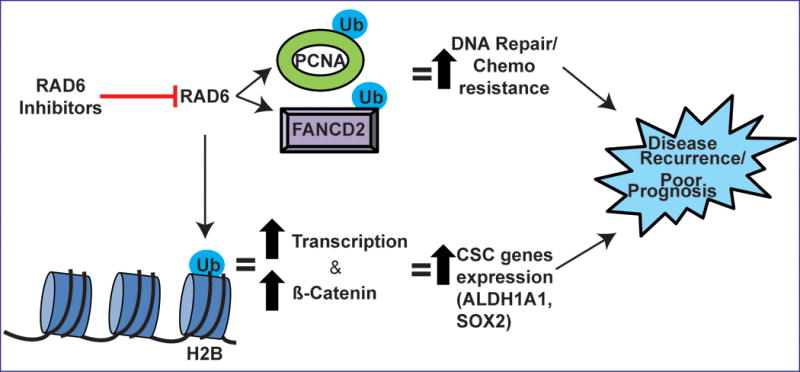
Model showing possible mechanisms by which RAD6-mediated ubiquitin signaling regulates acquired chemoresistance and stemness contributing to OC recurrence. RAD6 promotes expression of cancer stem cell genes by histone H2B ubiquitination and subsequent chromatin remodeling, as well as by enhancing activity of pro-stemness transcription factors like β-catenin. RAD6 protects OC cells from DNA damage caused by chemotherapy by activating multiple repair and damage tolerance pathways including TLS (PCNA-Ub) and FA/homologous recombination (FANCD2-Ub). The RAD6-mediated increase in stemness and DNA repair and damage tolerance allow some of the tumor cells to evade treatment and re-establish the tumor.
Materials and methods
Cell lines, culture methods and reagents
The OV90 and SKOV3 cells used in this study were acquired from ATCC and isogenic A2780 and A2780/CP70 cell lines were described previously35. Carboplatin-resistant SKOV3 cells were generated and utilized previously16. FTEC cells were a kind gift from Dr. Amir Jazaeri62. Commercially available cell lines utilized in this study were either validated or acquired in summer 2015. OV90 and SKOV3 cells were cultured in 1:1 DMEM/F12 (Corning, Manassas, VA) and A2780 and A2780/CP70 cells in RPMI (Corning). Cell lines were routinely tested for Mycoplasma contamination. All media were supplemented with 10% FBS (Atlanta Biologicals, Flowery Branch, GA) and 1× Penicillin/Streptomycin (Mediatech/Corning). The RAD6 SMI, TZ9, was synthesized according to methods described previously34,63. Carboplatin and other chemicals unless specified otherwise were purchased from Sigma (St Louis, MO). Multiple siRNAs were utilized for this work to confirm results and include RAD6A: 5′-GAACAAGCUGGCGUGAUUdTdT-3′64, RAD6B-1: 5′-CAAACGAGAAUAUGAGAAAdTdT-3′64, and RAD6B-2: 5′-UUUCUCAUAUUCUCGUUUGdTdT-3′. All siRNAs were purchased from Dharmacon (Lafayette, CO) and transfected using Lipofectamine 2000 (Invitrogen, Carlsbad, CA) according to the manufacturer’s instructions. Combination of RAD6A and RAD6B siRNAs (denoted siRAD6) were used for all experiments except figures 1C and 1D, where only RAD6B siRNA used. For this work, the following antibodies specific to these proteins were utilized: ALDH1A1 (sc-22589), β-catenin (sc-1496), FANCD2 (sc-20022), GAPDH (sc-32233), PCNA (sc-56), RAD18 (sc-52949) (Santa Cruz Biotechnology, Santa Cruz, CA); RAD6 (Bethyl Laboratories, Montgomery, TX); H2B (18977), H3K79me3 (2621), SOX2 (97959) (Abcam, Cambridge, MA); Ub-PCNA (13439) (Cell Signaling, Danvers, MA); γ-H2AX (05-636), H2B-Ub (51312) (Millipore, Billerica, MA).
Western blotting
Protein isolation and Western blots were performed as described previously65. Briefly, cells were washed thrice with ice-cold PBS and lysed in ice-cold cytoskeletal buffer [CSK: (10 mM PIPES (pH 6.8), 100 mM NaCl, 300 mM sucrose, 3 mM MgCl2, 1 mM EGTA, 1 mM dithiothreitol, 0.1 mM ATP, 1 mM Na3VO4, 10 mM NaF and 0.1% Triton X-100)] freshly supplemented with protease and phosphatase inhibitors (Roche, Indianapolis, IN). Protein concentrations were quantified using Bradford assay (Bio-Rad, Hercules, CA), and samples were normalized for equal loading. Samples were denatured by addition of 6× Laemmli buffer and heated at 100 °C for 15 minutes and resolved by SDS-PAGE and desired proteins identified by immunoblotting using specific antibodies.
Clonogenic Survival Assays
Cells were counted, and 500 cells were seeded per well in six well culture dishes. The following day, cells were treated with DMSO (vehicle control) or the indicated doses of carboplatin or TZ9. After twenty-four hours, cells were washed thrice with PBS and twice with growth medium. Colonies were allowed to form in complete growth medium for 10 days. After 10 days, colonies were fixed with methanol, stained with crystal violet (0.5% w/v) and counted as described previously66.
Cell Cycle Analysis
Cells were plated at 50% confluency in appropriate growth medium and allowed to attach overnight. The following day, cells were treated with the indicated concentrations of TZ9 or DMSO (vehicle control) for 24 hours. Cells were trypsinized, washed thrice with ice-cold PBS, and stained with PI solution (40 ug/ml Propidium Iodide in PBS, 10 ug/ml RNase A) at 37°C for 30 min and analyzed using a BD FACSCanto II (BD Biosciences, San Jose, CA).
Chromatin immunoprecipitation (ChIP)
SKOV3 cells transfected with control or RAD6B-specific siRNAs were grown for 48 hours after transfection and chromatin was isolated and subjected to ChIP using the Simple ChIP Enymatic Plus Kit following manufacturer’s suggested protocol (Cell Signaling). Rabbit IgG supplied with the kit was utilized as a negative control and chromatin containing monoubiquitinated H2B was immunoprecipitated using Ubiquityl-Histone H2B (Lys120) (D11) XP Rabbit mAb (Cell Signaling #5546) and chromatin bound to β-Catenin was immunoprecipitated with anti-β-Catenin antibody (Bethyl #A302-010A). Promoter regions (defined as 1000 bp upstream of the ATG) for ALDH1A1 (AY338497.1), SOX2 (NG_009080.1), and ACTB (β-actin, NG_007992.1, negative control) were analyzed for putative β-catenin/TCF binding sites using MatInspector (Genomatix, Munich, Germany). Consensus β-catenin/TCF [CTTTG(A/T)(A/T)] binding sites were identified in promoters of ALDH1A1 (2 sites 290-284 and 839-833 bases upstream of the ATG) and SOX2 (657-651 bases upstream of ATG). As anticipated no sites were detected in the β-actin gene promoter region. For the β-catenin ChIP, SKOV3 cells were treated with 2.5 μM carboplatin for 8 hours to stimulate expression of β-catenin, SOX2 and ALDH1A1. PCR was used to detect precipitated promoter regions as follows: ALDH1A1 (243 to 418 bp before ATG – forward: 5′-CTCTGATTCCAAGTCTGTCAG and reverse: 5′-CCAGTGCTCCAGCATCGAATT and 705 to 904 bp before ATG – forward: 5′-ATTGTCCTTCAGCTAAATATTA and reverse 5′-GCCAGCAGGACTTTTAGGGA) and SOX2 (542 to 778 bp upstream of ATG – forward: 5′-CCCTGCACCAAAAAGTAAATC and reverse: 5′-CCGGGTTTTGCATGAAAGG) and β-actin (607 to 851 bp upstream of ATG – forward: 5′-GCCAGGTAAGCCCGGCCA and reverse: 5′-GCCCGGCTCAGACAAAGA) was used as a control.
DNA Fiber Assay
SKOV3 cells were either treated with RAD6 siRNA for 48 hours or TZ9 (60μM) overnight followed by carboplatin (20μM) for 4 hours. Cells were then pulsed with 25μM CldU for 30 min (Invitrogen) followed by 30 min labeling with 250μM IdU (Invitrogen). Cells were trypsinized and re-suspended in ice cold PBS, and a small drop of cell suspension placed on glass slide and lysed for 5 minutes with 8μl of lysis buffer (0.5% SDS, 200 mMTris-HCl pH 7.4, 50 mM EDTA). DNA fibers were stretched by tilting slides to a 15° angle and air dried, fixed in 3:1 methanol:acetic acid, denatured in 2.5M HCL and blocked with 5% BSA. Next slides were incubated with rat anti-CldU and mouse anti-IdU for one hour followed by sheep anti-mouse Cy3 and goat anti-rat Alexa Fluor 488. To calculate fork velocity, the length of each replication region was measured and multiplied by DNA extension factor (2.59kbp/μm). This gives the DNA length in base pairs that was synthesized in 30 min labeling of CldU followed by 30 min labeling of IdU. Dividing the DNA length (kb) by 60 minutes will give the fork velocity achieved for a minute. For fork velocity, 100 replication fibers were measured and statistical significance were assessed using Mann-Whitney test and students “t” test was performed using Prism 5 (GraphPad Software, La Jolla, CA). To measure stalled/collapsed/terminal forks, a total of 100 replication structures (both red and green) from above experiment, red only tracks were counted and presented as stalled replication fork. The experiments were repeated thrice and mean values were presented with statistical significance.
Sphere formation assays
OV90 and SKOV3 cells were harvested in log phase, counted, and seeded in ultra-low attachment 6 well dishes (VWR, Radnor, PA) at 1000 to 10,000 cells/well. The cells were allowed to grow and form spheres as detailed earlier16. In brief, cells were carefully dispersed as single cells, cultured in stem cell-specific serum free media [2 mL; 1:1 DMEM/F-12 supplemented with 1× penicillin/streptomycin, B27 and N2 supplements (Gibco, Waltham, MA), and growth factors [recombinant human epidermal growth factor (EGF) and fibroblast growth factor (FGF), both from Invitrogen] in an ultra-low attachment six well plates for 10–12 days. After seeding the cells were observed daily to ensure that spheres were forming. Fresh media containing growth supplements EGF (20 ng/ml) and FGF (20 ng/ml) was added every 72 h. Spheres containing ≥50 cells were scored as large (true stem cell spheres), while spheres <50 cells were considered to be small spheres.
Immunohistochemistry
Ovarian tumor tissues collected before and after chemotherapy were stained for the expression of RAD6 and SOX2 proteins by immunohistochemistry. Antigen retrieval was carried out by heating the tissue specimens in 10mM sodium citrate buffer, pH 6.0. Endogenous peroxidase activity was inhibited by incubating the sections in 3% hydrogen peroxidase for 10 min. Tissue sections were incubated with specific primary antibodies, followed by a specific biotinylated secondary antibody, and then conjugated HRP streptavidin and DAB (3,3′-Diaminobenzidine) chromogen (Vector Laboratories, Burlingame, CA) and tissues were counterstained with hematoxylin (Vector Laboratories). Stained sections were analyzed by Zeiss Axioscope 2 microscope and images captured by AxioCam camera. DAB intensity was analyzed by Fiji Image J software. This process was carried out without any knowledge of the identity of each tissue sample to prevent bias in scoring the samples.
Tumor samples and RNA Isolation
A small pilot study was conducted with 26 advanced stage III/IV epithelial OC patients. After approval from the University of South Alabama Institutional Review Board, written consent from patients was obtained for acquisition of tumor during the surgical management of their disease. Tumor samples were collected at time of diagnosis (referred to as pre-chemo) using diagnostic laparoscopy, as well as during debulking surgery following 3 cycles of platinum / taxane based neoadjuvant chemotherapy (referred to as post-chemo). In order to provide adequate molecular data, all patients had at least 1 cubic centimeter of excess tumor tissue for analysis. Those without adequate tumor were not included in this analysis. Paraffin was removed from tumor samples and samples were lysed overnight. Total RNA was isolated from formalin-fixed, paraffin-embedded (FFPE) tissue samples by Trizol following manufacturer’s protocol (Invitrogen). Purity and quantity of total RNA in samples were determined using a NanoDrop ND-1000 UV spectrophotometer (NanoDrop, Wilmington, DE).
RT-PCR
Total RNA from adherent ovarian cancer cells and dispersed sphere cells was isolated by Trizol following manufacturer’s protocol (Invitrogen). Tumor sample RNA was prepared as outlined above. cDNA was prepared from total RNA using cDNA preparation kit (Applied Biosystems, Foster City, CA). Gene-specific primers (Prime PCR; Bio-Rad) were used for semi-quantitative RT-PCR analysis with 2μl cDNA in a total volume of 20μl using the following protocol: initial denaturation for 5min at 94°C; 30 cycles of denaturation for 30s at 94°C, annealing for 30s at 65°C and extension for 30s at 72°C; and final extension for 5min at 72°C.
Immunofluorescence (IF) staining
For immunofluorescence, cells were seeded in triplicate into 35mm glass bottom culture dishes (FluoroDish – World Precision Instruments, Sarasota, FL) for 48 hours. Next cells were treated with CPT or vehicle control (DMSO) as indicated and fixed with 4% formaldehyde at room temperature for 10 minutes and then in cold (−20°C) 100% methanol for 10 minutes. Fixed cells were then blocked in 10% goat serum for 30–60 minutes and washed 3 times with PBS. Cells were subsequently incubated overnight at 4°C with primary antibodies in PBS containing 5% BSA as described previously66. Cells were washed three or four times with PBS supplemented with 1% BSA and incubated with appropriate fluorophore-conjugated secondary antibody (IgG-Cy3, IgG-FITC or IgG-Cy5 – Invitrogen) for 2h at room temperature. Detection of protein foci was done using the Nikon Ti Eclipse confocal microscope (Nikon) and images were acquired at 100× and as described previously66.
Supplementary Material
Acknowledgments
This work was supported by Abraham Mitchell Cancer Research Scholar Endowment grant, and by National Institutes of Health grant [R01GM098956] to K.P. We would like to thank Joel Andrews, PhD and Steve McClellan for assistance with microscopy and flow cytometry, respectively.
Footnotes
Conflict of interest
The authors have no conflict of interest to report
Supplementary Information accompanies the paper on the Oncogene website (http://www.nature.com/onc).
References
- 1.Foley OW, Rauh-Hain JA, del Carmen MG. Recurrent epithelial ovarian cancer: an update on treatment. Oncol Williston Park N. 2013;27:288–294. 298. [PubMed] [Google Scholar]
- 2.Davis A, Tinker AV, Friedlander M. ‘Platinum resistant’ ovarian cancer: What is it, who to treat and how to measure benefit? Gynecol Oncol. 2014;133:624–631. doi: 10.1016/j.ygyno.2014.02.038. [DOI] [PubMed] [Google Scholar]
- 3.Kandalaft LE, Powell DJ, Singh N, Coukos G. Immunotherapy for ovarian cancer: what’s next? J Clin Oncol Off J Am Soc Clin Oncol. 2011;29:925–933. doi: 10.1200/JCO.2009.27.2369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Abdullah LN, Chow EK-H. Mechanisms of chemoresistance in cancer stem cells. Clin Transl Med. 2013;2:3. doi: 10.1186/2001-1326-2-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Khan IN, Al-Karim S, Bora RS, Chaudhary AG, Saini KS. Cancer stem cells: a challenging paradigm for designing targeted drug therapies. Drug Discov Today. 2015;20:1205–1216. doi: 10.1016/j.drudis.2015.06.013. [DOI] [PubMed] [Google Scholar]
- 6.Wicha MS, Liu S, Dontu G. Cancer stem cells: an old idea–a paradigm shift. Cancer Res. 2006;66:1883-1890–1896. doi: 10.1158/0008-5472.CAN-05-3153. [DOI] [PubMed] [Google Scholar]
- 7.Lawrence C. The RAD6 DNA repair pathway in Saccharomyces cerevisiae: What does it do, and how does it do it? BioEssays. 1994;16:253–258. doi: 10.1002/bies.950160408. [DOI] [PubMed] [Google Scholar]
- 8.Koken MH, Reynolds P, Jaspers-Dekker I, Prakash L, Prakash S, Bootsma D, et al. Structural and functional conservation of two human homologs of the yeast DNA repair gene RAD6. Proc Natl Acad Sci U S A. 1991;88:8865–8869. doi: 10.1073/pnas.88.20.8865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bailly V, Lamb J, Sung P, Prakash S, Prakash L. Specific complex formation between yeast RAD6 and RAD18 proteins: a potential mechanism for targeting RAD6 ubiquitin-conjugating activity to DNA damage sites. Genes Dev. 1994;8:811–820. doi: 10.1101/gad.8.7.811. [DOI] [PubMed] [Google Scholar]
- 10.Robzyk K, Recht J, Osley MA. Rad6-dependent ubiquitination of histone H2B in yeast. Science. 2000;287:501–504. doi: 10.1126/science.287.5452.501. [DOI] [PubMed] [Google Scholar]
- 11.Kao C-F, Hillyer C, Tsukuda T, Henry K, Berger S, Osley MA. Rad6 plays a role in transcriptional activation through ubiquitylation of histone H2B. Genes Dev. 2004;18:184–195. doi: 10.1101/gad.1149604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lyakhovich A, Shekhar MPV. RAD6B overexpression confers chemoresistance: RAD6 expression during cell cycle and its redistribution to chromatin during DNA damage-induced response. Oncogene. 2004;23:3097–3106. doi: 10.1038/sj.onc.1207449. [DOI] [PubMed] [Google Scholar]
- 13.Shekhar MPV, Lyakhovich A, Visscher DW, Heng H, Kondrat N. Rad6 overexpression induces multinucleation, centrosome amplification, abnormal mitosis, aneuploidy, and transformation. Cancer Res. 2002;62:2115–2124. [PubMed] [Google Scholar]
- 14.Shekhar MPV, Gerard B, Pauley RJ, Williams BO, Tait L. Rad6B is a positive regulator of beta-catenin stabilization. Cancer Res. 2008;68:1741–1750. doi: 10.1158/0008-5472.CAN-07-2111. [DOI] [PubMed] [Google Scholar]
- 15.Rosner K, Mehregan DR, Kirou E, Abrams J, Kim S, Campbell M, et al. Melanoma Development and Progression Are Associated with Rad6 Upregulation and β -Catenin Relocation to the Cell Membrane. J Skin Cancer. 2014;2014:439205. doi: 10.1155/2014/439205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Somasagara RR, Tripathi K, Spencer SM, Clark DW, Barnett R, Bachaboina L, et al. Rad6 upregulation promotes stem cell-like characteristics and platinum resistance in ovarian cancer. Biochem Biophys Res Commun. 2016;469:449–455. doi: 10.1016/j.bbrc.2015.11.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chirnomas D, Taniguchi T, de la Vega M, Vaidya AP, Vasserman M, Hartman A-R, et al. Chemosensitization to cisplatin by inhibitors of the Fanconi anemia/BRCA pathway. Mol Cancer Ther. 2006;5:952–961. doi: 10.1158/1535-7163.MCT-05-0493. [DOI] [PubMed] [Google Scholar]
- 18.Ferrer M, Span SW, Vischioni B, Oudejans JJ, van Diest PJ, de Winter JP, et al. FANCD2 expression in advanced non-small-cell lung cancer and response to platinum-based chemotherapy. Clin Lung Cancer. 2005;6:250–254. doi: 10.3816/CLC.2005.n.005. [DOI] [PubMed] [Google Scholar]
- 19.Geng L, Huntoon CJ, Karnitz LM. RAD18-mediated ubiquitination of PCNA activates the Fanconi anemia DNA repair network. J Cell Biol. 2010;191:249–257. doi: 10.1083/jcb.201005101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kannouche PL, Lehmann AR. Ubiquitination of PCNA and the polymerase switch in human cells. Cell Cycle Georget Tex. 2004;3:1011–1013. [PubMed] [Google Scholar]
- 21.Houghtaling S, Timmers C, Noll M, Finegold MJ, Jones SN, Meyn MS, et al. Epithelial cancer in Fanconi anemia complementation group D2 (Fancd2) knockout mice. Genes Dev. 2003;17:2021–2035. doi: 10.1101/gad.1103403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ng HH, Xu R-M, Zhang Y, Struhl K. Ubiquitination of histone H2B by Rad6 is required for efficient Dot1-mediated methylation of histone H3 lysine 79. J Biol Chem. 2002;277:34655–34657. doi: 10.1074/jbc.C200433200. [DOI] [PubMed] [Google Scholar]
- 23.Kim J, Guermah M, McGinty RK, Lee J-S, Tang Z, Milne TA, et al. RAD6-Mediated transcription-coupled H2B ubiquitylation directly stimulates H3K4 methylation in human cells. Cell. 2009;137:459–471. doi: 10.1016/j.cell.2009.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pavri R, Zhu B, Li G, Trojer P, Mandal S, Shilatifard A, et al. Histone H2B monoubiquitination functions cooperatively with FACT to regulate elongation by RNA polymerase II. Cell. 2006;125:703–717. doi: 10.1016/j.cell.2006.04.029. [DOI] [PubMed] [Google Scholar]
- 25.Zhang Z, Jones A, Joo H-Y, Zhou D, Cao Y, Chen S, et al. USP49 deubiquitinates histone H2B and regulates cotranscriptional pre-mRNA splicing. Genes Dev. 2013;27:1581–1595. doi: 10.1101/gad.211037.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Soares LM, Buratowski S. Histone Crosstalk: H2Bub and H3K4 Methylation. Mol Cell. 2013;49:1019–1020. doi: 10.1016/j.molcel.2013.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chandrasekharan MB, Huang F, Sun Z-W. Histone H2B ubiquitination and beyond: Regulation of nucleosome stability, chromatin dynamics and the trans-histone H3 methylation. Epigenetics. 2010;5:460–468. doi: 10.4161/epi.5.6.12314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nguyen AT, Zhang Y. The diverse functions of Dot1 and H3K79 methylation. Genes Dev. 2011;25:1345–1358. doi: 10.1101/gad.2057811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gerard B, Sanders MA, Visscher DW, Tait L, Shekhar MPV. Lysine 394 is a novel Rad6B-induced ubiquitination site on beta-catenin. Biochim Biophys Acta BBA – Mol Cell Res. 2012;1823:1686–1696. doi: 10.1016/j.bbamcr.2012.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yochum GS, Cleland R, Goodman RH. A Genome-Wide Screen for β-Catenin Binding Sites Identifies a Downstream Enhancer Element That Controls c-Myc Gene Expression. Mol Cell Biol. 2008;28:7368–7379. doi: 10.1128/MCB.00744-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Condello S, Morgan CA, Nagdas S, Cao L, Turek J, Hurley TD, et al. β-Catenin-regulated ALDH1A1 is a target in ovarian cancer spheroids. Oncogene. 2015;34:2297–2308. doi: 10.1038/onc.2014.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lyakhovich A, Shekhar MPV. Supramolecular complex formation between Rad6 and proteins of the p53 pathway during DNA damage-induced response. Mol Cell Biol. 2003;23:2463–2475. doi: 10.1128/MCB.23.7.2463-2475.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kothayer H, Spencer SM, Tripathi K, Westwell AD, Palle K. Synthesis and in vitro anticancer evaluation of some 4,6-diamino-1,3,5-triazine-2-carbohydrazides as Rad6 ubiquitin conjugating enzyme inhibitors. Bioorg Med Chem Lett. 2016;26:2030–2034. doi: 10.1016/j.bmcl.2016.02.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kothayer H, Elshanawani AA, Abu Kull ME, El-Sabbagh OI, Shekhar MPV, Brancale A, et al. Design, synthesis and in vitro anticancer evaluation of 4,6-diamino-1,3,5-triazine-2-carbohydrazides and -carboxamides. Bioorg Med Chem Lett. 2013;23:6886–6889. doi: 10.1016/j.bmcl.2013.09.087. [DOI] [PubMed] [Google Scholar]
- 35.Meng E, Mitra A, Tripathi K, Finan MA, Scalici J, McClellan S, et al. ALDH1A1 maintains ovarian cancer stem cell-like properties by altered regulation of cell cycle checkpoint and DNA repair network signaling. PloS One. 2014;9:e107142. doi: 10.1371/journal.pone.0107142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ginestier C, Hur MH, Charafe-Jauffret E, Monville F, Dutcher J, Brown M, et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell. 2007;1:555–567. doi: 10.1016/j.stem.2007.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Clark DW, Palle K. Aldehyde dehydrogenases in cancer stem cells: potential as therapeutic targets. Ann Transl Med. 2016;4:518. doi: 10.21037/atm.2016.11.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Haynes B, Zhang Y, Liu F, Li J, Petit S, Kothayer H, et al. Gold nanoparticle conjugated Rad6 inhibitor induces cell death in triple negative breast cancer cells by inducing mitochondrial dysfunction and PARP-1 hyperactivation: Synthesis and characterization. Nanomedicine Nanotechnol Biol Med. 2016;12:745–757. doi: 10.1016/j.nano.2015.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rosner K, Adsule S, Haynes B, Kirou E, Kato I, Mehregan DR, et al. Rad6 is a Potential Early Marker of Melanoma Development. Transl Oncol. 2014 doi: 10.1016/j.tranon.2014.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Djordjevic B, Stojanovic S, Conic I, Jankovic-Velickovic L, Vukomanovic P, Zivadinovic R, et al. Current approach to epithelial ovarian cancer based on the concept of cancer stem cells. J BUON Off J Balk Union Oncol. 2012;17:627–636. [PubMed] [Google Scholar]
- 41.Kobayashi S, Kasaishi Y, Nakada S, Takagi T, Era S, Motegi A, et al. Rad18 and Rnf8 facilitate homologous recombination by two distinct mechanisms, promoting Rad51 focus formation and suppressing the toxic effect of nonhomologous end joining. Oncogene. 2015;34:4403–4411. doi: 10.1038/onc.2014.371. [DOI] [PubMed] [Google Scholar]
- 42.Kannouche PL, Wing J, Lehmann AR. Interaction of human DNA polymerase eta with monoubiquitinated PCNA: a possible mechanism for the polymerase switch in response to DNA damage. Mol Cell. 2004;14:491–500. doi: 10.1016/s1097-2765(04)00259-x. [DOI] [PubMed] [Google Scholar]
- 43.Prakash S, Johnson RE, Prakash L. Eukaryotic translesion synthesis DNA polymerases: specificity of structure and function. Annu Rev Biochem. 2005;74:317–353. doi: 10.1146/annurev.biochem.74.082803.133250. [DOI] [PubMed] [Google Scholar]
- 44.Braun S, Madhani HD. Shaping the landscape: mechanistic consequences of ubiquitin modification of chromatin. EMBO Rep. 2012;13:619–630. doi: 10.1038/embor.2012.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hung S-H, Wong RP, Ulrich HD, Kao CF. Monoubiquitylation of histone H2B contributes to the bypass of DNA damage during and after DNA replication. Proc Natl Acad Sci U S A. 2017 doi: 10.1073/pnas.1612633114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Helchowski CM, Skow LF, Roberts KH, Chute CL, Canman CE. A small ubiquitin binding domain inhibits ubiquitin-dependent protein recruitment to DNA repair foci. Cell Cycle Georget Tex. 2013;12:3749–3758. doi: 10.4161/cc.26640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bi X, Barkley LR, Slater DM, Tateishi S, Yamaizumi M, Ohmori H, et al. Rad18 regulates DNA polymerase kappa and is required for recovery from S-phase checkpoint-mediated arrest. Mol Cell Biol. 2006;26:3527–3540. doi: 10.1128/MCB.26.9.3527-3540.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Palle K, Vaziri C. Rad18 E3 ubiquitin ligase activity mediates Fanconi anemia pathway activation and cell survival following DNA Topoisomerase 1 inhibition. Cell Cycle Georget Tex. 2011;10:1625–1638. doi: 10.4161/cc.10.10.15617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sanders MA, Brahemi G, Nangia-Makker P, Balan V, Morelli M, Kothayer H, et al. Novel inhibitors of Rad6 ubiquitin conjugating enzyme: design, synthesis, identification, and functional characterization. Mol Cancer Ther. 2013;12:373–383. doi: 10.1158/1535-7163.MCT-12-0793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wood A, Krogan NJ, Dover J, Schneider J, Heidt J, Boateng MA, et al. Bre1, an E3 ubiquitin ligase required for recruitment and substrate selection of Rad6 at a promoter. Mol Cell. 2003;11:267–274. doi: 10.1016/s1097-2765(02)00802-x. [DOI] [PubMed] [Google Scholar]
- 51.Hwang WW, Venkatasubrahmanyam S, Ianculescu AG, Tong A, Boone C, Madhani HD. A conserved RING finger protein required for histone H2B monoubiquitination and cell size control. Mol Cell. 2003;11:261–266. doi: 10.1016/s1097-2765(02)00826-2. [DOI] [PubMed] [Google Scholar]
- 52.Sun Z-W, Allis CD. Ubiquitination of histone H2B regulates H3 methylation and gene silencing in yeast. Nature. 2002;418:104–108. doi: 10.1038/nature00883. [DOI] [PubMed] [Google Scholar]
- 53.Lee J-S, Shukla A, Schneider J, Swanson SK, Washburn MP, Florens L, et al. Histone crosstalk between H2B monoubiquitination and H3 methylation mediated by COMPASS. Cell. 2007;131:1084–1096. doi: 10.1016/j.cell.2007.09.046. [DOI] [PubMed] [Google Scholar]
- 54.Chen S, Li J, Wang D-L, Sun F-L. Histone H2B lysine 120 monoubiquitination is required for embryonic stem cell differentiation. Cell Res. 2012;22:1402–1405. doi: 10.1038/cr.2012.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kühl SJ, Kühl M. On the role of Wnt/β-catenin signaling in stem cells. Biochim Biophys Acta BBA – Gen Subj. 2013;1830:2297–2306. doi: 10.1016/j.bbagen.2012.08.010. [DOI] [PubMed] [Google Scholar]
- 56.Miki T, Yasuda S, Kahn M. Wnt/β-catenin signaling in embryonic stem cell self-renewal and somatic cell reprogramming. Stem Cell Rev. 2011;7:836–846. doi: 10.1007/s12015-011-9275-1. [DOI] [PubMed] [Google Scholar]
- 57.Ring A, Kim Y-M, Kahn M. Wnt/Catenin Signaling in Adult Stem Cell Physiology and Disease. Stem Cell Rev. 2014;10:512–525. doi: 10.1007/s12015-014-9515-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cadigan KM. Wnt signaling–20 years and counting. Trends Genet TIG. 2002;18:340–342. doi: 10.1016/s0168-9525(02)02707-5. [DOI] [PubMed] [Google Scholar]
- 59.Shekhar MPV, Tait L, Gerard B. Essential role of T-cell factor/beta-catenin in regulation of Rad6B: a potential mechanism for Rad6B overexpression in breast cancer cells. Mol Cancer Res MCR. 2006;4:729–745. doi: 10.1158/1541-7786.MCR-06-0136. [DOI] [PubMed] [Google Scholar]
- 60.de la Rocha AMA, Sampron N, Alonso MM, Matheu A. Role of SOX family of transcription factors in central nervous system tumors. Am J Cancer Res. 2014;4:312–324. [PMC free article] [PubMed] [Google Scholar]
- 61.Ishiguro T, Sato A, Ohata H, Ikarashi Y, Takahashi R-U, Ochiya T, et al. Establishment and Characterization of an In Vitro Model of Ovarian Cancer Stem-like Cells with an Enhanced Proliferative Capacity. Cancer Res. 2016;76:150–160. doi: 10.1158/0008-5472.CAN-15-0361. [DOI] [PubMed] [Google Scholar]
- 62.Jazaeri AA, Bryant JL, Park H, Li H, Dahiya N, Stoler MH, et al. Molecular requirements for transformation of fallopian tube epithelial cells into serous carcinoma. Neoplasia N Y N. 2011;13:899–911. doi: 10.1593/neo.11138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kothayer H, Morelli M, Brahemi G, Elshanawani AA, Abu Kull ME, El-Sabbagh OI, et al. Optimised synthesis of diamino-triazinylmethyl benzoates as inhibitors of Rad6B ubiquitin conjugating enzyme. Tetrahedron Lett. 2014;55:7015–7018. [Google Scholar]
- 64.Zhang F, Yu X. WAC, a functional partner of RNF20/40, regulates histone H2B ubiquitination and gene transcription. Mol Cell. 2011;41:384–397. doi: 10.1016/j.molcel.2011.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Clark DW, Tripathi K, Dorsman JC, Palle K. FANCJ protein is important for the stability of FANCD2/FANCI proteins and protects them from proteasome and caspase-3 dependent degradation. Oncotarget. 2015;6:28816–28832. doi: 10.18632/oncotarget.5006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tripathi K, Mani C, Barnett R, Nalluri S, Bachaboina L, Rocconi RP, et al. Gli1 protein regulates the S-phase checkpoint in tumor cells via Bid protein, and its inhibition sensitizes to DNA topoisomerase 1 inhibitors. J Biol Chem. 2014;289:31513–31525. doi: 10.1074/jbc.M114.606483. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.